

Abstract
The most common cause of familial frontotemporal lobar degeneration with TAR DNA-binding protein-43 pathology (FTLD-TDP) has been found to be an expansion of a hexanucleotide repeat (GGGGCC) in a noncoding region of the gene C9ORF72. Hippocampal sclerosis (HpScl) is a common finding in FTLD-TDP. Our objective was to screen for the presence of C9ORF72 hexanucleotide repeat expansions in a pathologically-confirmed cohort of “pure” hippocampal sclerosis cases (n=33), outside the setting of FTLD-TDP and Alzheimer’s disease (AD). Using a recently described repeat-associated non-ATG (RAN) translation (C9RANT) antibody that was found to be highly specific for c9FTD/ALS, we identified a single “pure” HpScl autopsy case with a repeat expansion in C9ORF72 (c9HpScl). Mutation screening was also performed with repeat-primed polymerase chain reaction and further confirmed with southern blotting. The c9HpScl patient had a 14-year history of a slowly progressive amnestic syndrome and a clinical diagnosis of probable AD. Neuropsychological testing revealed memory impairment, but no deficits in other cognitive domains. Autopsy showed hippocampal sclerosis with TDP-43 immunoreactive neuronal inclusions relatively limited to limbic lobe structures. Neuritic pathology immunoreactive for p62 was more frequent than TDP-43 in amygdala and hippocampus. Frequent p62 positive neuronal inclusions were present in cerebellar granule neurons as is typical of C9ORF72 mutation carriers. There was no significant FTLD or motor neuron disease. C9RANT was found to be sensitive and specific in this autopsy-confirmed series of HpScl cases. The findings in this patient suggest that the clinical and pathologic spectrum of C9ORF72 repeat expansion is wider than frontotemporal dementia and motor neuron disease, including cases of progressive amnestic dementia with restricted TDP-43 pathology associated with HpScl.
Keywords: Hippocampus, C9ORF72, memory, neuropathology, frontotemporal lobar degeneration, C9RANT
INTRODUCTION
Hippocampal sclerosis (HpScl) in the elderly is characterized by selective neuronal loss and gliosis in CA1 sector and subiculum of the hippocampus, often associated with a slowly progressive amnestic syndrome [29]. HpScl is not uncommon in autopsy series of dementia of the elderly [10, 23]. The pathogenesis of HpScl is unknown, but it is hypothesized to be neurodegenerative and is often associated with neuronal inclusions containing TAR DNA binding protein-43 (TDP-43).[3] Neuronal loss in the same distribution can also be seen in patients with hypoxic-ischemic injury, but this is not associated with TDP-43 pathology [3, 14]. HpScl is frequent in other degenerative disorders, especially frontotemporal lobar degeneration with TDP-43 pathology (FTLD-TDP) [13]. When HpScl is the only neuropathologic finding sufficient to cause dementia, such that it cannot be explained by FTLD or Alzheimer’s disease (AD) pathology, it is considered “pure” HpScl [2].
Investigation of genetic correlates of HpScl is a relatively new area of research. Mutations in the gene for progranulin (GRN) cause FTLD-TDP [5], while a genetic variant in the 3’-UTR of GRN in a putative micro-RNA binding site (SNP rs5848, T minor allele) is a risk factor not only for FTLD-TDP [25], but also for HpScl [9]. Pathologically, many individuals harboring GRN mutations have concomitant HpScl pathology [16]. The most common genetic cause of FTLD-TDP has been found to be an expansion of a hexanucleotide repeat (GGGGCC) in a noncoding region of the gene C9ORF72 [8, 26]. The current recommended terminology for frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS) caused by mutations in C9ORF72 is c9FTD/ALS [8]. The clinical presentation of most patients with c9FTD/ALS is that of behavioral variant FTD, FTD with motor neuron disease or ALS, but other clinical phenotypes are also reported (e.g., psychosis or progressive aphasia) [12].
