

SUMMARY
Age-related macular degeneration (AMD) is a leading cause of visual impairment worldwide. Aberrant DNA methylation within the promoter of IL17RC in peripheral blood mononuclear cells has recently been reported in AMD. To validate this association, we examined DNA methylation of the IL17RC promoter in peripheral blood. First, we used Illumina Human Methylation450 Bead Arrays, a widely-accepted platform for measuring global DNA methylation. Second, methylation status at multiple sites within the IL17RC promoter was determined by bisulfite pyrosequencing in two cohorts. Third, a methylation-sensitive QPCR-based assay was performed on a subset of samples. In contrast to previous findings, we did not find evidence of differential methylation between AMD cases and age-matched controls. We conclude that hypomethylation within the IL17RC gene promoter in peripheral blood is not suitable for use as a clinical biomarker of AMD. This study highlights the need for considerable replication of epigenetic association studies prior to clinical application.
INTRODUCTION
Age-related macular degeneration (AMD) is the leading cause of irreversible loss of central vision in developed countries, affecting approximately 30–50 million people worldwide (Coleman et al., 2008). The disease severely impairs quality of life (Rung and Lovestam-Adrian, 2013) and is a substantial economic burden internationally. AMD is a complex disease involving an interaction between genetic and environmental risk factors. Numerous genes that confer a predisposition to AMD have now been identified, and together these account for 10–30% of the variability in disease risk (Fritsche et al., 2013). Strong associations have also been established between AMD and cigarette smoking (Delcourt et al., 2011; Delcourt et al., 1998) as well as, to some degree, sun exposure (Sui et al., 2013), whilst increased dietary antioxidants and fish consumption appear to confer a protective effect (Tan et al., 2009; van Leeuwen et al., 2005).
The mechanisms underlying the observed interplay of genes and the environment in the pathogenesis of AMD are poorly understood; however, cumulative and stable epigenetic variation represents a plausible and attractive model. Epigenetic modification of the human genome is an important mechanism mediating gene-environment interactions by modulating environmental effects on gene expression. For example, cigarette smoking has been shown to be associated with widespread changes in DNA methylation (Lee et al., 2013). Thus efforts have been begun to explore the role of epigenetics in AMD (Hunter et al., 2012; Baird & Wei, 2013).
It is widely accepted that highly expressed genes usually lack cytosine methylation within CpG-rich “islands” associated with many promoters; conversely, methylation within these regions is typically associated with suppression of transcription (Deaton and Bird, 2011). We have previously described the importance of DNA methylation in the regulation of retina-specific genes (Merbs et al., 2012; Oliver et al., 2013; Wan et al., 2013; Nasonkin et al. 2013). Aberrant CpG island hypermethylation leading to inactivation of tumor suppressor genes as well abnormal hypomethylation resulting in oncogene overexpression is characteristic of neoplasia (Jiang et al., 2013), including retinoblastoma (Choy et al., 2002) and uveal melanoma (Maat et al., 2007). Cytosine methylation may also influence the development of other age-related diseases (Adwan and Zawia, 2013; Miao et al., 2013), including AMD (Hunter et al., 2012; Wei et al., 2012). However, conclusive data in the field are scarce, primarily due to the lack of replication of original association studies. A recent report suggested that the promoter of the interleukin 17 receptor C (IL17RC) gene, which encodes a single-pass type I transmembrane protein involved in T-cell activation (Martin et al., 2011), is hypomethylated in peripheral blood samples from patients with AMD compared to unaffected controls (Wei et al., 2012). In addition, this report also showed that ILRC17 is over-expressed in the macula of AMD patients (Wei et al., 2012).
To further investigate the role of epigenetic modifications on gene regulation in AMD, we explored the methylation status of the CpG rich region within the IL17RC promoter in peripheral blood obtained from three independent cohorts of AMD patients and unaffected controls: the Michigan subset of the AMD-MMAP cohort, a Baltimore cohort and an Australian cohort. Initially, we examined the methylation status of probes adjacent to IL17RC from our ongoing genome-wide methylation analysis (using the Illumina Infinium Human Methylation450 [450K] Bead Array) in the Michigan AMD-MMAP cohort, comparing the peripheral whole blood profiles in patients with diagnosed AMD (either geographic atrophy [GA, dry AMD] or choroidal neovascularization [NV, wet AMD]) to that of healthy controls. We then specifically targeted the identical cytosine residues reported to be differentially methylated (Wei et al., 2012) and analyzed the methylation status of these residues in the Baltimore and Australian cohorts by direct bisulfite pyrosequencing and the methylation-sensitive, restriction enzyme based EpiTect Methyl II Polymerase Chain Reaction (PCR) assay. Combined, our analysis of the three cohorts revealed no evidence of disease association at this locus, with all peripheral whole blood samples examined showing very low levels of IL17RC promoter methylation. Hypomethylation at IL17RC was also evident in peripheral blood mononuclear cells (PBMCs) and granulocytes from control subjects with no evidence of any change in AMD individuals. Our results demonstrate that IL17RC hypomethylation in peripheral blood is unlikely to serve as a clinically useful biomarker for AMD.
RESULTS
Illumina Infinium Human Methylation450 Bead Arrays
We performed an epigenome-wide association study (EWAS) on peripheral whole blood from 100 AMD (GA and NV) case-control trios (a subset of the Michigan AMD-MMAP cohort; dbGaP phs000182) using the HM450 platform (unpublished data). Each trio (1 patient with bilateral GA, 1 patient with bilateral NV, 1 control) was matched for sex and for age (< 1 year difference between the 3 individuals in each trio). The sample group comprised 63% females, with a mean ± SD age of 79.3 ± 5.6. There was no significant difference in age between the AMD (79.3 ± 5.5, N=200) and control (79.3 ± 5.7, N=100) groups (P=0.93; unpaired Student’s t-test). We utilized this dataset to examine the methylation of IL17RC in a cohort independent of that previously described (Wei et al., 2012). The Illumina Infinium Human Methylation450 (450K) array included 35 probes located within 26 kb downstream and 8 kb upstream of the IL17RC transcription start site (TSS; chr3:9958758; hg19) (Table S1). The region interrogated using the Methyl-Profiler assay assesses methylation within a 80 bp region (chr3:9957001-9957080), ~1.7 kb upstream from the IL17RC TSS (Wei et al., 2012) and within the region covered interrogated in our analysis. One probe on the 450K array was contained within this region (probe cg19148440), located at chr3:9957031. This probe did not show a statistically significant difference in DNA methylation levels between the AMD and control groups (Figure 1, Table S1). Specifically, the AMD population (N=199) had a mean methylation level of 7.9% and the control population (N=99) had a mean methylation level of 6.9% (P=0.18) (Figure 1). A vast majority of the peripheral whole blood samples showed low levels of methylation of <10%, with only seven samples (one case and six controls of 198 total) having methylation levels >20% (Figure S1). Although five out of the 35 probes within 34 kb of IL17RC reached marginal statistical significance prior to multiple testing correction (P=<0.05), these differences had an absolute difference in methylation levels of <1% between the case and control groups, and represented a mix of both relative hypo- and hypermethylation of IL17RC in AMD cases; the remaining 30 probes did not show a difference in methylation between the AMD cases and controls (Table S1).
