

Abstract
Antioxidant defences are comparatively low during foetal development making the brain particularly susceptible to oxidative stress during antioxidant deficiencies. The brain is one of the organs containing the highest concentration of vitamin C (VitC) and VitC deficiency during foetal development may place the brain at risk of redox status imbalance. In the present study, we investigated the developmental pattern and effect of VitC deficiency on antioxidants, vitamin E and superoxide dismutase (SOD), assessed oxidative damage by measuring malondialdehyde (MDA), hydroxynonenal (HNE) and nitrotyrosine (NT) and analysed gene and protein expression of apoptosis marker caspase-3 in the guinea pig foetal brain at two gestational (GD) time points, GD 45/pre-term and GD 56/near term following either a VitC sufficient (CTRL) or deficient (DEF) maternal dietary regime. We show that except for SOD, antioxidants and oxidative damage markers are differentially expressed between the two GDs, with high VitC (p<0.0001), NT modified proteins (p<0.0001) and active caspase-3 levels (p<0.05) at pre-term and high vitamin E levels (p<0.0001), HNE (p<0.0001) and MDA (p<0.0001) at near term. VitC deficiency significantly increased SOD activity (p<0.0001) compared to CTRLs at both GDs indicating a compensatory response, however, low levels of VitC significantly elevated MDA levels (p<0.05) in DEF at near term. Our results show a differential regulation of the investigated markers during late gestation and suggest that immature brains are susceptible to oxidative stress due to prenatal vitC deficiency in spite of an induction of protective adaptation mechanisms.
Abbreviations: 1VitC, vitamin C; GD, gestational day; GPx, glutathione peroxidase; MDA, malondialdehyde; NT, nitrotyrosine; HNE, hydroxynonenal; SOD, superoxide dismutase; PCR, polymerase chain reaction; PFA, paraformaldehyde; s18, ribosomal protein 18S; CTRL, control; DEF, deficient
Keywords: Vitamin C, Deficiency, Brain, Oxidative stress, Guinea pig, Development
Graphical abstract
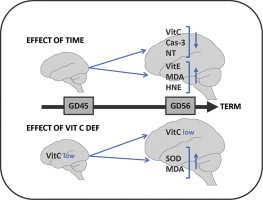
Highlights
-
•
Maternal vitamin C deficiency affects redox balance in the foetal brain.
-
•
Differential regulation during late gestation time-points.
-
•
The foetal brain is susceptible to oxidative stress imposed by vitamin C deficiency.
Introduction
The developing brain has a high metabolic activity making it vulnerable to oxidizing agents such as free radicals [55], a situation that is further exacerbated by a yet immature scavenging antioxidant system in early life [33], [48]. Although antioxidant defence-mechanisms in guinea pig brains during foetal development have been investigated [36], the manifestation of macromolecular modifications due to lipid peroxidation or by similar oxidizing agents and cell death in the near term brain is unknown. It is also not known if the pertinent oxidative changes during development are likely to increase in the brain when deprived of optimal antioxidant levels.
Guinea pigs are precocial rodents with a gestation length of approximately 66 days with the peak of exponential brain growth (‘the brain growth spurt’) being reached about 15 days before birth (i.e. around gestation day (GD) 50) [12]. Interference with optimal requirements for the foetus may impose several negative effects on developmental outcome such as lower body weight [11] and in the brain lead to reduction of cells in hippocampal morphogenesis [4] and cognitive alterations as observed in rats [38]. Oxidative stress caused by deficiency in antioxidants like glutathione in the developing brain has been shown to negatively affect mitochondria [21] and also to result in an age-dependent neurodegeneration in the hippocampus [1].
Activity of antioxidant enzymes in guinea pigs such as superoxide dismutase, catalase, glutathione peroxidase (GPx) and glutathione reductase have been demonstrated to remain constant from GD 30 to 45 and to increase from GD 45 to GD 60, with the exception of superoxide dismutase (SOD) that remained constant throughout the GD 30–60 period [36]. In rats, postnatal GPx activity in the brain has been found to remain constant from birth whereas the activity of cytoplasmic SOD increases from birth until 60 days after birth in rats [34]. Analysis of antioxidant protein levels in developing mice brains from embryonic day 18 to postnatal day 21 has shown that total SOD activity, catalase and GPx activity increases suggesting an increase in antioxidants in perinatal and neonatal brains relative to foetal development [25]. Thus, despite inter-species differences and differences in degree of postnatal development (i.e. altrician vs. precocial species), there seems to be a general trend of increasing antioxidant capacity in the brain during the course of development. Hence, a decrease in antioxidant capacity could potentially disturb the redox homeostasis in the brain leading to negative consequences on development.
Vitamin C (VitC) deficiency in humans has been associated with increases in premature births and predisposing newborns’ to oxidative stress [39], [44]. VitC is one of the primary antioxidants in the brain and selectively accumulates in high amounts also during foetal development [14], [26], [33]. Interruption of vitC transport to the brain in the svct2−/− mouse is detrimental to perinatal survival [13], [50], underlining the pivotal role of vitC in the developing central nervous system. Due to its one electron reduction potential, VitC effectively inhibits lipid peroxidation and scavenges several reactive oxidizing compounds such as superoxide, hydrogen peroxide and hydroxyl radicals as well as enabling the recycling of other antioxidant compounds such as vitamin E [7], [20], [30], [49]. In guinea pigs, VitC deficiency during both pre- and postnatal development has been linked to deviations in hippocampal development [51], [52]. Alteration of apoptotic mechanisms due to redox imbalance in the foetal brain has also been reported in several studies, however, the underlying mechanism and potential cause vs consequence remains to be disclosed [19], [43].
Like humans, guinea pigs cannot synthesize VitC due to a non-functional gulonolactone oxidase (Gulo) gene [40] and therefore rely on an adequate dietary supply. The present study investigated the developmental course of and potential effects of VitC deficiency on markers of antioxidants, redox imbalance and apoptosis signalling in foetal guinea pig brains at two different gestational time points at which brain growth is at a peak, GD 45/pre-term and GD 56/near term.