In an autopsy series of c9FTD/ALS, concomitant HpScl was frequent [21]. In addition, these cases presented with the characteristic c9FTD/ALS neuropathologic phenotype, namely, ubiquitin-positive, TDP-43-negative neuronal cytoplasmic inclusions in the cerebellum, hippocampus, and neocortex which are immunoreactive for ubiquitin-binding proteins sequestosome-1/p62 and ubiquilin-2 [6]. Recently, our group and a second independent group, uncovered an unconventional form of translation of the GGGGCC expanded repeat known as repeat-associated non-ATG (RAN) translation which leads to the generation of 3 alternating copolymer peptides [4, 20]. This pathology, termed C9RANT, is highly specific to the central nervous system of c9FTD/ALS [4].
In this study, our goals were to 1) screen pure HpScl cases with C9RANT immunohistochemistry [4] and 2) validate the results with genetic methods [8], in order to identify the frequency of pure HpScl cases harboring the C9ORF72 repeat expansion. The findings presented in this report contribute to increasing evidence that the clinical and pathologic spectrum of disorders in C9ORF72 is wider than previously suggested.
MATERIALS AND METHODS
Case material
The brain bank for neurodegenerative disorders at Mayo Clinic Jacksonville was queried for all cases with a pathologic diagnosis of HpScl with available frozen tissue for DNA extraction (n=280). Cases were excluded if there was a coexisting diagnosis of FTLD (n=73), AD (n=138), Lewy body disease (n=16). We also excluded other significant neurodegenerative diseases, such as progressive supranuclear palsy, multiple system atrophy, and corticobasal degeneration (n=22). A total of 33 cases met inclusion criteria and only one case was found to have a C9ORF72 repeat expansion (c9HpScl). The clinical information of the c9HpScl case was abstracted from the medical records.
Neuropathologic methods
At the time of neuropathologic examinations, brains were submitted for evaluation with the left hemibrain fixed in formalin and the right frozen at −80°. Fixed brain weight was calculated based upon doubling the weight of the left hemibrain. Using standardized dissection and sampling methods described previously [21], tissue samples were processed and embedded in paraffin blocks. The posterior hippocampus was used to evaluate C9RANT and TDP-43 immunoreactivity in all cases. Immunohistochemistry was performed on a DAKO Autostainer (Universal Staining System Carpinteria, California) with the pooled-peptide C9RANT antibody (Rb5823 1:5000, Mayo Clinic antibody)[4] and phospho-Serine 409/410 TDP-43 (1:5000 mouse monoclonal; Cosmo Bio Co., LTD.). Antigen retrieval was performed by steaming in deionized water for thirty minutes. To further characterize the C9ORF72 mutation case, sections of frontal, temporal, parietal and motor cortex, hippocampus, amygdala, basal ganglia, thalamus, midbrain, pons, medulla, and cerebellum were evaluated using additional histologic methods. Additional immunohistochemical evaluations included: IBA1 (1:3000 rabbit anti-ionized calcium binding adaptor molecule 1, Wako Chemicals), Ubi1 (1:40,000 mouse monoclonal anti-ubiquitin, Millipore), p62 (1:250 mouse monoclonal anti-p62/sequestosome; BD Transduction Laboratories), CP13 (1:1000 mouse monoclonal anti phospho-tau, gift from Peter Davies), collagen IV (1:1000 rabbit polyclonal anti-Collagen IV, Rockland Inc.), and alpha-synuclein (1:3000 rabbit polyclonal anti-alpha-synuclein, Mayo Clinic antibody). Steam was used as an antigen retrieval for each of the additional antibodies; except for alpha-synuclein, which was incubating slides for 30 minutes in 98% formic acid, followed by steaming in deionized water for thirty minutes. Senile plaques and neurofibrillary tangles (NFT) were counted and severity of amyloid angiopathy scored with thioflavin S fluorescent microscopy, as previously described [22]. At the time of diagnosis a Braak NFT stage was assigned using thioflavin S [7]. Thal Aβ phase was assigned by retrospectively assessing neuropathologic reports to determine senile plaque distribution on thioflavin S stains of the cortices, posterior hippocampus, basal ganglia, and cerebellum [27]. The maximum Thal Aβ phase was assigned if senile plaques were found in: Phase 1- neocortex (i.e. mid-frontal, inferior parietal, or superior temporal cortex); Phase 2 – entorhinal cortex or CA1/subiculum of the hippocampus; Phase 3 – basal ganglia; Phase 4 – CA4 of the hippocampus; and Phase 5 – cerebellar molecular layer.
