

Abstract
Prolonged exposure to hyperoxia results in acute lung injury (ALI), accompanied by a significant elevation in the levels of proinflammatory cytokines and leukocyte infiltration in the lungs. However, the mechanisms underlying hyperoxia-induced proinflammatory ALI remain to be elucidated. In this study, we investigated the role of the proinflammatory cytokine high mobility group box protein 1 (HMGB1) in hyperoxic inflammatory lung injury, using an adult mouse model. The exposure of C57BL/6 mice to ≥99% O2 (hyperoxia) significantly increased the accumulation of HMGB1 in the bronchoalveolar lavage fluids (BALF) prior to the onset of severe inflammatory lung injury. In the airways of hyperoxic mice, HMGB1 was hyperacetylated and existed in various redox forms. Intratracheal administration of recombinant HMGB1 (rHMGB1) caused a significant increase in leukocyte infiltration into the lungs compared to animal treated with a non-specific peptide. Neutralizing anti-HMGB1 antibodies, administrated before hyperoxia significantly attenuated pulmonary edema and inflammatory responses, as indicated by decreased total protein content, wet/dry weight ratio, and numbers of leukocytes in the airways. This protection was also observed when HMGB1 inhibitors were administered after the onset of the hyperoxic exposure. The aliphatic antioxidant, ethyl pyruvate (EP), inhibited HMGB1 secretion from hyperoxic macrophages and attenuated hyperoxic lung injury. Overall, our data suggest that HMGB1 plays a critical role in mediating hyperoxic ALI through the recruitment of leukocytes into the lungs. If these results can be translated to humans, they suggest that HMGB1 inhibitors provide treatment regimens for oxidative inflammatory lung injury in patients receiving hyperoxia through mechanical ventilation.
Abbreviations: ALI, acute lung injury; BALF, bronchoalveolar lavage fluids; EP, ethyl pyruvate; GST, gluthatione-s-transferase; HMGB1, high mobility group box protein 1; MV, mechanical ventilation; NLS, nuclear localization signal; PMNs, polymorphonuclear neutrophils; RA, room air; rHMGB1, recombinant HMGB1; ROS, reactive oxygen species
Keywords: Hyperoxia, Macrophage, HMGB1, Hyperacetylation, Redox state
Graphical abstract
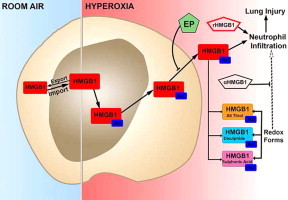
Highlights
-
•
Exposure to hyperoxia results in accumulation of high levels of airway HMGB1 that precede inflammatory acute lung injury (ALI).
-
•
Airway HMGB1 is critical in mediating hyperoxia-induced inflammatory ALI via recruiting leukocytes including neutrophils.
-
•
Extracellular HMGB1-accumulated upon prolonged exposure to hyperoxia is hyperacetylated, existing in different redox states.
-
•
Small molecule EP, administrated even after the onset of hyperoxic exposure, can mitigate hyperoxia-induced inflammatory ALI by inhibiting HMGB1 release into the extracellular milieu.
Introduction
Oxygen therapy with supraphysiological concentrations of oxygen (hyperoxia) is routinely administered during mechanical ventilation (MV) for the management of severe respiratory distress, such as acute respiratory distress syndrome [12,14,51]. However, oxygen therapy can also cause oxygen toxicity, inducing acute lung injury (ALI) [21,38]. Hyperoxia-induced ALI is characterized by excessive proinflammatory responses, endothelial and epithelial cell damage, and alveolar edema [9,13,20,35]. Although hyperoxia-induced ALI is mediated by excessive amounts of reactive oxygen species (ROS), the more severe ALI is associated with the influx of leukocytes including polymorphonuclear neutrophils (PMNs) into the lungs [6,47]. Both chemokines and pro-inflammatory cytokines are critical in mediating neutrophil recruitment into the lungs [5,34]. For example, chemokines, such as MIP-1, CXCL1, CXCL2 and IL-8, are involved in the regulation of neutrophil recruitment [18,34]. Although pro-inflammatory cytokines, including TNF-α and IL-1β have been implicated in mediating neutrophil influx into hyperoxic lungs [17,18], molecular mechanisms underlying PMNs recruitment under hyperoxic conditions remain to be elucidated. Clinically, there are no effective treatments that reduce the inflammatory lung injury of patients on mechanical ventilation.
High mobility group box 1 (HMGB1), originally identified as a nuclear DNA-binding protein [26], is critical for transcription regulation and gene expression [39,45]. Numerous studies have reported that extracellular HMGB1, as a pro-inflammatory cytokine, can trigger an overwhelming inflammatory response that promotes the progression of sepsis and ALI [1,24,29,43,49]. In addition, the intratracheal administration of recombinant HMGB1 (rHMGB1) to mice causes pulmonary pro-inflammatory responses, including neutrophil accumulation and release of cytokines such as IL-1β, TNF-α, MIP-2 and macrophage migration inhibitory factor [1,24]. Clinically, elevated levels of HMGB1, in both plasma and lung epithelial lining fluids, have been observed in patients with ALI [43,57]. Moreover, reduction of levels of extracellular HMGB1 results in reduced inflammatory responses and protection against organ failure in sepsis and endotoxemia [49,50,54]. Thus, there appears to be a link between extracellular HMGB1 and the pathogenesis of ALI, although little is known about the role of HMGB1 in oxidative stress-induced hyperoxic ALI.
High levels of airway HMGB1 were found in bronchoalveolar lavage fluids (BALF) of patients receiving MV and in animals subjected to high tidal volume ventilation [47]. Based on published studies, we hypothesize that extracellular HMGB1 is an important mediator of neutrophil infiltration in hyperoxic lungs and contributes to the development of hyperoxia-induced inflammatory ALI. In this report, we show that airway HMGB1 mediates the recruitment of neutrophils to the lungs upon prolonged exposure to hyperoxia, causing pronounced inflammatory ALI. The inhibition of HMGB1 by neutralizing anti-HMGB1 antibodies and the small molecule ethyl pyruvate (EP) significantly reduces HMGB1-induced neutrophil infiltration and attenuate ALI.
Materials and methods
Mice and treatments
Mice were housed and used in the experimental protocols in specific pathogen-free conditions in accordance with the Feinstein Institute for Medical Research and St. John׳s University׳s Institutional Animal Care and Use Committee Guidelines. Male C57BL/6 mice (6–8 weeks old) were purchased from Jackson Laboratories (Bar Harbor, Maine). Mice were exposed to ≥99% O2 at atmospheric pressure in a Plexiglass chamber as described previously [28,31] and the level of O2 in the chamber was constantly monitored using an oxygen analyzer (BioSpherix Ltd., Redfield, NY). The environmental temperature was maintained at 22±1 °C and relative humidity was 62±10%. Exposures were continuous for the time indicated except for 5 min twice daily after 24 h exposure, when the chamber was opened to allow intraperitoneal administrations.
To inhibit HMGB1, mice received either 360 μg/mouse of anti-HMGB1 IgGs or 40 mg/kg body weight EP (Sigma Aldrich, St. Louis, MO) by intraperitoneal injection, while the mice in the control groups received nonimmune IgGs or saline solution, respectively. Each group contained between 5 and 13 animals, and experiments were performed independently at least twice. After mice were euthanized by sodium pentobarbital (10 mg/kg intraperitoneal body weight), one lobe of the lungs was excised, weighed, and dried in an oven at 80 °C and weighed again; the other lobes of lung tissue were snap frozen in liquid nitrogen for molecular analyses as previously described [28]. To obtain BALF, the mice were anesthetized as described previously. A 1–2 cm incision was made on the front part of animals׳ neck as described previously [24]. The trachea was directly visualized, and a 20-gauge×1-in. intravenous catheter was inserted caudally into the lumen of exposed trachea and secured in place. The lungs were then gently lavaged with two aliquots of saline (total 2.5 ml), which were pooled and the total cell count and cell differential were assessed.
