

Abstract
The high mortality rates associated with ovarian cancer are largely due to a lack of highly effective treatment options for advanced stage disease; a time when initial diagnosis most commonly occurs. Recent evidence suggests that the steroid hormone, progesterone, may possess anti-tumorigenic properties. With the discovery of a new class of membrane-bound progesterone receptors (mPRs) belonging to the progestin and adipoQ receptor (PAQR) gene family in the ovary, there are undefined mechanisms by which progesterone may inhibit tumor progression. Therefore, our goal was to define potential mPR-dependent signaling mechanisms operative in ovarian cancer cells. We detected abundant mPRα (PAQR7), mPRβ (PAQR8), and mPRγ (PAQR5), but not classical nuclear PR (A or B isoforms) mRNA expression and mPRα protein expression in a panel of commonly used ovarian cancer cell lines. In contrast to mPR action in breast cancer cells, progesterone alone failed to induce changes in cyclic adenosine monophosphate (cAMP) levels in ovarian cancer cells. However, progesterone enhanced cAMP production by β1,2-adrenergic receptors and increased isoproterenol-induced transcription from a cAMP response element (CRE)-driven reporter gene. Independently of β-adrenergic signaling, we additionally observed activation of both JNK1/2 and p38 MAPK in response to progesterone alone. This finding was supported by the results of a screen for potential mPR gene targets. Progesterone induced a significant increase in transcription of the pro-apoptotic marker BAX, whose activity and expression has been linked to JNK1/2 and p38 signaling. Inhibitors of JNK, but not p38, blocked progesterone-induced BAX expression. Taken together, these observations implicate at least two distinct signaling pathways that may be utilized by mPRs in ovarian cancer cells that exhibit regulatory genomic changes. These studies on mPR signaling in ovarian cancer lay the foundation for future work aimed at understanding how progesterone exerts its anti-tumorigenic effects in the ovary and suggest that pharmacologic activation of mPRs, abundantly expressed in ovarian cancers, may provide a new treatment option for patients with advanced stage disease.
Keywords: Ovarian cancer, Progesterone, Membrane progesterone receptor (mPR), PAQR, cAMP, Jun kinase (JNK), p38 Mitogen-activated protein kinase, Apoptosis
Introduction
Ovarian cancer is the most lethal form of gynecologic malignancy, where US projections for 2009 alone estimated 14,600 ovarian cancer-related deaths compared to 21,550 newly diagnosed cases [1]. Thus, while ovarian cancer is only the ninth most common cancer diagnosed in women, it ranks fifth in terms of total cancer-related deaths [1]. Such high mortality is largely due to the fact that most ovarian cancers (approximately 70%) are diagnosed at an advanced stage where 5-year survival rates drop to 30% compared to 90% for women with localized disease [2]. The relative ineffectiveness of current therapy for advanced disease is complicated by a poor understanding of the etiology and molecular mechanisms governing ovarian cancer progression [3]. However, clinical observations made within the past 15 years suggest that the ovarian steroid hormone progesterone may play a protective role against ovarian cancer occurrence and progression.
Clinical studies have shown that multiparity, twin pregnancy, and pregnancy that occurs later in life all dramatically increase circulating progesterone levels and reduce a woman’s lifetime risk of developing ovarian cancer [4–7]. In contrast, the likelihood of developing ovarian cancer dramatically increases in women with inherent progesterone deficiency and after menopause when progesterone production decreases [8, 9]. Furthermore, multiple groups have shown that estrogen-progestin containing oral contraceptives are associated with a reduced incidence of ovarian cancer, which Rodriguez et al. demonstrate is mainly due to the effects of the progestin component [10–16]. However, while it is believed that much of the protective effects of progesterone are mediated by the nuclear progesterone receptor (n-PR), it is also well known that the expression of these receptors is progressively lost with increasing degrees of ovarian cancer malignancy [17–20]. Therefore, we focused our studies on the expression and function of a newly defined class of membrane-bound progesterone receptors (mPRs) in ovarian cancer cell line models [21].
A membrane progesterone receptor, mPRα (also known as PAQR7), was originally identified in seatrout ovaries as a novel ∼40 kDa seven transmembrane-spanning protein, and subsequently two other membrane progestin receptors, mPRβ (a.k.a. PAQR8) and mPRγ (a.k.a. PAQR5), were identified in other species belonging to the larger progestin and adipoQ receptor (PAQR) gene family [22–24]. The mPRs activate G proteins but are not members of the G protein-coupled receptor (GPCR) superfamily [23]. Typically, mPRs couple to the inhibitory G protein, 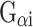 [24]. Stimulation of mPRs with endogenous progesterone has shown an ability of mPRs to decrease cyclic adenosine monophosphate (cAMP) synthesis, which is sensitive to pertussis toxin inhibition, and increase the activity of mitogen-activated protein kinases (MAPKs) [23, 25]. However, more recent studies have also linked mPRα to a stimulatory G protein, Golf, responsible for increasing cAMP levels in sperm cells of the Atlantic croaker [26]. These receptors bind progesterone with high affinity (K
d = 4.2–7.8 nM) and do not interact with the synthetic progestin R5020 or the n-PR antagonist RU486 [24]. The mPRs (α, β, and γ) are expressed in mammalian ovaries and their abundance changes during pregnancy, suggesting they have important roles in ovarian physiology [22, 27–29]. Originally seen in breast cancer biopsies and cell lines, mPR expression has also recently been observed in human ovarian cancer specimens originating from different histologic subtypes [30]. Therefore, the goal of our studies was to define the impact of progesterone stimulation on mPR signaling in n-PR-null ovarian cancer cell line models of advanced stage disease.
[24]. Stimulation of mPRs with endogenous progesterone has shown an ability of mPRs to decrease cyclic adenosine monophosphate (cAMP) synthesis, which is sensitive to pertussis toxin inhibition, and increase the activity of mitogen-activated protein kinases (MAPKs) [23, 25]. However, more recent studies have also linked mPRα to a stimulatory G protein, Golf, responsible for increasing cAMP levels in sperm cells of the Atlantic croaker [26]. These receptors bind progesterone with high affinity (K
d = 4.2–7.8 nM) and do not interact with the synthetic progestin R5020 or the n-PR antagonist RU486 [24]. The mPRs (α, β, and γ) are expressed in mammalian ovaries and their abundance changes during pregnancy, suggesting they have important roles in ovarian physiology [22, 27–29]. Originally seen in breast cancer biopsies and cell lines, mPR expression has also recently been observed in human ovarian cancer specimens originating from different histologic subtypes [30]. Therefore, the goal of our studies was to define the impact of progesterone stimulation on mPR signaling in n-PR-null ovarian cancer cell line models of advanced stage disease.