Figure 1.
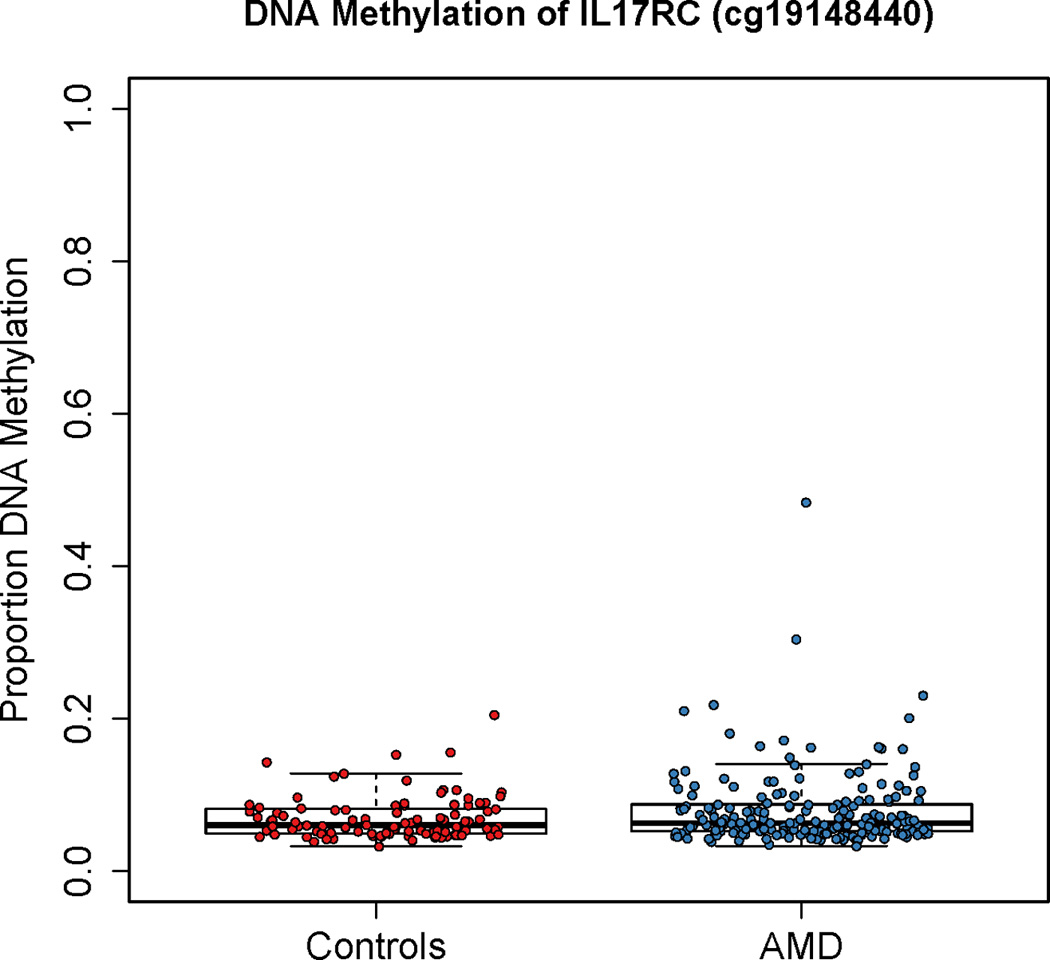
Methylation of the IL17RC promoter in peripheral blood from the Michigan AMD-MMAP cohort as measured by the Illumina 450K Bead Array at probe cg19148440. This region corresponds to the previously reported differentially methylated region in AMD (Wei et al., 2012). The proportion of DNA methylation from control (red, N = 99) and AMD (blue, N = 199) is plotted for each individual. There was no statistically significant difference in methylation between the AMD and control groups. See also Figure S1, Table S1.
Pyrosequencing of the IL17RC Promoter Region in Two Independent Cohorts
To explore the IL17RC methylation status in more detail, DNA methylation was measured using bisulfite pyrosequencing from samples derived from peripheral blood collected at Johns Hopkins University, Baltimore. The final cohort comprised 24 patients with GA (11 females), 15 patients with NV (10 females), and 23 controls (13 females). There was no significant age difference between the Baltimore control population (80.0 ± 7.4, mean ± SD; N=23) and the Baltimore AMD population (76.6 ± 10.2; N=39; unpaired Student’s t-test P=0.17). A total of 33 CpG sites were interrogated encompassing the region chr3:9956901-9957135, specifically including the region identified as hypomethylated in AMD (Wei et al., 2012). In accordance with the 450K data, both the AMD and the control samples from the Baltimore cohort showed very low levels of methylation overall (controls: 1.55% ± 0.07 [SEM], N=23; AMD: 2.11% ± 0.08, N=39) (Figure 2). This lack of methylation at the promoter region of IL17RC differed from the levels previously observed in peripheral blood mononuclear cells of 30% in AMD cases and 60% in controls [calculated as the average of 96 control individuals and 202 individuals with AMD from Table S4 in (Wei et al., 2012)]. We observed no significant hypomethylation of the promoter region of IL17RC in AMD cases relative to controls at any of the 33 CpG sites interrogated (Table S2). In fact, a slight but significant relative hypermethylation of AMD samples was observed (unpaired Student’s t-test, P=<0.001).