Wet/dry weight ratios
All mice used for lung wet/dry weight ratios were of identical ages. Lungs were excised, rinsed briefly in PBS, blotted, and then weighed to obtain the “wet” weight. Lungs were then dried in an oven at 80 °C for 7 days to obtain the “dry” weight.
Instillation of rHMGB1
The instillation of rHMGB1 was performed as described previously [24]. Mice were anesthetized by intraperitoneal injection of ketamine (75 mg/kg)/medetomidine (1.0 mg/kg). An incision (2–3 mm) was made to visualize the trachea. A 25-gauge needle was used to puncture the trachea, and the rHMGB1 was instilled using a 50-µl Hamilton syringe. Glutathion-S-transferase (GST) tag (Amersham Pharmacia Biotech, Piscataway, NJ) was similarly instilled in control animals.
Histopathology
Histopathological evaluation was performed on paraffin-embedded tissues as described previously [30]. Before removal from the animal, the lungs were rinsed with PBS and then instilled with 0.75 ml of buffered formalin through a 20-gauge angiocatheter placed in the trachea. The lungs were then immersed in buffered formalin overnight and processed for conventional paraffin histology. The sections were stained with hematoxylin and eosin and examined by light microscopy.
Cell culture
Murine macrophage-like RAW 264.7 cells (American Type Culture Collection, Boulevard Manassas, VA) were cultured in RPMI medium 1640 (Gibco BRL, Grand Island, NY) supplemented with 10% FBS (Gemini Biological Produces, Catabasas, CA), 1% penicillin and streptomycin (Life Technologies, Carlsbad, CA). Cells were maintained at 37 °C in 21% O2/5% CO2 (normoxia) and allowed to grow to 80–90% confluence. Hyperoxic exposure was performed in sealed humidified modular incubator chambers (Billups-Rothenberg Inc. Del Mar, CA) containing 95% O2/5% CO2 (hyperoxia) at 37 °C and medium was changed daily as previously described [31].
Western blot analysis
All procedures were performed as described previously [16]. Samples were ultra-filtered with Centricon 100 (Millpore, Billerica, MA). Protein from each samples were loaded on a 10% SDS-polyacrylamide gel transferred to a nitrocellulose membrane. After being blocked with 5% non-fat milk the membrane was incubated overnight at 4 °C with a specific polyclonal rabbit primary antibody to HMGB1 (kindly provided by Dr. Wang, from The Feinstein Institute for Medical Research, North Shore-LIJ Health System, Manhasset, NY) followed by incubation with an anti-rabbit horseradish peroxidase-coupled secondary antibody (Bio-Rad, Hercules, CA). After three washings, bands were detected using enhanced chemiluminescence plus Western blotting detection reagents (Amersham Pharmacia Biotech, Piscataway, NJ).
Immunohistochemical analysis
The localization of intracellular HMGB1 was assessed by immunohistochemical analysis as previously described [15]. RAW 264.7 cells were seeded onto chamber slides (BD Bioscience, Bedford, MA), fixed with phosphate-buffered formaldehyde (2%, pH 7.4, 15 min) after hyperoxic exposure. Immunohistochemical analysis of the expression of HMGB1 was performed on paraffin-embedded tissues as described above. The sections were de-waxed in xylene and rehydrated with ethanol arranged in a graded concentration. Then both fixed RAW cells and de-waxed tissues were permeabilized with 0.2% Triton X-100 (Sigma Aldrich, St. Louis, MO). After blocking the slides with 10% normal goat serum (Chemicon, Temecula, CA) cells were incubated with anti-HMGB1 antibodies for 1 h. Raw cells and lung tissues were incubated with anti-rabbit IgG (Molecular probes, Eugene, OR) and horseradish peroxidase conjugated secondary antibody, respectively, for 1 h. Normal non-specific serum was used as a negative control. Peroxidase-linked secondary antibody and diaminobenzidine (DAB) (Vectastain ABC elite kit; Vector Laboratories, Burlingame, CA) were used to detect specific binding. The sections were counterstained with hematoxylin as described previously [42] and examined using light microscopy (Olympus America Inc., Center Valley, PA). RAW cells were counterstained with 4,6-diamidine-2-phenylindole dihydrochloride (DAPI) (Roche Molecular Biochemicals, Indianapolis, IN) to visualize the nuclei using immunofluorescence microscopy (Nikon, Melville, NY).
Analysis of HMGB1 by liquid chromatography tandem mass spectrometry (LC–MS/MS)
All chemicals and solvents were of the highest available grade (Sigma-Aldrich, Poole, UK). Samples were pre-cleared with 50 µl protein G-Sepharose beads for 1 h at 4 °C. Supernatant HMGB1 was immunoprecipitated with 5 µg rabbit anti-HMGB1 (Abcam, Cambridge, UK) for 16 h at 4 °C as previously described [3]. Free thiol groups within HMGB1 were alkylated for 90 min with 10 mM iodoacetamide at 4 °C. Cysteine residues in disulfide bonds were then reduced with 30 mM dithiothreitol (DTT) at 4 °C for 1 h followed by alkylation of newly exposed thiol groups with 90 mM NEM at 4 °C for 10 min. Samples were subjected to GluC (New England Biolabs, Herts, UK) digestion according to manufacturer׳s instructions and de-salted using ZipTip C18 pipette tips (Millipore, Consett, UK). The characterization of acetylated lysine residues within HMGB1 was determined as described previously [2,32] using an AB Sciex TripleTOF 5600 (Sciex Inc.).
Statistical analysis
All experiments were performed independently twice. The data are presented as mean±standard error, and analyzed for statistical significance using the one-way analysis of variance (ANOVA). A P value of ≤0.05 was considered significant.
Result
Hyperoxia-induced inflammatory acute lung injury is associated with elevated levels of airway HMGB1
To determine whether extracellular HMGB1 may contribute to hyperoxia-induced ALI, markers of inflammatory ALI and levels of airway HMGB1 were assessed by Western blot analysis in the BALF of C57BL/6 mice that were exposed to hyperoxia (≥99% O2) for up to 4 days. As shown in Fig. 1A, airway HMGB1 became detectable in the BALF after 2 days of hyperoxic exposure and the signal became more pronounced after 3 and 4 days of exposure. Prolonged hyperoxic exposure (4 days) significantly increased markers of inflammatory ALI, including the levels of total protein content (Fig. 1B) and total PMNs count in BALF (Fig. 1C), as well as wet/dry weight ratio (Fig. 2B). The levels of total protein content in lung BALF were 0.42±0.003×103 µg/ml at day 1, 0.52±0.003×103 µg/ml at day 2, 1.91±0.03×103 µg/ml at day 3, and 4.62±0.06×103 µg/ml at day 4, compared to 0.45±0.003×103 µg/ml in animals remained at room air (RA, 21% O2) (Fig. 1B). There was a significant elevation of PMNs in the airways (0.24±0.02×104/ml BALF at day 3 and 2.47±0.6×104/ml BALF at day 4) (Fig. 1C). These data indicate a relationship between elevated levels of airway HMGB1 and significant inflammatory lung injury in mice subjected to prolonged hyperoxic exposure.
Fig. 1.
