

Abstract
Here, we review shortly the current knowledge on the regulation of the proteasomal system during and after oxidative stress. After addressing the components of the proteasomal system and the degradation of oxidatively damaged proteins in part I and II of this series, we address here which changes in activity undergo the proteasome and the ubiquitin-proteasomal system itself under oxidative conditions. While several components of the proteasomal system undergo direct oxidative modification, a number of redox-regulated events are modulating the proteasomal activity in a way it can address the major tasks in an oxidative stress situation: the removal of oxidized proteins and the adaptation of the cellular metabolism to the stress situation.
Keywords: 20S proteasome, Ubiquitin–proteasome-system, Nrf2, Jak/Stat, PARP1
Introduction
The main task of the ubiquitin-proteasomal-system (UPS) is the maintenance of a functional proteome of the cell. The UPS consist of two major pathways, the ATP-dependent 26S proteasome, degrading polyubiquitinated substrates formed by the polyubiquitinating enzymatic machinery and the ATP-independent 20S proteasome-catalyzed degradation pathway [1], [2]. While interestingly, the 26S proteasomal pathway is responsible for the degradation of a vast majority of regulator proteins, the 20S proteasome is responsible for the degradation of oxidized proteins [3]. Therefore, during oxidative stress the proteasomal system has to be adapted to the stress situation ensuring a cellular regulatory response and the degradation of oxidized proteins, as oxidative stress is not only involving cellular damage, but also redox-regulation [4], [5]. This damage and regulation is reflected also in proteasomal modifications (Fig. 1). One of the more prominent modifications is the glutathionylation of the proteasome. During oxidative stress, the cellular redox-state shifts and the ratio of cellular reduced and oxidized glutathione shifts leading to a modification of several proteasomal subunits. In addition to that several direct oxidative modifications of the proteasome are taking place, including protein oxidation leading to proteasome carbonylation, proteasomal glycoxidation and modification with lipid peroxidation products (Fig. 1). It is assumed, that these modifications modulate the proteasomal activity. However, to which extent these modifications impair the proteasomal activity remains unknown today. Several reports demonstrated that only high levels of damage to the 20S proteasome lead to a catalytic impairment [6], [7], whereas the 26S proteasome is more susceptible towards oxidative damage [7], [8].
Fig. 1.
Oxidative modifications of the proteasomal system.
Due to the requirement of an enhanced turnover of regulatory proteins in a stress situation and the necessity to degrade oxidized proteins, several components of the UPS are under the control of stress-related transcription factors. One of those is the Nrf2-Keap1-system [9]. So, oxidative stress is also able to induce de novo synthesis of proteasomal subunits and parts of the UPS (Fig. 2). Interestingly, the parts of the UPS are not only target genes of Nrf2, but the UPS itself is also involved in the degradation of Keap1. The Keap1-Cul3-Rbx-complex is catalyzing the polyubiqutination of Nrf2, making it a substrate for the 26S proteasome. During oxidative stress, Keap1 becomes oxidized and is unable to catalyze the polyubiquitination of Nrf2. This is an example for the redox sensitivity of the ubiquitination system, which was described earlier [10], [11]. Although a wide array of proteasomal subunits is regulated via Nrf2, for several proteasomal components other transcription factors are involved. Especially the inducible proteasomal subunits and the 11S proteasomal activator (also Pa28) [1], [2] are under the control of the Jak/Stat pathway (Fig. 3). It was thought traditionally, that this pathway is only induced by the cytokine interferon-γ (IFN-γ), but more recently also stress related factors – as glycoxidized proteins – are able to trigger this response, although via a different receptor. Therefore, in late phases after oxidative stress often an adaptive increase of proteasomal activity was observed.
Fig. 2.
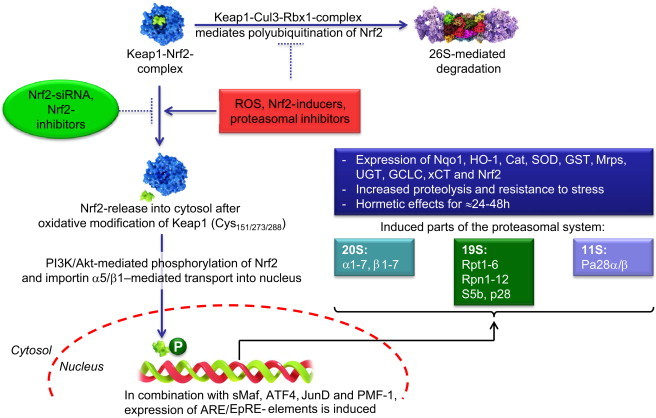
De novo synthesis of the proteasomal system via Nrf2-mediated stress-response.
Fig. 3.
