

Significance
Adsorption of proteins underlies the purification of biopharmaceuticals, as well as therapeutic apheresis, immunoassays, and biosensors. In particular, separation of proteins by interactions with charged ligands on surfaces (ion-exchange chromatography) is an essential tool of the modern pharmaceutical industry. By quantifying the interactions of single proteins with individual charged ligands, we demonstrate that clusters of charges are necessary to create functional adsorption sites and that even chemically identical ligands create sites of varying kinetic properties that depend on steric availability at the interface.
Keywords: ion-exchange chromatography, single-molecule kinetics, bioseparations, optical nanoscopy
Abstract
Chromatographic protein separations, immunoassays, and biosensing all typically involve the adsorption of proteins to surfaces decorated with charged, hydrophobic, or affinity ligands. Despite increasingly widespread use throughout the pharmaceutical industry, mechanistic detail about the interactions of proteins with individual chromatographic adsorbent sites is available only via inference from ensemble measurements such as binding isotherms, calorimetry, and chromatography. In this work, we present the direct superresolution mapping and kinetic characterization of functional sites on ion-exchange ligands based on agarose, a support matrix routinely used in protein chromatography. By quantifying the interactions of single proteins with individual charged ligands, we demonstrate that clusters of charges are necessary to create detectable adsorption sites and that even chemically identical ligands create adsorption sites of varying kinetic properties that depend on steric availability at the interface. Additionally, we relate experimental results to the stochastic theory of chromatography. Simulated elution profiles calculated from the molecular-scale data suggest that, if it were possible to engineer uniform optimal interactions into ion-exchange systems, separation efficiencies could be improved by as much as a factor of five by deliberately exploiting clustered interactions that currently dominate the ion-exchange process only accidentally.
The hundred-billion-dollar global pharmaceutical industry relies increasingly on the painstaking purification of therapeutic biomolecules such as proteins and nucleic acids (1). Separation of biologics is often performed using ion-exchange chromatography on stationary phases supporting singly charged ligands (2, 3) and constitutes an expensive, bottlenecking step in production. Improving bioseparations is thus highly desirable (4, 5); yet, a molecular-scale, mechanistic understanding is lacking, for ion-exchange chromatography in particular (6). Mechanistic detail is lost in ensemble analyses, reflecting the inherent heterogeneity of both the adsorbed biomolecules and the porous stationary phase supports (7). Ensemble adsorption isotherms, however, suggest the likelihood that protein and nucleic acid separations in ion-exchange columns may involve random ligand clustering (8–10). Additional support for such an assertion lies in the implementation of stationary phases of very high charge density by polymerization of charged monomers or layer-by-layer deposition (11–13), and in the demonstration that patches of high charge density on proteins often play a disproportionate role in their adsorption (4, 6, 14–17). In this work, we provide direct evidence of the importance of charge clustering in ion-exchange systems by direct observation of individual adsorption sites.
Although the role of multivalency is broadly accepted and exploited in a wide range of associative and adsorption processes, including some types of chromatography (18–25), the same level of understanding does not exist for clustered-charge interactions in ion exchange because, by definition, it is understood to be a straightforward electrostatic process, involving longer-range interactions. Our historic understanding of the role of cooperativity and geometric multivalency in adsorptive separations has been acquired from a combination of ensemble measurements and subsequent fitting with one of several empirical models, to acquire, for example, the appropriate isotherm coefficients, and such data usually is extended to the modeling of chromatographic processes by a purely phenomenological “theoretical plate” theory based on continuum mechanics (26–29).
Elucidating the true mechanistic details of adsorptive processes requires a move beyond empirical models and ensemble experimental information, and such details can be acquired via a combination of single-molecule spectroscopy and statistical theory. In particular, recently developed superresolution methods (30–43) offer the potential to considerably improve our understanding of adsorptive separations (as well as immunoassays, biosensors, and adsorption in general) at spatial resolutions far below the optical diffraction limit (down to ∼20 nm). Superresolution techniques have yet to be applied to molecular-scale investigations of separation methods, but recent efforts that identified heterogeneous interactions between proteins and biological membranes (44) demonstrate the potential. Pioneering diffraction-limited single-molecule studies of separations on relatively simple supports (45–52) and adsorption at interfaces (53, 54) provide incentive for applying higher-resolution spectroscopic analyses to a more realistic stationary phase. Because the ion-exchange process relies on maximizing the number of relatively weak interactions between the adsorbent and adsorbate within a finite volume, it is necessary to perform molecular-scale analysis of interactions at the porous interfaces used as supports, such as agarose. Correspondingly, a mechanistic, molecular theory that models chromatographic elution as resulting from the probabilistic adsorption of a single analyte on a single type of adsorption site has been available for many years in the form of Giddings and Eyring’s stochastic theory (55, 56).
Here, we apply superresolution single-molecule spectroscopic methods to study single-protein ion-exchange adsorption on a realistic support and combine the single-site kinetic data with the stochastic theory. We observe functional protein adsorption sites directly and demonstrate not only that clustered-charge ligands provide detectable, functional adsorption, but also show that even chemically homogeneous clusters of charge give rise to sites of varying kinetic properties due to steric factors. We observe that detectable localized adsorption of α-lactalbumin occurs only at clusters of charges whereas adsorption of proteins on individual singly charged ligands for longer than 3 ms is unobservable under our experimental conditions, unless the ligands are intentionally but randomly clustered at high ligand densities. The onset of clustered-ligand behavior and specific adsorption is shown to occur at lower total charge density with engineered multi-charge oligomers compared with random (stochastic) clustering of single charges. By extracting single-protein adsorption and desorption kinetics at individual ligands, direct experimental evidence in support of the stochastic theory is obtained. A mechanistic relationship between on- and off-rates is observed, due to the varying steric availability of the peptides. Finally, by extending the stochastic theory (57) to model macroscale observables, the single-molecule kinetics are used to simulate ensemble chromatographic elution profiles. The elution profiles establish that, if feasible, the engineering of ligand clustering could improve elution plate heights by a factor of five compared with randomly arranged single charges. More broadly, this work demonstrates the possibility of achieving a molecular-scale understanding of well-established processes such as protein chromatography, ELISA, and biosensing.