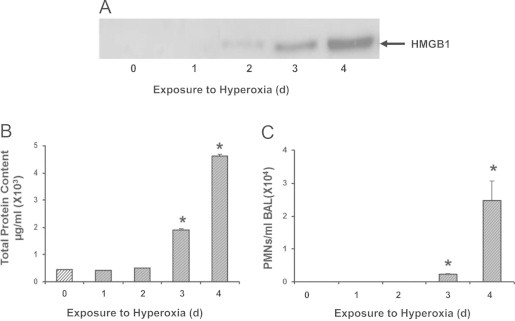
Hyperoxia-induced lung injury is associated with increased accumulation of HMGB1 in the airways. C57BL/6 mice were exposed to ≥99% O2 for indicated days (d) or remained at RA (Exposure to hyperoxia = 0 d). Levels of airway HMGB1 were analyzed by western blot analysis in mouse bronchoalveolar lavage fluids (BALF). Blots shown are representative of three independent experiments with similar results (A). Total protein content (B) and neutrophil (PMNs) infiltration (C) in the airway were analyzed as markers of inflammatory ALI. Data represent means±SE from two independent experiments, n=9 mice per group. ⁎, Statistically significant vs. the values of the control group that remained at RA (Exposure to hyperoxia = 0 d), P<0.05.
Fig. 2.
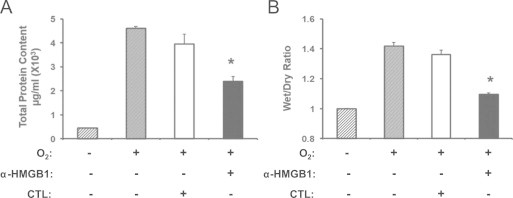
Pretreatment with anti-HMGB1 IgGs attenuates hyperoxia-induced inflammatory acute lung injury. Two hours prior to hyperoxic exposure, mice were treated intraperitonealy with either 360 μg/mouse anti-HMGB1 IgGs (α-HMGB1) or control IgGs (CTL). The animals were then exposed to ≥99% O2 for 4 days while receiving IgGs treatment every 12 h. Total protein content in BALF (A) and wet/dry weight ratio (B) were analyzed as markers of acute inflammatory lung injury. Data represent means±SE from two independent experiments, n=9 mice per group. ⁎, Statistically significant compared to that of mice either treated with control antibodies or exposed to hyperoxia alone, P<0.05.
Pretreatment with anti-HMGB1 antibodies protects against hyperoxia-induced inflammatory acute lung injury
To establish a causal relationship between elevated levels of airway HMGB1 and hyperoxia-induced inflammatory ALI, neutralizing polyclonal anti-HMGB1 IgGs [49] were administered to mice prior to exposure to hyperoxia. Mice pretreated with anti-HMGB1 IgGs had significantly decreased hyperoxia-induced protein leakage into the airways compared to mice that received control IgGs (2.4±0.25×103 µg/ml vs. 4.62±0.64×103 µg/ml; P<0.01) (Fig. 2A). In addition, mice receiving anti-HMGB1 IgGs had significantly less lung edema, as measured by the wet/dry weight ratio, compared to mice that received control IgGs (Fig. 2B, 1.1±0.02 vs. 1.36±0.04; P<0.005). In contrast, there was no statistically significant difference in these inflammatory ALI parameters between mice that received control IgGs and those exposed to hyperoxia alone (Fig. 2A and B). These data indicate that inhibiting airway HMGB1 attenuated lung injury, suggesting that HMGB1 plays a key role in mediating hyperoxic lung injury.
Hyperoxia induces hyperacetylation and translocation of nuclear HMGB1 to the cytoplasm
HMGB1, which plays an important role in the regulation of gene transcription in the nuclei [10], also contributes to the pathogenesis of various inflammatory diseases upon release into the extracellular milieu [7,19]. To determine whether hyperoxia-induced HMGB1 release into the airways is a result of the translocation of HMGB1 from the nuclei to the cytoplasm, an indicator of active release from cells not undergoing cell death, immunohistochemical analysis was performed in lung tissue sections of mice exposed to hyperoxia for 4 days. In mice that remained at RA (Fig. 3A, RA), HMGB1 was found predominantly in the nuclei of most lung cells. In contrast, HMGB1 was localized mainly in the cytoplasm of many lung cells in mice exposed to hyperoxia (Fig. 3A, O2). Thus, prolonged hyperoxic exposure resulted in HMGB1 translocation from the nuclei to the cytoplasm of lung cells. To confirm our in vivo findings, HMGB1 localization was characterized in murine macrophage-like RAW 264.7 cells exposed to 95% O2. HMGB1 was localized primarily in the nuclei of RAW cells that remained at RA. However, after 48 h hyperoxic exposure (O2), HMGB1 staining was observed predominantly in the cytoplasm of the RAW cells (Fig. 3A). This data indicates that hyperoxia induces the active release of HMGB1 from the nuclei to the cytoplasm of lung cells.
Fig. 3.
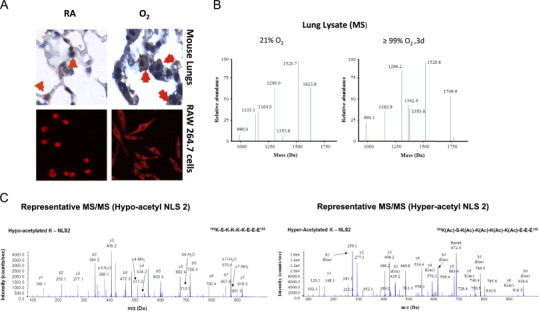
Hyperoxia induces hyperacetylation and translocation of nuclear HMGB1 to the cytoplasm. C57BL/6 mice were exposed to either 4 days of ≥99% O2 or remained at RA. (A) Images of lung tissue sections (original magnification, X1000) stained with anti-HMGB1 IgGs and DAB (3, 3'-diaminobenzidine) peroxidase substrate (brown color). The red signals showed immunofluoresence staining with anti-HMGB1 IgGs and Cy3 in RAW 264.7 macrophages that were either exposed to 95% O2 for 48 h or remained at RA (original magnification, X600). (B) Representative spectra of the liquid chromatography mass spectrometric (LC–MS) characterization of peptides produced from HMGB1 derived from mice lung tissue homogenate (Lung Lysate) enzymatically cleaved with endopeptidase GluC. The presence of the peptides with molecular weights 1624 and 1133 Da indicate the hypo-acetylation of lysine residues within NLS 1 and 2, respectively. The presence of the peptides with molecular weights 1750 and 1342 Da indicate the hyper-acetylation of lysine residues within NLS 1 and 2 respectively. (C) Representative spectra of the liquid chromatography tandem mass spectrometric (LC–MS/MS) characterization of a peptide (amino acids 180–188) covering the lysine (K) residues within NLS 2 of HMGB1 to confirm the presence or absence of acetyl modifications on specific K residues. Acetyl modifications are represented as (ac) on specific lysine residues (K181, K182, K183 and K184) when required and b and y ions are highlighted were appropriate.
An important factor in regulating the translocation of HMGB1 is its acetylation status. The lysine residues of two nuclear localization signal (NLS) sites on HMGB1 can be acetylated. Hyperacetylation of the lysine residues on NLS1 and NLS2 induces the release of HMGB1 from cells, whereas hypoacetylated HMGB1 tends to remain in the nuclei [56]. To determine the acetylation status of HMGB1 in hyperoxia, lung lysates and BALF of mice exposed to either 3 days of hyperoxia or remained at RA were analyzed using LC–MS/MS as previously described [2,32]. HMGB1 was primarily in the hypoacetylated form in animals remained at RA (Fig. 3B, 21% O2). However, HMGB1 was significantly hyperacetylated in mice exposed to hyperoxia (Fig. 3B, ≥99% O2). Fig. 3C shows representative images of the MS/MS analysis of hypoacetylation (left) and hyperacetylation (right) of HMGB1. These data suggest that hyperoxic exposure leads to hyperacetylation of HMGB1, resulting in its pronounced translocation to the cytoplasm and subsequent release.