Figure 2.

Comparison of DNA methylation levels within the IL17RC promoter in patients with AMD and age- and gender-matched controls with no current, past or family history of any ocular disease. (A) DNA methylation of IL17RC in peripheral blood DNA. Each data point represents the mean percentage methylation within individual AMD (circles; Baltimore N=39, Australia N=100) and control (squares; Baltimore N=23, Australia N=116) subjects. Methylation was measured across 33 CpG sites (Baltimore cohort) or 8 CpG sites (Australian cohort) which overlapped the region previously interrogated (Wei et al., 2012). All samples showed <6% methylation by bisulfite pyrosequencing. The Baltimore cohort indicated small yet significant hypomethylation of the control cases (P=<0.0001). There was no significant difference in mean methylation between cases and controls in the Australian cohort (P= 0.30). Note the split Y axis. See also Table S2. (B, C) Comparison of DNA methylation levels within the IL17RC promoter in donor retina/RPE tissue from individuals with age-related macular degeneration and controls with no reported history or pathological evidence of ocular disease. Each data point represents the mean percentage methylation within the individual tissue samples from AMD (circles) and control (squares) donors overlapping the region interrogated by Wei et al (Wei et al, 2012). (B) Methylation was measured across 19 CpG sites in retina from AMD (N=9) and control (N=6) donors. (C) Methylation was measured across 33 CpG sites in RPE/choroid from AMD (N=3) and control (N=3) donors. Hypomethylation was observed in both tissues at all CpG sites examined.
As comprehensive analysis in the Baltimore cohort revealed consistent hypomethylation of the IL17RC promoter in both AMD cases and normal controls, a second, independent cohort from Australia was examined. Bisulfite pyrosequencing was performed on 100 AMD patients and 118 controls (Figure 2A). The mean age of the AMD patients (61 females) was 72.1 ± 6.3 years and of the controls (54 females), 71.0 ± 7.8 years (Student t-test P=0.25). Following bisulfite pyrosequencing of eight CpG sites within the previously interrogated region (chr3:9957024–9957056) (Wei et al., 2012), we did not detect any significance differences in the mean methylation in AMD patients (1.66% ± 0.06, N=100) compared to controls (1.75% ± 0.06, N=118; P=0.30). The bisulfite pyrosequencing results indicated low levels of methylation (<6%) in all samples and was consistent with the results from the Baltimore cohort (Figure 2A). Only one individual (an AMD case) showed mildly elevated methylation (16%) of a CpG site (chr3:9957047) relative to the remainder of the cohort (Table S2).
Pyrosequencing of the IL17RC Promoter Region in Ocular Tissue
Retina samples were obtained from 9 AMD (M=6, 87.8 ± 5.8 [mean age ± SD]; F=3, 91.7 ± 0.6) and 6 controls (M=1, 91; F=5, 88.4 ± 3.6). Retinal pigment epithelium (RPE) samples were obtained from 3 AMD (M=3, 86.3 ± 8.0) and 3 control (M=3, 85.3 ± 5.5) donors. Bisulfite pyrosequencing was performed on the region interrogated by Wei et al (Wei et al, 2012), which showed low levels of methylation (<6%) in all samples examined (Figure 2B & C). There was no significant difference in the mean methylation of the retina samples between the control (4.23% ± 0.51, N=6) and AMD (3.49% ± 0.41, N=9; P=0.28; Student’s t-test) groups. The RPE samples also showed a lack of methylation, and no difference between the control (1.68% ± 0.08, N=3) and AMD (1.69% ± 0.03, N=3; P=0.88). The level of methylation within the ocular tissue samples was consistent with that observed in peripheral blood (Figure 2A).
EpiTect Methyl II PCR Assay of IL17RC Promoter Methylation in the Baltimore Samples
To determine whether the difference in overall peripheral blood methylation levels resulted from technical or biological differences, the commercial EpiTect Methyl II PCR assay for IL17RC (Qiagen) was performed on a subset of age/sex-matched Baltimore samples (AMD N=20, 12 females; controls N=10, 6 females). There was no significant age difference between the AMD samples (78.2 ± 5.7; mean ± SD) and the control samples (77.9 ± 6.2; unpaired Student’s t-test P=0.89). This assay measures the amount of methylated DNA in a 156 bp region centered around chr3:9957039 using methylation-sensitive and methylation-dependent restriction digests followed by QPCR as utilized previously (Wei et al., 2012). We did not detect any differences in the percentage of methylated DNA between the AMD (0.10% ± 0.05 [SEM]) and control groups (0.17% ± 0.03; P=0.34; unpaired Student’s t-test) using this restriction enzyme-based approach (Figure 3). Furthermore, all samples examined showed <1% methylated DNA using the EpiTect Methyl II PCR Assay for IL17RC. These results were entirely consistent with our bisulfite pyrosequencing data from this region. Incomplete digest by the methylation-dependent enzyme was excluded as a potentially confounding factor in the observation of hypomethylation by the inclusion of the methylation-dependent control assay (Table S3).
Figure 3.

EpiTect Methyl II PCR Assay results for the IL17RC promoter CpG island in peripheral blood from a subset of the Baltimore cohort. Each data point represents the percentage methylation from AMD (circles, N=20) and control (squares, N=10) subjects at chr3:9956961-9957116 (hg19). Data are plotted as the percentage of methylated DNA relative to the input. All samples showed hypomethylation by this assay. There was no significant difference in mean methylation between cases and controls using the EpiTect Methyl II PCR assay for IL17RC (P= 0.34). See also Table S3.
Comparison of IL17RC-promoter methylation between PBMCs and granulocytes
Given that PBMCs were investigated previously (Wei et al., 2012), we sought to directly compare the methylation profile of the IL17RC promoter region between PBMCs, granulocytes and whole blood. Additionally, given that hypermethylation of this region was previously identified in individuals without AMD, we analyzed fresh samples from elderly control subjects (N=18). Bisulfite pyrosequencing of the IL17RC promoter revealed hypomethylation of all individuals and cell populations examined (Figure 4, Table S4). While there was a significant difference in mean percentage methylation between the PBMCs (2.34% ± 0.16 [SEM]), granulocytes (1.71% ± 0.05) and whole blood (1.90% ± 0.05) (F(2,17)=13.92, P=0.0006), the overall methylation levels were low and are likely to be below the sensitivity level of the pyrosequencing assay. No significant cellular heterogeneity or cell type specific DNA methylation differences potentially accounting for the discrepancy with the previously published results could be identified at this locus.