Results and Discussion
Superresolution Imaging Distinguishes Between Single Proteins Adsorbing at Single Ligands and Nonspecific Interactions with the Porous Agarose Support.
Superresolution image analysis, with ∼30-nm precision, was performed on ligand-functionalized agarose supports spin-cast onto glass coverslips while the protein (Alexa Fluor 555-labeled α-lactalbumin, in 10 mM Tris·HCl, pH 8.0; 10 mM NaCl) diffused in solution over the interface (Fig. 1, SI Appendix, and Movies S1 and S2) (see SI Appendix for experimental details and controls). When a protein stochastically adsorbed to the interface, each diffraction-limited single-molecule fluorescence emission pattern was fitted by a 2D Gaussian function, and the centroid location was recorded (Fig. 1A). The interactions between the labeled protein and the stationary phase were tuned to be rare and reversible via low peptide loading on the agarose support, low protein concentration (500 pM), and use of a salt concentration chosen to make the average duration of adsorption events suitable for effective observation (10 mM Tris⋅HCl, pH 8.0; 10 mM NaCl; ionic strength 0.015 M; Debye length 24.5 Å). The diffusion constant (D ∼150 μm2/s) (58) of α-lactalbumin in bulk solution compared to the detector temporal resolution (16 Hz) allowed for the free protein to be unobservable. Thus, stochastic on/off behavior of the fluorescently labeled protein at the stationary phase interface allows for superresolution imaging of adsorption/desorption events.
Fig. 1.

Superresolution imaging procedure and pseudo images at respective interfaces. (A–C) Steps to obtain superresolution pseudo images. (A) Single frame with an adsorbed α-lactalbumin protein. The centroid location obtained by a 2D Gaussian fit of the point spread function is noted by “x”. (B) Centroid locations obtained from localizing 114 individual point-spread functions from 3,000 frames; location of centroid in A is indicated by “x”. (C) Superresolution pseudo image from 3,000 frames depicting location and number of individual adsorption events. Specific adsorption sites were identified by the detection of more than five adsorption events (threshold indicated by arrow below color bar). (D) Summed diffraction-limited image from 3,000 frames for comparison with superresolved image. (E–G) Schemes and (H–J) superresolution pseudo images for α-lactalbumin at three stationary phase interfaces: (E and H) pentaargininamide, (F and I) monoargininamide, and (G and J) agarose, respectively. Data collected from 3,000 frames over 3.1 min are shown. Protein structure from PDB based on Chandra, et al. (69). (Scale bars: 200 nm.) Intensity scales range from (A) 50–1,800 and (D) 2–4 × 105 counts.
The centroid locations of all adsorption events in all frames were summed, with no assumptions about protein adsorption/desorption history, resulting in a total centroid event map (Fig. 1B). We then renormalized to distinguish events persisting over multiple frames at the same location from events occurring in only a single frame. The resulting summed superresolution pseudo image in Fig. 1C clearly identifies three distinct sites with multiple adsorption events whereas the summed diffraction-limited image (Fig. 1D) cannot resolve these sites.
Superresolved event analysis distinguishes between specific protein adsorption at peptides and nonspecific steric interactions with the agarose support. Correlation analysis of the superresolution pseudo image selects Gaussian peaks that have maximum amplitude >5 adsorption events (colorbar, Fig. 1J). The threshold of five adsorption events was selected to satisfy 99% confidence that the adsorption sites were not due to the nonspecific interactions observed on the bare agarose control samples (SI Appendix, Table S1). Stationary phases compared in Fig. 1 include engineered clustered-charge pentaargininamide-functionalized agarose (pentaargininamide) (Fig. 1 E and H), traditional singly charged monoargininamide-functionalized agarose (monoargininamide) (Fig. 1 F and I), and unfunctionalized agarose support (Fig. 1 G and J). Monoargininamide and argininamide oligomers were used to compare ligands bearing isolated and clustered charges.
Superresolution Imaging at Single Ligands Demonstrates That Specific α-Lactalbumin Adsorption Is Detectable only at Clustered-Charge Peptides.
As shown in Fig. 1 H–J, only the clustered-charge pentaargininamide stationary phase induced specific protein adsorption within the time resolution of the experiment (see SI Appendix, Fig. S2 for discussion and simulations). α-Lactalbumin interacted repeatedly at individual locations on the clustered-charge stationary phase (Fig. 1H, SI Appendix, and Movie S1), indicating specific adsorption sites where pentaargininamide was present. Controls (SI Appendix, Fig. S3) showed that specific interactions were electrostatic, as expected for ion-exchange chromatography; this conclusion is further supported by the complete elution of α-lactalbumin from pentaargininamide adsorbents by 1 M NaCl in our previously reported ensemble studies of these systems (9). Nonspecific interactions with the porous agarose interface (distinguishable by statistical analysis as mentioned above) were observed with all supports because, at the low ligand loading used to achieve single-molecule-appropriate conditions, the majority of the surface is bare agarose.