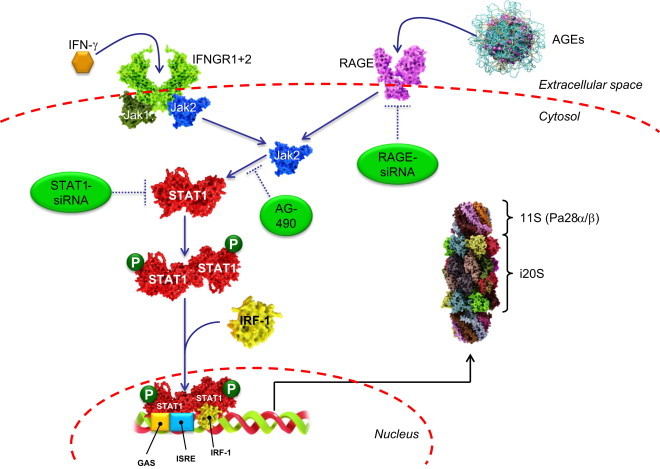
Jak/STAT-mediated induction of the inducible proteasome and PA28α/β.
However, in several situations also a quick activation of the proteasomal system seems to be needed. Especially in the nucleus a quick and effective degradation of damaged proteins is required, since such damaged proteins are likely to disturb DNA repair and transcription related stress responses. Interestingly a link between DNA repair and proteasomal activity was discovered, mediated by the poly-ADP-ribosyl transferase 1 (PARP1) [12]. Formation of poly-ADP-ribose is leading within minutes to a severalfold activation of the proteasomal activity (Fig. 4). This is enabling the cell to degrade rapidly damaged nuclear proteins and coordinate chromatin-reformation after stress events. Consequently, blocking of proteasomal activity and/or inhibition of PARP was leading to inhibition or slow down of DNA repair processes [13].
Fig. 4.
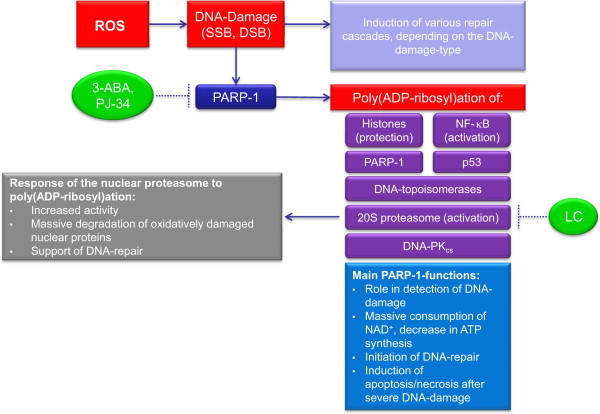
Nuclear PARP-1-mediated proteasomal activation.
Summary
The proteasomal system undergoes in the event of oxidative stress a series of regulators steps which are aiming to enable the proteasomal system in the early phases after stress to remove oxidized proteins, e.g. by activation of 20S proteasome by PARP1 and the inactivation of the 26S proteasome and the ubiqutination enzymes, and in later phases the reformation of the 26S protesome and UPS to address regulatory requirements in the cell accompanied by an activation of the 20S proteasome by PA28 (Fig. 5). The latter enables the proteasomal system further to cope with remaining damaged proteins. This is followed by adaptive responses enabling cell to cope with other oxidative stimuli by synthesizing higher levels of proteosomes.
Fig. 5.
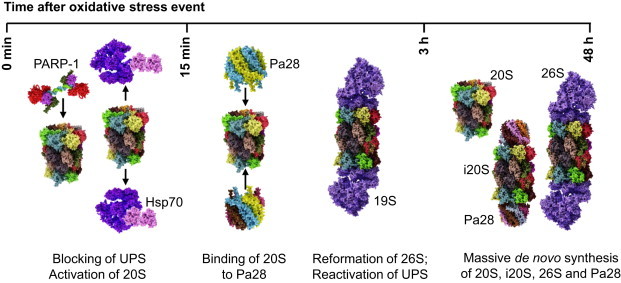
Response of the proteasomal system to oxidative stress.
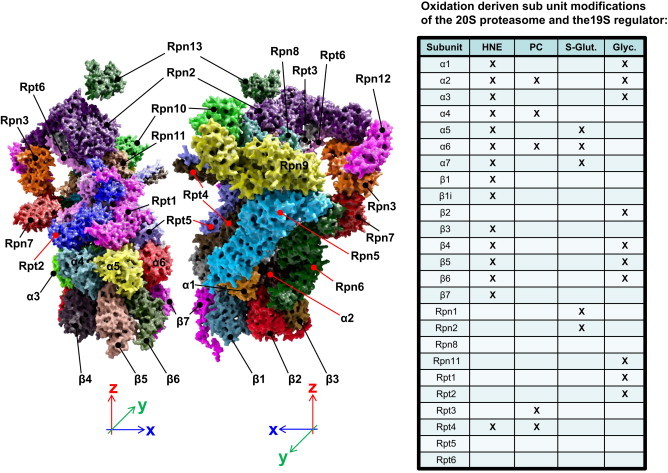
The left part of this figure shows a partial three-dimensional reconstruction of the 26S proteasome build of the 19S regulator protein (containing the 6 ATPases Rpt1–Rpt6 and the non-ATPase subunits Rpn2, Rpn3 and Rpn5–Rpn13), the attaching alpha-ring (subunits α1–α7) and one of the beta-rings (subunits β1–β7) of the 20S proteasome. This structure was described in detail in the first part of this series [1] and is here rendered according to the data published by Sledz et al. [14], gained via cryoelectron microscopy single-particle analysis.