Materials and methods
Animal experiment
The animal study adheres to the guidelines of EU Directive 2010/63/EU and was approved by Danish Animal Experimentation Inspectorate. Twenty pregnant guinea pigs between GD 6–10 were obtained from Charles Rivers Lab, Kieslegg, Germany. The animals were microchipped subcutaneously (PET-CHIP ID, Danworth farm, West Sussex, UK) and randomized according to GD and body weight into two groups receiving diets only differing in VitC content (specialized diets sniff, GmbH), Control: CTRL (900 mg/kg diet, n=10) and Deficient: DEF (100 mg/kg diet, n=10). It has previously been shown by us that the 100 mg VitC diet in guinea pigs results in non-scorbutic VitC deficiency [32], [53]. Each of the groups was further randomized to having caesarian section (followed by euthanasia) performed at GD 45 or GD 56. The animals were housed in floor pens with straw bedding and feed, hay and water were provided ad libitum. They were weighed once every week and blood was sampled (~300 µl) once in every two weeks from v. saphena at its superficial course on tibia to verify VitC status (data not shown).
At euthanasia three dams, one from CTRL group and two from DEF group were found not to have conceived. Necropsy revealed no signs of underlying disease and the animals were excluded from the study.
Euthanasia
Caesarean section was conducted on dams at GD 45 or GD 56. Ten to fifteen minutes prior to anaesthesia, dams were injected with 2 mg/kg body weight Torbugesic (10 mg/ml butorphanol, Scan Vet Animal Health, Fredensborg, Denmark) subcutaneously to achieve analgesia. Anaesthesia was achieved by inhalation of isoflurane (Isoba Vet 100%, Intervet International, Boxmeer, The Netherlands). After the disappearance of voluntary reflexes (interdigital and skin-pinch), caesarean was performed by laparotomy through linea alba exposing the uterus. Excision of fetuses was done one at a time starting from the apex of the left horn towards the basis and subsequently commencing at the apex of the right horn. Immediately following delivery of each pup, the body weight was recorded, an intracardial blood sample was taken and the pup was euthanized by decapitation, the procedure lasting no more than 2 minutes. In the event of a pup displaying reflexes, euthanization by intraperitoneal injection of 0.5 ml pentobarbital (200 mg/ml) supplemented with lidocaine (Veterinary Pharmacy, University of Copenhagen, Denmark) was performed. Gender was recorded and post mortem autopsy with tissue sampling was performed on each pup, tissues allocated either to fixative or frozen for later analysis. Blood samples were centrifuged, stabilized and frozen after the intracardial blood sampling of the final pup from each dam. Once all the pups were removed from the uterus, thoracotomy of the dam was performed and an intracardial blood sample was taken before sacrificing by decapitation and exsanguination.
The brain was removed and the left hemisphere frozen in liquid nitrogen and the right hemisphere fixated in 4% PFA (paraformaldehyde in phosphate buffered saline, 0.15 M, pH 7.5) for 48 h then transferred to 1%PFA for long term storage.
All frozen tissues were stored at −80 °C until further analysis. For the current study all foetal left brain hemispheres from a total of 85 fetuses, were blocked for gender and body weight and randomized to be used for gene and protein expression analysis (N values; 12 CTRL/GD 45, 9 DEF/GD 45, 10 CTRL/GD 56, 11 DEF/GD 56) or biochemistry (N Values; 12 CTRL/GD 45, 10 DEF/GD 45, 10 CTRL/GD 56, 11 DEF/GD 56).
Biochemistry
Analysis of VitC and malondialdehyde (MDA) in brain were performed as described previously [28], [29], [31]. Briefly, tissue samples (app. 0.5 g) were homogenized in PBS, centrifuged at 16,000×g for 1 min at 4 °C. For VitC analysis, an aliquot was stabilized with an equal volume of 10% meta-phosphoric acid containing 2 mM EDTA (Merck, Whitehouse Station, NJ, USA), centrifuged, and the supernatant analysed by high-performance liquid chromatography (HPLC) with colorimetric detection. Levels of MDA were assessed by thiobarbituric acid derivatization followed by specific quantification of the genuine MDA(TBA)2 adduct by HPLC with fluorescence detection.
Analysis of α-tocopherol and γ-tocopherol was performed by HPLC with coulometric detection as modified from Sattler et al. [46]. Briefly, to 100 µl of tissue homogenate was added 25 µl freshly prepared 2,6-di-tert-butyl-p-cresol (10 mg/ml; Sigma, Copenhagen, Denmark), 100 µl sodium dodecyl sulphate (29 mg/ml, Sigma), 800 µl H2O, 900 µl ethanol and 100 µl 2-propanol. The cold mixture was extracted with 1 ml of n-hexane (Merck, Damstadt, Germany) of which 500 µl of organic phase was reduced to dryness at 40 °C using an airstream and subsequently redissolved in 100 µl ethanol for 2 min using a vortex mixer. Following centrifugation, 20 µl of the supernatant was used for HPLC analysis. Superoxide dismutase activity (SOD) was analysed using the Ransod colorimetric assay (SD125, Randox Laboratories Limited, UK) on tissue lysates according to manufacturer’s instructions.
RNA extraction and RT-PCR
Approximately 25 mg of each of brain tissues was homogenized in trizol (InVitrogen, Merelbeke, Belgium) and precipitated with chloroform (Sigma Aldrich, Steinheim, Germany) and isopropanol (Merck, Darmstadt, Germany). Purified RNA (SV Total RNA Isolation System, Promega, Madison, WI, USA) was eluted with 50 µl nuclease free water and the purity of RNA was determined by absorbance ratios A260/A280 and A260/A230 (Nanodrop 2000; Thermo Scientific, Wilmington, DE, USA). cDNA synthesis was performed by RT-PCR with 2 µg of RNA (MmLV RT enzyme, 5×MmLV buffer and RNasin (Promega)); 10 mM dNTPs and Oligo (dT) primers (60 µg/120 µl) (Fermentas GmbH, St Leon Roth, Germany); Random hexamer primer (2 µg/µl) (GE Healthcare, Uppsala, Sweden).