Despite the fact that singly charged ligands are commonly used (at higher ligand densities) in ion-exchange adsorbents, the superresolved pseudo images in Fig. 1I, SI Appendix, and Movie S2 show that all α-lactalbumin interactions at the sparse monoargininamide interface were indistinguishable within our instrumental capabilities from the nonspecific steric interactions observed at the bare agarose interface in Fig. 1J. With the monoargininamide, out of 8,007 events analyzed, only three of the events were observed to have a single protein bound to a single location for longer than the five-frame threshold, similar to the frequency for the control agarose sample (SI Appendix, Table S1). Moreover, there were no repeated adsorption events at any of the 30-nm superresolved locations on monoargininamide supports. Overall, the comparison of Fig. 1 H–J, SI Appendix, and Movies S1 and S2 shows the importance of charge clustering to adsorption via superresolution image analysis.
Agarose was chosen as the stationary phase support because it is commonly used in protein separations (59). Agarose is a hydroxyl-containing polysaccharide resistant to nonspecific adsorption of proteins and is not spontaneously reactive toward any functional group, but can be activated for covalent coupling; we have used sodium periodate oxidation for later coupling of the primary amine groups of the peptide ligands via reductive amination (see SI Appendix, Fig. S3 for a control experiment on agarose stability). Additionally, although dye-labeled peptides were used to demonstrate single-peptide loading conditions (SI Appendix, Fig. S4), for quantifying adsorption dynamics, the peptides were not dye-labeled. The ion-exchange adsorption process relies on relatively weak charge–charge interactions, and the presence of a fluorescent labeling group on a relatively small ligand could substantially alter the equilibrium by introducing mixed-mode interactions that were not of current interest, especially when adsorption occurs on clusters of ligands. Further controls include labeled protein diffusion over the glass substrate (SI Appendix, Fig. S6), which yielded an insignificant number of identified events. This control demonstrates that there are no long-lived (>3 ms) (SI Appendix, Fig. S2) interactions between the glass substrate and the α-lactalbumin, consistent with previous observations (58, 60).
Comparison of the Onset of Specific Adsorption Investigated with Engineered Oligomer Clustered Charges vs. Stochastic Clustering of Single Charges.
The onset of detectable specific protein adsorption was tested as a function of increasing oligomer length (Fig. 2A) and was observed to occur with triargininamide, after which a trend of increasing affinity with oligomer length was observed. The number of specific adsorption sites as a function of oligomer length, along with their respective superresolution pseudo images, are reported in Fig. 2A. No specific adsorption events were observed for mono- or diargininamide. Only with triargininamide and higher oligo-peptide lengths were specific adsorption events identifiable; this result of course could vary with protein charge and solvent conditions. Based on the localized cation affinity site of aspartate residues between the two subdomains of α-lactalbumin (61) (SI Appendix, Fig. S6C), it is consistent that the onset of protein–ligand adsorption occurs between di- and tripeptides. The number of specific adsorption events increased with oligomer length from tri-, tetra-, to pentaargininamide due to stronger electrostatic interaction energy with oligomer length (62). It is important to note the large spread in detected adsorption sites for the pentaargininamide ligands among the multiple trials. There are several possible explanations for this observation, including variations in steric availability of the ligands, which is further addressed in Fig. 3C and SI Appendix, Fig. S5. As the maximum number of events was observed with pentaargininamide, the work presented on clustered-charge ligands in the remainder of this paper focuses on this ligand.
Fig. 2.

Onset of clustered-charge behavior, enabling specific adsorption of α-lactalbumin, via (A) synthetic peptide oligomer length and (B) increased grafting density of monoargininamide. The number of specific adsorption sites (per 160 µm2, 60 s) is reported on each plot with graphic representations of the ligands overlaid. (A) Peptide oligomer length on the functionalized agarose was varied. The preparation charge concentration (215 µM) was the same for all samples during substrate modification. (B) Monoargininamide density as a percent increase from the benchmark 215 µM shown in Fig. 1I was varied. Representative superresolution pseudo images are shown for each corresponding color-coded point. (Scale bar: 1 µm.) Dashed lines are shown as guide for the eye. Error bars represent the SD from five different 62-s trials taken from three different areas of the same sample.
Fig. 3.

Kinetics and elution profiles obtained from single-molecule data. Cumulative distributions of (A) desorption and (B) adsorption times and respective fits (solid lines) for the four individual pentaargininamide adsorption sites shown in the superresolution pseudo image in SI Appendix, Fig. S6. (C) Relationship between kd and ka for 53 individual sites, with displayed locations in A noted by colored circles. A linear trend is observed indicated by the dashed line, as opposed to a 2D normal distribution represented by the gray circle (mean ± SD). (D and E) Relative ensemble isocratic chromatographic elution profiles obtained from single-molecule data using stochastic theory. (D) Elution curves for the four individual pentaargininamide adsorption sites. (E) Elution curves for ensemble of specific adsorption events for pentaargininamide (n = 603) and clustered monoargininamide (Fig. 2B, light blue, 10 × 104%; n = 1,706). Data were obtained over 5.2 min.
If the hypothesis that clusters of charge are necessary for specific protein adsorption is correct, stochastic clustering of single charges must occur in commercial adsorbents bearing high concentrations of singly charged ligands. This hypothesis was tested by systematically increasing monoargininamide loading between factors of 2 and 1,000 times those used in the experiments described above (Fig. 2B). The numbers of specific adsorption sites and superresolution pseudo images were recorded. Protein interactions remained indistinguishable from the bare-agarose control as ligand densities increased by factors of 2–750, becoming specific only at a ligand charge density three orders of magnitude higher than the level initially used for the pentaargininamide samples in Fig. 1H. After the threshold was reached, many adsorption sites were observed, as shown in the superresolution pseudo image (Fig. 2, light blue). The onset of clustering and adsorption is consistent with previous ensemble studies at and above this threshold by Wu and Walters (10). Because these sites were due to stochastic clustering, it remains unknown how many monoargininamide ligands were present at each site. Heterogeneous protein adsorption at the stochastically distributed monopeptide clusters would be expected to yield a broad distribution of adsorption kinetics and a highly non-Gaussian column elution profile, as quantified in Fig. 3E. The data shown in Fig. 2B suggest that the formation of monoargininamide clusters is rare, likely due to electrostatic peptide–peptide repulsion, and explains the high ligand loading required for commercial stationary phases (∼100 mM). By comparing intentionally clustered (Figs. 1H and 2A) and stochastically clustered (Fig. 2B) peptide interactions, it can be concluded that the engineered clustering of peptide charges produces functional adsorption sites at much lower ligand concentrations than does stochastic arrangement of single charges.