Materials and Methods
Cell Culture
All cell lines were grown at 37°C under 5% CO2 in water-jacketed incubators (Forma Scientific, Marietta, OH). MCF-7 cells were kindly provided by Dr. Douglas Yee (University of Minnesota). 1816-575 cells were kindly provided by Dr. Patricia Kruk (University of South Florida). HEY, CAOV-3, and ES-2 cells were kindly provided by Dr. Amy Skubitz (University of Minnesota). OVCAR-3, OVCAR-5, and SKOV-3 cells were kindly provided by Dr. Sundaram Ramakrishnan (University of Minnesota). TOV-21 G cells (Cat. No. CRL-11730™) and TOV-112D cells (Cat. No. CRL-11731™) were purchased from American Type Culture Collection (ATCC®, Manassas, VA). The MCF-7 breast cancer cell line was maintained in DMEM cell culture media (cellgro®, Manassas, VA; 10-013-CV) supplemented with 5% FBS and 1% penicillin/streptomycin (GIBCO®, Carlsbad, CA; 15140122). The SKOV-3 ovarian cancer cell line was maintained in DMEM cell culture media (cellgro®, Manassas, VA; 10-013-CV) supplemented with 10% FBS and 1% penicillin/streptomycin. The ES-2 ovarian cancer cell line was maintained in McCoy’s 5A cell culture media (cellgro®, Manassas, VA; 10-050-CV) supplemented with 15% FBS and 1% penicillin/streptomycin. The OVCAR-3 ovarian cancer cell line was maintained in RPMI 1640 cell culture media (GIBCO®, Carlsbad, CA; 11875-093) supplemented with 15% FBS and 1% penicillin/streptomycin. With the exception of the experiments performed on growing cells in Fig. 1a and b, 24 h prior to all experiments, cells were washed with 1X PBS and placed in Modified IMEM (GIBCO®, Carlsbad, CA; A10488) supplemented with 1% charcoal-stripped serum (i.e., DCC; Hyclone, Fremont, CA; SH30068.03) in which the remainder of the experiment was carried out.
Fig. 1.

Ovarian cancer cell lines express membrane progesterone receptor (mPR) mRNA and protein. a qPCR identified and compared gene expression levels of each mPR isoform (α, β, and γ) in a panel of ovarian cancer cell lines relative to control MCF-7 breast cancer cells (mean ± SD, n = 3). b Immunoblot analysis defined protein expression of mPRα in the SKOV-3 and ES-2 ovarian cancer cell lines relative to control MCF-7 breast cancer cells. SKOV-3 and ES-2 cells do not express nuclear progesterone receptor (n-PR-A or -B) protein. c OVCAR-3 ovarian cancer cells transiently transfected with a n-PR-specific PRE-luciferase reporter construct only respond to stimulation with the n-PR specific ligand R5020 (10 nM) for 18 h when co-transfected with a n-PR-B overexpression construct (mean ± SD, n = 3; NS, not significant; asterisk, p ≤ 0.05; all firefly luciferase values are individually normalized to the expression of a Renilla-based luciferase reporter construct serving as an internal control)
Western Blotting
Total protein was extracted from whole cell lysates with a modified RIPA extraction buffer containing PMSF (1 mM), NaF (5 mM), Na3VO4 (10 mg/mL), β-glycerophosphate (25 mM), β-ME (14.3 mM), aprotonin (11,140 KIU/mL), and one Complete Mini-Protease Inhibitor Cocktail tablet (Roche, Indianapolis, IN; 1 836 153 001). Protein concentration was quantified with Bio-Rad Protein Assay (Bio-Rad, Hercules, CA; 500-0001), and protein was transferred to Immobilon-P™ PVDF membranes (Millipore, Billerica, MA; IPVH00010) after fractionation by 10% SDS-PAGE (20 μg of total protein for Fig. 1b and 30 μg of total protein for Fig. 3). All Western blots were blocked for 1 h at RT in phosphate-buffered saline/0.1% Tween-20 (PBS-T) containing 5% dried milk (Sanalac, Middleton, WI). mPRα Western blots were probed overnight at 4°C in PBS containing 5% milk using anti-human mPRα primary antibody (kindly provided by Dr. Peter Thomas as previously described [24, 25]). All other Western blots were probed overnight at 4°C in PBS-T containing 1% milk using the following primary antibodies: n-PR-A/B Ab-8 (NeoMarkers, Fremont, CA; MS-298-P), phospho-SAPK/JNK (Thr183/Tyr185; Cell Signaling Technology, Danvers, MA; 9255), SAPK/JNK (Cell Signaling Technology, Danvers, MA; 9258), phospho-p38 MAPK (Thr180/Tyr182; Cell Signaling Technology, Danvers, MA; 9211), p38 MAPK (Cell Signaling Technology, Danvers, MA; 9212), and Actin (Sigma–Aldrich, St. Louis, MO; A 4700). HRP-conjugated goat anti-rabbit (Bio-Rad, Hercules, CA; 170-6515) and rabbit anti-mouse (Bio-Rad, Hercules, CA; 170-6516) secondary antibodies were used to detect their respective primary antibodies, and immunoreactive proteins were visualized on radiographic film (Kodak, Rochester, NY) following ECL detection with Super Signal® West Pico Maximum Sensitivity Substrate (Pierce, Rockford, IL; 34080).
Fig. 3.

Stimulation of the mPR with progesterone increases JNK1/2 and p38 MAPK activity. Immunoblot analysis of SKOV-3 cells stimulated with progesterone (100 or 1,000 nM) demonstrated a time- and dose-dependent increase in phosphorylation of both JNK isoforms (p-JNK1 and p-JNK2) as well as p38 (p-p38). Total JNK1/2 (t-JNK1 and t-JNK2) and total p38 (t-p38) expression was unchanged with progesterone treatment
Quantitative PCR
Total RNA was extracted from whole cells using TRIzol® (Invitrogen, Carlsbad, CA; 15596-018) separation and isopropanol precipitation. A 1.0 μg of RNA was reverse transcribed to cDNA using random hexameric primers, dNTP nucleotides, RNaseOUT™ and SuperScript™ II reverse transcriptase (Invitrogen, Carlsbad, CA; 11904-018) as described by Invitrogen. Quantitative PCR (qPCR) was performed using Light Cycler® FastStart DNA Master SYBR Green I (Roche, Indianapolis, IN; 03 515 885 001) on a Mastercycler ep realplex4 S qPCR platform (eppendorf, Hauppage, NY; 63002 000.601). Primers (from Integrated DNA Technologies, Coralville, IA) were specific for h-mPRα (5′-cgctcttctggaagccgtacatctatg-3′ and 5′-cagcaggtgggtccagacattcac-3′), h-mPRβ (5′-agcctcctacatagatgctgccc-3′ and 5′-ggtgcctggttcacatgttcttca-3′), and h-mPRγ (5′-cagctgtttcacgtgtgtgtgatcctg-3′ and 5′-gcacagaagtatggctccagctatctgag-3′). qPCR cycling conditions used for the mPR isoforms were as follows: initial denature at 95°C for 10 min; 40 X’s: denature at 95°C for 10 s, anneal at 55°C for 10 s, and extension at 72°C for: 5 s (mPR-α), 10 s (mPR-β), and 8 s (mPR-γ). Primers were specific for h-n-PR-A/B (5′-aaatcattgccaggttttcg-3′ and 5′-tgccacatggtaaggcataa-3′), whose qPCR cycling conditions were as follows: initial denature at 95°C for 10 min; 40 X’s: denature at 95°C for 10 s, anneal at 54°C for 10 s, and extension at 72°C for 11 s. Primers were specific for h-BAX (5′-tctgacggcaacttcaactg-3′ and 5′-ttgaggagtctcacccaacc-3′), whose qPCR cycling conditions were as follows: initial denature at 95°C for 10 min; 35 X’s: denature at 95°C for 10 s, anneal at 58°C for 10 s, and extension at 72°C for 11 s. All qPCR results were normalized to expression of the housekeeping genes h-GAPDH (5′-ttgttgccatcaatgaccc-3′ and 5′-catcgccccacttgattt-3′) or h-β-actin (5′-tcagaaggattcctatgtgggc-3′ and 5′-atcttctcgcggttggcctt-3′). Conditions for h-GAPDH qPCR were as follows: initial denature at 95°C for 10 min; 35 X’s: denature at 95°C for 10 s, anneal at 52°C for 10 s, and extension at 72°C for 11 s. Conditions for h-β-actin qPCR were as follows: initial denature at 95°C for 10 min; 35 X’s: denature at 95°C for 10 s, anneal at 60°C for 10 s, and extension at 72°C for 11 s.
