

Abstract
The repair of free-radical oxidative DNA damage is carried out by lesion-specific DNA glycosylases as the first step of the highly conserved base excision repair (BER) pathway. In humans, three orthologs of the prototypical endonuclease VIII (Nei), the Nei-like NEIL1–3 enzymes are involved in the repair of oxidized DNA lesions. In recent years, several genome and cancer single-nucleotide polymorphic variants of the NEIL1 glycosylase have been identified. In this study we characterized four variants of human NEIL1: S82C, G83D, P208S, and ΔE28, and tested their ability to excise pyrimidine-derived lesions such as thymine glycol (Tg), 5-hydroxyuracil (5-OHU), and dihydrouracil (DHU) and the purine-derived guanidinohydantoin (Gh), spiroiminodihydantoin 1 (Sp1), and methylated 2,6-diamino-4-hydroxy-5-formamidopyrimidine (MeFapyG). The P208S variant has near wild-type activity on all substrates tested. The S82C and ΔE28 variants exhibit decreased Tg excision compared to wild-type. G83D displays little to no activity with any of the substrates tested, with the exception of Gh and Sp1. Human NEIL1 is known to undergo editing whereby the lysine at position 242 is recoded into an arginine. The non-edited form of NEIL1 is more efficient at cleaving Tg than the R242 form, but the G83D variant does not cleave Tg regardless of the edited status of NEIL1. The corresponding G86D variant in Mimivirus Nei1 similarly lacks glycosylase activity. A structure of a G86D-DNA complex reveals a rearrangement in the β4/5 loop comprising Leu84, the highly-conserved void-filling residue, thereby providing a structural rationale for the decreased glycosylase activity of the glycine to aspartate variant.
Keywords: Oxidized DNA lesions, base excision repair, DNA glycosylase, NEIL1, single nucleotide polymorphisms
1. INTRODUCTION
Reactive oxygen species (ROS) are generated in the cell as a result of byproducts of aerobic respiration, harmful chemicals found in cigarette smoke, chemotherapeutic drugs, ultraviolet light, and ionizing radiation [1, 2]. ROS lead to the generation of potentially lethal or mutagenic oxidative DNA damage, which if left unrepaired, results in deleterious mutations, cell death, and genomic instability [3, 4]. The base excision repair (BER) pathway is a highly coordinated sequential process, and is the major pathway involved in the repair of oxidative base lesions [5–8]. The pathway is initiated by a lesion-specific DNA glycosylase, which catalyzes the cleavage of the N-glycosidic bond, generating an apurinic/apyrimidinic (AP) site [9, 10]. Some glycosylases are bifunctional and have an associated lyase activity, which cleaves the DNA backbone 3′ to the generated AP site. In prokaryotes, removal of oxidized purines such as 8-oxo guanine (8-oxoG) is primarily carried out by formamidopyrimidine DNA glycosylase (Fpg), whereas oxidized pyrimidines such as thymine glycol (Tg) are cleaved by endonuclease III (Nth) and endonuclease VIII (Nei) [1, 11–13]. The Fpg and Nei enzymes share a common structural fold and are collectively termed the Fpg/Nei family [14, 15]. In humans, three Nei-like (NEIL) enzymes NEIL1, 2, and 3 have been characterized as structural homologs of Nei and Fpg [13, 16–20]. Human NEIL1 has a broad substrate specificity and can cleave lesions present in single-stranded (ss), double-stranded (ds), and bubble DNA structures [17, 21–24]. The best substrates for NEIL1 include the further oxidation products of 8-oxoG, spiroiminodihydantoin (Sp) and guanidinohydantoin (Gh), although 8-oxoG itself is a poor substrate for the enzyme [21],[25]. Gh- and Sp-containing substrates are cleaved ~100-fold faster than oxidized pyrimidine substrates containing Tg and 5-hydroxycytosine [21]. NEIL1 also excises 5-hydroxyuracil (5-OHU), the ring opened formamidopyrimidines (FapyG and FapyA), and dihydrouracil (DHU), among others (Fig. 1) [13, 17, 22, 24, 26].
Figure 1.