Gene expression analysis
Intron-spanning beta-actin primers were used on all cDNA (Table 1) prior to real time quantitative PCR (Q-PCR) to test for genomic contamination. None of the included samples displayed signs of contamination. PCR products of included genes were run on 2% agarose gels to confirm the product size and were then purified by PCR clean-up (PCR Clean Up System; Promega, Sweden) and subsequently submitted for sequencing (LGC genomics, Berlin, Germany) to confirm specificity.
Table 1.
Primer sequences applied in the gene expression analysis.
| Gene | Primer sequence | Product size (bp) | NCBI accession no. |
|---|---|---|---|
| Beta-actin | (F): gtaaggacctctatgccaacaca | 346 | AF508792 |
| (R): atgccaatctcatctcgttttct | |||
| s18 | (F): atgtggtgttgaggaaagcag | 195 | XM_003473925.1 |
| (R): gcttgttgtccagaccgttg | |||
| Caspase-3 | (F): ctggaatggcacctcgactt | 315 | XM_003469171.1 |
| (R): ccccggcaagcctgaataat |
Sequences are presented in the 5'–3' direction (F): forward, (R): reverse.
Q-PCR was conducted (SYBR Green I master LC480 and LC480, Roche, Basel, Switzerland) in 96-well white plates (Roche, Mannheim, Germany) with all samples in triplicates (in dilution 1:5), nuclease free water as negative control and calibrator as positive control. Gene expression analysis of Caspase 3 from brain samples was normalized to the reference gene, s18 (ribosomal protein S18). Primers are listed in Table 1.
Protein extraction and Western blot
Protein was extracted from brain tissue with radio-immuno-precipitation assay buffer (RIPA: 150 mM sodium chloride, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulphate, 50 mM Tris, pH 8) with 1:100 protease inhibitors (Sigma complete protease inhibitor cocktail). Estimation of protein was done by bicinchoninic acid assay (Calbiochem Novagen BCA Protein Assay kit), according to the manufacturer’s protocol, in triplicates at 562 nm on a SpectraMax Plus 384 UV/VIS plate reader (Molecular Devices Inc., CA, USA).
Approximately 100 µg of protein diluted with reducing agent and loading buffers (Invitrogen NuPAGE 4X LDS sample buffer, Invitrogen NuPAGE 10X sample reducing agent) were heated at 70 °C for 10 min before loading on precast gels (Invitrogen NuPAGE 4–12% Bis-Tris gels) in duplicates together with an internal standard comprising equal volumes of all samples. Gel to PVDF membrane transfer of protein was performed in a semi dry transfer unit (TE 77PWR, Amersham Biosciences) prior to 1 h blocking in 2% blocking buffer (Amersham ECL Prime Blocking Agent in PBS-T wash buffer,1XPBS and 0.1% Tween) before incubation with cleaved anti-caspase-3 (9661, Cell Signaling Technology, Danvers, MA, USA) or 4% blocking buffer before incubation with anti-nitrotyrosine (anti-NT, 06-284, Millipore, Temecula, CA) or anti-4-hydroxynonenal (anti-HNE, HNE 11-S, Alpha Diagnostic International, San Antonio, TX, USA). Antibody specificity of cleaved caspase-3 was tested by pre-absorption with equal volume of the blocking peptide (#1050, Cell Signaling Technology, Danvers, MA, USA) and specificity of anti-nitrotyrosine was tested by pre-absorption of 0.5 µg/ml primary antibody with 15 mM free NT (#89540, Cayman Chemical, Ann Arbor, MI, USA) with respective controls (#9663 Caspase-3 control Cell signalling technology, Danver, MA, USA); (#12–354, NT immunoblotting control, Chemicon, Temecula, CA, USA). Membranes were incubated with primary antibody overnight at 4 °C (1:1000, cleaved caspase-3 or 0.5 µg/ml anti-NT or 1:2000 anti-HNE), followed by secondary antibody(anti-rabbit IgG-HRP, 7074, Cell Signaling Technology, Danvers, MA, USA) 1: 1000, 1:6000 and 1:2000 for anti-caspase-3, anti-NT and anti-HNE proteins respectively. Imaging was achieved by enhanced chemiluminiscence (Amersham ECL Prime Western Blotting Reagent, UVP Biospectrum imaging system). Band-sizes were identified by Western blot protein standard (Magic MarkTM XP Western Protein Standard). Cleaved caspase-3 bands were located by an additional protein standard (ECL DualVue Western Blotting Markers, RPN810, Amersham Biosciences) with a specific secondary antibody (1:10000, HRP-S) diluted together with the secondary antibody. The NT and HNE blots were stripped with 0.5 M sodium hydroxide (Merck, Darmstad, Germany) before re-probing with loading control (mouse anti-actin, 1:20000, MAB 1501, Millipore, Temecula, CA, USA). Target bands of cleaved caspase-3 or lanes in case of NT and HNE blots were normalized to the loading control, detected by anti-mouse IgG-HRP secondary antibody (1:10000, SC-2005, Santa Cruz Biotechnology, CA, USA). UVP Life Science Series Image Acquisition and Analysis software was used for densitometry and blots were normalized to an internal control.
Statistics
SAS/JMP statistical version 8.0 was used to analyse all data. Analysis of Q-PCR expression data was performed by Student’s t-test. Two-way ANOVA was used to analyse biochemistry and Western blot densitometry. Interaction between factors, diet and GD, was reported together with main effects when statistically significant (p<0.05) followed by Tukeys HSD posthoc comparisons.
Results
Brain antioxidant status
As shown in Table 2, VitC concentration in the brain was significantly higher in the CTRL groups of both gestational days compared to DEF counterparts (p<0.0001). Two-way ANOVA showed effects of both diet and GD (p<0.0001) on VitC levels as well as an interaction between diet and GD (p<0.0001). A decrease of approximately 20% in VitC was observed in CTRL GD 56 relative to GD 45 (p<0.0001).This was not found in the DEF group, where VitC levels did not differ between gestational dates. In contrast to total VitC in the brain, α-tocopherol levels were high on GD 56 relative to GD 45 (p<0.0001) with no significant effect of diet. Conversely, two-way ANOVA showed a significant effect of diet on γ-tocopherol (p<0.05), the DEF-group displaying overall increased levels. However, tocopherols were not significantly different between CTRL and DEF groups at the individual time-points. SOD was significantly increased by VitC deficiency (p<0.0001) but was not affected by GD.