Hyperoxia increases oxidative states of airway HMGB1 and induces leukocyte infiltration into the lungs
Inflammatory ALI is defined by the infiltration of leukocytes, especially PMNs into the lungs [37]. To gain insights into the mechanism underlying how HMGB1 contributes to the development of hyperoxia-induced lung injury, we evaluated the effect of HMGB1 on leukocyte infiltration in mice exposed to hyperoxia. After 24 h of hyperoxic exposure, mice were intratracheally instilled with 10 µg of either recombinant HMGB1 with GST-tag or GST-tag alone [54] and exposed to ≥99% O2 for an additional 24 h. Recombinant HMGB1 instillation significantly increased the infiltration of leukocytes compared to hyperoxia, control GST-tag and RA (34±3.93×104/ml BALF vs. 19.75±2.75×104/ml BALF, 16±1.8×104/ml BALF, and 11.1±1.5×104/ml BALF; P<0.01). HMGB1 significantly increased the infiltration of PMNs into the airways, compared to either hyperoxia, GST-tag alone or RA (2.42±0.41×104/ml BALF vs. 0/ml BALF and 0.35±0.12×104/ml BALF, and 0/ml BALF; P<0.001) (Fig. 4A and B). In contrast, the administration of anti-HMGB1 IgGs significantly attenuated the infiltration of leukocytes compared to those receiving vehicle or control IgGs (39.8±5.8×104/ml BALF vs. 87.1±7.1×104/ml BALF and 69.45±8.7×104/ml BALF; P<0.01, respectively), as well as PMNs (0.37±0.08×104/ml BALF vs. 2.86±0.7×104/ml BALF and 2.21±0.6×104/ml BALF; P<0.01) in mice exposed to ≥99% O2 for an additional 24 h (Fig. 4C and D). Overall, these data suggest that HMGB1 plays a critical role in mediating hyperoxia-induced inflammatory ALI through evoking leukocyte infiltration into the lungs.
Fig. 4.
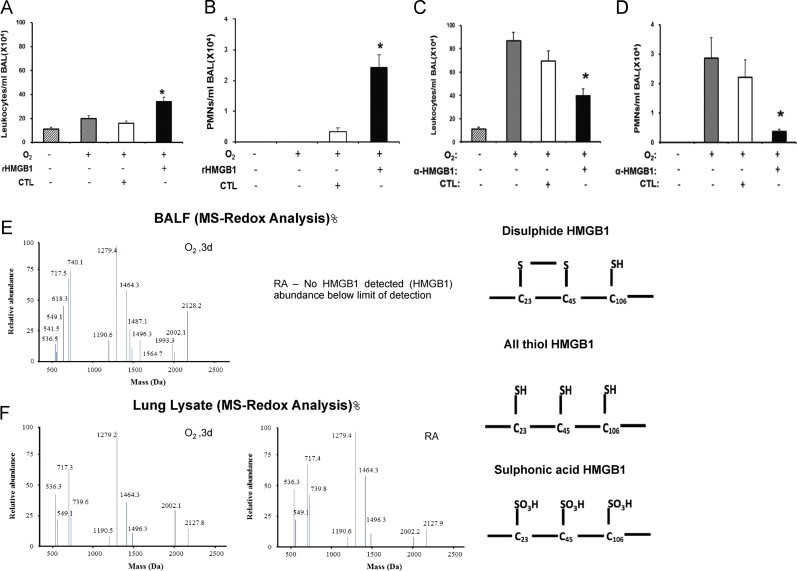
Hyperoxia increases oxidative states of airway HMGB1 that induces leukocyte infiltration into the lungs. C57BL/6 mice were given 10 μg rHMGB1- or gluthatione-s-transferase (GST-tag) as control (CTL) via intratracheal instillation 24 h post-hyperoxic (≥99% O2) exposure. Mice were then exposed to hyperoxia for an additional 24 h. Total numbers of leukocytes and PMNs infiltrated into the airways were analyzed as markers of inflammatory ALI. Data represent means±SE from two independent experiments, n=9 mice per group. ⁎, Statistically significant vs. the values of the control group which received GST-tag, P<0.05 (A and B). Mice were treated intraperitonealy with either 360 μg/mouse anti-HMGB1 IgGs (α-HMGB1) or control IgGs (CTL) 2 h prior to hyperoxic exposure. Mice were then exposed to ≥99% O2 and received either α-HMGB1 IgGs or control IgGs intraperitonealy every 12 h for 4 days while still being exposed to ≥99% O2. Total numbers of leukocytes and PMNs infiltrated into the airways were analyzed. Data represent means±SE from two independent experiments, n=9 mice per group. ⁎, Statistically significant vs. mice treated with control IgGs, P<0.05 (C and D). BALF and lung tissue from mice exposed to ≥99% O2 for 3 days or that remained at RA, were analyzed for the presence of three redox forms of HMGB1: disulfide, all thiol and sulfonic acid using LC–MS (E and F).
Three critical cysteine residues (C23, C45 and C106) of HMGB1 are subjected to redox modifications, which affect its chemotactic activity [52]. To determine the redox state of extracellular HMGB1 in hyperoxia exposed animals, LC–MS/MS was used to analyze BALF of mice exposed to ≥99% O2 for 3 days. MS/MS analysis indicated that all three redox forms of HMGB1 were present in BALF of hyperoxia exposed mice, including all reduced and oxidized thiols, and a disulfide bond between C23 and C45 with C106 reduced (Fig. 4E). However, no extracellular HMGB1 was detected in the BALF of mice that were remained at RA (Fig. 4E). In the lungs of both hyperoxia exposed mice and mice that remained at RA, HMGB1 was in the reduced form (Fig. 4F). These data suggest that hyperoxia-induced oxidative modifications of HMGB1 occurred after its release into the airways.
Administration of anti-HMGB1 IgGs post-onset of hyperoxic exposure attenuates hyperoxia-induced inflammatory acute lung injury
Although the administration of anti-HMGB1 IgGs prior to exposing animals to hyperoxia was an important proof of concept, strategies aimed at inhibiting HMGB1 after the onset of hyperoxic exposure are more clinically relevant. Therefore, we assessed the efficacy of administrating anti-HMGB1 IgGs to animals post-hyperoxic exposure on inflammatory ALI. As shown in Fig. 5, the administration of anti-HMGB1 IgGs 24 h post-exposure to hyperoxia significantly attenuated hyperoxia-induced inflammatory ALI, as indicated by a significant decrease in total protein content in BALF (2.4±0.2×103 µg/ml vs. 4.2±0.6×103 µg/ml; P<0.01), wet/dry weight ratio (1.01±0.02 vs. 1.36±0.04; P<0.05), leukocyte infiltration (39.8±5.8×104/ml BALF vs. 69.45±8.7×104/ml BALF; P<0.01), and PMNs infiltration (0.37±0.08×104/ml BALF vs. 2.21±0.6×104/ml BALF; P<0.01) (Fig. 5A). The attenuation of inflammatory ALI by anti-HMGB1 IgGs was confirmed by histologic examination of lung tissue sections (Fig. 5B). Alveolar hemorrhage and thickening of the interalveolar septum were significantly decreased in lung tissues of animals treated with anti-HMGB1 IgGs compared to those treated with control IgGs or those exposed to hyperoxia alone (Fig. 5B). These results indicate that blocking HMGB1 after the onset of hyperoxia exposure can alleviate hyperoxia-induced inflammatory ALI.