Many of the 20S/26S proteasomal subunits have been shown to be modified in different reactions, forming the according adducts or specific modifications, as HNE-induced adducts (HNE), protein carbonyls (PC), products of S-glutathionylation (S-Glut.) or glycoxidation (Glyc.) as shown in the table on the right. Certainly more subunits of both 20S and 19S can be modified by ROS resulting in the induction of protein carbonyls, but until now, the according products have not been shown experimentally. The inhibitory effects of HNE-modification on the proteasome have already been described in detail [6], [15], [16], [17], [18], [19]. Almost, every single alpha- and beta-subunits can be modified by HNE, as well as Rpt4 of the 19S regulator. Formation of HNE-adducts in proteins is often not a reversible modification and it can be assumed that massive HNE-adduct formation will probably decrease proteasomal activity. Besides of HNE-modification, many ROS are able to induce the formation of protein carbonyls, the most common product of protein oxidation, in different proteasomal subunits. The 19S-subunit Rpt3 (ATPase-subunit) was shown by Ishii to be very susceptible to oxidation, in this case induction of protein carbonyls via a copper-catalyzed Fenton-system [20]. The formation of protein carbonyls is not a reversible modification, so the 26S-mediated degradation of proteins is significantly decreased [20] as was also shown in other stress models [7]. A very important reversible posttranslational modification of proteasomal subunits is the S-glutathionylation of certain cysteine-residues, found in both the 20S proteasome and the 19S regulator. S-Glutathionylation is the addition of glutathione to a cysteine-residue that can be reversed in an enzymatic way. S-glutathionylation of 20S proteasomal subunits affects mainly alpha subunits as shown predominantly in yeast, directly influencing their gating function and thus proteasomal activity. The residues Cys76 and Cys221 of the α5-subunit of the 20S in yeast were shown to be S-glutathionylated in an ROS-dependent manner, increasing the proteasomal activity via opening of the annulus [21]. The same modification was found in subunits of the 19S regulator: after H2O2-mediated S-glutathionylation of the 26S proteasome, 26S proteasomal activity decreased significantly, while 20S activity remained unchanged [22], as found in isolated samples, as well as in HEK 293-cells and neutrophils. The affected 19S subunits were Rpn1 and Rpn2, which play both a role in recognition and binding of polyubiquitinated 26S-substrates as well as in the transfer of those substrates to the 20S core for terminal degradation [23]. Interestingly, also other components of the UPS have been shown to be S-glutathionylated, so some E1 and E2 enzymes, responsible for polyubiquitination of 26S proteasomal substrates. All these enzymes contain a functional sulfhydryl, than can be S-glutathionylated under oxidative stress, decreasing the polybiquitinylation of native proteins [10], [24]. Another predominant modification is glycoxidation of proteasomal subunits. This was found for the first time in the subunits α1–α3, as well as in β4–β6 of murine cardiac proteasomes [25]. Other studies found glycoxidation of the subunits β2, β4 and β5 after incubation of proteasomes with methyglyoxal (MGO), while no modifications in the alpha-subunits were detected [26]. The same study revealed a significantly decreased chymotrypsin-like activity (β5-subunit) in the endothelial cells after incubation with high glucose, as well as a reduction of the 19S regulator complex; in diabetic mice the proteasomal activity was shown to be decreased, without any change in the proteasomal protein levels detected [26].