Table 2.
Brain antioxidant status in prenatally VitC deficient and control guinea pigs at GD 45 and GD 56.
| CTRL GD 45 | CTRL GD 56 | DEF GD 45 | DEF GD 56 | Effect of VitC | Effect of GD | |
|---|---|---|---|---|---|---|
| VitC (nmol/g tissue) | 3043±285a | 2351±137b | 1016±270c | 966±242c | ⁎⁎⁎ | ⁎⁎⁎ |
| α-Tocopherol (nmol/g tissue) | 5.5±0.8b | 9.0±1.0a | 4.6±1.0b | 9.4±1.5a | NS | ⁎⁎⁎ |
| γ-Tocopherol (nmol/g tissue) | 0.52±0.46 | 0.50±0.36 | 0.80±0.5 | 0.89±0.39 | ⁎ | NS |
| Superoxide dismutase (U/g tissue) | 90±2a | 92±2a | 133±3b | 137±4b | ⁎⁎⁎ | NS |
CTRL: controls; DEF: deficient. GD: gestational day. Values are presented as means±SD. Differences between groups were assessed using two-way ANOVA with diet and GD as factors followed by Tukeys HSD test for individual comparisons. NS: not significant. Different superscript letters indicate that groups are significantly different.
⁎p<0.05.
⁎⁎p<0.001.
⁎⁎⁎p<0.0001.
Brain oxidative damage
Western blot of HNE modified proteins ranged between 20 and 120 kDa in guinea pig brain lysates (data not shown). Two-way ANOVA of densitometry data showed that the intensity of HNE modifications at GD 56 was twice as high as GD 45 with a significant effect of GD (p<0.0001) (Fig. 1A). However, no significant differences were found between CTRL and DEF groups at either gestational day. MDA was measured in brain homogenate as a marker of lipid oxidation. Two-way ANOVA showed significant effects of both diet (p<0.001) and gestational age (p<0.0001). In agreement with the anti-HNE densitometry data, MDA was significantly elevated at GD 56 compared to 45 for both dietary regimens separately (p<0.0001). Moreover, VitC deficiency resulted in increased brain MDA compared to controls at GD 56 (p<0.05) but at GD45, the increase did not reach statistical significance (Fig. 1B).
Fig. 1.
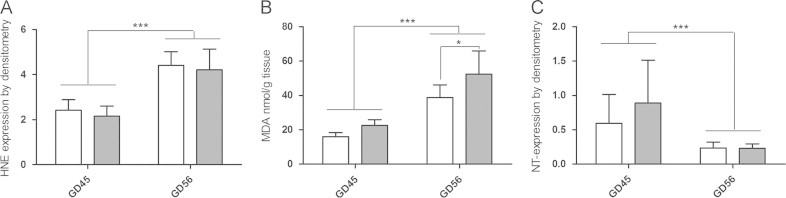
Oxidative damage in foetal guinea pig brains. Markers of increased oxidation and suggestive oxidative damage in the brain of guinea pigs subjected to prenatal vitC deficiency (DEF) (dark bars) or control diet (CTRL) (light bars) during development. Foetuses were excised by caesarean section at GD 45 and GD 56. (A) Densitometric analysis of HNE modified proteins in brain homogenate; results are normalized to actin; (B) Malondialdehyde in brain homogenate, measured by HPLC; (C). Densitometric analysis of nitrotyrosine modified proteins normalized to actin. Data are expressed as means±SD. ⁎p<0.05; ⁎⁎⁎p<0.0001 by ANOVA.
NT modified proteins were detected by Western blotting between 35 and 15 kDa in brain lysates. Pre-absorption of primary antibody with free NT abolished all the bands in guinea pig brain lysate and almost all bands in the positive control confirming specificity of the antibody (data not shown). A significant main effect of GD (p<0.0001) was detected with highest levels of NT modified proteins in GD 45 brains (Fig. 1C). There was no detectable effect of diet on the level of NT modified proteins at either gestational day.
Caspase-3 expression
Q-PCR analysis of caspase-3 gene-expression in the brain was not significantly different between CTRL and DEF groups in either gestational day (Fig. 2A). Cleaved caspase-3 Western blotting showed a single band just below 15 kDa. Pre-absorption of primary antibody with blocking peptide abolished the specific bands from guinea pig brain lysate and the positive control confirming the specificity of the antibody (data not shown). Two-way ANOVA of densitometry data showed that GD had a significant effect on cleaved caspase-3 (p<0.05) with higher levels at GD 45 compared to GD 56 (Fig. 2B). Densitometry analysis at either gestational day did not show significant differences between CTRL and DEF groups (Fig. 2B).
Fig. 2.
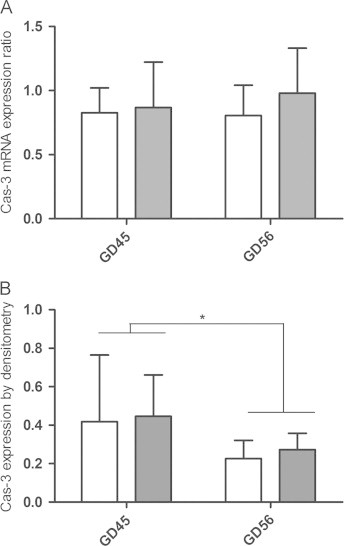
Expression analysis of caspase-3 in the brain. (A) Quantitative PCR analysis of caspase-3 in guinea pig foetal brains comparing vitC deficient (dark bars) to controls (light bars) on two gestational times points (GD45 and 56). Expressed values are normalized to the reference gene s18 and displayed as mean±SD. (B) Densitometric analysis of levels of cleaved caspase-3 obtained by Western blot and normalized to actin levels. Data are expressed as means±SD. ⁎p<0.05 by ANOVA.
Discussion
In the present study, we show that immature brains are susceptible to oxidative stress in spite of an induction of protective adaptation mechanisms. Oxidative stress and damage markers in guinea pig cerebral cortex were assessed at preterm and near term gestational time points with and without maternal VitC deficiency as a potential oxidative insult.