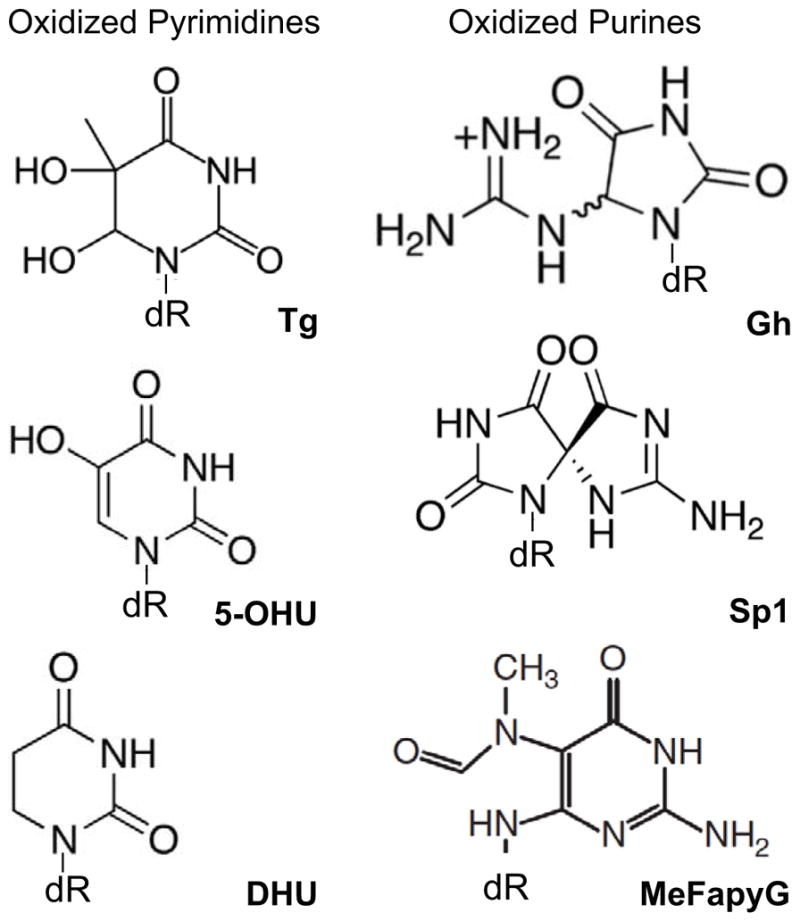
Human NEIL1 substrates used in this study. The oxidized pyrimidines include Tg, thymine glycol; 5-OHU, 5-hydroxyuracil; and DHU, dihydrouracil. The oxidized purines include Gh, guanidinohydantoin; Sp1, spiroiminodihydantoin 1; and MeFapyG, methylated 2,6-diamino-4-hydroxy-5-formamidopyrimidine.
In recent years, there has been increasing evidence that sequence variants and single nucleotide polymorphisms (SNPs) in DNA repair genes modulate DNA repair capacity and predisposition to various disease states including cancer [27–29]. This is based on numerous reports in the literature regarding the involvement of variant BER enzymes such as MUTYH, NEIL2, OGG1, APE1, and Pol β in tumorigenesis [30], [31]. Over the past decade, several polymorphic variants of the human NEIL1 enzyme have been identified in the germline as well as in some cancers [32–35]. Roy et al. conducted studies on four variants of NEIL1 including S82C, G83D, C136R, and D252N using Tg- and AP-containing substrates. Their analysis indicated that while S82C and D252N had near wild-type activity on Tg, G83D and C136R were devoid of glycosylase activity, and C136R also had reduced AP-lyase activity [32]. The G83D variant also showed a lack of activity on 8-oxoG, Tg, and dihydrothymine substrates [35]. Another variant of NEIL1, lacking a glutamate at position 28 (ΔE28), displayed a reduced activity with a Tg substrate [33].
We selected four genome and cancer variants, S82C, G83D, P208S, and ΔE28, and further characterized them by measuring their glycosylase and lyase activity using a substrate panel of six purine or pyrimidine-based lesions and an AP site (Fig. 1). The latter three variants were identified in patients with primary sclerosing cholangitis/ cholangiocarcinoma, colon, and gastric cancers, respectively [33–35]. RNA editing of human NEIL1 from lysine to arginine at position 242 was shown to modulate its glycosylase activity: the non-edited K242 form cleaves Tg 30–40 fold faster than the edited R242 form [36]. We show here that the G83D variant has little to no activity on oxidized pyrimidine substrates, regardless of the edited status of NEIL1. The sequence conservation of the glycine residue at position 83 led us to solve the crystal structure of a viral ortholog of human NEIL1, Nei1 from the Acanthamoeba polyphaga mimivirus (MvNei1), bearing the corresponding G86D mutation [37, 38]. The structure of the G86D variant bound to DNA reveals a shift in a loop involved in lesion stabilization, thereby providing a structural rationale for the decreased glycosylase activity of the variant. Our structural and biochemical data are supported by a rifampicin resistance assay, which shows that NEIL1-G83D has little to no glycosylase activity in vivo.