Kinetic Characterization of Individual Sites Reveals Kinetic Heterogeneity of Chemically Identical Charge Clusters.
A kinetic analysis of single α-lactalbumin adsorption at single pentaargininamide peptides (Fig. 3 A–C) offers a direct experimental test of the physical model proposed in the stochastic theory (55, 56, 63): single analyte adsorption to a single ligand. For each identified adsorption site, the desorption time was defined as the dwell time from when a protein was first observed until it was not whereas the adsorption time was the time between the end of one adsorption event and the beginning of the next adsorption event. Desorption and adsorption times for individual protein–ligand adsorption events at each specific adsorption site (identified in the superresolution pseudo image in SI Appendix, Fig. S6A) were extracted as reported in Fig. 3 A and B. To maximize the resolution of each stochastic adsorption event, the frequency of events per site was controlled to be low by maintaining a low protein concentration in solution (SI Appendix, Table S2). The distributions of these times were plotted as cumulative distributions, for increased sensitivity to rare events (64) compared with population density (histogram) distributions (Fig. 3 A and B). Control experiments (SI Appendix, Fig. S4) confirmed that photophysical effects and protein oligomerization were negligible, despite their presence in other systems (64).
Each specific adsorption site exhibited single-exponential decay kinetics, and a least squares fit extracted the respective desorption (kd) and adsorption (ka) rate constants (SI Appendix, Table S2). That each individual site exhibits a single decay (higher-order decay fits resulted in a single decay time to, verifying a single component) (SI Appendix, Table S3) (65) validates the second assumption of the stochastic theory, namely, that a nonvarying rate constant describes each individual adsorption site (56). In addition, this result supports the stochastic model of a Poisson-distributed process for α-lactalbumin adsorption/desorption at each site, with each site corresponding to an individual pentaargininamide ligand (i.e., our distribution of pentaargininamide molecules is not ≥2 sites per 30 nm). Further, these results confirm that diffusion is not the rate-limiting step within the adsorption/desorption process. Additional controls supporting these assertions include desorption kinetics comparisons at varied flow rates, which are included in SI Appendix, Fig. S5.
Comparing the kinetics among distinct adsorption sites, however, reveals heterogeneity, as depicted in Fig. 3 A and B by the distribution of decay kinetics at the four different sites. To understand the origin of this adsorption/desorption heterogeneity, the distribution of kd and ka for 53 individual pentaargininamide adsorption sites is shown in Fig. 3C on a double logarithmic scale. If kd and ka were truly independent for a single type of adsorption site, as described by the pure stochastic theory (55), the comparison depicted in Fig. 3C would yield a 2D Gaussian with a spread indicative of the experimental noise and uncertainty in rate constant fitting (Fig. 3C, gray oval). Instead, an inverse relationship is observed, indicated by the least squares fit shown as a dashed line in Fig. 3C, which is inconsistent with the theory.
There are several possible explanations for the intersite adsorption/desorption heterogeneity. The possibility of clustering of peptides was ruled out by both kinetic (Fig. 3 A and B) and single-event analyses (SI Appendix, Fig. S4). Also, because the variation occurs only between sites, and not at the same site, any variability in valence and geometry from the analyte (see SI Appendix, Fig. S3 for structural stability control experiment) or mobile phase can be ruled out. Further evidence in support of this assertion, in the form of an investigation into the role of potential flow-induced shear force, is presented in SI Appendix, Fig. S5.
Experiments performed by varying the agarose concentration from 1% to 3%, and thus the agarose pore size (66), suggest that steric availability of the peptides is the most likely source of the observed intersite heterogeneity (SI Appendix, Fig. S5). As pore size decreases, fewer adsorption events are observed, and the events last for shorter periods of time. Steric screening of peptides impairs protein interaction with the charged ligand, reducing interaction energy, leading to shorter desorption times and longer adsorption times, and the inverse relationship between ka and kd observed. The importance of agarose sterics in α-lactalbumin separations is consistent with our previously reported findings (58) and is one reason why affinity chromatography supports often use a “spacer arm” between ligand and matrix (67). Future studies using the single-molecule technology developed in this work are needed to investigate the use of spacer arms with clustered-charge peptides to control steric effects.
Single-Molecule Kinetics Included in the Stochastic Theory Inform Ensemble Elution Profiles Indicating That Molecular-Scale Control Can Improve Macroscale Protein Separations.