Brain VitC levels in DEF groups were significantly lower than CTRL as expected, and while VitC levels in the DEF group remained similar at the two gestational days, VitC in GD 56 CTRL was significantly lower than GD 45 CTRL. This finding indicates a developmental requirement of increased VitC during the preterm period possibly as a response to lower protective mechanisms against oxidizing agents as has previously been suggested [36]. However, in contrast to CTRLs, the DEF group did not display a near-term drop in concentration but rather showed retention of VitC, possibly to avoid any further loss than the already existing low level. The decrease in VitC toward term in CTRL animals is in line with findings in humans reporting a late gestation decrease of VitC in all foetal tissues including the brain [58], [59]. This decrease in the brain has been suggested to be due to growth and maturation of non-neuronal cells and maturation and myelination of neurones [16], [59]. Recent studies by us have shown that a pre-natal low VitC causes a reduction in foetal body and brain weight at GD 45 but not at GD 56 (unpublished results), indicating that a developing foetus is able to compensate for a pre-imposed negative effect of VitC deficiency during the final weeks of gestation.
Our α-tocopherol data implies that it increases with progression in development during late gestation. Interaction between cytosolic VitC and membrane bound vitamin E regenerates oxidized vitamin E and results in an efficient antioxidant mechanism [9], which suggests that VitC deficiency may alter vitamin E levels hereby possibly disrupting membrane integrity. Some in-vivo studies have shown that intake of high VitC results in an increased vitamin E in tissues [5], [17], [18]. This was not the case when evaluating α-tocopherol in CTRL vs. DEF groups of this study, however, VitC showed a main effect on γ-tocopherol levels in the brain. γ-Tocopherol has been associated with anti-inflammatory properties, decreased superoxide anion generation and inhibition of lipid peroxidation, hereby reducing adverse effects of oxidative imbalance [23], [45]. Our present findings suggest an increase in γ-tocopherol in response to VitC deficiency, whereas levels during late gestational progression were relatively constant in both CTRL and DEF groups.
In the current study, VitC deficiency increased SOD activity during late gestation. SOD activity in CTRL groups at both GD is in agreement with a previous report of constant SOD levels in guinea pig foetal brains throughout gestation from GD 30 to GD 60 [36]. As a consequence of low VitC levels, increases in SOD activity in DEF compared to CTRLs at both GD indicates foetal ability to induce a compensatory mechanism, likely due to increased production of superoxide radicals [24]. Various studies have reported an increase in SOD activity in the brain as an adaptive mechanism to tolerate oxidative damage in neurodegenerative conditions [41], stress [10] and oxidant induced neurotoxicity [6]. Studies like these with additional assessment of antioxidants like catalase, glutathione peroxidase or oxidative damage markers helps in understanding whether such adaptive mechanisms are able to preserve the redox status [3], [57].
HNE modified proteins, which indicate oxidative damage due to lipid peroxidation, were significantly high at GD 56, regardless of VitC status, likely due to the increase in poly-unsaturated fatty acids in the brain at near term. An increased susceptibility of lipid peroxidation in near term brains of guinea pigs has previously been reported [37]. Although HNE modified proteins were not significantly different between CTRL and DEF, a trend similar to the obtained MDA levels was observed between the two GD, with significantly higher MDA levels at near term further supporting the maturational related increase of lipid peroxidation in the brain as reported by others in guinea pigs brain [37]. Significant increase of MDA levels in DEF at GD 56 and not in DEF at GD45 compared to their CTRL suggests that compensatory mechanisms that may have been present at earlier gestation may not be enough to protect from lipid peroxidation due to low levels of VitC in the brain. Earlier studies by us and others have shown a similar inverse relation of VitC levels to lipid peroxidation in the brain [15], [33] and the vital role of ascorbate has previously been demonstrated to inhibit lipid peroxidation in rat brain microsomes [47]. Conversely, no significant differences in NT modified proteins between CTRL and DEF suggests that posttranslational modification of proteins by NT is not effected by VitC deficiency and instead, as proposed by others, NT may be involved in mediating cell signalling at its basal levels during development [27], [35].
Results from oxidative stress based in-vivo studies have suggested an increased free radical production or decreased antioxidant mechanisms in the immature brain [36], [56], which in turn may initiate cellular events to initiate mechanisms of apoptosis [8], [42] like caspase-3 activation [22]. Our finding of high levels of cleaved caspase-3 in GD 45 compared to GD 56 indicates a developmental phenomenon in agreement with what has been shown by others [2], placing cleaved caspase-3 as playing an important role in reducing neuronal overproduction in the developing brain [54]. The guinea pig is a precocial species and neuronal number relative to adult stage are achieved by GD 48 [12]. Hence, although a gestational effect was seen for cleaved caspase-3, the difference was small implying that most of the cell death associated with development is likely to have occurred before the investigated time-points. No significant differences in caspase-3 gene expression or cleaved caspase-3 protein levels between CTRL and DEF groups at any of the GDs indicates that VitC deficiency does not modulate caspase-3 mediated apoptosis and may not be the underlying mechanism associated with hippocampal impairment observed in our earlier findings [51], [52].
Conclusion
Guinea pig foetal brains have differential requirements of antioxidants with high VitC levels at pre-term, a higher requirement of α-tocopherol at near term and constant SOD activity. Oxidative NT modifications and active caspase-3 levels are higher at pre-term and are prone to increased lipid peroxidation at near term signifying differential expression of oxidative damage markers associated with brain maturity. Prenatal VitC deficiency in the guinea pig foetal brain does not modulate levels of vitamin E, NT or HNE protein modifications or caspase-3 however, increases SOD activity as compensation although this is not adequate to prevent increased lipid peroxidation at the investigated time points.
Acknowledgements
Joan Frandsen, Elisabeth Veyhe Andersen, Belinda Bringtoft and Annie Bjergby Kristensen are thanked for excellent technical assistance. The present work was supported in part by the Danish Research Councils, University of Copenhagen and the LIFEPHARM Centre for In Vivo Pharmacology. All authors declare no conflicts of interest that could influence the present work.