Fig. 5.
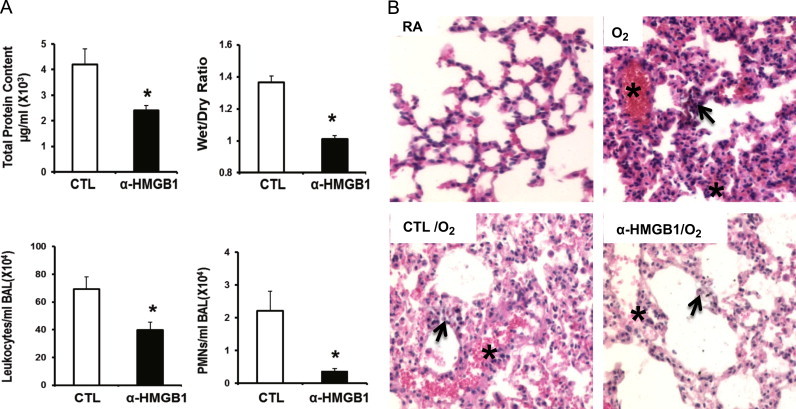
Administration of anti-HMGB1 IgGs post-onset of hyperoxic exposure attenuates hyperoxia-induced inflammatory acute lung injury. After 24 h of exposure to ≥99% O2 mice were treated intraperitonealy with 360 μg/mouse of either anti-HMGB1 IgGs (α-HMGB1) or control IgGs (CTL) every 12 h for 3 days while still being exposed to ≥99% O2. Total protein content, wet/dry weight ratio, and total numbers of leukocytes and PMNs infiltrated in airways were analyzed as markers of inflammatory ALI. Data represent means±SE from two independent experiments, n=13 mice per group. ⁎, Statistically significant vs. mice treated with control IgGs, P<0.05 (A). Hematoxylin–eosin-stain of the lung tissues from control and experimental groups of mice (original magnification 200×). Images are representative lung sections from 5 mice per group. Asterisks (⁎) show the alveolar hemorrhage and arrows show the thickening of the interalveolar septum in lung tissues of animals (B).
Ethyl pyruvate (EP) attenuates hyperoxia-induced inflammatory acute lung injury via inhibiting HMGB1 release
Ethyl pyruvate (EP) has been reported to inhibit LPS-induced HMGB1 secretion [44]. To investigate the effects of EP on hyperoxia-induced lung injury, mice were given 40 mg/kg intraperitoneal of EP 24 h post-hyperoxic exposure. The administration of EP significantly decreased the inflammatory ALI in hyperoxic animals as compared to animals administered vehicle control alone, as indicated by a decreased total protein content in BALF (1.87±0.34×103 µg/ml vs. 3.71±0.69×103 µg/ml; P<0.01) (Fig. 6A) and wet/dry weight ratio (0.98±0.02 vs. 1.3±0.04; P<0.01) (Fig. 6B), and significantly lower levels of airway leukocytes (29±4.9×104/ml BALF vs. 70.7±5.8×104/ml BALF; P<0.01) (Fig. 6C) and PMNs (0.96±0.3×104/ml BALF vs. 3±0.42×104/ml BALF; P<0.01) (Fig. 6D).
Fig. 6.
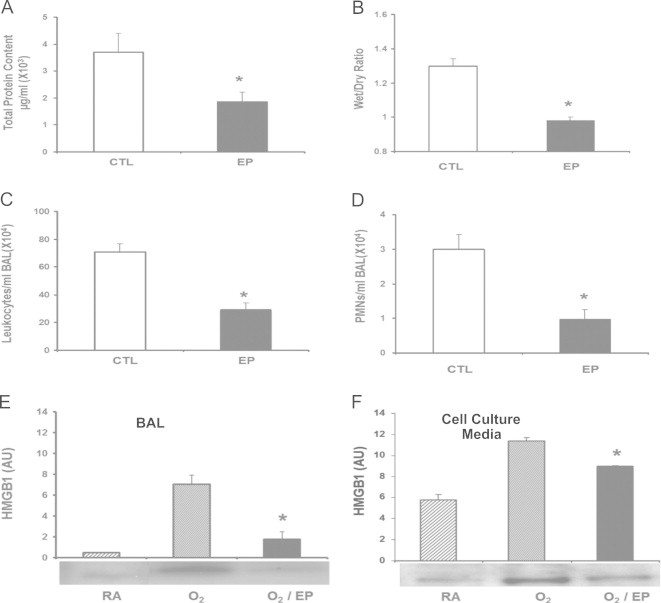
Ethyl pyruvate (EP) attenuates hyperoxia-induced inflammatory acute lung injury via inhibiting HMGB1 release. After 24 h of exposure to ≥99% O2 mice were treated intraperitonealy with 40 mg/kg of either ethyl pyruvate (EP) or saline as a control (CTL) every 12 h for 3 days while still being exposed to ≥99% O2. Total protein (A), wet/dry weight ratio (B), total numbers of leukocytes (C), and PMNs (D) infiltrated in the airways were analyzed as markers of acute inflammatory lung injury. Data represent means±SE from two independent experiments, n=9 mice per group. *, Statistically significant vs. mice treated with saline, P<0.005. Levels of airway HMGB1 in the BALF were analyzed by western blot. (A) The relative optical intensity in arbitrary units (AU) of the immunoreactive bands on Western blots expressed as mean±SE of two independent experiments (n=9). *, Statistically significant vs. the group that was exposed to ≥99% O2 alone for the same period of time; P<0.05. Raw 264.7 macrophages were exposed to 95% O2 for 24 h with or without the addition of 5 mM ethyl pyruvate (EP). (B) Levels of extracellular HMGB1 were measured in culture media by Western blots. The relative optical intensity in arbitrary units (AU) of the immunoreactive bands on Western blots expressed as mean±SE of three independent experiments (n=6). ⁎, Statistically significant vs. the group that was exposed to hyperoxia alone; P<0.05.
To determine the mechanism underlying the effects of EP on hyperoxic ALI, levels of airway HMGB1 were determined in the BALF of animals exposed to hyperoxia and treated with EP. As shown in Fig. 6E, the levels of airway HMGB1 were significantly lower in mice treated with EP compared to those exposed to vehicle control alone (1.78±0.71 vs. 7.6±0.85 AU; P<0.01). The inhibitory effect of EP on hyperoxia-induced HMGB1 release was further confirmed in RAW 264.7 cells exposed to 95% O2. EP treatment (5 mM) significantly reduced HMGB1 release into the culture media of RAW cells (8.98±0.06 AU vs. 11.35±0.31 AU; P<0.001) (Fig. 6F). These results demonstrate that EP protects animals against hyperoxia-induced inflammatory ALI by inhibiting HMGB1 release into the airways.
Discussion
High levels of airway HMGB1 were found in patients receiving MV, although the contribution of HMGB1 to hyperoxia-induced ALI remains largely undefined. In the present study, we provide evidence of HMGB1 as a critical mediator for developing ALI upon prolonged hyperoxia exposure via recruiting PMNs. Our results manifest that increased levels of airway HMGB1 during hyperoxia are linked to the induction of inflammatory ALI, while inhibition of HMGB1 protects lung tissue against hyperoxia-induced injury through decreasing pulmonary neutrophil accumulation. Our data also show that EP has protective effects against hyperoxia-induced inflammatory ALI by inhibiting hyperoxia-induced HMGB1 release into the airways.