2. MATERIALS AND METHODS
2.1. Cloning, Overexpression, and Purification
The full-length human NEIL1 construct was cloned and purified as described previously [39]. Briefly, the protein was expressed in Rosetta2 (DE3) pLysS Escherichia coli cells (Novagen), followed by IPTG induction either for 4 hrs at 30°C or overnight at 12 – 16°C. The cell pellet was resuspended in a buffer containing 50 mM sodium phosphate, pH 8.0, 150 mM NaCl, 10 mM imidazole pH 8.0, 10% (v/v) glycerol, 5 mM β-Me, and 1 mM PMSF and sonicated. The clarified cell lysate was added to pre-equilibrated TALON cobalt resin (Clontech). The proteins were eluted using 250 mM imidazole in the above buffer and dialyzed overnight into a buffer containing 20 mM Tris-HCl, pH 7.5, 300 mM NaCl, 10% (v/v) glycerol, and 1 mM DTT. A HiTrap SP-FF column (GE healthcare) was used to further purify the enzymes to homogeneity. A linear NaCl gradient (300 mM – 1 M) was used to elute the enzymes and the resultant fractions were pooled and dialyzed into a buffer containing 20 mM Tris-HCl, pH 7.5, 300 mM NaCl, 10% (v/v) glycerol, and 1 mM DTT. The QuikChange XL II site-directed mutagenesis kit (Stratagene) was used to introduce the S82C, G83D, P208S, ΔE28, and K242 point mutations into NEIL1 in the pET30(a) (Novagen) expression vector. The resulting variants were completely sequenced to ensure the correct mutation was present. Circular dichroism spectra using a C-terminal truncation mutant of NEIL1 (NEIL1Δ56) at varying temperatures were acquired at the CD facility at Robert Wood Johnson Medical School (Piscataway, NJ).
Cloning, expression, and purification of MvNei1 have been described previously [37, 40]. After purification, MvNei1 was dialyzed into crystallization buffer (20 mM HEPES, pH 7.5, 300 mM NaCl, 10% (v/v) glycerol, and 1 mM DTT) and concentrated to 10 mg/ml. The G86D variant was introduced by site directed mutagenesis using the QuikChange XL II site-directed mutagenesis kit (Stratagene) and the protein was purified in a similar manner as wild-type MvNei1 and concentrated to ~2 mg/ml.
2.2. DNA Preparation and Complex Formation
The 35-mer oligodeoxynucleotides used for the glycosylase/lyase activity assays were purchased from Midland Certified Reagent Co. (Midland, TX) and purified by urea PAGE. The sequence of the damage-containing strand was 5′-TGTCAATAGCAAG(X)GGAGAAGTCAATCGTGAGTCT-3′, where X was Tg, 5-OHU, DHU, or uracil, which was used to create an apurinic/apyrimidic site (AP-site). The complementary oligonucleotide had the following sequence: 5′-AGACTCACGATTGACTTCTCC(C/G/A)CTTGCTATTGACA-3′, in which (C/G/A) denotes the base opposite the damaged lesion. Gh, Sp1, and 2,6-diamino-4-hydroxy-5N-methyl-formamidopyrimidine (MeFapyG) were synthesized as described previously [41],[42] in the following sequence context: 5′-TGTTCATCATGCGTC(Y)TCGGTATATCCCAT-3′, with Y being either Gh or Sp1, and 5′-GGTGTGAGTGTTA[MeFapyG]GGTGAG-AG-3′. The complementary strands for Gh/Sp1- and MeFapyG-containing DNA were 5′-ATGGGATATACCGA(C)GACGCATGATGAACA-3′ and 5′-CTCTCACC(C)T AACACTCACACC-3′. End labeling of substrates was carried out on 1 pmole of each damage-containing strand using T4 polynucleotide kinase (New England Biolabs, Beverly, MA) in presence of [γ-32P] dATP, for 30 min at 37 °C. The phosphorylation reaction was terminated by addition of 1 mM EDTA and heat inactivation. The end-labeled DNA was separated from the [γ-32P] dATP by ethanol precipitation and diluted in 9 pmoles of the appropriate non-labeled damage-containing oligodeoxynucleotide and 10 pmoles of the complementary oligodeoxynucleotide to a final concentration of 250 nM in 10 mM Tris–HCl (pH 8.0) and 50 mM NaCl. In order to create an AP site, a double-stranded uracil-containing oligodeoxynucleotide was treated with 2 units of uracil DNA glycosylase (New England Biolabs) for 30 min at 37 °C.