Figure 4.

Comparison of DNA methylation levels within the IL17RC promoter in Ficoll-separated PBMCs and granulocytes or unsorted whole blood from patients with no current or past ocular disease or known family history of AMD. Methylation was measured in 18 individuals across 14 CpG sites encompassing the region interrogated by Wei et al. (Wei et al., 2012). Each data point represents the mean percentage methylation across this region with the lines joining samples obtained from the same individual. No individual CpG sites had >6% methylation by bisulfite pyrosequencing in any sample. There was a small but significant difference in methylation between the cell populations (P<0.001). Note the split Y axis. See also Table S4.
DISCUSSION
A recent study reported hypomethylation of the IL17RC promoter in PBMCs of AMD patients and a corresponding increase in IL17RC gene expression in macular tissue of AMD cadaver eyes (Wei et al., 2012). These results suggested IL17RC as a potential biomarker for AMD diagnosis. To explore these reported findings further, we examined the IL17RC promoter region in circulating peripheral whole blood (leukocytes), using three distinct techniques in three independent cohorts (summarized in Table 1). Our results indicate that IL17RC is essentially unmethylated in peripheral blood of both AMD and control populations, and that the methylation level of IL17RC is unlikely to serve as a useful clinical biomarker of AMD.
Table 1.
Summary of our present IL17RC promoter methylation results in comparison to the previously published AMD dataset.
| Cohort | Technique | Target Chromosome Location (hg19) |
Mean Methylation Level (%) |
|
|---|---|---|---|---|
| Case | Control | |||
| Michigan AMD-MMAP | Illumina Infinium Human Methylation450 (450K) Bead Arrays | chr3:9957031 | 7.9 | 6.9 |
| Baltimore | Bisulfite Pyrosequencing | chr3:9956901-9957135 | 2.1 | 1.6 |
| Methylation-Sensitive QPCR (EpiTect Methyl II PCR Assay) | chr3:9956961-9957117 | 0.1† | 0.2† | |
| Australia | Bisulfite Pyrosequencing | chr3:9957024-9957056 | 1.7 | 1.8 |
| NEI (Wei et al., 2012) | Methylation-Sensitive QPCR (Methyl-Profiler) | chr3:9957001-9957080 | 30.0# | 60.0# |
percentage of methylated DNA relative to input
Calculated from Supplemental Table 4 (Wei et al., 2012)
Using two methods that interrogate methylation changes at individual CpG dinucleotides (bisulfite pyrosequencing and the 450K methylation array), we were unable to reproduce the high percentage of methylated DNA (60%) in the IL17RC promoter previously seen in control individuals (Wei et al., 2012). To ensure that our findings did not reflect a population-specific effect, we bisulfite pyrosequenced the region in a second independent cohort from Australia, confirming a low level of methylation in both the AMD and control peripheral whole blood samples (1.7% and 1.8%, respectively). Unexpectedly, the difference in our results is not due to a difference in the clinical definition of the AMD phenotype, as our results differed with respect to the unaffected control population, which contained over two and a half times the number of controls compared with the Wei et al. study (263 vs. 96) (Wei et al., 2012). The average age of our Michigan AMD-MMAP control individuals is 79 years, and that of our Baltimore controls is 80 years. Although these are not statistically different from the average age of our AMD patients (79 years and 75 years, respectively; P=0.93), they are slightly older than the average age of the controls (70 years) presented in the study by Wei et al. (Wei et al., 2012). However, it seems unlikely that the younger age in the NEI cohort (Wei et al., 2012) alone explains the higher level of methylation, as the average age of controls within the Australian cohort is 71 years and each of these individuals showed a methylation level of <6%. In the previous report, control individuals had methylation levels covering the entire spectrum of 0–100% methylation (Wei et al., 2012).
The previous study (Wei et al., 2012) used an enzymatic method to measure methylation that relies on effective digestion of high-quality genomic DNA with methylation-dependent and methylation-independent restriction enzymes (Methyl-Profiler). It is designed to detect global methylation changes, rather than site-specific methylation changes. To verify that our observation did not result from differences in the methylation analysis techniques, we performed the same restriction enzyme-based analysis using the rebranded EpiTect Methyl Restriction kit (previously Methyl-Profiler) followed by the EpiTect Methyl II PCR Assay for IL17RC on a subset of the Baltimore peripheral whole blood samples.
Using the commercially available IL17RC primers, we again found that the percentage of methylated DNA was very low in both cases and controls. Wei et al. used primers that differed from those provided in the commercially available EpiTect Methyl II PCR assay (Supplemental Information 2 in Wei et al., 2012), although the same (but not identical) genetic location was targeted for amplification and interrogation by the enzyme digestion. This technique is susceptible to giving falsely high methylation measurements if incomplete digestion of unmethylated DNA with the methylation-dependent enzyme occurs. Sodium bisulfite conversion-based methods are widely considered the gold-standard for measuring DNA methylation, and enable quantitative measurements from a single CpG-site. Both of the techniques utilized in our study (Illumina Infinium Human Methylation450 [450K] Bead Array and bisulfite pyrosequencing) are based on bisulfite converted DNA.
Cellular heterogeneity is widely regarded as a potential confounding factor in case:control studies aimed at linking epigenetic disruption to specific phenotypes. As such, we considered this as another potential explanation for the discrepant findings. Our collection of peripheral blood samples contained the entire leukocyte (white blood cell) component of blood, unlike the PBMC fraction previously interrogated, which lacks polymorphonuclear (granulocyte) cells (Wei et al., 2012). To address this issue, we isolated PBMCs and granulocytes from peripheral blood and compared the methylation status of 14 previously analyzed CpG sites in each cell type as well as in peripheral whole blood from the same control individual. All three cell populations had methylation levels of <5%, thereby eliminating the possibility that cell composition differences could account for the discrepancy. As Ficoll-separated samples were processed within 2 hours of phlebotomy and still yielded low methylation levels, we also excluded the possibility that processing time may have altered the DNA methylation in our cohorts.