Hyperoxia-induced leukocyte infiltration plays a critical role in the development of ALI [4]. Several lines of evidence presented in this report demonstrate that elevated levels of airway HMGB1 mediate leukocyte infiltration under hyperoxic conditions. First, levels of airway HMGB1 increased time-dependently upon hyperoxic exposure for up to 4 days (Fig. 1A). The elevated levels of airway HMGB1 were preceding the recruitment of neutrophils into the airways and lung injury (Fig. 1C and B). In addition, instillation of 10 μg of HMGB1, amount that is similar to those found in patients with ALI [40], significantly increased leukocyte infiltration into the airways of mice exposed to hyperoxia (Fig. 4A and B). Furthermore, pretreatment of mice exposed to hyperoxia with neutralizing anti-HMGB1 antibodies ameliorated hyperoxia-induced neutrophil infiltration and lung injury (Figs. 2 and 4C and D). Thus, our results are similar to those of other studies indicating that HMGB1 is a critical mediator of ALI in various pathologic conditions [1,23,26,27]. The inactivation of HMGB1 with specific antibodies improved survival of animals with severe polymicrobial sepsis, reduced high tidal volume ventilation-induced lung injury, and diminished endotoxin and hemorrhage-induced increases in pulmonary level of inflammatory cytokines [1,27,33,43,49]. Together, these studies indicate that HMGB1 plays an important role in neutrophil-mediated lung injury induced by either oxidative stress or reagents.
In this report, we demonstrated that hyperacetylation of HMGB1 may be a critical mechanism underlying hyperoxia-induced HMGB1 release from lung cells. Fig. 3 shows that hyperoxia-induced HMGB1 hyperacetylation corresponds to the cytoplasmic translocation and release of nuclear HMGB1. Mass spectrometric analysis of lung lysates shows that four lysine residues on NLS1 and five on NLS2 motifs of HMGB1 were hyperacetylated after 3 days of exposure to hyperoxia, while no hyperacetylation of HMGB1 was observed in lung lysates of mice remained at RA (Fig. 3B and C). These results suggest a correlation between hyperoxia-induced HMGB1 translocation and subsequent release with hyperacetylation of lysine residues. Under normoxic conditions, acetylation of lysine residues of HMGB1 has also been implicated to regulate this nucleocytoplsamic shuttling and its subsequent active release from LPS-activated monocytes and macrophages [8]. These results suggest the involvement of hyperacetylation in hyperoxia-induced HMGB1 release, although further studies are needed to establish a causal relation between hyperacetylation of HMGB1 and its release under hyperoxic conditions.
Post-translational redox modifications of three critical thiol groups of HMGB1 are important in determining its bioactivity [41]. Although no detectable extracellular HMGB1 was found in the airways of mice exposed to RA, airway HMGB1 molecules in mice exposed to prolonged hyperoxia were found in different redox states, including all reduced thiol groups and disulfide forms (Fig. 4E and F). Two forms of HMGB1, all reduced and disulfide (C23 and C45), possess chemoattractant and cytokine-inducing activities, respectively, while terminally oxidized forms lack any inflammatory activity [25,41,48,53]. The presence of the two not-terminally oxidized forms of HMGB1 in the airways of hyperoxia-exposed mice supports HMGB1׳s role in hyperoxia-induced inflammatory responses. Further quantification of each redox form of HMGB1 in hyperoxic airways and characterization of their specific roles in the inflammatory response still remain to be determined. Interestingly, airway HMGB1 molecules present in mice exposed to hyperoxia exist only in its hyperacetylated form, even though they are in different redox states (Figs. 4 and 5). Currently, however, it is not clear and needs to be clarified whether hyperacetylation of the different redox forms of HMGB1 affect its inflammatory activities.
Pretreatment with antibodies provides proof of the concept that airway HMGB1 mediates hyperoxia-induced ALI. However, administration of reagents to inhibit the accumulation or the activity of airway HMGB1 post-onset of hyperoxic exposure would be more clinically relevant. Our results shown in Figs. 5 and 6 demonstrate that this strategy is effective in ameliorating hyperoxia-induced both number of neutrophils in the lungs and ALI. Though highly effective, high production costs and large molecular sizes, which affect pharmacokinetics, may limit clinical use of antibody therapies [11]. Thus, the use of EP, a small molecule, in reducing hyperoxia-induced inflammatory ALI (Fig. 6), may circumvent the limitations associated with antibody therapies. The amelioration of lung injury by EP was most likely due to a decrease in hyperoxia-induced accumulation of airway HMGB1 (Fig. 6). Others have shown that EP treatment also decreased LPS-induced HMGB1 release, even with delayed treatment, 2 h after LPS administration and 24 h after cecal ligation and puncture surgery [36,46,54]. Similarly, EP attenuated high tidal volume induced HMGB1 expression and ameliorated lung injury [22].
In conclusion, this study provides a novel mechanism underlying hyperoxia-induced inflammatory ALI and potential therapeutic approaches to mitigate such injury. We find that HMGB1 mediates, at least in part, hyperoxia-induced neutrophil infiltration into the lungs. Post-translational modifications of HMGB1 such as hyperacetylation may play a critical role in translocation of nuclear HMGB1 to the cytoplasm and subsequent release into the airways. Inhibition of extracellular HMGB1 and/or its accumulation in the airways, with either antibodies or small molecules, provides significant protection against hyperoxic lung injury. Thus, therapeutic approaches targeting airway HMGB1, using small inhibitory molecules, such as EP, may attenuate lung injury in patients on oxygen therapy.
Acknowledgments
This work was supported by Grants (LLM) National Heart and Blood Institute (HL093708) and St. John׳s University. Author DJA would like to acknowledge financial support from the Medical Research Council and a Wellcome Trust Research fellowship. The authors would like to thank Dr. Louis Trombetta for his insightful discussion, Dr. Edmund Miller and Ms. Shloka Reddy for their help in editing the manuscript.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
This work was supported by grants (LLM) from National Heart and Blood Institute (HL093708), Feinstein Institute for Medical Research and St. John׳s University.