cAMP EIA
SKOV-3 cells were plated at a density of 100,000 cells/100 mm plate and the following day they were starved for 24 h in 1% DCC (as described earlier). Two days after plating, cells were treated for 10 min as described in the results section with vehicle control, progesterone (Calbiochem, Los Angeles, CA; 5341), and/or (−)-isoproterenol hydrochloride (Sigma–Aldrich, Indianapolis, IN; I 6504). All samples were co-treated at the same time with the phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine (IBMX; 1 mM; Sigma–Aldrich, St. Louis, MO; I 5879). Cells were immediately washed with ice-cold PBS and harvested with 1 mL of 0.1 N HCl. Cellular debris was removed by centrifugation and the remainder of the cAMP enzyme immunoassay (EIA) was performed according to the manufacturer’s recommendations (Cayman Chemical, Ann Arbor, MI; 581001). Individual absorbance readings taken at 405 nm were read in duplicate and converted to cAMP concentrations via an online Xcel worksheet provided by Cayman Chemical (http://www.caymanchem.com).
Luciferase Assays
ES-2 cells were plated at a density of 100,000 cells/well of a six-well plate. The following day, cells were co-transfected overnight with 0.9 μg of a cAMP response element (CRE)-firefly luciferase reporter construct (kindly provided by Dr. Paul Mermelstein as previously described [31]) and 0.1 μg of a constitutively active pRL-TK-Renilla luciferase construct (Promega; E2241) using FuGene HD® transfection reagent (Roche, Indianapolis, IN; 04 709 713 001) according to the manufacturer’s recommendations. The next day, cells were washed with PBS and starved for 24 h in 1% DCC as previously described. After starvation, cells were treated for 24 h with vehicle control, progesterone, and/or isoproterenol at which point they were washed with ice-cold PBS and harvested in 200 μL of 1X passive lysis buffer (Promega, Madison, WI; E1941). All samples were co-treated at the same time with IBMX (1 mM). Cellular debris was removed by centrifugation and samples were analyzed for 10.0 s each by a Monolight™ 3010 luminometer (PharMingen, San Diego, CA) that injected 80 μL of firefly luciferase substrate followed by 100 μL of Stop-N-Glo® Renilla luciferase substrate (Promega, Madison, WI; E1960). Progesterone response element (PRE)-luciferase assays were carried out as described for the CRE-luciferase assays using a previously described wild-type n-PR-B overexpression construct and PRE-luciferase reporter construct [32].
qPCR Superarray
SKOV-3 cells were plated at a density of 1,000,000 cells/150 mm plate and allowed to grow to approximately 75% confluency at which point they were washed with PBS and starved for 24 h with un-supplemented modified IMEM. Cells were treated for 24 h with vehicle control or progesterone, washed with ice-cold PBS, and RNA was isolated using TRIzol® (Invitrogen, Carlsbad, CA; 15596-018) and isopropanol extraction. After isolation, 12.0 μg of RNA was converted to cDNA as previously described (see qPCR section). Samples were aliquoted onto the Human Cancer PathwayFinder™, RT2 Profiler™ PCR Array (SABiosciences™, Frederick, MD; PAHS-033A). qPCR was performed using Light Cycler® FastStart DNA Master SYBR Green I (Roche, Indianapolis, IN; 03 515 885 001) on a Mastercycler ep realplex4 S qPCR platform (eppendorf, Hauppage, NY; 63002 000.601). Gene expression changes were determined by normalizing the value of each target gene in the progesterone-treated group to its corresponding value from the vehicle control-treated group using on-line analysis software provided by SABiosciences™ (http://www.sabiosciences.com/pcr/arrayanalysis.php).
Statistical Analysis
All reported values represent the mean ± the standard deviation (SD). Statistical significance was determined with 95% confidence (asterisk, p ≤ 0.05) using a Student’s t test for two-group comparisons and ANOVA followed by Tukey’s post-hoc analysis for between group comparisons.
Results
mPRs Expressed in Ovarian Cancer Cells May Indirectly Modulate cAMP Levels
Expression of mPRα in humans is limited to the kidney, placenta, testis, and ovary [22]. Membrane-PRα expression has also been observed in human breast and ovarian cancer specimens and human breast cancer cell lines [30, 33]. Therefore, we measured basal mRNA expression of each mPR isoform (mPRα/PAQR7, mPRβ/PAQR8, and mPRγ/PAQR5) in a panel of eight distinct ovarian cancer cell line models and one non-tumorigenic immortalized ovarian surface epithelial cell line (1816-575) using quantitative PCR (qPCR; Fig. 1a). We also confirmed the absence of protein or mRNA-encoding nuclear-PR-A and -B isoforms from all eight ovarian cancer cell lines using n-PR positive MCF-7 breast cancer cells as a positive control (not shown). Based on these observations, we focused our attention on two ovarian cancer cell lines demonstrating differences in mPR isoform transcript levels that were derived from aggressive human tumors of different histologic subtypes. SKOV-3 ovarian cancer cells were originally isolated from a metastatic adenocarcinoma of the ovary and ES-2 ovarian cancer cells were established from a poorly differentiated ovarian clear cell carcinoma. Using MCF-7 breast cancer cells as a reference for mPRα expression, we positively identified mPRα protein expression by immunoblot analysis in SKOV-3 and ES-2 cells (Fig. 1b). Although n-PR-A/B expression appears to be limited to MCF-7 breast cancer cells relative to the ovarian cancer cell lines in our panel, the possibility of endogenous n-PR activity was further excluded, as OVCAR-3 (Fig. 1c) and ES-2 (not shown) ovarian cancer cells failed to elicit a transcriptional response from a PRE-driven luciferase reporter gene unless cells were also co-transfected with a human PR-B expression vector. Importantly, these cells also failed to respond to synthetic progestins in soft-agar and MTT growth assays and mPR mRNA expression did not change in response to treatment (24 h) of cells with either progesterone or estrogen (not shown).