Under normal conditions (no oxidative stress), the transcription factor Nuclear factor (erythroid-derived 2)-like 2 (Nrf2) is kept in complex and thus inactive by the kelch-like ECH-associated protein 1 (Keap1) [9]. Keap1 forms a complex with Cul3 and Rbx1, which function as an E3-ligase and mediates the polyubiquitination of Nrf2, thus, labeling it as a substrate for 26S proteasomal degradation. Consequently, the intracellular half-life of Nrf2 is very short (less than 30 min [27]). In phases of oxidative stress, Keap1 may become (oxidatively) damaged and Nrf2 is released into the cytosol. The cysteine residues of Keap1 Cys151/273/288 [28] function as “redox-sensors” inducing a shift in the tertiary structure of Keap1 after oxidation that results in a release of Nrf2 from Keap1. The same effect can be produced by Nrf2-inducers (like lipoic acid, curcumine or sulforaphane [28]), that trigger the release of Nrf2 from Keap1 as well as by proteasomal inhibition, preventing Nrf2 from proteolytic degradation. After being released into the cytosol, Nrf2 becomes quickly phosphorylated by phosphatidylinositol-3-kinase (PI3K) or protein kinase B (PKB, also termed as Akt) and is then transported into the cellular nucleus via the importins α5 and β1 [29]. Once in the nucleus, it induces in combination with other transcription factors (sMaf, ATF4, JunD and PMF-1 [30]) the expression of genes harboring the ARE/EpRE-element. Thus, Nrf2 is one of the major factors in the antioxidative response of mammalian cells. This response can be suppressed via Nrf2-siRNA or Nrf2-inhibitors, as retinoic acid [32]. That antioxidative response includes the expression of different enzymes like NAD(P)H quinone oxidoreductase 1 (Nqo1), heme oxigenase-1 (HO-1), catalase (Cat), Cu/Zn-superoxide dismutase (SOD), glutathione S-transferase (GST), multidrug resistance-associated proteins (Mrps), UDP-glucuronosyltransferases (UGTs), glutamate-cysteine ligase (GCLC, the rate- limiting enzyme in the synthesis of the antioxidant glutathione), the cysteine/glutamate antiporter (xCT) and Nrf2 itself [28], [31]. Overall, these enzymes increase the cellular resistance to oxidative and chemical stress. In addition to those also several subunits of the proteasomal system are induced. A study using microarrays identified the following subunits induced: α1 and α2, as well as α3–α7 and β1–β6 of the 20S proteasome, together with the 19S-subunits Rpt1/2/5, Rpn2/5/6/8/9/10/11/12 and S5b, while both subunits of Pa28 (also termed as 11S proteasome regulator) and the inducible subunits of the proteasome were not detected [31]. Another source mentions the alpha-subunits 1 and 2, the beta-subunits 3, 5 and 6, the 19S-subunits Rpt2, Rpt5, and Rpn11 all increase, as well as no Pa28 or inducible 20S-subunits [28]. However, Pickering et al. instead lists up 20S proteasomal subunits, as well as Pa28α and Pa28β [32]. So the role of Nrf2 in the induction of Pa28 remains to be tested.
As already mentioned in part I of this series [1], [2] several 20S proteasomal subunits are inducible. The traditional inducers were cytokines as γ-interferon [2]. However, as shown by Grimm et al. [33], both interferon-gamma (IFN-γ) and advanced glycation end products (AGEs) are able to induce the formation of both inducible proteasomal subunits (β1i/2i/5i) and α- and β-subunits of the proteasomal regulator protein Pa28 (also named 11S regulator). IFN-γ is a cytokine involved in the activation of macrophages in adaptive immune-response and is a recognized agent for the induction of both the inducible proteasome and Pa28-subunits [34], hence the name immunoproteasome. AGEs are glycoxidized proteins that result from the non-enzymatic reaction of lysine residues with different carbonyls. The common link between IFN-γ and AGEs is the Janus kinase (Jak2) that can be induced by both IFN-γ and AGEs. Binding of IFN-γ to the according receptor (IFNGR1+2, left part of the figure), induces phosphorylation and release of Jak1 and Jak2 (blue) from the membrane-bound receptor-complex into the cytosol. AGEs bind to the receptor for advanced glycation end products (RAGE) that belongs to the superfamily of immunoglobulins (right part of this figure), that is formed of three immunoglobulin-like domains (two constant and one variable type). The variable type is the critical one for the recognition of AGEs [35]. The RAGE-mediated activation of Jak2 was shown to be inhibited with RAGE-siRNA, as well as both the AGE and the IFN-γ mediated formation of immunoproteasomal and Pa28-subunits via AG-490 or STAS-1-siRNA [33]. Jak2 activates STAT1 (red) via a phosphorylation of its Tyr701 residue. After AGE-treatment the amount of cellular phosphorylated STAT1 showed a significant increase [33]. Phosphorylated STAT1 forms homodimers and is transported with the interferon regulatory factor-1 (IRF-1, yellow) into the nucleus. This cascade can be inhibited effectively with STAT1 siRNA, too. After transport into the nucleus, a complex of two phosphorylated STAT-1 proteins and interferon regulatory factor-1 (IRF-1) bind to the interferon gamma activated site (GAS), a short stretch of DNA, that functions as the promotor of IFN-gamma-activated genes, in this case the interferon-stimulated response element (ISRE) [36], that expresses (amongst others) the inducible proteasomal subunits (β1i, β2i and β5i), that are incorporated into de novo synthesized 20S proteasomes, and the Pa28α- and Pa28β-subunits of the 11S regulator.