References
- 1.Abraham E., Arcaroli J., Carmody A., Wang H., Tracey K.J. HMG-1 as a mediator of acute lung inflammation. J. Immunol. 2000;165:2950–2954. doi: 10.4049/jimmunol.165.6.2950. [DOI] [PubMed] [Google Scholar]
- 2.Antoine D.J., Jenkins R.E., Dear J.W., Williams D.P., McGill M.R., Sharpe M.R., Craig D.G., Simpson K.J., Jaeschke H., Park B.K. Molecular forms of HMGB1 and keratin-18 as mechanistic biomarkers for mode of cell death and prognosis during clinical acetaminophen hepatotoxicity. J. Hepatol. 2012;56(5):1070–1079. doi: 10.1016/j.jhep.2011.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 3.Antoine D.J., Williams D.P., Kipar A., Jenkins R.E., Regan S.L., Sathish J.G. High-mobility group box-1 protein and keratin-18, circulating serum proteins informative of acetaminophen-induced necrosis and apoptosis in vivo. Toxicol. Sci. 2009;112(2):521–531. doi: 10.1093/toxsci/kfp235. [DOI] [PubMed] [Google Scholar]
- 4.Auten R.L., Whorton M.H., Nicholas Mason S. Blocking neutrophil influx reduces DNA damage in hyperoxia-exposed newborn rat lung. Am. J. Respir. Cell Mol. Biol. 2002;26(4):391–397. doi: 10.1165/ajrcmb.26.4.4708. [DOI] [PubMed] [Google Scholar]
- 5.Belperio J.A. The role of cytokines during the pathogenesis of ventilator-associated and ventilator-induced lung injury. Semin. Respir. Crit. Care Med. 2006;27:350–364. doi: 10.1055/s-2006-948289. [DOI] [PubMed] [Google Scholar]
- 6.Bhandari V. Molecular mechanisms of hyperoxia-induced acute lung injury. Front. Biosci. 2008;13:6653–6661. doi: 10.2741/3179. [DOI] [PubMed] [Google Scholar]
- 7.Bianchi M.E. DAMPs, PAMPs and alarmins: all we need to know about danger. J. Leukoc. Biol. 2007;81(1):1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 8.Bonaldi T., Talamo F., Scaffidi P., Ferrera D., Porto A., Bachi A., Rubartelli A., Agresti A., Bianchi M.E. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003;22(20):5551–5560. doi: 10.1093/emboj/cdg516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Budinger G.R. Epithelial cell death is an important contributor to oxidant-mediated acute lung injury. Am. J. Respir. Crit. Care Med. 2011;183:1043–1054. doi: 10.1164/rccm.201002-0181OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bustin M. Regulation of DNA-dependent activities by the functional motifs of the high-mobility-group chromosomal proteins. Mol. Cell. Biol. 1999;19(8):5237–5246. doi: 10.1128/mcb.19.8.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chames P., Van Regenmortel M., Weiss E., Baty D. Therapeutic antibodies: successes, limitations and hopes for the future. Br. J. Pharmacol. 2009;157(2):220–233. doi: 10.1111/j.1476-5381.2009.00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cordingley J.J., Keogh B.F. The pulmonary physician in critical care. 8: Ventilatory management of ALI/ARDS. Thorax. 2002;8:729–734. doi: 10.1136/thorax.57.8.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crapo J.D., Barry B.E., Foscue H.A., Shelburne J. Structural and biochemical changes in rat lungs occurring during exposures to lethal and adaptive doses of oxygen. Am. Rev. Respir. Dis. 1980;122:123–143. doi: 10.1164/arrd.1980.122.1.123. [DOI] [PubMed] [Google Scholar]
- 14.Esteban A. How is mechanical ventilation employed in the intensive care unit? An international utilization review. Am. J. Respir. Crit. Care Med. 2000;161:1450–1458. doi: 10.1164/ajrccm.161.5.9902018. [DOI] [PubMed] [Google Scholar]
- 15.Franek W.R., Horowitz S., Stansberry L., Kazzaz J.A., Koo H.C., Li Y., Arita Y., Davis J.M., Mantell A.S., Scott W., Mantell L.L. Hyperoxia inhibits oxidant-induced apoptosis in lung epithelial cells. J. Biol. Chem. 2001;276:569–575. doi: 10.1074/jbc.M004716200. [DOI] [PubMed] [Google Scholar]
- 16.Franek W.R., Morrow D.M., Zhu H., Vancurova I., Miskolci V., Darley-Usmar K., Simms H.H., Mantell L.L. NF-kappaB protects lung epithelium against hyperoxia-induced nonapoptotic cell death-oncosis. Free Radic. Biol. Med. 2004;37:1670–1679. doi: 10.1016/j.freeradbiomed.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 17.Goodman R.B., Strieter R.M., Martin D.P., Steinberg K.P., Milberg J.A., Maunder R.J., Kunkel S.L., Walz A., Hudson L.D., Martin T.R. Inflammatory cytokines in patients with persistence of the acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 1996;154:602–611. doi: 10.1164/ajrccm.154.3.8810593. [DOI] [PubMed] [Google Scholar]
- 18.Grommes J., Soehnlein O. Contribution of neutrophils to acute lung injury. Mol. Med. 2011;34:293–307. doi: 10.2119/molmed.2010.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harris H.E., Raucci A. Alarmin(g) news about danger: workshop on innate danger signals and HMGB1. EMBO Rep. 2006;7(8):774–778. doi: 10.1038/sj.embor.7400759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jackson R.M. Molecular, pharmacologic, and clinical aspects of oxygen-induced lung injury. Clin. Chest Med. 1990;11:73–86. [PubMed] [Google Scholar]
- 21.Kallet R.H., Matthay M.A. Hyperoxic acute lung injury. Respir. Care. 2013;58:123–141. doi: 10.4187/respcare.01963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li L.F., Kao K.C., Yang C.T., Huang C.C., Liu Y.Y. Ethyl pyruvate reduces ventilation-induced neutrophil infiltration and oxidative stress. Exp. Biol. Med. 2012;237(6):720–727. doi: 10.1258/ebm.2012.011184. [DOI] [PubMed] [Google Scholar]
- 23.Li L.F., Yang C.T., Huang C.C., Liu Y.Y., Kao K.C., Lin H.C. Low-molecular-weight heparin reduces hyperoxia-augmented ventilator-induced lung injury via serine/threonine kinase-protein kinase B. Respir. Res. 2011;12:90. doi: 10.1186/1465-9921-12-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin X., Yang H., Sakuragi T., Hu M., Mantell L.L., Hayashi S., Al-Abed Y., Tracey K.J., Ulloa L., Miller E.J. Alpha-chemokine receptor blockade reduces high mobility group box 1 protein-induced lung inflammation and injury and improves survival in sepsis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005;289(L5):83–90. doi: 10.1152/ajplung.00091.2005. [DOI] [PubMed] [Google Scholar]
- 25.Liu G., Wang J., Park Y.J., Tsuruta Y., Lorne E.F., Zhao X., Abraham E. High mobility group protein-1 inhibits phagocytosis of apoptotic neutrophils through binding to phosphatidylserine. J. Immunol. 2008;181(6):4240–4246. doi: 10.4049/jimmunol.181.6.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lotze M.T., Tracey K.J. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 2005;5:331–342. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- 27.Lutz W., Stetkiewicz J. High mobility group box 1 protein as a late-acting mediator of acute lung inflammation. Int. J. Occup. Med. Environ. Health. 2004;17:245–254. [PubMed] [Google Scholar]
- 28.Mantell L.L., Kazzaz J.A., Xu J., Palaia T.A., Piedboeuf B., Hall S., Rhodes G.C., Niu G., Fein A.F., Horowitz S. Unscheduled apoptosis during acute inflammatory lung injury. Cell Death Differ. 1997;4:600–607. doi: 10.1038/sj.cdd.4400278. [DOI] [PubMed] [Google Scholar]
- 29.Mantell L.L., Parrish W.R., Ulloa L. Hmgb-1 as a therapeutic target for infectious and inflammatory disorders. Shock. 2006;25:4–11. doi: 10.1097/01.shk.0000188710.04777.9e. [DOI] [PubMed] [Google Scholar]