After confirming abundant mPR isoform expression in the absence of n-PR, we sought to examine progesterone-mediated signaling events in ovarian cancer cells. Characterization of mPRα activity in a variety of cell types demonstrated the coupling of mPRα to an inhibitory G protein 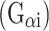 [21, 24, 25]. However, in contrast to a progesterone-mediated reduction in cAMP levels, we observed no significant alterations of intracellular cAMP concentration in SKOV-3 ovarian cancer cells stimulated with progesterone alone or upon the inclusion of IBMX to block phosphodiesterase activity (Fig. 2a). One interpretation of this surprising observation is that in these cells, mPRs are not functionally linked (i.e. at least directly) to inhibitory or stimulatory G proteins. Signal transduction from the cell surface through the G protein alpha subunit may be functionally impaired, as observed in MDA-MB-231 breast cancer cells not overexpressing mPRs [24]. Notably, however, treatment of SKOV-3 cells with isoproterenol, a well-characterized β1,2-adrenergic receptor agonist resulted in robust production of cAMP (Fig. 2a).
[21, 24, 25]. However, in contrast to a progesterone-mediated reduction in cAMP levels, we observed no significant alterations of intracellular cAMP concentration in SKOV-3 ovarian cancer cells stimulated with progesterone alone or upon the inclusion of IBMX to block phosphodiesterase activity (Fig. 2a). One interpretation of this surprising observation is that in these cells, mPRs are not functionally linked (i.e. at least directly) to inhibitory or stimulatory G proteins. Signal transduction from the cell surface through the G protein alpha subunit may be functionally impaired, as observed in MDA-MB-231 breast cancer cells not overexpressing mPRs [24]. Notably, however, treatment of SKOV-3 cells with isoproterenol, a well-characterized β1,2-adrenergic receptor agonist resulted in robust production of cAMP (Fig. 2a).
Fig. 2.

Membrane progesterone receptor (mPR) activity enhances β1,2-adrenergic receptor stimulated cAMP levels and cAMP-mediated transcription. a Stimulating SKOV-3 cells with progesterone (Prog.; 1 μM) for 10 min did not alter cAMP levels relative to vehicle control. Treatment with the β1,2-adrenergic agonist isoproterenol (Iso.; 100 nM) served as a positive control for the cAMP EIA (mean ± SD, n = 4; NS, not significant; asterisk, p ≤ 0.05). b Co-treating SKOV-3 cells with progesterone (Prog.; 1 μM) for 10 min significantly increased cAMP induction by isoproterenol (Iso.; 100 nM; mean ± SD, n = 3; asterisk, p ≤ 0.05). c ES-2 cells transiently transfected with a CRE-luciferase reporter demonstrated a significant increase in transcription in the presence of isoproterenol (Iso.; 100 nM) but not progesterone (Prog.; 1 μM) when compared to vehicle treated cells (mean ± SD, n = 6; asterisk, p ≤ 0.05). CRE-luciferase expression was significantly increased when cells were treated with progesterone (Prog.; 1 μM) plus isoproterenol (Iso.; 100 nM) for 24 h versus cells exposed to isoproterenol alone (mean ± SD, n = 6; number sign p ≤ 0.05; all firefly luciferase values are individually normalized to the expression of a Renilla-based luciferase reporter construct serving as an internal control)
Stimulation of mPRs in human myometrial cells inhibited isoproterenol-induced cAMP synthesis [25]. We therefore examined the impact of mPR activation on isoproterenol-induced β1,2-adrenergic signaling in ovarian cancer cells. SKOV-3 cells were co-treated with either isoproterenol and vehicle or isoproterenol and progesterone (Fig. 2b). In contrast to results in human myometrial cells acting through an inhibitory G protein 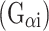 [24], progesterone significantly enhanced isoproterenol-induced cAMP levels. Similar augmentation of isoproterenol-induced cAMP levels by progesterone was also observed in ES-2 ovarian cancer cells (not shown). To test whether mPR-mediated increases in isoproterenol-induced cAMP synthesis also translate to changes in gene expression, we transfected ES-2 cells with a cAMP response element (CRE)-driven luciferase reporter construct. Cells were treated for 24 h with either vehicle, progesterone alone, isoproterenol alone, or both agents. Just as progesterone alone failed to alter cAMP levels (Fig. 2a), stimulation of reporter gene-transfected ES-2 cells with progesterone for 24 h failed to elicit an appreciable transcriptional response as measured by CRE-luciferase expression (Fig. 2c). However, co-treatment of cells with progesterone and the β1,2-adrenergic agonist, isoproterenol, led to a significant increase in CRE-luciferase activity relative to isoproterenol alone (Fig. 2c). In contrast, stimulation of either follicle-stimulating hormone (FSH) or luteinizing hormone (LH) receptors with their respective ligands failed to augment isoproterenol-induced cAMP levels (not shown).
[24], progesterone significantly enhanced isoproterenol-induced cAMP levels. Similar augmentation of isoproterenol-induced cAMP levels by progesterone was also observed in ES-2 ovarian cancer cells (not shown). To test whether mPR-mediated increases in isoproterenol-induced cAMP synthesis also translate to changes in gene expression, we transfected ES-2 cells with a cAMP response element (CRE)-driven luciferase reporter construct. Cells were treated for 24 h with either vehicle, progesterone alone, isoproterenol alone, or both agents. Just as progesterone alone failed to alter cAMP levels (Fig. 2a), stimulation of reporter gene-transfected ES-2 cells with progesterone for 24 h failed to elicit an appreciable transcriptional response as measured by CRE-luciferase expression (Fig. 2c). However, co-treatment of cells with progesterone and the β1,2-adrenergic agonist, isoproterenol, led to a significant increase in CRE-luciferase activity relative to isoproterenol alone (Fig. 2c). In contrast, stimulation of either follicle-stimulating hormone (FSH) or luteinizing hormone (LH) receptors with their respective ligands failed to augment isoproterenol-induced cAMP levels (not shown).
mPRs Expressed in Ovarian Cancer Cells Rapidly Activate JNK and p38 MAPKs
In addition to its effects on intracellular cAMP concentrations, progesterone-mediated mPR activity has been shown to alter other key intracellular signaling pathways [21]. In particular, progesterone stimulation of mPRs in breast and human myometrial cells caused an increase in the activity of ERK1/2 and p38, both members of the mitogen-activated protein kinase (MAPK) family of signaling intermediates [23, 25]. Similar to these findings, progesterone-treated SKOV-3 ovarian cancer cells demonstrated a significant increase in phosphorylation of JNK1/2 and p38 MAPKs, as measured by Western blotting using phospho-specific antibodies (Fig. 3). JNK1/2 activation occurred in a time- and dose-dependent manner, where treatment with progesterone concentrations of 100 and 1,000 nM lead to increased JNK1/2 phosphorylation at both 5 and 15 min. JNK1/2 activity was sustained for 30 min with the higher dose of progesterone (Fig. 3). Similar to JNK1/2, p38 MAPK was transiently activated by both low and high concentrations of progesterone. However, p38 phosphorylation increased at 5 and 15 min, but returned to baseline by 30 min (Fig. 3). Lower concentrations of progesterone (1, 10, and 50 nM) failed to reproducibly activate these kinases under similar conditions and when cells were co-treated with both progesterone (1 μM) and isoproterenol (100 nM), there was no further increase in either JNK1/2 or p38 MAPK activity as measured by phosphorylation (not shown). These results are consistent with the activation of an alternate mPR signaling pathway (possibly mediated by the βγ subunits of heterotrimeric G proteins); mPR-mediated increases in MAPK activity appear to be independent of changes in cAMP signaling.