Exposure of cells to highly reactive oxidants from different sources (in this figure summarized as “ROS”), can damage various cellular structures as proteins, lipids or RNA/DNA. Even if the bulk of oxidative modification is found in the cytosolic protein pool [37], [38], [39], [40], the nuclear DNA is not completely spared. Since the proteins are subject to a constant turnover, moderate oxidative modification of proteins is not an acute threat for cellular viability or functionality. In contrast, even slight oxidative damage to a virtual static structure as the DNA, that is construed for perdurability and not subjected to constant turnover, is very well compromising cellular viability or functionality. Considering the life span of the DNA in mammalian cells (and thus in whole organisms), powerful systems are needed, that are able to recognize and repair any DNA-damage immediately. One of the first “detectors” of oxidative DNA-damage like single-(SSB) and double-strand breaks (DSB) is the nuclear poly(ADP-ribose)-polymerase-1 (PARP-1). Besides of PARP-1; PARP-2 and PARP-3 fulfill similar tasks in recognition of DNA-damage and induction of repair-cascades, but PARP-1 is the most active one of this group [41]. After binding to SSB/DSB, PARP-1 is actively producing poly-ADP-ribose. PARP-1s main targets of poly(ADP-ribosyl)ation are histones. Further targets are NF-κB, p53, DNA-topoisomerases, DNA-PKcs, PARP-1 itself [42] and the nuclear 20S proteasome [13], [43], [44]. Interestingly, the poly(ADP-ribosyl)ation of histones seems to be restricted to the non-damaged ones, while oxidatively damaged histones are not modified by PARP-1. Therefore, PARP-1-mediated modification of histones seems to have a protective, selection-like effect preserving functional histones from proteasomal degradation [13]. While PARP-1 modifies its targets, large amounts of NAD+ are consumed. In the presence of DNA-damage, the PARP-1 activity can increase very quickly, and thus, the limiting substrate of mitochondrial ATP-synthesis (NAD+) is depleted. Recently PARP-1 is considered as the link between the DNA damage repair machinery and the remove of oxidatively damaged nuclear proteins [13]. Poly(ADP-ribosyl)ation of PARP-1-targets can be prevented effectively with specific inhibitors like 2-ABA or PJ-34 [13], while the PARP-1-mediated proteasomal activation can be suppressed effectively with both proteasomal inhibitors like lactacystin (LC) and PARP-1 inhibitors [13], [44]. As mentioned in the first part of this series [1], [2], the proteasomal activity declines in aged cells, even if there is no change in the amount of proteasome that may be due to proteasomal inhibition via protein aggregates [45], [46], [47], [48], [49]. The decline of the nuclear proteasomal fraction turned out to be less distinct, even if the amount of protein oxidation in the nucleus is higher in aged than in young cells, though no large covalently cross-linked protein aggregates are found there [37], [40]. Experiments to clarify those questions revealed that the nuclear proteasomal PARP-1-mediated activation in senescent mammalian cells seems to be decreased due to a decrease in the amount of PARP-1 [44]. This PARP-1-mediated proteasomal activation has been shown in different cell types [12], [50], [51], [52], [53], plays an essential role in the recognition and removal of oxidized histones and thus contributes to restore of DNA integrity. Furthermore, decreased PARP-1-function results in an increased DNA-damage as well as lowered amount of proteolytic degradation of damaged histones. Consequently, repair of hydrogen peroxide induced DNA-damage (quantified via 8-OHdG formation) was effectively abolished via proteasomal inhibitors, as well as by inhibition of PARP-1 [13].
One of the first stress-responses of the UPS is the proteasomal activation by nuclear PARP-1 as described in detail above (see Fig. 4). Furthermore, a rapid decline of the 26S proteasomal activity takes place, which is driven by an Hsp70-mediated detachment of the 19S regulator from the 26S proteasome [54]. In the cytosol, additional stress-induced modifications like S-glutathionylation and phosphorylation may play additional roles in the activation of free 20S “core” proteasome as well as in inactivation/disassembly of the 26S proteasome. All this takes place in a time frame of minutes (5–30 min, depending on the cell type). By liberation of the 20S proteasome from the 26S proteasomal particle an enhanced binding of the Pa28 proteasome activator takes place, leading to an effective breakdown of oxidized proteins [3]. This degradation of oxidized proteins is completed between 3 and 24 h after the stress. After coping with the most dramatic redox-related cellular changes within a few hours the 26S proteasome re-assembles again releasing Hsp70 and the ubiquitin-proteasomal-system (UPS) is reactivated [54]. In later hours after the stress exposure (some 12–72 h) a de novo synthesis of proteasome, inducible proteasome and Pa28- and Pa700-subunits is induced, significantly increasing the proteolytic capacity of the cell. Consequently, low amounts of oxidative stress (like “pretreatment” with low amounts of oxidants) can induce an increased resistance of the cell to following events of oxidative stress [55], an effect termed as ‘adaptation’ or ‘hormesis’.