Genes expressed in many different tissues typically have an unmethylated CpG island upstream from their TSS, and methylation of such islands is strongly associated with decreased gene expression (Jones, 2012). The region in question is ~1700 bp upstream from the IL17RC TSS and located in a CpG island. Consistent with our finding of a low methylation level is the fact that IL17RC is widely expressed in a range of human tissues (Haudenschild et al., 2002). We have shown comparably low levels of methylation at Rho and Rbp3 in expressing cells, whilst non-expressing cells showed >60% methylation (Merbs et al., 2012). Many factors other than DNA methylation could explain the expression difference between AMD patients and controls seen by Wei et al. Our bisulfite pyrosequencing also indicated that the IL17RC promoter is lowly methylated in the retina and RPE of both AMD cases and controls. Wei et al previously reported increased expression of IL17RC in the macula of patients with AMD. Whilst our data does not dispute these findings, it does suggest that the regulation of IL17RC expression is unlikely to be the result of differential methylation of the promoter region in AMD individuals. While we believe that the level of IL17RC DNA methylation is not a good marker for AMD, it is possible that the expression and protein levels of IL17RC might still be useful biomarkers.
In summary, our comprehensive analysis of DNA methylation of the IL17RC promoter in peripheral blood samples from three independent AMD cohorts was unable to replicate the reported differential methylation using three different techniques, including the gold standard of bisulfite pyrosequencing. Examination of retinal tissue also indicated low levels of methylation in all samples examined. Furthermore, we observed very little methylation in all of our control samples, compared to the relatively high levels previously reported. Consistent with our finding of a low methylation level is the fact that IL17RC is widely expressed in a range of human tissues (Haudenschild et al., 2002). The difference in the percentage of methylation previously reported is surprisingly large. A 30% methylation difference between cases and controls is much greater than methylation differences typically seen in other complex diseases such as autism (7–16% in brain samples; Ladd-Acosta et al., 2013) and obesity (1–16% in blood; Almén et al., 2012) and is also higher than many methylation differences seen between cancer and normal tissues (Hansen et al., 2011; Irizarry et al., 2009). Our findings suggest that hypomethylation within the IL17RC gene promoter is not suitable for use as a clinical biomarker of AMD and highlight the need for considerable replication of epigenetic association studies prior to clinical application.
EXPERIMENTAL PROCEDURES
Ethical approval
All aspects of this project were conducted in accordance with the principles of the Declaration of Helsinki, with informed consent being obtained from all participants. This project was approved by the Human Research Ethics Committees of the Royal Victorian Eye and Ear Hospital and the University of Western Australia and with Institutional Review Board (IRB) approval from the Johns Hopkins University School of Medicine.
Illumina Infinium Human Methylation450 Bead Array
The 450K methylation array is widely accepted as a robust platform to assess DNA methylation, showing a high concordance with bisulfite pyrosequencing (r = >0.80) (Roessler et al., 2012). DNA samples from peripheral blood of 298 age- and sex-matched samples (comprising 100 bilateral geographic atrophy [GA], 99 bilateral neovascularization [NV] and 99 controls) were obtained from a subset of the Michigan patients from the Age-related Macular Degeneration – Michigan, Mayo, AREDS, Pennsylvania (AMD-MMAP) study cohort (dbGaP: phs000182). DNA was extracted from whole blood using the Puregene Blood Core Kit C (Qiagen). Both the AMD and the control groups contained 63% females, with a mean ± SD age of 79.3 ± 5.7 in the AMD group and 79.3 ± 5.5 in the control group. All samples were derived from the Kellogg Eye Center (KEC) at the University of Michigan (Chen et al., 2010). From the original KEC cohort, those patients with bilateral GA or NV were selected. Control patients were examined and accepted if they had small drusen and pigment changes in one eye only with no family history of AMD. All cases and controls were ≥60 years of age. DNA samples (1.3 µg) were processed for the HM450 platform (Illumina) at the Center for Inherited Disease Research (CIDR; Johns Hopkins University) as per the manufacturers’ protocols. All matched samples (GA-NV-control) were run within a single BeadChip array.
Illumina Infinium Human Methylation450 Bead Array data preprocessing
We normalized the intensity data using a modified version of quantile normalization in the minfi Bioconductor package (http://bioconductor.org/packages/devel/bioc/html/minfi.html), which, briefly, forces the distribution of type I and type II probes on the Illumina 450K microarray to be the same. Quantile normalization is performed on the type II probes and then interpolated to a reference distribution to which the type I probes are normalized. This normalization is performed separately on the methylated (M) and unmethylated (U) channel intensities. The logit-transformed DNA methylation (DNAm) is y=log2(M/U), and the proportion methylation (“beta” scale, as reported by Illumina) can be obtained by: 2^y/(1+2^y). The “beta” values for probes in and around IL17RC (N=35) were selected for subsequent statistical analyses.
Sample selection from the Baltimore cohort
DNA samples derived from peripheral whole blood were obtained from a cohort collected at Johns Hopkins University in Baltimore as previously described (Yang et al., 2008; Yang et al., 2010). In short, diagnosis of advanced AMD was based on the presence of GA or CNV (equivalent to AREDS category 4 or 5). Controls were identified as >60 years of age, having fewer than five small drusen (<63 µm) and no RPE abnormalities. The final cohort comprised of 24 patients with GA (mean ± SD age at interview of 78.5 ± 7.6; males = 13, females = 11; European = 24), 15 patients with NV (73.7 ± 13.1; males = 5, females = 10; European = 14, Ashkenazi Jew = 1), and 23 controls (80.0 ± 7.4; males = 10, females = 13; European = 23). Genomic DNA was isolated from peripheral whole blood using Puregene Blood Kit chemistry on an Autopure LS automated DNA purification instrument (Qiagen, Valencia, CA). DNA concentrations are determined by spectrophotometry using a DU 530 Life Science UV/Vis Spectrophotometer (Beckman Coulter, Inc., Fullerton, CA).