It is now well documented that JNK1/2 and p38 have the ability to significantly impact gene expression, often by altering the activation of transcription factor targets or their co-regulators that act as nuclear substrates for these kinases. To identify gene targets downstream of mPR signaling, we analyzed progesterone-dependent changes in the expression level of selected genes commonly associated with cellular transformation and tumorigenesis (qPCR Superarray; SABiosciences™). cDNA was made using mRNA isolated from vehicle or progesterone-treated (24 h) SKOV-3 ovarian cancer cells. Following qPCR reactions, we identified a significant increase in BAX gene expression. Bax is a prominent member of the pro-apoptotic Bcl-protein family. Validation of BAX as a mPR gene target demonstrated a significant dose-dependent upregulation of BAX mRNA in SKOV-3 ovarian cancer cells treated with 50- and 100-nM concentrations of progesterone but not 10-nM levels (Fig. 4a). To further link BAX regulation to mPR signaling, we tested the requirement of JNK1/2 and p38 MAPK activities for progesterone-induced upregulation of BAX mRNA. Cells were again treated with progesterone, but in this set of experiments, we included small-molecule inhibitors for either JNK1/2 or p38 MAPKs. Notably, BAX mRNA expression was significantly decreased in SKOV-3 ovarian cancer cells exposed to both progesterone (100 nM) and the JNK1/2 small-molecule inhibitor SP 600125 (10 μM), but not the p38 small-molecule inhibitor SB 203580 (2.6 μM; Fig. 4b). Taken together, these observations identify a known pro-apoptotic mediator (BAX)-regulated downstream of mPR signaling by JNK1/2 and distinct from cAMP signaling.
Fig. 4.

mPR activity in ovarian cancer cells increases BAX gene expression in a JNK1/2-dependent manner. a SKOV-3 ovarian cancer cells treated for 24 h with progesterone (50 nM and 100 nM) demonstrated a significant increase in BAX gene expression versus vehicle treated cells but not when compared against each other as determined by quantitative PCR (qPCR) relative to the GAPDH housekeeping gene (mean ± SD, n = 4; NS, not significant; asterisk, p ≤ 0.05). b The JNK1/2 inhibitor SP 600125 (10 μM), but not the p38 inhibitor SB 203580 (2.6 μM), significantly decreased BAX gene expression in SKOV-3 ovarian cancer cells stimulated with progesterone (100 nM) for 24 h as determined by qPCR relative to the β-ACTIN housekeeping gene (mean ± SD, n = 3; asterisk, p ≤ 0.05)
Discussion
Mounting evidence suggests that the ovarian steroid hormone, progesterone, plays both a protective role in preventing ovarian cancer development and a therapeutic role acting against further tumor progression. Unfortunately, advanced ovarian tumors lose expression of the well-characterized classical nuclear progesterone receptors (n-PR-A/B), and are thus assumed to be entirely resistant to the potential benefits of progesterone’s anti-tumorigenic effects. However, recent work demonstrating the presence of a newly defined family of membrane progesterone receptors (mPRs) in human ovarian tumor specimens raises the possibility that progesterone, acting through this class of novel receptors unrelated to n-PRs, may continue to limit ovarian cancer progression in tumors of all stages and independently of n-PR expression.
Notably, mPRs were recently discovered in ovarian cancer tissue samples [30]. Our goal was to begin defining how these novel GPCR-like steroid hormone receptors might alter intracellular signaling networks in ovarian cancer cells. We first confirmed robust expression of each mPR isoform in normal (non-tumorigenic, immortalized) ovarian surface epithelial cells and ovarian cancer cell lines that model advanced stage disease (Fig. 1). Importantly, these cell lines lack n-PR, making them excellent model systems in which to study mPRs. Based on current literature [24, 25], we assumed mPRs would primarily negatively regulate intracellular cAMP levels. Surprisingly, our results demonstrated that mPRs, when stimulated with progesterone alone, do not appreciably alter cAMP concentrations either positively or negatively in SKOV-3 and ES-2 cells as they do in other cell types [24]. Instead, however, we found that progesterone-induced mPR activity enhanced β1,2-adrenergic receptor-mediated increases in cAMP levels (Fig. 2). This may occur by receptor crosstalk, such as via GPCR heterodimerization of adrenergic receptors with mPRs [34], although there are currently no reports of mPRs capable of forming heterodimers with other receptors. Moreover, the formation of adrenergic receptor/mPR heterodimers is not consistent with the observation that progesterone-induced activation of MAP kinase is not influenced by isoproterenol. Nonetheless, the observation that mPR activity increases isoproterenol-induced cAMP levels in ovarian cancer cells suggests a means for limiting their proliferative potential; increased cAMP levels cause up to a 62% decrease of cellular proliferation in HEY ovarian cancer cells [35]. Thus, mPR-dependent augmentation of cAMP levels in ovarian cancer cells could function to limit proliferation in response to other signals, possibly by increasing the expression of pro-apoptotic markers. For example, activating transcription factor-3 (ATF-3), a known gene target of the cAMP-CREB signaling pathway has been shown to be significantly upregulated in progesterone-treated ovarian cancer cells, and when overexpressed in vitro, causes ovarian cancer cell apoptosis [36, 37]. These studies were performed prior to the discovery of mPRs in human ovarian cancer cells [30]. However, the ability of mPRs to augment the natural induction of cAMP by other hormones provides a rationale to consider adding progesterone to current therapies for the treatment of advanced stage ovarian cancer.