References
- 1.Jung T., Grune T. The proteasome and the degradation of oxidized proteins: Part I-Structure of proteasomes. Redox Biol. 2013;1:178–182. doi: 10.1016/j.redox.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jung T., Catalgol B., Grune T. The proteasomal system. Mol. Aspects Med. 2009;30:191–296. doi: 10.1016/j.mam.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 3.Jung T., Höhn A., Grune T. The proteasome and the degradation of oxidized proteins: Part II – protein oxidation and proteasomal degradation. Redox Biol. 2014;2:99–104. doi: 10.1016/j.redox.2013.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sies H. Oxidative stress: from basic research to clinical application. Am. J. Med. 1991;91:31S–38. doi: 10.1016/0002-9343(91)90281-2. [DOI] [PubMed] [Google Scholar]
- 5.Jones D.P. Redefining oxidative stress. Antioxid. Redox Signal. 2006;8:1865–1879. doi: 10.1089/ars.2006.8.1865. [DOI] [PubMed] [Google Scholar]
- 6.Okada K., Wangpoengtrakul C., Osawa T., Toyokuni S., Tanaka K., Uchida K. 4-Hydroxy-2-nonenal-mediated impairment of intracellular proteolysis during oxidative stress. Identification of proteasomes as target molecules. J. Biol. Chem. 1999;274:23787–23793. doi: 10.1074/jbc.274.34.23787. [DOI] [PubMed] [Google Scholar]
- 7.Reinheckel T., Sitte N., Ullrich O., Kuckelkorn U., Davies K.J., Grune T. Comperative resistance of the 20S and 26S proteasome to oxidative stress. Biochem. J. 1998;335:637–642. doi: 10.1042/bj3350637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reinheckel T., Ullrich O., Sitte N., Grune T. Differential impairment of 20S and 26S proteasome activities in human hematopoietic K562 cells during oxidative stress. Arch. Biochem. Biophys. 2000;377:65–68. doi: 10.1006/abbi.2000.1717. [DOI] [PubMed] [Google Scholar]
- 9.Kansanen E., Kuosmanen S.M., Leinonen H., Levonen A.L. The Keap1-Nrf2 pathway: mechanisms of activation and dysregulation in cancer. Redox Biol. 2013;1:45–49. doi: 10.1016/j.redox.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Obin M., Shang F., Gong X., Handelman G., Blumberg J., Taylor A. Redox regulation of ubiquitin-conjugating enzymes: mechanistic insights using the thiol-specific oxidant diamide. FASEB J. 1998;12:561–569. doi: 10.1096/fasebj.12.7.561. [DOI] [PubMed] [Google Scholar]
- 11.Shang F., Taylor A. Ubiquitin–proteasome pathway and cellular responses to oxidative stress. Free Radical Biol. Med. 2011;51:5–16. doi: 10.1016/j.freeradbiomed.2011.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ullrich O., Reinheckel T., Sitte N., Hass R., Grune T., Davies K.J. Poly-ADP ribose polymerase activates nuclear proteasome to degrade oxidatively damaged histones. Proc. Nat. Acad. Sci. U.S.A. 1999;96:6223–6228. doi: 10.1073/pnas.96.11.6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Catalgol B., Wendt B., Grimm S., Breusing N., Ozer N.K., Grune T. Chromatin repair after oxidative stress: role of PARP-mediated proteasome activation. Free Radical Biol. Med. 2010;48:673–680. doi: 10.1016/j.freeradbiomed.2009.12.010. [DOI] [PubMed] [Google Scholar]
- 14.Sledz P., Unverdorben P., Beck F., Pfeifer G., Schweitzer A., Forster F. Structure of the 26S proteasome with ATP-gammaS bound provides insights into the mechanism of nucleotide-dependent substrate translocation. Proc. Nat. Acad. Sci. U.S.A. 2013;110:7264–7269. doi: 10.1073/pnas.1305782110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Farout L., Mary J., Vinh J., Szweda L.I., Friguet B. Inactivation of the proteasome by 4-hydroxy-2-nonenal is site specific and dependant on 20S proteasome subtypes. Arch. Biochem. Biophys. 2006;453:135–142. doi: 10.1016/j.abb.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 16.Ferrington D.A., Kapphahn R.J. Catalytic site-specific inhibition of the 20S proteasome by 4-hydroxynonenal. FEBS Lett. 2004;578:217–223. doi: 10.1016/j.febslet.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 17.Hyun D.H., Lee M.H., Halliwell B., Jenner P. Proteasomal dysfunction induced by 4-hydroxy-2,3-trans-nonenal, an end-product of lipid peroxidation: a mechanism contributing to neurodegeneration? J. Neurochem. 2002;83:360–370. doi: 10.1046/j.1471-4159.2002.01125.x. [DOI] [PubMed] [Google Scholar]
- 18.Okada K., Wangpoengtrakul C., Osawa T., Toyokuni S., Tanaka K., Uchida K. 4-Hydroxy-2-nonenal-mediated impairment of intracellular proteolysis during oxidative stress. Identification of proteasomes as target molecules. J. Biol. Chem. 1999;274:23787–23793. doi: 10.1074/jbc.274.34.23787. [DOI] [PubMed] [Google Scholar]