Sample collection in an Australian cohort
Patients with AMD were recruited from ophthalmology clinics at Launceston Eye Institute, Australia. The clinical criteria for the diagnosis of AMD included the presence of extensive drusen and associated pigmentary abnormalities of the RPE layer or evidence of advanced disease by presence of GA or NV. Age- and sex-matched controls were recruited from adjunct genetic studies. All control subjects underwent a comprehensive examination including fundus photography as well as macular assessment using optical coherence tomography. To be included as a control, subjects required no evidence of ophthalmic pathology as well as no prior or known family history of ocular disease. In total, we recruited 100 patients with AMD and 120 age-matched individuals with no signs of AMD. Two control samples failed DNA extraction from whole blood. All participants were of reported Northern European ancestry. The mean ± SD age of the AMD patients and controls (passing whole blood DNA QC) was 72.1 ± 6.3 years and 71.0 ± 7.8 years, respectively. 39% of the AMD patients and 54% of controls were male. Approximately 20 mL EDTA peripheral whole blood was collected via venipuncture from all participants and genomic DNA was extracted from circulating leukocytes by the QIAcube platform (Qiagen, Maryland, USA) through the facilities at the Western Australian DNA Bank.
Bisulfite pyrosequencing of the IL17RC promoter
Bisulfite pyrosequencing is widely considered the gold-standard approach for quantitatively measuring DNA methylation across targeted regions (<400 bp) of the genome. Pyrosequencing assays were designed using algorithms built into the PyroMark Assay Design Software (Version 2.0.1, Qiagen, Maryland USA). Briefly, primers designed to target specific CpG sites within the IL17RC promoter were chosen from a list generated by the software on the basis of the algorithms’ predicted assay quality. Genomic DNA (500 ng) was bisulfite converted using the EZ DNA Methylation Gold kit (Zymo Research) according to the manufacturer’s instructions. Following bisulfite treatment all previously unmethylated cytosine residues are converted to uracil, whereas methylated cytosine residues remain unconverted. DNA containing the segment of the IL17RC promoter analyzed by Wei and colleagues (Wei et al., 2012) was amplified from the bisulfite-treated DNA using PCR primer set #1 (Australian samples) and PCR primer sets #1 & 3 (Baltimore samples) as shown in Table S5.
PCR products were bound to Streptavidin Sepharose High Performance beads (GE Healthcare Life Sciences, Rydalmere, Australia) and a single-strand template was isolated using the Pyrosequencing Vacuum Prep Tool (Qiagen, Maryland USA). The beads were transferred to an optically clear, 24 well sequencing plate in 0.3 (<63 µm) and no RPE abnormalities. The final cohort comprised of 24 M of the sequencing primer. Serial pyrosequencing was performed where multiple sequencing primers were contained within a single PCR amplicon (Tost et al., 2006). Pyrosequencing was performed on a PyroMark 24 Pyrosequencing System (Qiagen, Maryland USA) as per the manufacturer’s instructions. Data were analyzed on the PyroMark Q24 software (Qiagen, Maryland USA). This software calculates the C peak as a percentage of the T plus C peak at each CpG site taking into account sequence length and signal strength. The mean methylation for each individual was calculated. Initially eight CpG sites overlapping the region specifically targeted previously (Wei et al., 2012) were investigated in the Baltimore and Australian cohorts. To further explore additional regions at the locus an additional 25 CpGs were sequenced in the Baltimore cohort alone. An unpaired Student’s t-test was used to compare the methylation difference between the matched case-control groups.
Collection of retina/RPE from cadaver eyes
Cadaver eyes with a known medical history of AMD and unaffected controls were obtained from the National Disease Research Interchange (NDRI) with ethical approval from the Johns Hopkins University School of Medicine IRB. In brief, the anterior segment was removed, along with the pupil and lens. Four cuts were made down the eye cup to expose the macula. The flower-cut eye cup was exposed to a sucrose gradient as previously described (Hackler et al., 2012). The macula and optic nerve head were removed by 6 mm biopsy punches, and the remaining peripheral retina and retina pigment epithelium (RPE)/choroid were stored at −80°C. AMD was confirmed by assessment of macula photos taken prior to the sucrose treatment. DNA was extracted from the retina/RPE using the DNEasy Blood & Tissue kit (Qiagen) as per the manufacturer’s protocol. Retina samples were obtained from 9 AMD (M=6, 87.8 ± 5.8 [mean age ± SD]; F=3, 91.7 ± 0.6) and 6 controls (M=1, 91; F=5, 88.4 ± 3.6). RPE samples were obtained from 3 AMD (M=3, 86.3 ± 8.0) and 3 control (M=3, 85.3 ± 5.5) donors. The bisulfite conversion and subsequent pyrosequencing of IL17RC was performed as described above.
Application of the EpiTect Methyl II PCR Assay for IL17RC to Baltimore samples
DNA samples from ten age/sex-matched GA/NV/control trios (derived from peripheral whole blood) were selected from the Baltimore samples previously used for bisulfite pyrosequencing. The mean age of the subset of Baltimore AMD samples was 78.2 ± 5.7 (mean ± SD), N=20, 12 females. The mean age of the Baltimore controls subgroup was 77.9 ± 6.2, N=10 of whom six were female. The MethylProfiler restriction assay kit (Cat, #MeA-01, SABiosciences) has been rebranded as the EpiTect Methyl II Restriction kit (Cat. #335452, Qiagen). The EpiTect Methyl II restriction digest was set up according to the manufacturer’s protocol. Briefly, 230 ng of DNA was used in each of the following restriction enzyme digest reactions: 1) methylation-sensitive, 2) methylation-dependent, 3) methylation-sensitive and methylation-dependent double digest or 4) mock digest. Q-PCR was performed as per the manufacturer’s protocol, using commercially available primers for IL17RC (Cat. #EPHS110029-1A, Qiagen) in RT2 SYBR Green qPCR Mastermix (Cat. #330500, Qiagen) on a CFX96 real-time PCR machine (Bio-Rad). The commercially validated IL17RC EpiTect Methyl II PCR primers interrogated methylated DNA in a 156 bp region centered around chr3:9957039, targeting the same region reported by Wei et al. (Wei et al., 2012). Methylation-sensitive (Cat. #EPHS115450-1A) and methylation-dependent (Cat. #EPHS115451-1A) digest control assays were performed on a subset (N=7) of samples. Samples were analyzed as recommended by the manufacturer (http://www.sabiosciences.com/dna_methylation_data_analysis.php).