Direct measurement of kinetic rate constants for single proteins adsorbing to individual ligands allows us to relate the microscopic stochastic theory to a macroscopic observable, the chromatographic retention curve (Fig. 3 D and E). By converting the Poisson distribution of the duration of protein adsorption/desorption events to the frequency domain (ω) and accounting for the discontinuous distribution of single-molecule desorption times using the Lévy representation, the distributions of protein desorption times, ΔF(τs ,i), were related to the characteristic function formalism of the distribution, ϕ(ts ;ω|tm) (68), by:
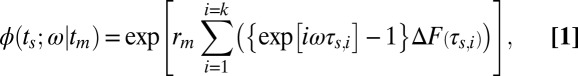 |
where ts and tm are the times spent in the stationary phase and mobile phase, respectively, for an analyte that has adsorbed to the stationary phase rm times during a given tm, and k is the index of the discrete set of desorption times (57, 68). By performing a Fourier transform of Eq. 1 to the time domain, the chromatographic peak for each adsorption site was simulated using the cumulative distributions of desorption times to directly relate experimental single-molecule measurements to theory. A fixed value of rm (the number of stochastic adsorption events for a given time in the mobile phase) based on experimental conditions was used to simulate relative elution curves. It is therefore valid to consider the resulting curves to be relative to one another but individually unique to the specific, low-concentration, single-molecule conditions used.
Desorption cumulative distributions for the four individual sites depicted in Fig. 3 A and B were used to simulate the characteristic elution curves for hypothetical columns populated purely with one of the four adsorption sites (Fig. 3D). The variability of the elution curves in Fig. 3D demonstrates the macroscopic effects of the intersite heterogeneity due to the steric variation discussed earlier. Overall, however, the influence of intersite heterogeneity was drastically reduced when all 603 events for the pentaargininamide support are considered as an ensemble (Fig. 3E, red curve). The pentaargininamide curve is dominated by a Gaussian distribution, with only slight fronting due to the heterogeneity in adsorption/desorption kinetics depicted in Fig. 3D.
In contrast, the simulated elution profile for high-density monoargininamide shown in the light blue curve in Fig. 3E demonstrates that elution peak broadening and asymmetry result when stochastic clustering dominates the adsorption kinetics. The kinetics of events (n = 1,706) at all identified specific adsorption sites for the high loading density-induced stochastically clustered single-charge monoargininamide (Fig. 2B) (see SI Appendix, Fig. S6D for an example of stochastically clustered monoargininamide single site kinetics) were used to extract the light blue elution curve in Fig. 3E. This result is consistent with the notion that stochastic clustering is by definition heterogeneous in comparison with engineered clustering.
The potential practical improvement in separations efficiency provided by engineered vs. stochastically clustered-charge ligands can be calculated by relating the elution curves shown in Fig. 3E to the plate theory of chromatography (26). A relative comparison of the plate heights (H) between the two samples presented in Fig. 3E can be made by taking the ratio of the square of the SDs of Gaussian fits to the peaks. The resulting Hmono/Hpenta = 4.55, shows that plate heights potentially could be greatly reduced if it were possible to optimize the nature of the functional sites in ion-exchange chromatographic matrices and also illustrates the potential for modeling macroscopic separations behavior from the fundamental properties of individual functional sites. Further improvements in this system also could be achieved by reducing the sterically induced heterogeneity in adsorption/desorption kinetics of functional adsorption sites. Although the very low ligand and protein concentrations used in the present study to prevent optical or functional overlaps are poorly compatible with standard column chromatography, the approach presented here is applicable to the characterization of higher-density conventional adsorbents usable in columns, as well as to other protein–surface interactions.
Conclusions.
Several important conclusions can be drawn from this first molecular-scale investigation into protein ion-exchange chromatography by superresolution techniques and the stochastic theory. i) It is possible to apply the latest optical-imaging methodology to map functional adsorption sites in realistic agarose adsorbents with a resolution at least an order of magnitude smaller than the wavelength of light. ii) Clustering of charges is necessary for detectable ion-exchange adsorption under the conditions tested. iii) Engineering clusters is potentially more effective than relying on stochastic clustering. To create adsorption sites that specifically retain α-lactalbumin proteins on the agarose substrate, 215-µM preparation charge concentration is enough for the argininamide oligomers that have lengths of three or more monomers whereas 1,000 times as much, at least 200 mM, is required if monomers are used. iv) In a previously unidentified finding, the second assumption of the stochastic theory was confirmed to be valid by an experimental protein–ligand adsorption system: a nonvarying rate constant describes each individual adsorption site. v) Even chemically identical charged ligands give rise to kinetically heterogeneous single-protein adsorption/desorption kinetics, which we ascribe to steric effects. This heterogeneity between adsorption sites helps explain the unwanted fronting observed in bulk chromatographic protein separations. vi) The simulated elution profiles produced by combining the extracted single-molecule results and the stochastic theory lead us to speculate that a fivefold improvement in separations efficiency, as defined by theoretical plate height, could result if it were possible to extend the single-molecule results to a realistic separations medium. In summary, the current work offers a molecular-scale method to fundamentally understand the mechanisms of protein chromatography, as well as other processes involving protein adsorption, such as immunoassays and biosensing.
Methods
A detailed description of the methods and analysis is included in SI Appendix. Briefly, for sample preparation, 1% agarose was spin-cast on O2 plasma-cleaned glass coverslips. Peptide-functionalized samples were prepared by introducing 43 µM pentaargininamide or 215 µM monoargininamide over aldehyde-activated agarose. α-Lactalbumin was preferentially labeled at the N terminus with Alexa Fluor 555 (estimated final charge at pH 8.0, −14 to −15) and studied at 500 pM in buffer (10 mM Tris·HCl, pH 8.0; 10 mM NaCl). A flow system (Genie Plus; Kent Scientific) at 1 µL/min was used to introduce new proteins to the sample throughout the measurement. The low flow rate ensured negligible net diffusion of the protein. For fluorescence imaging, data were collected on a home-built total internal reflectance fluorescence microscope with 532 nm excitation and electron multiplying charge-coupled device detection. Analysis was performed using MATLAB R2011b and OriginPro 8.6.