The C9RANT antibody detects discrete neuronal inclusions, which were used to screen for C9RANT positivity throughout hippocampal subsectors and medial-temporal cortex. The TDP-43 antibody was used to screen for positivity, as well as classify previously described harmonized TDP-43 subtypes A–C [17].
Genetic methods
Genomic DNA was extracted from frozen brain tissue and screened for the C9ORF72 expanded hexanucleotide repeat using repeat-primed PCR and southern blotting techniques as previously described [8]. Briefly, an initial assay of GGGGCC hexanucleotide repeat expansion was amplified for each case (n=33) using one fluorescently labeled PCR primer. Next, using an automated ABI3730 DNA analyzer, a fragment length analysis was performed using GeneMapper software (Applied Biosystems). If a case appears homozygous in the initial assay, they were then analyzed using a repeat primed PCR method. An individual with a repeat expansion will show a characteristic stutter amplification pattern on electropherogram.
Results
Pathogenetic screen
Of the 280 cases identified, 33 cases (12%) were not found to have coexisting FTLD, AD, or other significant neurodegenerative diseases. The demographics and pathologic information are summarized in Table 1. Of the final 33 HpScl there were 22 TDP-43 positive cases, 10 TDP-43 negative cases, and one case that lacked fixed tissue. Regardless of TDP-43 positivity, each case was screened with C9RANT in the hippocampus. One case was found to have C9RANT positivity with the highest burden of inclusions found in the dentate fascia and endplate (CA4). Occasional neuronal cytoplasmic inclusions (NCI) and “pre-inclusions” with granular somatodendritic C9RANT immunoreactivity could be found in the medial temporal lobe cortex (Fig. 1a, b).
Table 1.
Summary of demographics and neuropathology in hippocampal sclerosis
| c9HpScl | HpScl TDP-43 positive | HpScl TDP-43 negative | |
|---|---|---|---|
| Sample size, n. | 1 | 21 | 10 |
| Sex, Males (%) | 1 (100) | 9 (43) | 3 (30) |
| Age at death, years | 76 | 91 (87, 94) | 88 (84, 93) |
| Brain weight, g | 1140 | 1160 (1095, 1220) | 1091 (978, 1250) |
| Braak NFT stage | III | II (II, III) | III (I, IV) |
| Thal amyloid phase | 0 | 2 (2, 3) | 2 (0, 2) |
| TDP-43 positivity | Positive | Positive | Negative |
| Type A (1), n. (%) | -- | 17 (81) | -- |
| Type B (3), n. (%) | 1 (100) | 4 (19) | -- |
| Type C (2), n. (%) | -- | -- | -- |
All data is presented as median (25th, 75th interquartile range) unless noted otherwise. One case lacked fixed tissue, therefore could not be evaluated for TDP-43 and is not presented in this table. Acronyms: HpScl - hippocampal sclerosis, TDP-43 – TAR DNA binding protein-43, g – grams.
Figure 1.

C9RANT immunohistochemistry in the hippocampus (A) shows the highest number of neuronal inclusions (arrows) in the dentate fascia. (B) Occasional neuronal cytoplasmic inclusions (NCI, inset) and “pre-inclusions” with granular somatodendritic C9RANT immunoreactivity (inset) could be found in the medial temporal lobe cortex. TDP-43 immunohistochemistry in the CA1 sector of the hippocampus shows (C) synaptic or fine neurites in areas of relative neuronal preservation, as well as (D) TDP-43-positive perivascular glial inclusions. They can be seen as a bi-lobed inclusion (inset) or as dense perivascular deposits (arrowhead). (bar 50μm A–D, insets 100μm)
Blinded to the C9RANT results, we found one out of the 33 cases with the characteristic stutter pattern in the repeat-primed PCR assay, indicative of an expansion in C9ORF72 (Fig. 2a). Southern blot using DNA extracted from cerebellum was performed to confirm the presence of a pathogenic repeat expansion, which highlighted a smear of high-molecular weight bands (Fig. 2b). The C9RANT antibody identified the same case that was found to have a C9ORF72 repeat expansion using genetic methods. In this series of HpScl cases that lacked neuropathology consistent with FTLD or advanced AD diagnoses, the C9RANT antibody was 100% sensitive and specific.
Figure 2.