Contributor Information
Maya D. Paidi, Email: maypa@sund.ku.dk.
Janne G. Schjoldager, Email: jannes@sund.ku.dk.
Jens Lykkesfeldt, Email: jopl@sund.ku.dk.
Pernille Tveden-Nyborg, Email: ptn@sund.ku.dk.
References
- 1.Aoyama K., Suh S.W., Hamby A.M., Liu J., Chan W.Y., Chen Y., Swanson R.A. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat. Neurosci. 2005;9:119–126. doi: 10.1038/nn1609. [DOI] [PubMed] [Google Scholar]
- 2.Baehrecke E.H. How death shapes life during development. Nat. Rev. Mol. Cell Biol. 2002;3:779–787. doi: 10.1038/nrm931. [DOI] [PubMed] [Google Scholar]
- 3.Baker K., Marcus C.B., Huffman K., Kruk H., Malfroy B., Doctrow S.R. Synthetic combined superoxide dismutase/catalase mimetics are protective as a delayed treatment in a rat stroke model: a key role for reactive oxygen species in ischemic brain injury. J. Pharmacol. Exp. Ther. 1998;284:215–221. [PubMed] [Google Scholar]
- 4.Bedi K. Effects of undernutrition during early life on granule cell numbers in the rat dentate gyrus. J. Comp. Neurol. 1991;311:425–433. doi: 10.1002/cne.903110311. [DOI] [PubMed] [Google Scholar]
- 5.Bendich A., D’Apolito P., Gabriel E., Machlin L.J. Interaction of dietary vitamin C and vitamin E on guinea pig immune responses to mitogens. J. Nutr. 1984;114:1588–1593. doi: 10.1093/jn/114.9.1588. [DOI] [PubMed] [Google Scholar]
- 6.Bordet R., Deplanque D., Maboudou P., Puisieux F., Pu Q., Robin E., Martin A., Bastide M., Leys D., Lhermitte M. Increase in endogenous brain superoxide dismutase as a potential mechanism of lipopolysaccharide-induced brain ischemic tolerance. J. Cereb. Blood Flow Metab. 2000;20:1190–1196. doi: 10.1097/00004647-200008000-00004. [DOI] [PubMed] [Google Scholar]
- 7.Buettner G.R. The pecking order of free radicals and antioxidants: lipid peroxidation, -tocopherol, and ascorbate. Arch. Biochem. Biophys. 1993;300:535–543. doi: 10.1006/abbi.1993.1074. [DOI] [PubMed] [Google Scholar]
- 8.Buttke T.M., Sandstrom P.A. Oxidative stress as a mediator of apoptosis. Immunol. Today. 1994;15:7–10. doi: 10.1016/0167-5699(94)90018-3. [DOI] [PubMed] [Google Scholar]
- 9.Chan A.C. Partners in defense, vitamin E and vitamin C. Can. J. Physiol. Pharmacol. 1993;71:725–731. doi: 10.1139/y93-109. [DOI] [PubMed] [Google Scholar]
- 10.Chan P., Yang G., Chen S., Carlson E., Epstein C. Cold‐induced brain edema and infarction are reduced in transgenic mice overexpressing CuZn‐superoxide dismutase. Ann. Neurol. 1991;29:482–486. doi: 10.1002/ana.410290506. [DOI] [PubMed] [Google Scholar]
- 11.Dobbing J. 2008. Vulnerable periods of brain development, in: Ciba Foundation Symposium 3—Lipids, Malnutrition and the Developing Brain. p 9–20. [PubMed]
- 12.Dobbing J., Sands J. Growth and development of the brain and spinal cord of the guinea pig. Brain Res. 1970;17:115–123. doi: 10.1016/0006-8993(70)90311-2. [DOI] [PubMed] [Google Scholar]
- 13.Harrison F., Dawes S., Meredith M., Babaev V., Li L., May J. Low vitamin C increased oxidative stress cell death in mice that lack the sodium-dependent vitamin C transporter SVCT2. Free Radic. Biol. Med. 2010;49:821–829. doi: 10.1016/j.freeradbiomed.2010.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harrison F., Green R., Dawes S., May J. Vitamin C distribution and retention in the mouse brain. Brain Res. 2010;1348:181–186. doi: 10.1016/j.brainres.2010.05.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harrison F., Meredith M., Dawes S., Saskowski J., May J. Low ascorbic acid and increased oxidative stress in gulo-/-mice during development. Brain Res. 2010;1349:143–152. doi: 10.1016/j.brainres.2010.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hasselholt S., Tveden-Nyborg P.Y., Lykkesfeldt J. Vitamin C: its role in brain development and cognition. Nutr. Cogn. Perform.: Dev. Perspect. 2011:29–52. [Google Scholar]
- 17.Huang H.-Y., Appel L.J., Croft K.D., Miller E.R., Mori T.A., Puddey I.B. Effects of vitamin C and vitamin E on in vivo lipid peroxidation: results of a randomized controlled trial. Am. J. Clin. Nutr. 2002;76:549–555. doi: 10.1093/ajcn/76.3.549. [DOI] [PubMed] [Google Scholar]
- 18.Igarashi O., Yonekawa Y., Fujiyama-Fujihara Y. Synergistic action of vitamin E and vitamin C in vivo using a new mutant of Wistar-strain rats, ODS, unable to synthesize vitamin C. J. Nutr. Sci. Vitaminol. 1991;37:359. doi: 10.3177/jnsv.37.359. [DOI] [PubMed] [Google Scholar]
- 19.Ikonomidou C., Kaindl A.M. Neuronal death and oxidative stress in the developing brain. Antioxidants Redox Signaling. 2011;14:1535–1550. doi: 10.1089/ars.2010.3581. [DOI] [PubMed] [Google Scholar]
- 20.Iyanagi T., Yamazaki I., Anan K.F. One-electron oxidation–reduction properties of ascorbic acid. Biochim. Biophys. Acta, Bioenerg. 1985;806:255–261. [Google Scholar]
- 21.Jain A., Mårtensson J., Stole E., Auld P., Meister A. Glutathione deficiency leads to mitochondrial damage in brain. Proc. Nat. Acad. Sci. 1991;88:1913–1917. doi: 10.1073/pnas.88.5.1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jänicke R.U., Sprengart M.L., Wati M.R., Porter A.G. Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J. Biol. Chem. 1998;273:9357–9360. doi: 10.1074/jbc.273.16.9357. [DOI] [PubMed] [Google Scholar]