Supplementary Material
Acknowledgments
We thank E. Kulla and J. McDevitt for images, M. Kang and J. Hartgerink for instrument use, and S. Link, P. Wolynes, N. Halas, T. J. Ha, M. Pasquali, B. Weisman, H. Jennissen, and A. Yethiraj for discussions. C.F.L. thanks the National Science Foundation (NSF) (Grants CBET-1134417 and CHE-1151647), the Welch Foundation (Grant C-1787), and the National Institutes of Health (Grant GM94246-01A1) for support of this work. R.C.W. thanks the NSF for Grant CBET-1133965, the Welch Foundation for Grant E-1264, and the Huffington-Woestemeyer Professorship. L.K. thanks the NSF for Graduate Research Fellowship 0940902.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1318405111/-/DCSupplemental.
References
- 1.Walsh G. Biopharmaceutical benchmarks 2010. Nat Biotechnol. 2010;28(9):917–924. doi: 10.1038/nbt0910-917. [DOI] [PubMed] [Google Scholar]
- 2.Regnier FE. High-performance liquid chromatography of biopolymers. Science. 1983;222(4621):245–252. doi: 10.1126/science.6353575. [DOI] [PubMed] [Google Scholar]
- 3.Dziennik SR, et al. Nondiffusive mechanisms enhance protein uptake rates in ion exchange particles. Proc Natl Acad Sci USA. 2003;100(2):420–425. doi: 10.1073/pnas.0237084100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chung WK, Freed AS, Holstein MA, McCallum SA, Cramer SM. Evaluation of protein adsorption and preferred binding regions in multimodal chromatography using NMR. Proc Natl Acad Sci USA. 2010;107(39):16811–16816. doi: 10.1073/pnas.1002347107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Novotny MV. Recent developments in analytical chromatography. Science. 1989;246(4926):51–57. doi: 10.1126/science.246.4926.51. [DOI] [PubMed] [Google Scholar]
- 6.Freed AS, Garde S, Cramer SM. Molecular simulations of multimodal ligand-protein binding: Elucidation of binding sites and correlation with experiments. J Phys Chem B. 2011;115(45):13320–13327. doi: 10.1021/jp2038015. [DOI] [PubMed] [Google Scholar]
- 7.Schellekens H. How similar do ‘biosimilars’ need to be? Nat Biotechnol. 2004;22(11):1357–1359. doi: 10.1038/nbt1104-1357. [DOI] [PubMed] [Google Scholar]
- 8.Chen WH, Fu JY, Kourentzi K, Willson RC. Nucleic acid affinity of clustered-charge anion exchange adsorbents: Effects of ionic strength and ligand density. J Chromatogr A. 2011;1218(2):258–262. doi: 10.1016/j.chroma.2010.11.024. [DOI] [PubMed] [Google Scholar]
- 9.Fu JY, Balan S, Potty A, Nguyen V, Willson RC. Enhanced protein affinity and selectivity of clustered-charge anion-exchange adsorbents. Anal Chem. 2007;79(23):9060–9065. doi: 10.1021/ac070695n. [DOI] [PubMed] [Google Scholar]
- 10.Wu D, Walters RR. Effects of stationary phase ligand density on high-performance ion-exchange chromatography of proteins. J Chromatogr A. 1992;598(1):7–13. [Google Scholar]
- 11.Müller W. New ion exchangers for the chromatography of biopolymers. J Chromatogr A. 1990;510:133–140. [Google Scholar]
- 12.Atkinson A, Jack GW. Precipitation of nucleic acids with polyethyleneimine and the chromatography of nucleic acids and proteins on immobilised polyethyleneimine. Biochim Biophys Acta. 1973;308(7):41–52. doi: 10.1016/0005-2787(73)90120-2. [DOI] [PubMed] [Google Scholar]
- 13.Millot M-C, et al. Ion-exchange chromatographic supports obtained by formation of polyelectrolyte multi-layers for the separation of proteins. Chromatographia. 2003;58(5-6):365–373. [Google Scholar]
- 14.Daly SM, Przybycien TM, Tilton RD. Adsorption of poly(ethylene glycol)-modified lysozyme to silica. Langmuir. 2005;21(4):1328–1337. doi: 10.1021/la048316y. [DOI] [PubMed] [Google Scholar]
- 15.Gill DS, Roush DJ, Willson RC. Presence of a preferred anion-exchange binding site on cytochrome b5: Structural and thermodynamic considerations. J Chromatogr A. 1994;684(1):55–63. doi: 10.1016/s0021-9673(94)89132-x. [DOI] [PubMed] [Google Scholar]
- 16.Ramsden J, et al. Protein adsorption kinetics drastically altered by repositioning a single charge. J Am Chem Soc. 1995;117(33):8511–8516. [Google Scholar]
- 17.Asthagiri D, Lenhoff AM. Influence of structural details in modeling electrostatically driven protein adsorption. Langmuir. 1997;13(25):6761–6768. [Google Scholar]
- 18.Jennissen HP. Hydrophobic interaction chromatography: Harnessing multivalent protein-surface interactions for purification procedures. Methods Mol Biol. 2005;305:81–99. [PubMed] [Google Scholar]
- 19.Jennissen HP. Multivalent adsorption of proteins on hydrophobic agaroses. Hoppe Seylers Z Physiol Chem. 1976;357(8):1201–1203. [PubMed] [Google Scholar]
- 20.Hogg PJ, Winzor DJ. Effects of solute multivalency in quantitative affinity chromatography: Evidence for cooperative binding of horse liver alcohol dehydrogenase to blue Sepharose. Arch Biochem Biophys. 1985;240(1):70–76. doi: 10.1016/0003-9861(85)90009-8. [DOI] [PubMed] [Google Scholar]
- 21.Johnson RD, Arnold FH. Review: Multipoint binding and heterogeneity in immobilized metal affinity chromatography. Biotechnol Bioeng. 1995;48(5):437–443. doi: 10.1002/bit.260480505. [DOI] [PubMed] [Google Scholar]