(A) The electropherogram plot of repeat-primed PCR reaction products zoomed to 2,000 relative fluorescence units to show stutter amplification. Our case and a control (C9ORF72-negative) with HpScl are graphically represented in the upper and lower panel, respectively. (B) Southern blotting of two expanded repeat carriers and one noncarrier using genomic DNA extracted from cerebellum. Lane 1 shows DIG-labeled DNA Molecular Weight Marker II (Roche) with fragments of 2,027; 2,322; 4,361; 6,557; 9,416; 23,130 bp. Lane 2 shows DIG labeled DNA Molecular Weight Marker VII (Roche) with fragments of 1,882; 1,953; 2,799; 3,639; 4,899; 6,106; 7,427; and 8,576 bp. Lane 3 shows a positive control using DNA extracted from an expanded repeat lymphoblast cell line shows the expected 2.3 kb wild-type allele with expanded repeats showing an additional allele around 5.5 kb (arrow). The HpScl noncarrier control only shows the 2.3 kb wild-type allele (lane 4), while the chromosome 9 frontotemporal dementia positive-control and chromosome 9 HpScl case show an additional allele from at around 8.5 kb (lanes 5 & 6). Note the smear of high molecular weight bands in DNA extracted from cerebellum suggesting somatic instability of the repeat.
Clinicopathological Summary
Here, we give a clinicopathologic summary of the c9HpScl case identified in the pathogenetic screen. The c9HpScl case was a 76-year-old man with a final diagnosis prior to death of clinically probable AD, initially evaluated in 1995 at 72-years-of-age with a 10-year history of slowly progressive episodic memory impairment (age of onset = 62 years). The wife reported no change in personality or behavior, “He was socially active and plays piano, bridge, reads, walks, and shoots pool”. His mother had slowly progressive amnestic dementia with onset in her 70s and death at age 96. There was no other family history of dementia or ALS. His past medical history was notable for coronary artery disease with a 4-vessel coronary artery bypass graft in 1987. Cranial nerves were unremarkable; his palate moved symmetrically; and his tongue was midline. Although there were no EMG studies conducted, motor testing revealed preserved strength in upper and lower extremities. Word fluency was within normal limits, naming 13 words with the letter “F.” A Mini-Mental State Examination [11] revealed difficulties with the date, month, and day of the week. He also had mild difficulties with recall and scored a total of 24 out of 30. There were no signs of paranoia or psychosis. Neuropsychological evaluation showed executive/frontal functioning within normal limits and severe impairment in memory, especially for delayed recall (Table 2). Specific neuropsychological tests designed for the diagnosis of FTD were not conducted. Magnetic resonance imaging in 1995 showed diffuse cerebral volume loss and white matter changes consistent with age. The patient was further lost to follow-up in the five years preceding his death.
Table 2.
Neuropsychological evaluation of C9ORF72-associated HpScl case (September 1995)
| Test/Subtest | Percentile | Score |
|---|---|---|
| Wechsler Adult Intelligence Scale – Revised* | ||
| Digit span | 98 | 16 |
| Vocabulary | 95 | 15 |
| Comprehension | 84 | 13 |
| Similarities | 75 | 12 |
| Block design | 37 | 9 |
| Digit symbol | 25 | 8 |
| Wechsler Memory Scale – Revised | ||
| Verbal Memory | 2 | 70 |
| Visual Memory | 7 | 77 |
| General Memory | 1 | 68 |
| Attention/Concentration | 91 | 120 |
| Delayed Recall | 1 | 67 |
| Controlled Word Association Test | ||
| Verbal Fluency | 60 | N/A |
| Trail Making Test B | 75 | N/A |
| Beck Depression Inventory | N/A | 4 |
Acronyms: HpScl – hippocampal sclerosis, n/a – test not available in clinical records.