- 23.Jiang Q., Elson-Schwab I., Courtemanche C., Ames B.N. γ-Tocopherol and its major metabolite, in contrast to α-tocopherol, inhibit cyclooxygenase activity in macrophages and epithelial cells. Proc. Nat. Acad. Sci. 2000;97:11494–11499. doi: 10.1073/pnas.200357097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keller J.N., Kindy M.S., Holtsberg F.W., Clair D.K.S., Yen H.-C., Germeyer A., Steiner S.M., Bruce-Keller A.J., Hutchins J.B., Mattson M.P. Mitochondrial manganese superoxide dismutase prevents neural apoptosis and reduces ischemic brain injury: suppression of peroxynitrite production, lipid peroxidation, and mitochondrial dysfunction. J. Neurosci. 1998;18:687–697. doi: 10.1523/JNEUROSCI.18-02-00687.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khan J.Y., Black S.M. Developmental changes in murine brain antioxidant enzymes. Pediatr. Res. 2003;54:77–82. doi: 10.1203/01.PDR.0000065736.69214.20. [DOI] [PubMed] [Google Scholar]
- 26.Kratzing C., Kelly J., Kratzing J. Ascorbic acid in fetal rat brain. J. Neurochem. 1985;44:1623–1624. doi: 10.1111/j.1471-4159.1985.tb08804.x. [DOI] [PubMed] [Google Scholar]
- 27.Levonen A.-L., Patel R.P., Brookes P., Go Y.-M., Jo H., Parthasarathy S., Anderson P.G., Darley-Usmar V.M. Mechanisms of cell signaling by nitric oxide and peroxynitrite: from mitochondria to MAP kinases. Antioxidants Redox Signaling. 2001;3:215–229. doi: 10.1089/152308601300185188. [DOI] [PubMed] [Google Scholar]
- 28.Lykkesfeldt J. Determination of ascorbic acid and dehydroascorbic acid in biological samples by high-performance liquid chromatography using subtraction methods: reliable reduction with tris [2-carboxyethyl] phosphine hydrochloride. Anal. Biochem. 2000;282:89–93. doi: 10.1006/abio.2000.4592. [DOI] [PubMed] [Google Scholar]
- 29.Lykkesfeldt J. Determination of malondialdehyde as dithiobarbituric acid adduct in biological samples by HPLC with fluorescence detection: comparison with ultraviolet-visible spectrophotometry. Clin. Chem. 2001;47:1725–1727. [PubMed] [Google Scholar]
- 30.Lykkesfeldt J. Increased oxidative damage in vitamin C deficiency is accompanied by induction of ascorbic acid recycling capacity in young but not mature guinea pigs. Free Radical Res. 2002;36:567–574. doi: 10.1080/1071576022411256. [DOI] [PubMed] [Google Scholar]
- 31.Lykkesfeldt J. Ascorbate and dehydroascorbic acid as reliable biomarkers of oxidative stress: analytical reproducibility and long-term stability of plasma samples subjected to acidic deproteinization. Cancer Epidemiol. Biomarkers Prev. 2007;16:2513–2516. doi: 10.1158/1055-9965.EPI-07-0639. [DOI] [PubMed] [Google Scholar]
- 32.Lykkesfeldt J., Moos T. Age-dependent change in Vitamin C status: a phenomenon of maturation rather than of ageing. Mech. Ageing Dev. 2005;126:892–898. doi: 10.1016/j.mad.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 33.Lykkesfeldt J., Trueba G.P., Poulsen H.E., Christen S. Vitamin C deficiency in weanling guinea pigs: differential expression of oxidative stress and DNA repair in liver and brain. Br. J. Nutr. 2007;98:1116–1119. doi: 10.1017/s0007114507787457. [DOI] [PubMed] [Google Scholar]
- 34.Mavelli I., Rigo A., Federico R., Ciriolo M., Rotilio G. Superoxide dismutase, glutathione peroxidase and catalase in developing rat brain. Biochem. J. 1982;204:535. doi: 10.1042/bj2040535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Minetti M., Mallozzi C., Di Stasi A. Peroxynitrite activates kinases of the src family and upregulates tyrosine phosphorylation signaling<sup> 1, 2</sup>. Free Radic. Biol. Med. 2002;33:744–754. doi: 10.1016/s0891-5849(02)00891-2. [DOI] [PubMed] [Google Scholar]
- 36.Mishra O.P., Delivoria-Papadopoulos M. Anti-oxidant enzymes in fetal guinea pig brain during development and the effect of maternal hypoxia. Dev. Brain Res. 1988;42:173–179. doi: 10.1016/0165-3806(88)90235-0. [DOI] [PubMed] [Google Scholar]
- 37.Mishra O.P., Delivoria-Papadopoulos M. Lipid peroxidation in developing fetal guinea pig brain during normoxia and hypoxia. Dev. Brain Res. 1989;45:129–135. doi: 10.1016/0165-3806(89)90014-x. [DOI] [PubMed] [Google Scholar]
- 38.Morley R., Lucas A. Nutrition and cognitive development. Br. Med. Bull. 1997;53:123–134. doi: 10.1093/oxfordjournals.bmb.a011595. [DOI] [PubMed] [Google Scholar]
- 39.Negi R., Pande D., Kumar A., Khanna R.S., Khanna H. Evaluation of biomarkers of oxidative stress and antioxidant capacity in the cord blood of preterm low birth weight neonates. J. Mater. Fetal Neonatal Med. 2012;25:1338–1341. doi: 10.3109/14767058.2011.633672. [DOI] [PubMed] [Google Scholar]
- 40.Nishikimi M., Kawai T., Yagi K. Guinea pigs possess a highly mutated gene for l-gulono-gamma-lactone oxidase, the key enzyme for l-ascorbic acid biosynthesis missing in this species. J. Biol. Chem. 1992;267:21967–21972. [PubMed] [Google Scholar]