Cell-separation for comparison of PBMCs and granulocyte cell IL17RC-promoter methylation status
PBMCs and granulocytes were isolated from whole blood collected from 20 Australian controls (a subset of those described above, mean age 82, range 77–90, 65% male) using Ficoll-Plaque Premium (GE Healthcare Lifesciences) as per the manufacturer’s protocol. Blood samples were all processed within 2 hours of phlebotomy. The granulocyte fraction was extracted from the bottom of the tubes, with the PBMC fraction being extracted from the serum/medium boundary. Genomic DNA isolated from each cell type was bisulfite treated as described above. Two samples failed DNA extraction from whole blood. The methylation status of 14 previously analyzed CpG sites within the IL17RC promoter was compared between PBMCs, granulocytes (polymorphonuclear leukocytes, PMLs) and unsorted whole blood using bisulfite pyrosequencing primer sets 1 & 2 (Table S5). Groups of paired measurements were compared using a repeated measure ANOVA.
Supplementary Material
HIGHLIGHTS.
IL17RC is hypomethylated (<6%) in all peripheral blood samples examined
IL17RC is not differentially methylated between cases & controls in 3 AMD cohorts
IL17RC promoter shows hypomethylation in both mononuclear cells and granulocytes
IL17RC hypomethylation in peripheral blood is not a suitable biomarker of AMD
ACKNOWLEDGEMENTS
The authors gratefully acknowledge the technical advice provided by Dr David Chandler, the assistance of the Western Australian DNA Bank and the Center for Inherited Disease Research (CIDR, Johns Hopkins University). This work was supported by funding from the Ophthalmic Research Institute of Australia (ORIA) and the American Health Assistance Foundation to AWH as well as from an Australian National Health and Medical Research Council (NHMRC) Centres of Research Excellence Grant 1023911 (2012–2016) to JEC, DAM and AWH. CERA receives operational infrastructure support from the Victorian government. Collection of the Michigan AMD-MMAP cohort was enabled by funding from the National Eye Institute (NEI) (EY016862) to AS and JRH. Funding from the NEI (X01HG006605) to SLM, VFO, AEJ, KEB, JRH, AS and DJZ was used for the analysis of samples on the Illumina Human Methylation 450K platform at CIDR. Pyrosequencing validation was funded by NEI R01EY020406 and the generosity of Agnes Nixon to SLM, and aided by NEI Core Grant P30EY001765.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Adwan L, Zawia NH. Epigenetics: a novel therapeutic approach for the treatment of Alzheimer's disease. Pharmacol. Ther. 2013;139:41–50. doi: 10.1016/j.pharmthera.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almén MS, Jacobsson JA, Moschonis G, Benedict C, Chrousos GP, Fredriksson R, Schiöth HB. Genome wide analysis reveals association of a FTO gene variant with epigenetic changes. Genomics. 2012;99:132–137. doi: 10.1016/j.ygeno.2011.12.007. [DOI] [PubMed] [Google Scholar]
- Baird PN, Wei L. Age-related macular degeneration and DNA methylation. Epigenomics. 2013;5:239–241. doi: 10.2217/epi.13.19. [DOI] [PubMed] [Google Scholar]
- Chen W, Stambolian D, Edwards AO, Branham KE, Othman M, Jakobsdottir J, Tosakulwong N, Pericak-Vance MA, Campochiaro PA, Klein ML, et al. Genetic variants near TIMP3 and high-density lipoprotein-associated loci influence susceptibility to age-related macular degeneration. Proc. Natl. Acad. Sci. U. S. A. 2010;107:7401–7406. doi: 10.1073/pnas.0912702107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy KW, Pang CP, To KF, Yu CB, Ng JS, Lam DS. Impaired expression and promotor hypermethylation of O6-methylguanine-DNA methyltransferase in retinoblastoma tissues. Invest. Ophthalmol. Visual Sci. 2002;43:1344–1349. [PubMed] [Google Scholar]
- Coleman HR, Chan CC, Ferris FL, Chew EY., 3rd Age-related macular degeneration. Lancet. 2008;372:1835–1845. doi: 10.1016/S0140-6736(08)61759-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011;25:1010–1022. doi: 10.1101/gad.2037511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcourt C, Delyfer MN, Rougier MB, Amouyel P, Colin J, Le Goff M, Malet F, Dartigues JF, Lambert JC, Korobelnik JF. Associations of complement factor H and smoking with early age-related macular degeneration: the ALIENOR study. Invest. Ophthalmol. Visual Sci. 2011;52:5955–5962. doi: 10.1167/iovs.10-6235. [DOI] [PubMed] [Google Scholar]
- Delcourt C, Diaz JL, Ponton-Sanchez A, Papoz L. Smoking and age-related macular degeneration. The POLA Study. Pathologies Oculaires Liees a l'Age. Archives Ophthalmol. 1998;116:1031–1035. doi: 10.1001/archopht.116.8.1031. [DOI] [PubMed] [Google Scholar]
- Fritsche LG, Chen W, Schu M, Yaspan BL, Yu Y, Thorleifsson G, Zack DJ, Arakawa S, Cipriani V, Ripke S, et al. Seven new loci associated with age-related macular degeneration. Nat. Genet. 2013;45:433–439. 439e431–439e432. doi: 10.1038/ng.2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackler L, Jr, Masuda T, Oliver VF, Merbs SL, Zack DJ. Use of laser capture microdissection for analysis of retinal mRNA/miRNA expression and DNA methylation. Methods Mol. Biol. 2012;884:289–304. doi: 10.1007/978-1-61779-848-1_21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KD, Timp W, Bravo HC, Sabunciyan S, Langmead B, McDonald OG, Wen B, Wu H, Liu Y, Diep D, Briem E, Zhang K, Irizarry RA, Feinberg AP. Increased methylation variation in epigenetic domains across cancer types. Nat. Genet. 2011;43:768–775. doi: 10.1038/ng.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haudenschild D, Moseley T, Rose L, Reddi AH. Soluble and transmembrane isoforms of novel interleukin-17 receptor-like protein by RNA splicing and expression in prostate cancer. J. Biol. Chem. 2002;277:4309–4316. doi: 10.1074/jbc.M109372200. [DOI] [PubMed] [Google Scholar]