2.3. Glycosylase/Lyase Activity Assays
Glycosylase/lyase assays were performed with 20 nM lesion-containing substrate and increasing concentrations of enzyme in 20 mM HEPES, pH 7.5, 150 mM NaCl, 2 mM EDTA (pH 8.0) with 200 μg/ml BSA at 25 °C for 30 minutes. The reactions were stopped with the addition of an equal volume of formamide loading buffer (98% formamide, 5 mM EDTA, 0.1% xylene cyanol and 0.1% bromophenol blue) to assay for both glycosylase and lyase activities. The reaction products were separated from the uncleaved substrates with a 12% (w/v) denaturing polyacrylamide gel and quantified with an isotope imaging system (Molecular Imaging System, Bio-Rad).
2.4. Spontaneous Mutation Frequency Assays
E. coli KL16 Δfpg::Amp, Δnei::Cam, ΔmutY::Tet, and KL16 Δnei::Cam, Δnth::Kan were created as described earlier [16, 43]. The pET30(a) (Kan) and pET22(b) (Amp) vectors containing WT-NEIL1 or the G83D variant were electroporated into KL16 Δfpg::Amp, Δnei::Cam, ΔmutY::Tet (DE3), and KL16 Δnei::Cam, Δnth::Kan (DE3), respectively. A low-level of heterologous expression was maintained with 0.1 mM IPTG. Overnight cultures of each strain containing 0.1 mM IPTG and appropriate antibiotics were diluted 50-fold into fresh LB medium and further grown at 37 °C until an OD600 of 0.6. The cultures were plated onto LB-agar plates with and without 100 μg/mL of rifampin (Rif) and 0.1 mM IPTG. The Rif-resistant (RifR) mutation rates were calculated using the Ma-Sandri-Sarkar Maximum Likelihood Estimator (MSS-MLE) [44].
2.5. Crystallization: Conditions and Complex Formation
Sitting drop crystallization experiments with human NEIL1Δ56-K242 were set up as described previously [39]. Crystals belonging to the R3 space group in the hexagonal setting grew in 0.25 M lithium sulfate and 35% polyethylene glycol (PEG) 3350 (w/v) and data were collected using a rotating anode X-ray generator (Rigaku) and MAR image plate detector (MarResearch, Hamburg, Germany). The data collected were processed with Denzo and reduced with Scalepack [45]. The data indicated one molecule per asymmetric unit (asu) with a calculated solvent content of 44 % [46]. Data collection statistics are summarized in Table S2.
The DNA oligonucleotide containing tetrahydrofuran (THF) was purchased from Midland Certified Reagent Co. The 13-mer oligonucleotides 5′-CGTCCAXGTCTAC-3′ (X = THF) and 5′-GTAGACCTGGACG-3′ were annealed in a 1:1 ratio after PAGE purification. The protein-DNA complex was prepared by adding 1.2 molar excess of DNA to MvNei1 in the crystallization buffer. After buffer exchange into 20 mM HEPES, pH 7.5, 50 mM NaCl, 10% (v/v) glycerol, and 5 mM β-Me, the protein-DNA complex was concentrated to an A280 of 5.5–6 mg/ml as measured by a NanoDrop instrument (NanoDrop Technologies, Wilmington, DE). Crystals of MvNei1-G86D in complex with THF-containing DNA were obtained at 12 °C by the hanging drop, vapor-diffusion method by mixing 0.5 μl of protein-DNA complex in a 1:1 ratio with a reservoir solution made of 0.125 M potassium chloride, 0.05 M magnesium sulfate, 4% PEG 8000 (w/v), and 100 mM MES, pH 5.6. The crystals of MvNei1 in complex with THF were transferred to a solution of mother liquor supplemented with 50% (w/v) glycerol, followed by flash cooling. The monoclinic crystals diffract to a resolution of 2.85 Å using a rotating anode X-ray generator (Rigaku) and MAR image plate detector (MarResearch, Hamburg, Germany). The data set collected was processed with Denzo and reduced with Scalepack [45]. The data indicated two molecules per asu with a calculated solvent content of 55.5% [46]. Data collection statistics are summarized in Table S2.