The calculated fixed brain weight was 1140 grams (Table 1). There was mild cortical atrophy and ventricular dilation, especially the temporal horn of the lateral ventricle. The hippocampal formation, but not the amygdala was atrophic (Fig. 3a). The cerebellum was unremarkable. The substantia nigra and locus coeruleus had visible neuromelanin pigment. Microscopic inspection of the substantia nigra and locus coeruleus showed a normal neuronal population with no NFTs or Lewy bodies. There was severe neuronal loss and gliosis that was selective for CA1, with less neuronal loss in the subiculum and preservation of CA2/3, endplate and dentate fascia (Fig. 3d). Senile plaques were not detected in any cortical region, hippocampus, amygdala, or cerebellum. There were only a few isolated NFTs in the frontal and temporal cortices and in the hippocampus and amygdala, but more numerous NFTs in the entorhinal cortex. As expected, no NFT pathology was found in the cerebellum. Mild amyloid angiopathy was detected in frontal and occipital cortices. Phospho-TDP-43 immunohistochemistry showed NCIs in the dentate gyrus of the hippocampus (Fig. 3b), but very sparse NCIs in frontal and temporal cortices and no significant cortical neuronal loss or gliosis. There were sparse TDP-43-positive dystrophic neurites and no neuronal intranuclear inclusions (NII) in the hippocampus, consistent with Type B [17]. The presence of hippocampal synaptic or fine neurites in the CA1 (Fig. 1c) was also noted in areas of relative neuronal preservation, a previously reported feature of many C9ORF72-associated FTLD cases [21]. TDP-43-positive perivascular glial inclusions (Fig. 1d), considered to be abnormal inclusions in astrocytic end-feet processes [15], were present in the hippocampus and entorhinal cortex, but very sparse in frontal and temporal cortices, nucleus accumbens, and amygdala. No evidence of motor neuron degeneration was present. We did not find TDP-43 pathology, neuronal loss, or Bunina bodies in the spinal cord.
Figure 3.

(A) Macroscopic findings in c9HpScl shows atrophic hippocampus (arrow) in the CA1 region. (B) TDP-43 staining of the hippocampus shows few neuronal cytoplasmic inclusions (NCIs) and no intranuclear inclusions (NIIs). (C) p62 immunohistochemistry of the same region shows many NCIs and scattered NIIs in dentate fascia. (D) An H&E of the anterior hippocampus shows focal CA1 and subiculum pathology, while CA2/3 pyramidal neurons remain resistant (arrow shows transition to neuronal dropout). Macroscopic findings correlate with underlying neuronal loss and gliosis (inset shows higher magnification of neuronal loss and gliosis characteristic of HpScl in of CA1). (bar 45μm for B&C at 40× magnification, 1150μm for D at 1× magnification, and 90μm for inset at 20× magnification)
In addition to TDP-43 positive pathology, there were also TDP-43-negative, p62/sequestosome-1-positive NCIs and dystrophic neurites (Fig. 3c). In all cortical and subcortical regions evaluated, p62-positive pathologic inclusions were more numerous than TDP-43-positive inclusions, as reported recently by others in c9FTD/ALS [1]. TDP-43 pathology was absent in the cerebellum, but C9RANT immunopositivity showed many NCIs in granule cells and occasionally in Purkinje cells (Fig. 4 a, b). As in other previously reported C9ORF72 repeat expansion cases [1], p62 and ubiquitin immunohistochemistry revealed NCIs in both granule cells and Purkinje cells of the cerebellum (Fig. 4 c, d).
Figure 4.