Based on our work herein (Fig. 3), and previous observations, progesterone-mediated mPR activity clearly leads to the activation of downstream MAPK family members [25]. In response to mPR stimulation, we observed activation of JNK1/2 and p38 MAPKs and genomic changes (i.e., in BAX expression) associated with JNK1/2 signaling. While the mechanism(s) behind progesterone-dependent JNK1/2 and p38 phosphorylation remains unknown in ovarian cancer cells, we hypothesize that mPRs may be linked to these downstream kinases by increasing the activity of apoptosis signal-regulating kinase 1 (ASK1). Previous work has shown that GPCR signaling can directly activate ASK1, which in turn leads to the phosphorylation of both JNK1/2 and p38 via their upstream kinases, MKK4/7 and MKK3/6, respectively [38]. Furthermore, each of these kinases may be physically linked to GPCRs by the β-arrestin family of scaffolding proteins, which have also been shown to uncouple heterotrimeric G proteins from their cognate GPCRs. This mode of signal transduction (i.e., uncoupling from G protein effectors) may explain our modest cAMP results (Fig. 2 and discussed above) [39]. However, it is not yet known whether similar interactions occur between mPRs and these macromolecules.
Of great interest is the linkage of progesterone/mPR-mediated JNK1/2 activity with BAX gene expression. BAX is a known pro-apoptotic effector of ovarian cancer cells both in vitro and in vivo [40]. Prior work has demonstrated that (1) human ovarian cancer tissue samples possess higher expression levels of BAX mRNA compared to normal tissues, (2) the p53 tumor suppressor protein is a substrate that is phosphorylated and activated by JNK1/2 signaling, and (3) activated p53 directly increases BAX expression, a transcriptional response often disrupted in cisplatin-resistant ovarian cancer cells [41–43]. Additionally, increased JNK1/2 and p38 MAPK activities have been shown to induce ovarian cancer cell apoptosis; these kinases possess the ability to stimulate the activation and translocation of BAX to the outer mitochondrial membrane leading to cell death via the intrinsic pathway [44, 45]. Furthermore, in ovarian cancer, JNK1/2 and p38 have been shown to mediate both cisplatin- and paclitaxel-induced cytotoxicity, which may explain the improved prognosis of ovarian cancer patients whose tumors demonstrate increased MAPK activity [46–48]. Taken together, these exciting observations implicate a complete signaling pathway leading to ovarian cancer cell death that can be induced in part by the input of activated mPRs to pro-apoptotic MAPK signaling modules; we are currently pursuing this idea.
Over the past decade, numerous in vitro studies have documented the ability of progesterone to inhibit ovarian cancer cell proliferation and promote ovarian cancer cell death by a variety of different mechanisms including: p53 upregulation [49], differential regulation of TGF-β [16], increased FasL expression [50], and enhancing TRAIL-induced cell death [51]. It has also been shown that exposing ovarian cancer cells to the high levels of progesterone achieved during pregnancy decreases ovarian cancer cell proliferation, while treating n-PR negative ovarian cancer cells with progesterone decreases cell survival [52]. Additionally, patient clinical trials using medroxyprogesterone acetate (MPA) to treat ovarian cancer demonstrated up to 16 months of disease stabilization and improved the prognosis for advanced stage ovarian cancer patients also receiving platinum-based chemotherapy [53, 54]. The relative contributions of mPRs and classical nuclear-PRs to these effects remain to be determined. However, the success of these clinical trials, and prior in vitro studies using progesterone and progesterone analogues combined with the potentially anti-tumorigenic mPR-mediated signaling events discussed herein, suggest that high-dose progesterone therapy (or newly defined mPR agonists) may improve upon current chemotherapeutic approaches. In sum, the presence of abundant mPR and progesterone-dependent signaling events in ovarian cancer cells described herein suggest that progesterone-based hormone therapy, alone or in conjunction with other standard regimens, may provide an effective strategy for treating advanced stage ovarian cancer.
Acknowledgements
This work was supported by a grant from the Minnesota Ovarian Cancer Alliance. From the University of Minnesota Masonic Cancer Center, we would like to thank Dr. Douglas Yee, Dr. Amy Skubitz, and Dr. Sundaram Ramakrishnan for providing us with ovarian cancer cell line models and Dr. Paul Mermelstein for the CRE-luciferase reporter construct. We would also like to thank Dr. Patricia Kruk (University of South Florida) for providing us with the 1816-575 cell line.
Conflict of Interest
The authors declare that they have no conflict of interest.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Cho KR, Shih Ie M. Ovarian cancer. Annu Rev Pathol. 2009;4:287–313. doi: 10.1146/annurev.pathol.4.110807.092246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leung PC, Choi JH. Endocrine signaling in ovarian surface epithelium and cancer. Hum Reprod Update. 2007;13:143–162. doi: 10.1093/humupd/dml002. [DOI] [PubMed] [Google Scholar]
- 4.Adami HO, Hsieh CC, Lambe M, Trichopoulos D, Leon D, Persson I, et al. Parity, age at first childbirth, and risk of ovarian cancer. Lancet. 1994;344:1250–1254. doi: 10.1016/S0140-6736(94)90749-8. [DOI] [PubMed] [Google Scholar]
- 5.Thomas HV, Murphy MF, Key TJ, Fentiman IS, Allen DS, Kinlen LJ. Pregnancy and menstrual hormone levels in mothers of twins compared to mothers of singletons. Ann Hum Biol. 1998;25:69–75. doi: 10.1080/03014469800005432. [DOI] [PubMed] [Google Scholar]
- 6.Salazar-Martinez E, Lazcano-Ponce EC, Gonzalez Lira-Lira G, Escudero-De los Rios P, Salmeron-Castro J, Hernandez-Avila M. Reproductive factors of ovarian and endometrial cancer risk in a high fertility population in Mexico. Cancer Res. 1999;59:3658–3662. [PubMed] [Google Scholar]
- 7.Tambyraja AL, Sengupta F, MacGregor AB, Bartolo DC, Fearon KC. Patterns and clinical outcomes associated with routine intravenous sodium and fluid administration after colorectal resection. World J Surg. 2004;28:1046–1051. doi: 10.1007/s00268-004-7383-7. [DOI] [PubMed] [Google Scholar]