2.6. Structure Determination
The structure of human NEIL1Δ56-K242 was solved using difference Fourier methods using phases from the previously reported NEIL1Δ56 crystal structure devoid of all non-protein atoms (protein data bank (PDB) code 1TDH [47]). An overall cross-R value on amplitudes of 0.208 was obtained [48] (personal communication with Dr. Mark Rould).
The structure of DNA-bound G86D MvNei1 was solved by molecular replacement methods using Phaser-MR from the PHENIX package suite [49, 50]. This was performed using the MvNei1-THF complex as a search model (PDB code 3A46 [37]) lacking all non-protein atoms. The resulting solution had a final translation function Z-score of 31.4 and a log-likelihood gain of 2616. Both the structures were refined using phenix.refine [50–52] with good geometry (Table S2), and Ramachandran analysis yielded no outliers in the disallowed region [53, 54]. The resulting coordinates and structure factors are deposited in the protein data bank as 4RNV (NEIL1-K242) and 4RNW (MvNei1-G86D-THF).
3. RESULTS
3.1. Human NEIL1 Genomic and Cancer Variants
Over the past few years, the search for SNPs in the human genome has been widely assisted by publically available databases such as the 1000 Genomes Project (1KG) and The Cancer Genome Atlas (TCGA) [55–57]. These databases provide a means to screen for low frequency, common variants with potentially important functional consequences obtained from individuals of different populations. In the current study we selected four polymorphic variants of the human NEIL1 enzyme, which occur at a frequency of 1–2% in the normal population and in tumors [55–57]. These include S82C, G83D, P208S, and ΔE28 [32–35]. Ser82 and Gly83 are in the vicinity of the void-filling Met81 residue (Fig. S1). This highly conserved methionine residue (or leucine residue as in the viral ortholog, MvNei1 [37]) is one of three void-filling residues (or is the only residue in the case of Neil3 [58]) that fill the void created upon lesion extrusion into the glycosylase active site. The other two residues, typically a phenylalanine and an arginine, cause buckling of the DNA at the damaged base pair and stabilize the orphaned base opposite the lesion, respectively [15, 59, 60]. P208S precedes αF, a helix that is part of an insertion sequence element that is unique to the human NEIL1 enzyme (Fig. S1). ΔE28 is present in β-strand 1 in the N-terminal domain of the enzyme (Fig. S1). All these variants of full-length NEIL1 were expressed in E. coli and purified to homogeneity (Fig. S2). In the sections below, we test the glycosylase activity of these variants using the panel of lesions shown in Fig. 1.
3.2. Catalytic Activity of the NEIL1 Variants
Although NEIL1 does not usually cleave 8-oxoG, it recognizes the further oxidation products of 8-oxoG, i.e. Gh and Sp, along with several pyrimidine-derived lesions including Tg [21, 25]. To compare the activity of the polymorphic variants with wild-type (WT) NEIL1, we selected the following lesions and opposite bases: Tg:A, 5-OHU:G, DHU:A, Gh:C, Sp1:C, MeFapyG:C, and AP:C. We tested the glycosylase and lyase activity of all the NEIL1 variants with each of the lesions at varying substrate:enzyme ratios (Fig. 2) and quantified the percentage of product detected for each reaction (Table S1). The most drastic change in glycosylase activity was observed with the G83D variant. This variant, unlike the other two cancer variants (P208S and ΔE28), displays a markedly decreased glycosylase activity compared to WT enzyme. The G83D variant has no detectable activity with the Tg lesion (Fig. 2A, Table S1), as shown earlier [32, 35]. A gel-shift assay indicates that, even though G83D does not excise Tg, the variant does bind to the oligonucleotide containing Tg:A (Fig. S3). We further show here that G83D has 3 – 8 fold reduced activity on the pyrimidine-derived lesions 5-OHU and DHU (Fig. 2B, 2C, Table S1), as well as MeFapyG (Fig. 2F, Table S1). However, it can cleave the Gh and Sp1 lesions (Fig. 2 D, E, Table S1), albeit to a lesser extent than WT. G83D and all other variants exhibit robust lyase activity on AP-site-containing DNA oligonucleotides (Fig. 2G, Table S1), with over 90% product detected at the higher enzyme concentrations.