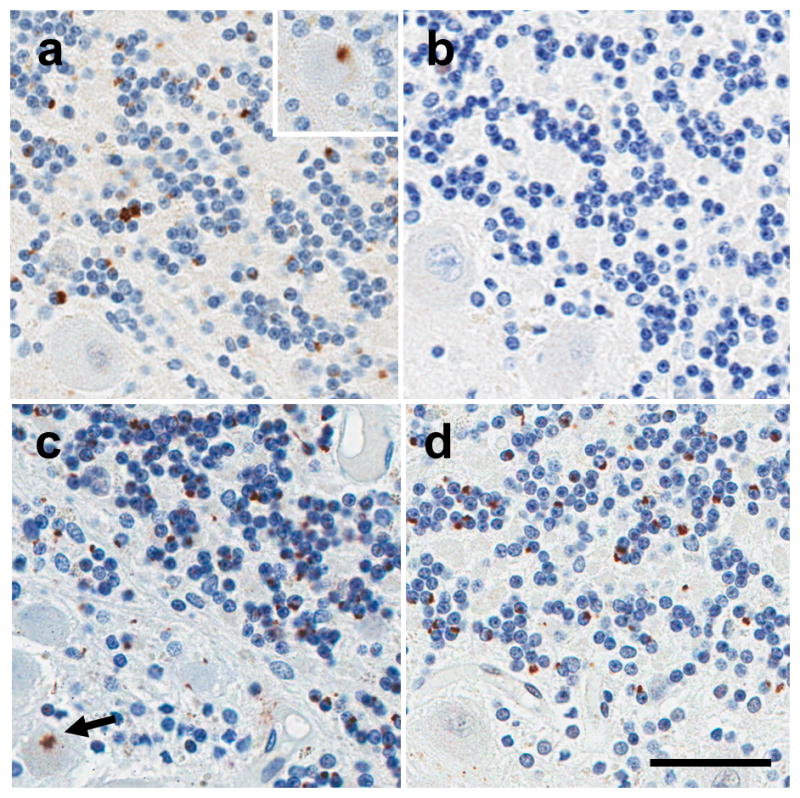
Immunohistochemistry of the cerebellum in c9HpScl shows (A) C9RANT immunopositivity shows many NCIs in granule cells, although occasional purkinje cells can be observed (inset). As expected (B), the cerebellum lacks TDP-43 pathology. (C) p62 immunohistochemistry shows many NCIs and occasional NIIs of the granule cell layer. A star-shaped NCI (arrow) was observed in the purkinje cell layer of the cerebellum with p62 staining, but not with (D) Ub1 staining. Ub1-positive NCIs were seen at moderate frequency, and no NII were observed. (bar 45μm)
Discussion
This study identified a hippocampal sclerosis case with an expansion in C9ORF72 that lacked FTLD, AD, or other significant neuropathology. The case was identified using a recently described antibody generated against an unconventional mechanism of translation-repeat-associated non-ATG translation that was validated using repeat primed PCR and southern blotting techniques, which was sensitive and specific in this series of HpScl cases. The clinical phenotype in the c9HpScl patient, namely, slowly progressive amnestic syndrome without behavioral or motor neuron signs, is distinctly different from the most common presentations in c9FTD/ALS [12]. Although the final clinical phenotype of this individual was unknown in the five years prior to his death, it is possible he developed FTD, but unlikely given the slowly progressive nature of the disease course and sparing of frontal cognitive domains at the last neuropsychological examination (i.e. attention/concentration). HpScl is more apt to be clinically misdiagnosed as AD than behavioral variant FTD [24, 29]. Pathologically, there was selective neuronal loss and gliosis in the hippocampus consistent with HpScl of the elderly, which fits well with the clinical presentation [23, 29]. TDP-43 inclusions were most abundant in limbic structures and sparse in the cortex. The cortical lesions were not associated with cortical neuronal loss or gliosis. Immunohistochemistry for ubiquitin and p62 in C9ORF72-associated HpScl of the elderly show that pathology can be more widespread than previously suspected based upon TDP-43 immunohistochemistry, with NCIs even in the cerebellar cortex. The greater propensity of p62 pathology compared to TDP-43 pathology in C9ORF72-associated HpScl suggests that TDP-43 may not be central to the C9ORF72 disease mechanism, but rather another unknown polyubiquitinated substrate bound by p62.
In our previous clinicopathologic characterization of c9FTD/ALS, we reported that HpScl was common in c9FTLD-TDP (12 of 15), and some patients in this series also presented with amnestic dementia that lead to clinical consideration of AD [21]. A large series of familial late-onset clinical AD patients screened for mutations in C9ORF72, found 6 of 771 patients with mutations [18]. Postmortem analysis of two patients showed pathology consistent with FTLD, with inconsistent concurrent Alzheimer type neuropathology. There is no mention in this report about whether the two patients with C9ORF72 mutations and Alzheimer-like clinical presentation had HpScl. The reported frequency of 0.8% clinical AD patients with C9ORF72 mutations was consistent with a recent report on a clinical AD patient series from our group, which found 0.9% of AD patients with an expansion mutation [28]. The frequency of C9ORF72 in our pathologically-confirmed HpScl series is 3.0% if all cases are considered and 4.5% if only TDP-43 positive cases are included.