- 19.Shringarpure R., Grune T., Sitte N., Davies K.J. 4-Hydroxynonenal-modified amyloid-beta peptide inhibits the proteasome: possible importance in Alzheimer's disease. Cell. Mol. Life Sci. 2000;57 doi: 10.1007/PL00000660. (1802-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ishii T., Sakurai T., Usami H., Uchida K. Oxidative modification of proteasome: identification of an oxidation-sensitive subunit in 26S proteasome. Biochemistry. 2005;44:13893–13901. doi: 10.1021/bi051336u. [DOI] [PubMed] [Google Scholar]
- 21.Silva G.M., Netto L.E., Simoes V., Santos L.F., Gozzo F.C., Demasi M.A. Redox control of 20S proteasome gating. Antioxid. Redox Signal. 2012;16:1183–1194. doi: 10.1089/ars.2011.4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zmijewski J.W., Banerjee S., Abraham E. S-glutathionylation of the Rpn2 regulatory subunit inhibits 26S proteasomal function. J. Biol. Chem. 2009;284:22213–22221. doi: 10.1074/jbc.M109.028902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rosenzweig R., Bronner V., Zhang D., Fushman D., Glickman M.H. Rpn1 and Rpn2 coordinate ubiquitin processing factors at proteasome. J. Biol. Chem. 2012;287:14659–14671. doi: 10.1074/jbc.M111.316323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jahngen-Hodge J., Obin M.S., Gong X., Shang F., Nowell T.R., Jr., Gong J. Regulation of ubiquitin-conjugating enzymes by glutathione following oxidative stress. J. Biol. Chem. 1997;272:28218–28226. doi: 10.1074/jbc.272.45.28218. [DOI] [PubMed] [Google Scholar]
- 25.Zong C., Young G.W., Wang Y., Lu H., Deng N., Drews O. Two-dimensional electrophoresis-based characterization of post-translational modifications of mammalian 20S proteasome complexes. Proteomics. 2008;8:5025–5037. doi: 10.1002/pmic.200800387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Queisser M.A., Yao D., Geisler S., Hammes H.P., Lochnit G., Schleicher E.D. Hyperglycemia impairs proteasome function by methylglyoxal. Diabetes. 2010;59:670–678. doi: 10.2337/db08-1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Katoh Y., Iida K., Kang M.I., Kobayashi A., Mizukami M., Tong K.I. Evolutionary conserved N-terminal domain of Nrf2 is essential for the Keap1-mediated degradation of the protein by proteasome. Arch. Biochem. Biophys. 2005;433:342–350. doi: 10.1016/j.abb.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 28.Chapple S.J., Siow R.C., Mann G.E. Crosstalk between Nrf2 and the proteasome: therapeutic potential of Nrf2 inducers in vascular disease and aging. Int. J. Biochem. Cell Biol. 2012;44:1315–1320. doi: 10.1016/j.biocel.2012.04.021. [DOI] [PubMed] [Google Scholar]
- 29.Theodore M., Kawai Y., Yang J., Kleshchenko Y., Reddy S.P., Villalta F. Multiple nuclear localization signals function in the nuclear import of the transcription factor Nrf2. J. Biol. Chem. 2008;283:8984–8994. doi: 10.1074/jbc.M709040200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Giudice A., Arra C., Turco M.C. Review of molecular mechanisms involved in the activation of the Nrf2-ARE signaling pathway by chemopreventive agents. Methods Mol. Biol. 2010;647:37–74. doi: 10.1007/978-1-60761-738-9_3. [DOI] [PubMed] [Google Scholar]
- 31.Kwak M.K., Wakabayashi N., Greenlaw J.L., Yamamoto M., Kensler T.W. Antioxidants enhance mammalian proteasome expression through the Keap1-Nrf2 signaling pathway. Mol. Cell. Biol. 2003;23:8786–8794. doi: 10.1128/MCB.23.23.8786-8794.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pickering A.M., Linder R.A., Zhang H., Forman H.J., Davies K.J. Nrf2-dependent induction of proteasome and Pa28alphabeta regulator are required for adaptation to oxidative stress. J. Biol. Chem. 2012;287:10021–10031. doi: 10.1074/jbc.M111.277145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grimm S., Ott C., Horlacher M., Weber D., Hohn A., Grune T. Advanced-glycation-end-product-induced formation of immunoproteasomes: involvement of RAGE and Jak2/STAT1. Biochem. J. 2012;448:127–139. doi: 10.1042/BJ20120298. [DOI] [PubMed] [Google Scholar]
- 34.Bose S., Stratford F.L., Broadfoot K.I., Mason G.G., Rivett A.J. Phosphorylation of 20S proteasome alpha subunit C8 (alpha7) stabilizes the 26S proteasome and plays a role in the regulation of proteasome complexes by gamma-interferon. Biochem. J. 2004;378:177–184. doi: 10.1042/BJ20031122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schmidt A.M., Yan S.D., Yan S.F., Stern D.M. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J. Clin. Invest. 2001;108:949–955. doi: 10.1172/JCI14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gomez-Lucia E., Collado V.M., Miro G., Domenech A. Effect of type-I interferon on retroviruses. Viruses. 2009;1:545–573. doi: 10.3390/v1030545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jung T., Hohn A., Catalgol B., Grune T. Age-related differences in oxidative protein-damage in young and senescent fibroblasts. Arch. Biochem. Biophys. 2009;483:127–135. doi: 10.1016/j.abb.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 38.Jung T., Bader N., Grune T. Oxidized proteins: intracellular distribution and recognition by the proteasome. Arch. Biochem. Biophys. 2007;462:231–237. doi: 10.1016/j.abb.2007.01.030. [DOI] [PubMed] [Google Scholar]