Figure 2.

Glycosylase and lyase activity panel for human NEIL1 and its variants. Glycosylase assays were performed by incubating 20 nM of double-stranded substrates with increasing amounts of enzyme with the following substrate:enzyme ratios: 1:0.5, 1:1, 1:4, and 1:16. “∅” indicates no enzyme and F indicates E. coli Fpg used as a control. Assays were performed at room temperature for 30 minutes. Panel (A) Tg:A; (B) OHU:G; (C) DHU:A; (D) Gh:C; (E) Sp1:C; (F) MeFapyG:C; (G) AP:C. S and P indicate substrates and products respectively. Data shown are representative of two repeat experiments.
The S82C and ΔE28 variants excise Tg:A (Fig. 2A, Table S1) with ~2–3 fold reduction in product formation compared to WT, but retain the ability to cleave 5-OHU-, DHU-, Gh-, Sp1-, and MeFapyG-containing substrates (Fig. 2B–F, Table S1). Previously, S82C was reported to cleave a Tg substrate at near WT level whereas ΔE28 had a decreased Tg excision activity [32, 33]. We show that P208S possesses near WT activity for all lesions tested here (Fig. 2 A – G, Table S1).
3.3. Effects of Editing on the NEIL1-G83D Variant
The identification of RNA editing sites using a genome-wide sequencing approach indicated several hundred adenosine to inosine (A to I) editing sites within non-repetitive RNA sequences [61]. One such site predicted to cause recoding by the adenosine deaminase acting on RNA (ADAR1) enzyme is the mRNA for human NEIL1 where the AAA codon for Lys242 is edited to AIA, which encodes an Arg [36]. The edited NEIL1-R242 form removes a Tg lesion from DNA substrates ~30 – 40 fold more slowly than the NEIL1-K242 enzyme encoded in the genome [36]. In contrast, repair of Gh-containing substrates is enhanced by editing [36].
In an effort to analyze the structural consequences of editing on the NEIL1 enzyme, we solved the crystal structure of NEIL1-K242 and compared it to the previously reported structure of the R242 edited form of the enzyme [47]. The protein sequence originally reported for the NEIL1 enzyme was derived from cDNA and happened to contain an arginine at position 242. The published structure was thus that of the edited form of the glycosylase [47]. The structure of NEIL1-K242 was solved using difference Fourier methods [48] to a resolution of 2.6 Å (see Table S2 for statistics) and was globally very similar to the Arg242 structure (with a root mean square deviation (r.m.s.d.) of 0.22 Å calculated on 289 aligned Cα, Fig. S4). Arg242 is located in a short, flexible glycine-rich loop (GGK/R242GYG) connecting αG and αH. Bacterial Fpg glycosylases bear a longer loop at the same location, called the αF-β9/10 loop or the 8-oxoG capping loop [62, 63]. In the bacterial enzymes this loop is specific for the cleavage of the 8-oxoG lesion [63]. In human NEIL1 [47], MvNei1 [37], and mouse Neil3 [58], this loop is truncated which correlates with the fact that 8-oxoG is not a preferred substrate for any of these glycosylases. In the human NEIL1-R242 structure, the carboxylate moiety of Glu16 from helix A would be positioned to make a H-bond interaction with nitrogen Nε of Arg242 (Fig. 3A). In addition, the main-chain oxygen of Gly241 forms an H-bond interaction with the side-chain Nε2 of Gln12. In the K242 structure the electron density of the residues surrounding the mutation (i.e. 240–243) is weak indicating inherent flexibility and disorder, and as a result only the main-chain atoms of lysine were built into the model. The stabilizing H-bond interaction between Arg242 and Glu16 is lost and Gln12 moves further away from Gly241 (Fig. 3B). Thus, in the K242 form the loop region appears to be more flexible. How the increased flexibility of this loop translates into an enzyme that removes Tg lesions 30–40 fold faster than the edited R242 form is unclear and will likely require the structure of a NEIL1-Tg complex.