Mutations in C9ORF72 have been reported in seemingly sporadic cases of FTLD and ALS [8]. The family history of this patient included amnestic disorder in her mother (presenting with symptoms in her 70s), but there was no history of dementia or motor neuron disease in other family members. Although there was no autopsy confirmation, the patient’s mother died in her late 90s, which is more than a decade later than our patient. While it is impossible to make any conclusions from these two individuals, it is of interest that the possibility of anticipation, with earlier age of onset in subsequent generations, has been raised in other c9FTD/ALS series [12].
The pathogenesis of HpScl in our patient is unknown, but most evidence favors a neurodegenerative disease process. He had no history of seizures. He had coronary artery disease and had a 4-vessel coronary artery bypass graft, but there was no history of cognitive deficits after surgery. The presence of TDP-43 and p62 NCIs and dystrophic neurites in the hippocampus and to a lesser extent in other brain regions supports a neurodegenerative origin for HpScl. It is of some interest that microvascular TDP-43-related pathology was also present [15]. The significance of TDP-43 inclusions in astrocytic end-feet of microvessels remains to be determined; however, we speculate that this pathology may compromise microvascular function and contribute to neuronal loss in regions known to be vulnerable to hypoxic-ischemic injury.
This case adds increasing evidence that the clinical and pathologic spectrum of disorders associated with mutations in C9ORF72 is wider than merely FTD and ALS. Screening for mutations in C9ORF72 should be considered in patients with slowly progressive amnestic dementia who have a positive family history, especially if other biomarkers (e.g., apolipoprotein E genotype, neuroimaging, or cerebrospinal fluid analyses) do not support a diagnosis of AD [19]. Further studies are needed to identify the underlying molecular mechanisms responsible for clinical and pathologic heterogeneity of c9FTLD/ALS.
Acknowledgments
We are grateful to all patients, family members, and caregivers who agreed to brain donation, without which these studies would have been impossible. We also acknowledge expert technical assistance of Linda Rousseau and Virginia Phillips for histology, and Monica Castanedes-Casey for immunohistochemistry. We would like to thank Martha Purdy, John Gonzalez, and Beth Marten for brain bank coordination. This research was funded by Mayo Foundation (Jacoby Professorship of Alzheimer Research); National Institutes of Health (P50-AG16574, P50-NS72187, P01-AG03949, R01-AG37491, R01-NS080882 and R01-AG026251); The ALS Therapy Alliance; and the State of Florida Alzheimer Disease Initiative. MEM is supported by a fellowship from the Robert and Clarice Smith and Abigail Van Buren Alzheimer’s Disease Research Program.
Footnotes
Competing Interests
Dr. Rademakers and Mrs. Dejesus-Hernandez have a patent on the clinical testing and therapeutic intervention for the hexanucleotide repeat expansion of C9ORF72.
References
- 1.Al-Sarraj S, King A, Troakes C, et al. p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathologica. 2011;122:691–702. doi: 10.1007/s00401-011-0911-2. [DOI] [PubMed] [Google Scholar]
- 2.Ala TA, Beh GO, Frey WH., 2nd Pure hippocampal sclerosis: a rare cause of dementia mimicking Alzheimer's disease. Neurology. 2000;54:843–848. doi: 10.1212/wnl.54.4.843. [DOI] [PubMed] [Google Scholar]
- 3.Amador-Ortiz C, Ahmed Z, Zehr C, Dickson DW. Hippocampal sclerosis dementia differs from hippocampal sclerosis in frontal lobe degeneration. Acta Neuropathologica. 2007;113:245–252. doi: 10.1007/s00401-006-0183-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ash PE, Bieniek KF, Gendron TF, et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013;77:639–646. doi: 10.1016/j.neuron.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baker M, Mackenzie IR, Pickering-Brown SM, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–919. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- 6.Bieniek KF, Murray ME, Rutherford NJ, et al. Tau pathology in frontotemporal lobar degeneration with C9ORF72 hexanucleotide repeat expansion. Acta Neuropathol. 2013;125:289–302. doi: 10.1007/s00401-012-1048-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 8.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dickson DW, Baker M, Rademakers R. Common variant in GRN is a genetic risk factor for hippocampal sclerosis in the elderly. Neurodegener Dis. 2010;7:170–174. doi: 10.1159/000289231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dickson DW, Davies P, Bevona C, et al. Hippocampal sclerosis: a common pathological feature of dementia in very old (> or = 80 years of age) humans. Acta Neuropathologica. 1994;88:212–221. doi: 10.1007/BF00293396. [DOI] [PubMed] [Google Scholar]