- 30.Mantell L.L., Shaffer T.H., Horowitz S., Foust R., 3rd, Wolfson M.R., Cox C., Khullar P., Zakeri Z., Lin L., Kazzaz J.A., Palaia T., Scott W., Davis J.M. Distinct patterns of apoptosis in the lung during liquid ventilation compared with gas ventilation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002;283:L31–L41. doi: 10.1152/ajplung.00037.2001. [DOI] [PubMed] [Google Scholar]
- 31.Morrow D.M., Entezari-Zaher T., Romashko J., 3rd, Azghani A.O., Javdan M., Ulloa L., Miller E.J., Mantell L.L. Antioxidants preserve macrophage phagocytosis of Pseudomonas aeruginosa during hyperoxia. Free Radic. Biol. Med. 2007;42:1338–1349. doi: 10.1016/j.freeradbiomed.2007.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nyström S., Antoine D.J., Lundbäck P., Lock J.G., Nita A.F., Högstrand K., Grandien A., Erlandsson-Harris H., Andersson U., Applequist S.E. TLR activation regulates damage-associated molecular pattern isoforms released during pyroptosis. EMBO J. 2013;32(1):86–99. doi: 10.1038/emboj.2012.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ogawa E.N., Ishizaka A., Tasaka S., Koh H., Ueno H., Amaya F., Ebina M., Yamada S., Funakoshi Y., Soejima J., Moriyama K., Kotani T., Hashimoto S., Morisaki H., Abraham E., Takeda J. Contributions of High Mobility Group Box-1 to the development of VILI. Am. J. Respir. Crit. Care Med. 2006;174:400–407. doi: 10.1164/rccm.200605-699OC. [DOI] [PubMed] [Google Scholar]
- 34.Olson T.S., Ley K. Chemokines and chemokine receptors in leukocyte trafficking. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002;283(1):R7–R28. doi: 10.1152/ajpregu.00738.2001. [DOI] [PubMed] [Google Scholar]
- 35.Pagano A., Barazzone-Argiroffo C. Alveolar cell death in hyperoxia-induced lung injury. Ann. N. Y. Acad. Sci. 2003;1010:405–416. doi: 10.1196/annals.1299.074. [DOI] [PubMed] [Google Scholar]
- 36.Shang G.H., Lin D.J., Xiao W., Jia C.Q., Li Y., Wang A.H., Dong L. Ethyl pyruvate reduces mortality in an endotoxin-induced severe acute lung injury mouse model. Respir. Res. 2009;2:10–91. doi: 10.1186/1465-9921-10-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sibille Y., Reynolds H.Y. Macrophages and polymorphonuclear neutrophils in lung defense and injury. Am. Rev. Respir. Dis. 1990;141(2):471–501. doi: 10.1164/ajrccm/141.2.471. [DOI] [PubMed] [Google Scholar]
- 38.Sinclair S.E. Augmented lung injury due to interaction between hyperoxia and mechanical ventilation. Crit. Care Med. 2004;32:2496–2501. doi: 10.1097/01.ccm.0000148231.04642.8d. [DOI] [PubMed] [Google Scholar]
- 39.Stros M. HMGB proteins: interactions with DNA and chromatin. Biochim. Biophys. Acta. 2010;1799:101–113. doi: 10.1016/j.bbagrm.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 40.Talwar S.F.C., Black K., Yates C.R., Meduri G.U., Suffredini A.F. High mobility group protein-1 (HMGB1) is increased in bronchoalveolar lavage (BAL) after lung endotoxin challenge and in patients with acute respiratory distress syndrome (ARDS) (Abstract) Am. J. Respir. Crit. Care Med. 2003;167:A661. [Google Scholar]
- 41.Tang D., Billiar T.R., Lotze M.T.A. Janus tale of two active high mobility group box 1 (HMGB1) redox states. Mol. Med. 2012;18:1360–1362. doi: 10.2119/molmed.2012.00314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsung A., Sahai R., Tanaka H., Nakao A., Fink M.P., Lotze M.T., Yang H., Li J., Tracey K.J., Geller D.A., Billiar T.R. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J. Exp. Med. 2005;201(7):1135–1143. doi: 10.1084/jem.20042614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ueno H., Matsuda T., Hashimoto S., Amaya F., Kitamura Y., Tanaka M., Kobayashi A., Maruyama I., Yamada S., Hasegawa N., Soejima J., Koh H., Ishizaka A. Contributions of high mobility group box protein in experimental and clinical acute lung injury. Am. J. Respir. Crit. Care Med. 2004;170:1310–1316. doi: 10.1164/rccm.200402-188OC. [DOI] [PubMed] [Google Scholar]
- 44.Ulloa L., Tracey K.J. The “cytokine profile”: a code for sepsis. Trends Mol. Med. 2005;11:56–63. doi: 10.1016/j.molmed.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 45.Ulloa L., Batliwalla F.M., Andersson U., Gregersen P.K., Tracey K.J. High mobility group box chromosomal protein 1 as a nuclear protein, cytokine, and potential therapeutic target in arthritis. Arthritis Rheumatol. 2003;48:876–881. doi: 10.1002/art.10854. [DOI] [PubMed] [Google Scholar]
- 46.Ulloa L., Ochani M., Yang H., Tanovic M., Halperin D., Yang R., Czura C.J., Fink M.P., Tracey K.J. Ethyl pyruvate prevents lethality in mice with established lethal sepsis and systemic inflammation. Proc. Natl. Acad. Sci. USA. 2002;99:12351–12356. doi: 10.1073/pnas.192222999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Van Zoelen M.A., Ishizaka A., Wolthuls E.K., Choi G., van der Poll T., Schultz M.J. Pulmonary levels of high-mobility group box 1 during mechanical ventilation and ventilator-associated pneumonia. Shock. 2008;4:441–445. doi: 10.1097/SHK.0b013e318157eddd. [DOI] [PubMed] [Google Scholar]
- 48.Venereau E., Schiraldi M., Uguccioni M., Bianchi M.E. HMGB1 and leukocyte migration during trauma and sterile inflammation. Mol. Immunol. 2013;55(1):76–82. doi: 10.1016/j.molimm.2012.10.037. [DOI] [PubMed] [Google Scholar]
- 49.Wang H., Bloom O., Zhang M., Vishnubhakat J.M., Ombrellino M., Che J., Frazier A., Yang H., Ivanova S., Borovikova L., Manogue K.R., Faist E., Abraham E., Andersson J., Andersson U., Molina P.E., Abumrad N.N., Sama A., Tracey K.J. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–251. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 50.Wang H., Liao H., Ochani M., Justiniani M., Lin X., Yang L., Al-Abed Y., Metz C., Miller E.J., Tracey K.J., Ulloa L. Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat. Med. 2004;10:1216–1221. doi: 10.1038/nm1124. [DOI] [PubMed] [Google Scholar]
- 51.Wheeler A.P., Bernard G.R. Acute lung injury and the acute respiratory distress syndrome: a clinical review. Lancet. 2007;369:1553–1564. doi: 10.1016/S0140-6736(07)60604-7. [DOI] [PubMed] [Google Scholar]
- 52.Yang H., Antoine D.J., Andersson U., Tracey K.J. The many faces of HMGB1: molecular structure–functional activity in inflammation, apoptosis, and chemotaxis. J. Leukoc. Biol. 2013;93(6):865–873. doi: 10.1189/jlb.1212662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang H., Lundbäck P., Ottosson L., Erlandsson-Harris H., Venereau E., Bianchi M.E., Al-Abed Y., Andersson U., Tracey K.J., Antoine D.J. Redox modification of cysteine residues regulates the cytokine activity of high mobility group box-1 (HMGB1) Mol. Med. 2012;18:250–259. doi: 10.2119/molmed.2011.00389. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 54.Yang H., Ochani M., Li J., Qiang X., Tanovic M., Harris H.E., Susarla S.M., Ulloa L., Wang H., Diraimo R., Czura C.J., Roth J., Warren H.S., Fink M.P., Fenton M.J., Andersson U., Tracey K.J. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc. Natl. Acad. Sci. USA. 2004;101:296–301. doi: 10.1073/pnas.2434651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Youn J.H., Shin J.S. Nucleocytoplasmic shuttling of HMGB1 is regulated by phosphorylation that redirects it toward secretion. J. Immunol. 2006;177(11):7889–7897. doi: 10.4049/jimmunol.177.11.7889. [DOI] [PubMed] [Google Scholar]
- 57.Zaher T.E., Miller E.J., Morrow D.M., Javdan M., Mantell L.L. Hyperoxia-induced signal transduction pathways in pulmonary epithelial cells. Free Radic. Biol. Med. 2007;42(7):897–908. doi: 10.1016/j.freeradbiomed.2007.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]