- 39.Jung T., Engels M., Klotz L.O., Kroncke K.D., Grune T. Nitrotyrosine and protein carbonyls are equally distributed in HT22 cells after nitrosative stress. Free Radical Biol. Med. 2007;42:773–786. doi: 10.1016/j.freeradbiomed.2006.11.029. [DOI] [PubMed] [Google Scholar]
- 40.Jung T., Engels M., Kaiser B., Poppek D., Grune T. Intracellular distribution of oxidized proteins and proteasome in HT22 cells during oxidative stress. Free Radical Biol. Med. 2006;40:1303–1312. doi: 10.1016/j.freeradbiomed.2005.11.023. [DOI] [PubMed] [Google Scholar]
- 41.De V.M., Schreiber V., Dantzer F. The diverse roles and clinical relevance of PARPs in DNA damage repair: current state of the art. Biochem. Pharmacol. 2012;84:137–146. doi: 10.1016/j.bcp.2012.03.018. [DOI] [PubMed] [Google Scholar]
- 42.Abeti R., Duchen M.R. Activation of PARP by oxidative stress induced by beta-amyloid: implications for Alzheimer's disease. Neurochem. Res. 2012;37:2589–2596. doi: 10.1007/s11064-012-0895-x. [DOI] [PubMed] [Google Scholar]
- 43.Arnold J., Grune T. PARP-mediated proteasome activation: a co-ordination of DNA repair and protein degradation? Bioessays. 2002;24:1060–1065. doi: 10.1002/bies.10179. [DOI] [PubMed] [Google Scholar]
- 44.Bakondi E., Catalgol B., Bak I., Jung T., Bozaykut P., Bayramicli M. Age-related loss of stress-induced nuclear proteasome activation is due to low PARP-1 activity. Free Radical Biol. Med. 2011;50:86–92. doi: 10.1016/j.freeradbiomed.2010.10.700. [DOI] [PubMed] [Google Scholar]
- 45.Sitte N., Huber M., Grune T., Ladhoff A., Doecke W.D., von Z.T. Proteasome inhibition by lipofuscin/ceroid during postmitotic aging of fibroblasts. FASEB J. 2000;14:1490–1498. doi: 10.1096/fj.14.11.1490. [DOI] [PubMed] [Google Scholar]
- 46.Hohn A., Grune T. Lipofuscin: formation, effects and role of macroautophagy. Redox Biol. 2013;1:140–144. doi: 10.1016/j.redox.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hohn A., Jung T., Grimm S., Catalgol B., Weber D., Grune T. Lipofuscin inhibits the proteasome by binding to surface motifs. Free Radical Biol. Med. 2011;50:585–591. doi: 10.1016/j.freeradbiomed.2010.12.011. [DOI] [PubMed] [Google Scholar]
- 48.Grune T., Jung T., Merker K., Davies K.J. Decreased proteolysis caused by protein aggregates, inclusion bodies, plaques, lipofuscin, ceroid, and ‘aggresomes’ during oxidative stress, aging, and disease. Int. J. Biochem. Cell Biol. 2004;36:2519–2530. doi: 10.1016/j.biocel.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 49.Grune T., Reinheckel T., Davies K.J.A. Degradation of oxidized proteins in mammalian cells. FASEB J. 1997;11:526–534. [PubMed] [Google Scholar]
- 50.Ullrich O., Ciftci O., Hass R. Proteasome activation by poly-ADP-ribose-polymerase in human myelomonocytic cells after oxidative stress. Free Radical Biol. Med. 2000;29:995–1004. doi: 10.1016/s0891-5849(00)00399-3. [DOI] [PubMed] [Google Scholar]
- 51.Ciftci O., Ullrich O., Schmidt C.A., Diestel A., Hass R. Regulation of the nuclear proteasome activity in myelomonocytic human leukemia cells after adriamycin treatment. Blood. 2001;97:2830–2838. doi: 10.1182/blood.v97.9.2830. [DOI] [PubMed] [Google Scholar]
- 52.Mayer-Kuckuk P., Ullrich O., Ziegler M., Grune T., Schweiger M. Functional interaction of poly(ADP-ribose) with the 20S proteasome in vitro. Biochem. Biophys. Res. Commun. 1999;259:576–581. doi: 10.1006/bbrc.1999.0824. [DOI] [PubMed] [Google Scholar]
- 53.Ullrich O., Diestel A., Bechmann I., Homberg M., Grune T., Hass R. Turnover of oxidatively damaged nuclear proteins in BV-2 microglial cells is linked to their activation state by poly-ADP-ribose polymerase. FASEB J. 2001;15:1460–1462. doi: 10.1096/fj.00-0540fje. [DOI] [PubMed] [Google Scholar]
- 54.Grune T., Catalgol B., Licht A., Ermak G., Pickering A.M., Ngo J.K. HSP70 mediates dissociation and reassociation of the 26S proteasome during adaptation to oxidative stress. Free Radical Biol. Med. 2011;51:1355–1364. doi: 10.1016/j.freeradbiomed.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pickering A.M., Davies K.J. Differential roles of proteasome and immunoproteasome regulators Pa28alphabeta, Pa28gamma and Pa200 in the degradation of oxidized proteins. Arch. Biochem. Biophys. 2012;523:181–190. doi: 10.1016/j.abb.2012.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]