- 11.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 12.Hodges J. Familial frontotemporal dementia and amyotrophic lateral sclerosis associated with the C9ORF72 hexanucleotide repeat. Brain. 2012;135:652–655. doi: 10.1093/brain/aws033. [DOI] [PubMed] [Google Scholar]
- 13.Josephs KA, Jones AG, Dickson DW. Hippocampal sclerosis and ubiquitin-positive inclusions in dementia lacking distinctive histopathology. Dement Geriatr Cogn Disord. 2004;17:342–345. doi: 10.1159/000077168. [DOI] [PubMed] [Google Scholar]
- 14.Lee EB, Lee VM, Trojanowski JQ, Neumann M. TDP-43 immunoreactivity in anoxic, ischemic and neoplastic lesions of the central nervous system. Acta Neuropathologica. 2008;115:305–311. doi: 10.1007/s00401-007-0331-5. [DOI] [PubMed] [Google Scholar]
- 15.Lin WL, Castanedes-Casey M, Dickson DW. Transactivation response DNA-binding protein 43 microvasculopathy in frontotemporal degeneration and familial Lewy body disease. J Neuropathol Exp Neurol. 2009;68:1167–1176. doi: 10.1097/NEN.0b013e3181baacec. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mackenzie IR, Baker M, Pickering-Brown S, et al. The neuropathology of frontotemporal lobar degeneration caused by mutations in the progranulin gene. Brain. 2006;129:3081–3090. doi: 10.1093/brain/awl271. [DOI] [PubMed] [Google Scholar]
- 17.Mackenzie IR, Neumann M, Baborie A, et al. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 2011;122:111–113. doi: 10.1007/s00401-011-0845-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Majounie E, Abramzon Y, Renton AE, et al. Repeat expansion in C9ORF72 in Alzheimer's disease. N Engl J Med. 2012;366:283–284. doi: 10.1056/NEJMc1113592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:263–269. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mori K, Weng SM, Arzberger T, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339:1335–1338. doi: 10.1126/science.1232927. [DOI] [PubMed] [Google Scholar]
- 21.Murray ME, DeJesus-Hernandez M, Rutherford NJ, et al. Clinical and neuropathologic heterogeneity of c9FTD/ALS associated with hexanucleotide repeat expansion in C9ORF72. Acta Neuropathologica. 2011;122:673–690. doi: 10.1007/s00401-011-0907-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murray ME, Graff-Radford NR, Ross OA, et al. Neuropathologically defined subtypes of Alzheimer's disease with distinct clinical characteristics: a retrospective study. Lancet Neurol. 2011;10:785–796. doi: 10.1016/S1474-4422(11)70156-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nelson PT, Schmitt FA, Lin Y, et al. Hippocampal sclerosis in advanced age: clinical and pathological features. Brain. 2011;134:1506–1518. doi: 10.1093/brain/awr053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pao WC, Dickson DW, Crook JE, et al. Hippocampal sclerosis in the elderly: genetic and pathologic findings, some mimicking Alzheimer disease clinically. Alzheimer Dis Assoc Disord. 2011;25:364–368. doi: 10.1097/WAD.0b013e31820f8f50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rademakers R, Eriksen JL, Baker M, et al. Common variation in the miR-659 binding-site of GRN is a major risk factor for TDP43-positive frontotemporal dementia. Hum Mol Genet. 2008;17:3631–3642. doi: 10.1093/hmg/ddn257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thal DR, Rub U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791–1800. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- 28.Wojtas A, Heggeli KA, Finch N, et al. C9ORF72 repeat expansions and other FTD gene mutations in a clinical AD patient series from Mayo Clinic. Am J Neurodegener Dis. 2012;1:107–118. [PMC free article] [PubMed] [Google Scholar]
- 29.Zarow C, Sitzer TE, Chui HC. Understanding hippocampal sclerosis in the elderly: epidemiology, characterization, and diagnostic issues. Curr Neurol Neurosci Rep. 2008;8:363–370. doi: 10.1007/s11910-008-0057-3. [DOI] [PubMed] [Google Scholar]