- Hunter A, Spechler PA, Cwanger A, Song Y, Zhang Z, Ying GS, Hunter AK, Dezoeten E, Dunaief JL. DNA methylation is associated with altered gene expression in AMD. Invest. Ophthalmol. Visual Sci. 2012;53:2089–2105. doi: 10.1167/iovs.11-8449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, Ji H, Potash JB, Sabunciyan S, Feinberg AP. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009;41:178–186. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Liu S, Chen X, Cao Y, Tao Y. Genome-wide distribution of DNA methylation and DNA demethylation and related chromatin regulators in cancer. Biochim. Biophys. Acta. 2013;1835:155–163. doi: 10.1016/j.bbcan.2012.12.003. [DOI] [PubMed] [Google Scholar]
- Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012;13:484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- Ladd-Acosta C, Hansen KD, Briem E, Fallin MD, Kaufmann WE, Feinberg AP. Common DNA methylation alterations in multiple brain regions in autism. Mol. Psychiatry. 2013 doi: 10.1038/mp.2013.114. Published online September 3, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KW, Pausova Z. Cigarette smoking and DNA methylation. Front. Genet. 2013;4:132–143. doi: 10.3389/fgene.2013.00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maat W, van der Velden PA, Out-Luiting C, Plug M, Dirks-Mulder A, Jager MJ, Gruis NA. Epigenetic inactivation of RASSF1a in uveal melanoma. Invest. Ophthalmol. Visual Sci. 2007;48:486–490. doi: 10.1167/iovs.06-0781. [DOI] [PubMed] [Google Scholar]
- Martin DF, Maguire MG, Ying GS, Grunwald JE, Fine SL, Jaffe GJ. Ranibizumab and bevacizumab for neovascular age-related macular degeneration. N. Engl. J. Med. 2011;364:1897–1908. doi: 10.1056/NEJMoa1102673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merbs SL, Khan MA, Hackler L, Jr, Oliver VF, Wan J, Qian J, Zack DJ. Cell-specific DNA methylation patterns of retina-specific genes. PloS One. 2012;7:e32602. doi: 10.1371/journal.pone.0032602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao CG, Yang YY, He X, Li J. New advances of DNA methylation and histone modifications in rheumatoid arthritis, with special emphasis on MeCP2. Cell. Signalling. 2013;25:875–882. doi: 10.1016/j.cellsig.2012.12.017. [DOI] [PubMed] [Google Scholar]
- Oliver VF, Wan J, Agarwal S, Zack DJ, Qian J, Merbs SL. A novel methyl-binding domain protein enrichment method for identifying genome-wide tissue-specific DNA methylation from nanogram DNA samples. Epigenet. Chromatin. 2013;6:17. doi: 10.1186/1756-8935-6-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasonkin IO, Merbs SL, Lazo K, Oliver VF, Brooks M, Patel K, Enke RA, Nellissery J, Jamrich M, Le YZ, Bharti K, Fariss RN, Rachel RA, Zack DJ, Rodriguez-Boulan EJ, Swaroop A. Conditional knockdown of DNA methyltransferase 1 reveals a key role of retinal pigment epithelium integrity in photoreceptor outer segment morphogenesis. Development. 2013;140:1330–1341. doi: 10.1242/dev.086603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler J, Ammerpohl O, Gutwein J, Hasemeier B, Anwar SL, Kreipe H, Lehmann U. Quantitative cross-validation and content analysis of the 450K DNA methylation array from Illumina, Inc. BMC Res. Notes. 2012;5:210–217. doi: 10.1186/1756-0500-5-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rung L, Lovestam-Adrian M. Three-year follow-up of visual outcome and quality of life in patients with age-related macular degeneration. Clin. Ophthalmol. 2013;7:395–401. doi: 10.2147/OPTH.S41585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui GY, Liu GC, Liu GY, Gao YY, Deng Y, Wang WY, Tong SH, Wang L. Is sunlight exposure a risk factor for age-related macular degeneration? A systematic review and meta-analysis. Br. J. Ophthalmol. 2013;97:389–394. doi: 10.1136/bjophthalmol-2012-302281. [DOI] [PubMed] [Google Scholar]
- Tan JS, Wang JJ, Flood V, Mitchell P. Dietary fatty acids and the 10-year incidence of age-related macular degeneration: the Blue Mountains Eye Study. Archives Ophthalmology. 2009;127:656–665. doi: 10.1001/archophthalmol.2009.76. [DOI] [PubMed] [Google Scholar]
- Tost J, El abdalaoui H, Gut IG. Serial pyrosequencing for quantitative DNA methylation analysis. BioTechniques. 2006;40:721–722. 724, 726. doi: 10.2144/000112190. [DOI] [PubMed] [Google Scholar]
- Tost J, Gut IG. DNA methylation analysis by pyrosequencing. Nat. Protoc. 2007;2:2265–2275. doi: 10.1038/nprot.2007.314. [DOI] [PubMed] [Google Scholar]
- van Leeuwen R, Boekhoorn S, Vingerling JR, Witteman JC, Klaver CC, Hofman A, de Jong PT. Dietary intake of antioxidants and risk of age-related macular degeneration. JAMA. 2005;294:3101–3107. doi: 10.1001/jama.294.24.3101. [DOI] [PubMed] [Google Scholar]
- Wan J, Oliver VF, Zhu H, Zack DJ, Qian J, Merbs SL. Integrative analysis of tissue-specific methylation and alternative splicing identifies conserved transcription factor binding motifs. Nucleic Acids Res. 2013 Jul 24; doi: 10.1093/nar/gkt652. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L, Liu B, Tuo J, Shen D, Chen P, Li Z, Liu X, Ni J, Dagur P, Sen HN, et al. Hypomethylation of the IL17RC promoter associates with age-related macular degeneration. Cell Rep. 2012;2:1151–1158. doi: 10.1016/j.celrep.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Stratton C, Francis PJ, Kleinman ME, Tan PL, Gibbs D, Tong Z, Chen H, Constantine R, Yang X, et al. Toll-like receptor 3 and geographic atrophy in age-related macular degeneration. N. Engl. J. Med. 2008;359:1456–1463. doi: 10.1056/NEJMoa0802437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Tong Z, Chen Y, Zeng J, Lu F, Sun X, Zhao C, Wang K, Davey L, Chen H, et al. Genetic and functional dissection of HTRA1 and LOC387715 in age-related macular degeneration. PLoS Genet. 2010;6:e1000836. doi: 10.1371/journal.pgen.1000836. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.