- 8.Modan B, Ron E, Lerner-Geva L, Blumstein T, Menczer J, Rabinovici J, et al. Cancer incidence in a cohort of infertile women. Am J Epidemiol. 1998;147:1038–1042. doi: 10.1093/oxfordjournals.aje.a009397. [DOI] [PubMed] [Google Scholar]
- 9.Syed V, Ulinski G, Mok SC, Yiu GK, Ho SM. Expression of gonadotropin receptor and growth responses to key reproductive hormones in normal and malignant human ovarian surface epithelial cells. Cancer Res. 2001;61:6768–6776. [PubMed] [Google Scholar]
- 10.Gross TP, Schlesselman JJ, Stadel BV, Yu W, Lee NC. The risk of epithelial ovarian cancer in short-term users of oral contraceptives. Am J Epidemiol. 1992;136:46–53. doi: 10.1093/oxfordjournals.aje.a116419. [DOI] [PubMed] [Google Scholar]
- 11.Rosenberg L, Palmer JR, Zauber AG, Warshauer ME, Lewis JL, Jr, Strom BL, et al. A case-control study of oral contraceptive use and invasive epithelial ovarian cancer. Am J Epidemiol. 1994;139:654–661. doi: 10.1093/oxfordjournals.aje.a117055. [DOI] [PubMed] [Google Scholar]
- 12.Ness RB, Grisso JA, Vergona R, Klapper J, Morgan M, Wheeler JE. Oral contraceptives, other methods of contraception, and risk reduction for ovarian cancer. Epidemiology. 2001;12:307–312. doi: 10.1097/00001648-200105000-00010. [DOI] [PubMed] [Google Scholar]
- 13.Greer JB, Modugno F, Allen GO, Ness RB. Short-term oral contraceptive use and the risk of epithelial ovarian cancer. Am J Epidemiol. 2005;162:66–72. doi: 10.1093/aje/kwi162. [DOI] [PubMed] [Google Scholar]
- 14.Beral V, Doll R, Hermon C, Peto R, Reeves G. Ovarian cancer and oral contraceptives: collaborative reanalysis of data from 45 epidemiological studies including 23, 257 women with ovarian cancer and 87, 303 controls. Lancet. 2008;371:303–314. doi: 10.1016/S0140-6736(08)60167-1. [DOI] [PubMed] [Google Scholar]
- 15.Rodriguez GC, Walmer DK, Cline M, Krigman H, Lessey BA, Whitaker RS, et al. Effect of progestin on the ovarian epithelium of macaques: cancer prevention through apoptosis? J Soc Gynecol Investig. 1998;5:271–276. doi: 10.1016/S1071-5576(98)00017-3. [DOI] [PubMed] [Google Scholar]
- 16.Rodriguez GC, Nagarsheth NP, Lee KL, Bentley RC, Walmer DK, Cline M, et al. Progestin-induced apoptosis in the Macaque ovarian epithelium: differential regulation of transforming growth factor-beta. J Natl Cancer Inst. 2002;94:50–60. doi: 10.1093/jnci/94.1.50. [DOI] [PubMed] [Google Scholar]
- 17.Lee BH, Hecht JL, Pinkus JL, Pinkus GS. WT1, estrogen receptor, and progesterone receptor as markers for breast or ovarian primary sites in metastatic adenocarcinoma to body fluids. Am J Clin Pathol. 2002;117:745–750. doi: 10.1309/QLV6-HH0H-UCTF-WEF6. [DOI] [PubMed] [Google Scholar]
- 18.Noguchi T, Kitawaki J, Tamura T, Kim T, Kanno H, Yamamoto T, et al. Relationship between aromatase activity and steroid receptor levels in ovarian tumors from postmenopausal women. J Steroid Biochem Mol Biol. 1993;44:657–660. doi: 10.1016/0960-0760(93)90275-2. [DOI] [PubMed] [Google Scholar]
- 19.Akahira J, Suzuki T, Ito K, Kaneko C, Darnel AD, Moriya T, et al. Differential expression of progesterone receptor isoforms A and B in the normal ovary, and in benign, borderline, and malignant ovarian tumors. Jpn J Cancer Res. 2002;93:807–815. doi: 10.1111/j.1349-7006.2002.tb01323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lau KM, Mok SC, Ho SM. Expression of human estrogen receptor-alpha and -beta, progesterone receptor, and androgen receptor mRNA in normal and malignant ovarian epithelial cells. Proc Natl Acad Sci USA. 1999;96:5722–5727. doi: 10.1073/pnas.96.10.5722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thomas P. Characteristics of membrane progestin receptor alpha (mPRalpha) and progesterone membrane receptor component 1 (PGMRC1) and their roles in mediating rapid progestin actions. Front Neuroendocrinol. 2008;29:292–312. doi: 10.1016/j.yfrne.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu Y, Bond J, Thomas P. Identification, classification, and partial characterization of genes in humans and other vertebrates homologous to a fish membrane progestin receptor. Proc Natl Acad Sci USA. 2003;100:2237–2242. doi: 10.1073/pnas.0436133100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu Y, Rice CD, Pang Y, Pace M, Thomas P. Cloning, expression, and characterization of a membrane progestin receptor and evidence it is an intermediary in meiotic maturation of fish oocytes. Proc Natl Acad Sci USA. 2003;100:2231–2236. doi: 10.1073/pnas.0336132100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thomas P, Pang Y, Dong J, Groenen P, Kelder J, de Vlieg J, et al. Steroid and G protein binding characteristics of the seatrout and human progestin membrane receptor alpha subtypes and their evolutionary origins. Endocrinology. 2007;148:705–718. doi: 10.1210/en.2006-0974. [DOI] [PubMed] [Google Scholar]
- 25.Karteris E, Zervou S, Pang Y, Dong J, Hillhouse EW, Randeva HS, et al. Progesterone signaling in human myometrium through two novel membrane G protein-coupled receptors: potential role in functional progesterone withdrawal at term. Mol Endocrinol. 2006;20:1519–1534. doi: 10.1210/me.2005-0243. [DOI] [PubMed] [Google Scholar]
- 26.Tubbs C, Thomas P. Progestin signaling through an olfactory G protein and membrane progestin receptor-alpha in Atlantic croaker sperm: potential role in induction of sperm hypermotility. Endocrinology. 2009;150:473–484. doi: 10.1210/en.2008-0512. [DOI] [PubMed] [Google Scholar]
- 27.Cai Z, Stocco C. Expression and regulation of progestin membrane receptors in the rat corpus luteum. Endocrinology. 2005;146:5522–5532. doi: 10.1210/en.2005-0759. [DOI] [PubMed] [Google Scholar]
- 28.Ashley RL, Arreguin-Arevalo JA, Nett TM. Binding characteristics of the ovine membrane progesterone receptor alpha and expression of the receptor during the estrous cycle. Reprod Biol Endocrinol. 2009;7:42. doi: 10.1186/1477-7827-7-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ashley RL, Clay CM, Farmerie TA, Niswender GD, Nett TM. Cloning and characterization of an ovine intracellular seven transmembrane receptor for progesterone that mediates calcium mobilization. Endocrinology. 2006;147:4151–4159. doi: 10.1210/en.2006-0002. [DOI] [PubMed] [Google Scholar]
- 30.Romero-Sanchez M, Peiper SC, Evans B, Wang Z, Catasus L, Ribe A, et al. Expression profile of heptahelical putative membrane progesterone receptors in epithelial ovarian tumors. Hum Pathol. 2008;39:1026–1033. doi: 10.1016/j.humpath.2007.11.007. [DOI] [PubMed] [Google Scholar]