- 22.Arnold FH. Metal-affinity separations: A new dimension in protein processing. Biotechnology (N Y) 1991;9(2):151–156. doi: 10.1038/nbt0291-151. [DOI] [PubMed] [Google Scholar]
- 23.Jendeberg L, et al. Kinetic analysis of the interaction between protein A domain variants and human Fc using plasmon resonance detection. J Mol Recognit. 1995;8(4):270–278. doi: 10.1002/jmr.300080405. [DOI] [PubMed] [Google Scholar]
- 24.Mammen M, Choi S-K, Whitesides GM. Polyvalent interactions in biological systems: Implications for design and use of multivalent ligands and inhibitors. Angew Chem Int Ed. 1998;37(20):2754–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 25.Mourez M, et al. Designing a polyvalent inhibitor of anthrax toxin. Nat Biotechnol. 2001;19(10):958–961. doi: 10.1038/nbt1001-958. [DOI] [PubMed] [Google Scholar]
- 26.Martin AJ, Synge RL. A new form of chromatogram employing two liquid phases: A theory of chromatography. 2. Application to the micro-determination of the higher monoamino-acids in proteins. Biochem J. 1941;35(12):1358–1368. doi: 10.1042/bj0351358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamamoto S, Yoshimoto N, Nishizumi Y. Theoretical background of monolithic short layer ion-exchange chromatography for separation of charged large biomolecules or bioparticles. J Chromatogr A. 2009;1216(13):2612–2615. doi: 10.1016/j.chroma.2009.01.059. [DOI] [PubMed] [Google Scholar]
- 28.Gétaz D, Butté A, Morbidelli M. Model-based design space determination of peptide chromatographic purification processes. J Chromatogr A. 2013;1284:80–87. doi: 10.1016/j.chroma.2013.01.117. [DOI] [PubMed] [Google Scholar]
- 29.Gallant SR, Kundu A, Cramer SM. Modeling non-linear elution of proteins in ion-exchange chromatography. J Chromatogr A. 1995;702(1):125–142. [Google Scholar]
- 30.Moerner WE. New directions in single-molecule imaging and analysis. Proc Natl Acad Sci USA. 2007;104(31):12596–12602. doi: 10.1073/pnas.0610081104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klar TA, Jakobs S, Dyba M, Egner A, Hell SW. Fluorescence microscopy with diffraction resolution barrier broken by stimulated emission. Proc Natl Acad Sci USA. 2000;97(15):8206–8210. doi: 10.1073/pnas.97.15.8206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gustafsson MGL. Nonlinear structured-illumination microscopy: Wide-field fluorescence imaging with theoretically unlimited resolution. Proc Natl Acad Sci USA. 2005;102(37):13081–13086. doi: 10.1073/pnas.0406877102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Westphal V, et al. Video-rate far-field optical nanoscopy dissects synaptic vesicle movement. Science. 2008;320(5873):246–249. doi: 10.1126/science.1154228. [DOI] [PubMed] [Google Scholar]
- 34.Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nat Methods. 2006;3(10):793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Betzig E, et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313(5793):1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- 36.Burnette DT, Sengupta P, Dai Y, Lippincott-Schwartz J, Kachar B. Bleaching/blinking assisted localization microscopy for superresolution imaging using standard fluorescent molecules. Proc Natl Acad Sci USA. 2011;108(52):21081–21086. doi: 10.1073/pnas.1117430109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharonov A, Hochstrasser RM. Wide-field subdiffraction imaging by accumulated binding of diffusing probes. Proc Natl Acad Sci USA. 2006;103(50):18911–18916. doi: 10.1073/pnas.0609643104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou X, et al. Quantitative super-resolution imaging uncovers reactivity patterns on single nanocatalysts. Nat Nanotechnol. 2012;7(4):237–241. doi: 10.1038/nnano.2012.18. [DOI] [PubMed] [Google Scholar]
- 39.Jungmann R, et al. Single-molecule kinetics and super-resolution microscopy by fluorescence imaging of transient binding on DNA origami. Nano Lett. 2010;10(11):4756–4761. doi: 10.1021/nl103427w. [DOI] [PubMed] [Google Scholar]
- 40.Wu D, Liu Z, Sun C, Zhang X. Super-resolution imaging by random adsorbed molecule probes. Nano Lett. 2008;8(4):1159–1162. doi: 10.1021/nl0733280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walder R, Nelson N, Schwartz DK. Super-resolution surface mapping using the trajectories of molecular probes. Nat Commun. 2011;2:515. doi: 10.1038/ncomms1530. [DOI] [PubMed] [Google Scholar]
- 42.Skaug MJ, Schwartz DK. Using the dynamics of fluorescent cations to probe and map charged surfaces. Soft Matter. 2012;8(48):12017–12024. [Google Scholar]
- 43.Kastantin M, Keller TF, Jandt KD, Schwartz DK. Single-molecule tracking of fibrinogen dynamics on nanostructured poly(ethylene) films. Adv Funct Mater. 2012;22(12):2617–2623. [Google Scholar]