Figure 3.
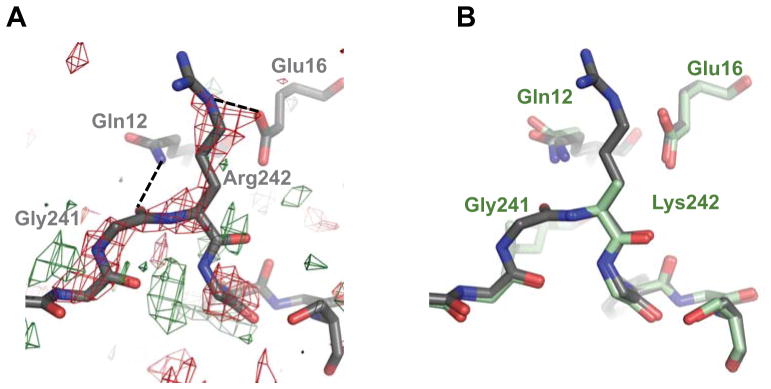
Effects of RNA editing on the structure of NEIL1. (A) Difference Fourier (Fo-Fo) map of NEIL1-K242 – NEIL1-R242, created in PHENIX [50], contoured at 2.5σ. Red and green coloring indicate negative and positive density, respectively. (B) Superposition of NEIL1-WT (gray) with K242 (pale green) indicates subtle differences in this region. The density for Lys242 was weak and hence the side-chain was not built in the model. Dotted lines indicate H-bonds.
The glycosylase assays reported here and earlier were obtained with the edited form of NEIL1 (R242) [40]. In order to evaluate the possible effects of RNA editing on the activity of the G83D variant, we measured the glycosylase activity of the non-edited form of NEIL1 (i.e. NEIL1-K242) and compared it to the activity of the K242-G83D variant (Fig. 4, Table S3). K242 possesses robust activity with all the lesions tested (Fig. 4 A–F) and appears to cleave Tg more efficiently than its edited counterpart, as previously reported by the David and Beal groups [36]. The K242-G83D variant has little to no detectable glycosylase activity on the pyrimidine lesions (Fig. 4 A–C, Table S3), and 2–20% reduced activity on the purine lesions (Fig. 4 D–F, Table S3). It retains lyase activity, however (>90%, Fig. 4G, Table S3), which is an indication that the loss of glycosylase activity is not due to improper folding of the enzyme. The R242 edited form of G83D appears to have greater excision activity with Gh and Sp1 than the K242 non-edited form encoded in the genome, as reported earlier [36]. Tg:A, however, remains a poor substrate for G83D regardless of the edited status of the enzyme.
Figure 4.

Glycosylase and lyase activity panel for the non-edited K242 form of human NEIL1. Glycosylase assays were performed as described in Fig. 2 using K242 and K242-G83D. Panel (A) Tg:A; (B) OHU:G; (C) DHU:A; (D) Gh:C; (E) Sp1:C; (F) MeFapyG:C; (G) AP:C. S and P indicate substrates and products respectively. Data shown are representative of two repeat experiments.
3.4. Substrate Preference of the G86D Variant in a Viral Ortholog of NEIL1
Although the three-dimensional structure of human NEIL1 has been solved by X-ray crystallography, there is no structural data of NEIL1 bound to DNA. However, several crystal structures of a viral ortholog of NEIL1, MvNei1, have been reported, both unliganded and in a complex with lesion-containing DNA [37, 38]. Gly83 in NEIL1 is highly conserved among members of the Fpg/Nei family and is present at the N-terminal tip of β-strand 5, in close proximity to the void-filling Met81 residue (Fig. 5 A, B). We performed glycosylase assays on the various substrates listed in Fig. 1 on MvNei1-G86D, the variant corresponding to NEIL1 G83D, and WT MvNei1. The latter cleaves the pyrimidine derived Tg and OHU lesions, and DHU to a lesser extent (Fig. 6A–C, Table S4) whereas G86D has no detectable activity on these lesions (Fig. 6A–C, Table S4). The best substrates previously noted for MvNei1 are the Gh and Sp1 lesions as well as MeFapyG to a lesser extent and this finding is confirmed here (Fig. 6 D–F, Table S4) [37, 38, 58]. The G86D variant exhibited diminished activity on Gh, Sp1 and MeFapyG substrates (Fig. 6D–F, Table S4). This variant possesses robust lyase activity as demonstrated by cleavage of AP:C even at very low enzyme concentrations (>90% product formed, Fig. 6G, Table S4), indicating that the glycosylase is folded properly.