- 31.Boulware MI, Weick JP, Becklund BR, Kuo SP, Groth RD, Mermelstein PG. Estradiol activates group I and II metabotropic glutamate receptor signaling, leading to opposing influences on cAMP response element-binding protein. J Neurosci. 2005;25:5066–5078. doi: 10.1523/JNEUROSCI.1427-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pierson-Mullany LK, Lange CA. Phosphorylation of progesterone receptor serine 400 mediates ligand-independent transcriptional activity in response to activation of cyclin-dependent protein kinase 2. Mol Cell Biol. 2004;24:10542–10557. doi: 10.1128/MCB.24.24.10542-10557.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dressing GE, Thomas P. Identification of membrane progestin receptors in human breast cancer cell lines and biopsies and their potential involvement in breast cancer. Steroids. 2007;72:111–116. doi: 10.1016/j.steroids.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 34.Milligan G. G protein-coupled receptor hetero-dimerization: contribution to pharmacology and function. Br J Pharmacol. 2009;158:5–14. doi: 10.1111/j.1476-5381.2009.00169.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shaw TJ, Keszthelyi EJ, Tonary AM, Cada M, Vanderhyden BC. Cyclic AMP in ovarian cancer cells both inhibits proliferation and increases c-KIT expression. Exp Cell Res. 2002;273:95–106. doi: 10.1006/excr.2001.5426. [DOI] [PubMed] [Google Scholar]
- 36.Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol. 2001;2:599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- 37.Syed V, Mukherjee K, Lyons-Weiler J, Lau KM, Mashima T, Tsuruo T, et al. Identification of ATF-3, caveolin-1, DLC-1, and NM23-H2 as putative antitumorigenic, progesterone-regulated genes for ovarian cancer cells by gene profiling. Oncogene. 2005;24:1774–1787. doi: 10.1038/sj.onc.1207991. [DOI] [PubMed] [Google Scholar]
- 38.Matsukawa J, Matsuzawa A, Takeda K, Ichijo H. The ASK1-MAP kinase cascades in mammalian stress response. J Biochem. 2004;136:261–265. doi: 10.1093/jb/mvh134. [DOI] [PubMed] [Google Scholar]
- 39.Miller WE, Lefkowitz RJ. Expanding roles for beta-arrestins as scaffolds and adapters in GPCR signaling and trafficking. Curr Opin Cell Biol. 2001;13:139–145. doi: 10.1016/S0955-0674(00)00190-3. [DOI] [PubMed] [Google Scholar]
- 40.Arafat WO, Gomez-Navarro J, Xiang J, Barnes MN, Mahasreshti P, Alvarez RD, et al. An adenovirus encoding proapoptotic Bax induces apoptosis and enhances the radiation effect in human ovarian cancer. Mol Ther. 2000;1:545–554. doi: 10.1006/mthe.2000.0071. [DOI] [PubMed] [Google Scholar]
- 41.Marone M, Scambia G, Mozzetti S, Ferrandina G, Iacovella S, De Pasqua A, et al. bcl-2, bax, bcl-XL, and bcl-XS expression in normal and neoplastic ovarian tissues. Clin Cancer Res. 1998;4:517–524. [PubMed] [Google Scholar]
- 42.Wu GS. The functional interactions between the p53 and MAPK signaling pathways. Cancer Biol Ther. 2004;3:156–161. doi: 10.4161/cbt.3.2.614. [DOI] [PubMed] [Google Scholar]
- 43.Perego P, Giarola M, Righetti SC, Supino R, Caserini C, Delia D, et al. Association between cisplatin resistance and mutation of p53 gene and reduced bax expression in ovarian carcinoma cell systems. Cancer Res. 1996;56:556–562. [PubMed] [Google Scholar]
- 44.Fister S, Gunthert AR, Aicher B, Paulini KW, Emons G, Grundker C. GnRH-II antagonists induce apoptosis in human endometrial, ovarian, and breast cancer cells via activation of stress-induced MAPKs p38 and JNK and proapoptotic protein Bax. Cancer Res. 2009;69:6473–6481. doi: 10.1158/0008-5472.CAN-08-4657. [DOI] [PubMed] [Google Scholar]
- 45.Kim BJ, Ryu SW, Song BJ. JNK- and p38 kinase-mediated phosphorylation of Bax leads to its activation and mitochondrial translocation and to apoptosis of human hepatoma HepG2 cells. J Biol Chem. 2006;281:21256–21265. doi: 10.1074/jbc.M510644200. [DOI] [PubMed] [Google Scholar]
- 46.Mansouri A, Ridgway LD, Korapati AL, Zhang Q, Tian L, Wang Y, et al. Sustained activation of JNK/p38 MAPK pathways in response to cisplatin leads to Fas ligand induction and cell death in ovarian carcinoma cells. J Biol Chem. 2003;278:19245–19256. doi: 10.1074/jbc.M208134200. [DOI] [PubMed] [Google Scholar]
- 47.Lee LF, Li G, Templeton DJ, Ting JP. Paclitaxel (Taxol)-induced gene expression and cell death are both mediated by the activation of c-Jun NH2-terminal kinase (JNK/SAPK) J Biol Chem. 1998;273:28253–28260. doi: 10.1074/jbc.273.43.28253. [DOI] [PubMed] [Google Scholar]
- 48.Givant-Horwitz V, Davidson B, Lazarovici P, Schaefer E, Nesland JM, Trope CG, et al. Mitogen-activated protein kinases (MAPK) as predictors of clinical outcome in serous ovarian carcinoma in effusions. Gynecol Oncol. 2003;91:160–172. doi: 10.1016/S0090-8258(03)00434-7. [DOI] [PubMed] [Google Scholar]
- 49.Bu SZ, Yin DL, Ren XH, Jiang LZ, Wu ZJ, Gao QR, et al. Progesterone induces apoptosis and up-regulation of p53 expression in human ovarian carcinoma cell lines. Cancer. 1997;79:1944–1950. doi: 10.1002/(SICI)1097-0142(19970515)79:10<1944::AID-CNCR15>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 50.Syed V, Ho SM. Progesterone-induced apoptosis in immortalized normal and malignant human ovarian surface epithelial cells involves enhanced expression of FasL. Oncogene. 2003;22:6883–6890. doi: 10.1038/sj.onc.1206828. [DOI] [PubMed] [Google Scholar]
- 51.Syed V, Mukherjee K, Godoy-Tundidor S, Ho SM. Progesterone induces apoptosis in TRAIL-resistant ovarian cancer cells by circumventing c-FLIPL overexpression. J Cell Biochem. 2007;102:442–452. doi: 10.1002/jcb.21304. [DOI] [PubMed] [Google Scholar]
- 52.Chao KC, Wang PH, Yen MS, Chang CC, Chi CW. Role of estrogen and progesterone in the survival of ovarian tumors–a study of the human ovarian adenocarcinoma cell line OC-117-VGH. J Chin Med Assoc. 2005;68:360–367. doi: 10.1016/S1726-4901(09)70176-5. [DOI] [PubMed] [Google Scholar]
- 53.Belinson JL, McClure M, Badger G. Randomized trial of megestrol acetate vs. megestrol acetate/tamoxifen for the management of progressive or recurrent epithelial ovarian carcinoma. Gynecol Oncol. 1987;28:151–155. doi: 10.1016/0090-8258(87)90208-3. [DOI] [PubMed] [Google Scholar]
- 54.Chen X, Feng Y. Effect of progesterone combined with chemotherapy on epithelial ovarian cancer. Chin Med J (Engl) 2003;116:388–391. [PubMed] [Google Scholar]