- 41.Noack H., Lindenau J., Rothe F., Asayama K., Wolf G. Differential expression of superoxide dismutase isoforms in neuronal and glial compartments in the course of excitotoxically mediated neurodegeneration: relation to oxidative and nitrergic stress. Glia. 1998;23:285–297. doi: 10.1002/(sici)1098-1136(199808)23:4<285::aid-glia1>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 42.Orrenius S., Gogvadze V., Zhivotovsky B. Mitochondrial oxidative stress: implications for cell death. Annu. Rev. Pharmacol. Toxicol. 2007;47:143–183. doi: 10.1146/annurev.pharmtox.47.120505.105122. [DOI] [PubMed] [Google Scholar]
- 43.Rees S., Inder T. Fetal and neonatal origins of altered brain development. Early Hum. Dev. 2005;81:753–761. doi: 10.1016/j.earlhumdev.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 44.Rumbold A., Crowther C.A. Vitamin C supplementation in pregnancy. Cochrane Database Syst. Rev. 2005;2 doi: 10.1002/14651858.CD004069.pub2. [DOI] [PubMed] [Google Scholar]
- 45.Saldeen T., Li D., Mehta J.L. Differential effects of α- and γ-tocopherol on low-density lipoprotein oxidation, superoxide activity, platelet aggregation and arterial thrombogenesis. J. Am. Coll. Cardiol. 1999;34:1208–1215. doi: 10.1016/s0735-1097(99)00333-2. [DOI] [PubMed] [Google Scholar]
- 46.Sattler W., Mohr D., Stocker R. Rapid isolation of lipoproteins and assessment of their peroxidation by high-performance liquid chromatography postcolumn chemiluminescence. Methods Enzymol. 1994;233:469–489. doi: 10.1016/s0076-6879(94)33053-0. [DOI] [PubMed] [Google Scholar]
- 47.Seregi A., Schaefer A., Komlós M. Protective role of brain ascorbic acid content against lipid peroxidation. Cell. Mol. Life Sci. 1978;34:1056–1057. doi: 10.1007/BF01915344. [DOI] [PubMed] [Google Scholar]
- 48.Shim S.-Y., Kim H.-S. Oxidative stress and the antioxidant enzyme system in the developing brain. Korean J. Pediatr. 2013;56:107–111. doi: 10.3345/kjp.2013.56.3.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sies H., Stahl W. Vitamins E and C, beta-carotene, and other carotenoids as antioxidants. Am. J. Clin. Nutr. 1995;62:1315S–1321S. doi: 10.1093/ajcn/62.6.1315S. [DOI] [PubMed] [Google Scholar]
- 50.Sotiriou S., Gispert S., Cheng J., Wang Y., Chen A., Hoogstraten-Miller S., Miller G., Kwon O., Levine M., Guttentag S. Ascorbic-acid transporter Slc23a1 is essential for vitamin C transport into the brain and for perinatal survival. Nat. Med. 2002;8:514–517. doi: 10.1038/0502-514. [DOI] [PubMed] [Google Scholar]
- 51.Tveden-Nyborg P., Johansen L.K., Raida Z., Villumsen C.K., Larsen J.O., Lykkesfeldt J. Vitamin C deficiency in early postnatal life impairs spatial memory and reduces the number of hippocampal neurons in guinea pigs. Am. J. Clin. Nutr. 2009;90:540–546. doi: 10.3945/ajcn.2009.27954. [DOI] [PubMed] [Google Scholar]
- 52.Tveden-Nyborg P., Vogt L., Schjoldager J.G., Jeannet N., Hasselholt S., Paidi M.D., Christen S., Lykkesfeldt J. Maternal Vitamin C Deficiency during Pregnancy Persistently Impairs Hippocampal Neurogenesis in Offspring of Guinea Pigs. PLoS One. 2012;7:e48488. doi: 10.1371/journal.pone.0048488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tveden‐Nyborg P., Hasselholt S., Miyashita N., Moos T., Poulsen H.E., Lykkesfeldt J. Chronic vitamin C deficiency does not accelerate oxidative stress in ageing brains of Guinea Pigs. Basic Clin. Pharmacol. Toxicol. 2012;110:524–529. doi: 10.1111/j.1742-7843.2011.00852.x. [DOI] [PubMed] [Google Scholar]
- 54.Urase K., Fujita E., Miho Y., Kouroku Y., Mukasa T., Yagi Y., Momoi M.Y., Momoi T. Detection of activated caspase-3 (CPP32) in the vertebrate nervous system during development by a cleavage site-directed antiserum. Dev. Brain Res. 1998;111:77–87. doi: 10.1016/s0165-3806(98)00124-2. [DOI] [PubMed] [Google Scholar]
- 55.Uttara B., Singh A.V., Zamboni P., Mahajan R. Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol. 2009;7:65. doi: 10.2174/157015909787602823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Welin A.-K., Sandberg M., Lindblom A., Arvidsson P., Nilsson U.A., Kjellmer I., Mallard C. White matter injury following prolonged free radical formation in the 0.65 gestation fetal sheep brain. Pediatr. Res. 2005;58:100–105. doi: 10.1203/01.PDR.0000163388.04017.26. [DOI] [PubMed] [Google Scholar]
- 57.Yusa T., Crapo J.D., Freeman B.A. Liposome-mediated augmentation of brain SOD and catalase inhibits CNS O2 toxicity. J. Appl. Physiol. 1984;57:1674–1681. doi: 10.1152/jappl.1984.57.6.1674. [DOI] [PubMed] [Google Scholar]
- 58.Zalani S., Bharaj B.S., Rajalakshmi R. Ascorbic acid and reduced glutathione concentration of human fetal tissues in relation to gestational age, fetal size and maternal nutritional status. Int. J. Vitam. Nutr. Res. 1987;57:411. [PubMed] [Google Scholar]
- 59.Zalani S., Rajalakshmi R., Parekh L. Ascorbic acid concentration of human fetal tissues in relation to fetal size and gestational age. Br. J. Nutr. 1989;61:601–606. doi: 10.1079/bjn19890147. [DOI] [PubMed] [Google Scholar]