- 44.Lippincott-Schwartz J, Manley S. Putting super-resolution fluorescence microscopy to work. Nat Methods. 2009;6(1):21–23. doi: 10.1038/nmeth.f.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kang SH, Shortreed MR, Yeung ES. Real-time dynamics of single-DNA molecules undergoing adsorption and desorption at liquid-solid interfaces. Anal Chem. 2001;73(6):1091–1099. doi: 10.1021/ac0013599. [DOI] [PubMed] [Google Scholar]
- 46.Xu X-HN, Yeung ES. Long-range electrostatic trapping of single-protein molecules at a liquid-solid interface. Science. 1998;281(5383):1650–1653. doi: 10.1126/science.281.5383.1650. [DOI] [PubMed] [Google Scholar]
- 47.Cuppett CM, Doneski LJ, Wirth MJ. Irreversible adsorption of lysozyme to polishing marks on silica. Langmuir. 2000;16(18):7279–7284. [Google Scholar]
- 48.Ludes MD, Wirth MJ. Single-molecule resolution and fluorescence imaging of mixed-mode sorption of a dye at the interface of C18 and acetonitrile/water. Anal Chem. 2002;74(2):386–393. doi: 10.1021/ac010943u. [DOI] [PubMed] [Google Scholar]
- 49.Wirth MJ, Legg MA. Single-molecule probing of adsorption and diffusion on silica surfaces. Annu Rev Phys Chem. 2007;58:489–510. doi: 10.1146/annurev.physchem.58.032806.104427. [DOI] [PubMed] [Google Scholar]
- 50.Wirth MJ, Swinton DJ, Ludes MD. Adsorption and diffusion of single molecules at chromatographic interfaces. J Phys Chem B. 2003;107(26):6258–6268. [Google Scholar]
- 51.Zhong Z, Lowry M, Wang G, Geng L. Probing strong adsorption of solute onto C18-silica gel by fluorescence correlation imaging and single-molecule spectroscopy under RPLC conditions. Anal Chem. 2005;77(8):2303–2310. doi: 10.1021/ac048290f. [DOI] [PubMed] [Google Scholar]
- 52.Fang N, Zhang H, Li J, Li HW, Yeung ES. Mobility-based wall adsorption isotherms for comparing capillary electrophoresis with single-molecule observations. Anal Chem. 2007;79(16):6047–6054. doi: 10.1021/ac071009n. [DOI] [PubMed] [Google Scholar]
- 53.Myers GA, Gacek DA, Peterson EM, Fox CB, Harris JM. Microscopic rates of peptide-phospholipid bilayer interactions from single-molecule residence times. J Am Chem Soc. 2012;134(48):19652–19660. doi: 10.1021/ja306074k. [DOI] [PubMed] [Google Scholar]
- 54.Skaug MJ, Mabry J, Schwartz DK. Intermittent molecular hopping at the solid-liquid interface. Phys Rev Lett. 2013;110(25):256101. doi: 10.1103/PhysRevLett.110.256101. [DOI] [PubMed] [Google Scholar]
- 55.Giddings JC. Dynamics of Chromatography. Boca Raton, FL: CRC; 1965. [Google Scholar]
- 56.Giddings JC, Eyring H. A molecular dynamic theory of chromatography. J Phys Chem. 1955;59(5):416–421. [Google Scholar]
- 57.Pasti L, Cavazzini A, Felinger A, Martin M, Dondi F. Single-molecule observation and chromatography unified by Lévy process representation. Anal Chem. 2005;77(8):2524–2535. doi: 10.1021/ac0482514. [DOI] [PubMed] [Google Scholar]
- 58.Daniels CR, et al. Fluorescence correlation spectroscopy study of protein transport and dynamic interactions with clustered-charge peptide adsorbents. J Mol Recognit. 2012;25(8):435–442. doi: 10.1002/jmr.2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Boschetti E. Advanced sorbents for preparative protein separation purposes. J Chromatogr A. 1994;658(2):207–236. [Google Scholar]
- 60.Daniels CR, Reznik C, Landes CF. Dye diffusion at surfaces: Charge matters. Langmuir. 2010;26(7):4807–4812. doi: 10.1021/la904749z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stuart DI, et al. Alpha-lactalbumin possesses a novel calcium binding loop. Nature. 1986;324(6092):84–87. doi: 10.1038/324084a0. [DOI] [PubMed] [Google Scholar]
- 62.Wirth MJ, Fairbank RW, Fatunmbi HO. Mixed self-assembled monolayers in chemical separations. Science. 1997;275(5296):44–47. doi: 10.1126/science.275.5296.44. [DOI] [PubMed] [Google Scholar]
- 63.McQuarrie DA. On the stochastic theory of chromatography. J Chem Phys. 1963;38(2):437–445. [Google Scholar]
- 64.Kastantin M, Walder R, Schwartz DK. Identifying mechanisms of interfacial dynamics using single-molecule tracking. Langmuir. 2012;28(34):12443–12456. doi: 10.1021/la3017134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zielesny A. From Curve Fitting to Machine Learning: An Illustrative Guide to Scientific Data Analysis and Computational Intelligence. Berlin: Springer; 2011. [Google Scholar]
- 66.Pernodet N, Maaloum M, Tinland B. Pore size of agarose gels by atomic force microscopy. Electrophoresis. 1997;18(1):55–58. doi: 10.1002/elps.1150180111. [DOI] [PubMed] [Google Scholar]
- 67.Cuatrecasas P. Protein purification by affinity chromatography: Derivatizations of agarose and polyacrylamide beads. J Biol Chem. 1970;245(12):3059–3065. [PubMed] [Google Scholar]
- 68.Dondi F, Cavazzini A, Pasti L. Chromatography as Lévy stochastic process. J Chromatogr A. 2006;1126(1-2):257–267. doi: 10.1016/j.chroma.2006.06.030. [DOI] [PubMed] [Google Scholar]
- 69.Chandra N, Brew K, Acharya KR. Structural evidence for the presence of a secondary calcium binding site in human α-lactalbumin. Biochemistry. 1998;37(14):4767–4772. doi: 10.1021/bi973000t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.