Figure 5.

Glycine 83 is conserved among members of the Fpg/Nei family. (A) Superposition of the human NEIL1 structure (1TDH [47], gray) with that of MvNei1 bound to THF-containing DNA (3A46 [37], blue). The MvNei1-DNA is colored in blue and THF is flipped out into the active site. Human NEIL1-Gly83 corresponds to MvNei1-Gly86. (B) Sequence alignment of human NEIL1 with other members of the Fpg/Nei family. * Indicates the conserved Met/Leu void-filling residue in the β4/5 loop. The residues colored in red are highly conserved among members of the Fpg/Nei family.
Figure 6.

Glycosylase and lyase activity assays of MvNei1-WT compared with the G86D variant. Assays were performed as described in Fig. 2. Panel (A) Tg:A; (B) OHU:G; (C) DHU:A; (D) Gh:C; (E) Sp1:C; (F) MeFapyG:C; (G) AP:C. S and P indicate substrates and products, respectively.
3.5. Crystal Structure of MvNei1-G86D Bound to THF-containing DNA
The crystal structures of human NEIL1 and MvNei1 are very similar and share many of the same features, including a zincless-finger motif and thus the viral glycosylase has proven to be a useful tool to obtain complexes of an Nei glycosylase with DNA lesions [37, 47]. Since we could not get a complex of human NEIL1-G83D with DNA we grew crystals of MvNei1-G86D in complex with DNA containing THF. The structure was solved by molecular replacement using the MvNei1-THF complex (PDB 3A46 [37]) (see Table S2 for statistics). The 2.85 Å structure of MvNei1-G86D-THF was refined to Rwork and Rfree values of 26% and 31.45%, respectively.
Overall the structures of WT MvNei1 and the G86D variant bound to DNA are very similar, with an r.m.s.d. of 0.33 Å calculated on 286 aligned Cα with a maximum deviation of 2 Å (Fig. 7A) [54]. Although there are two protein-DNA complex molecules in the asymmetric unit of the crystal, the discussion below will focus mainly on chain A since both molecules are comparable and have similar B-factor values.
Figure 7.
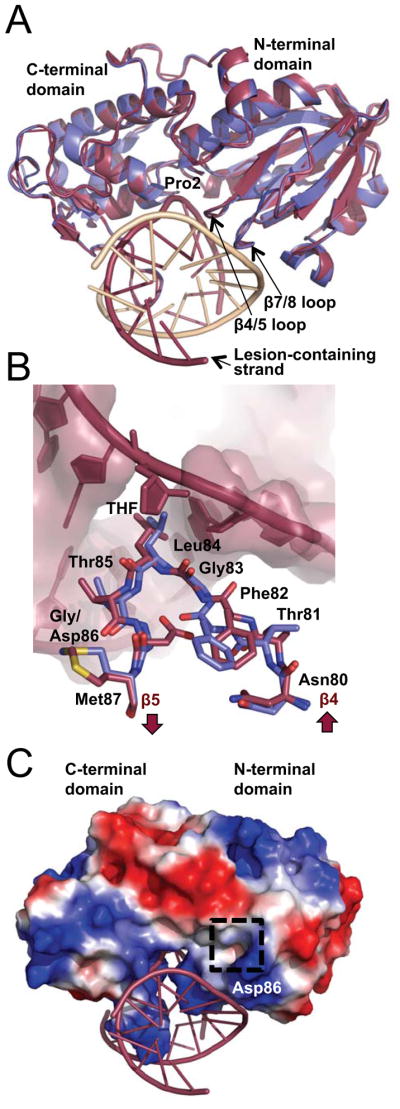
Structure of the MvNei1-G86D variant. (A) Superposition of MvNei1-G86D-THF (burgundy) with MvNei1-THF (blue, PDB code 3A46 [37]) illustrates the overall structural similarity between the two enzymes. (B) Superposition of residues 80–87 comprising the β4/5 loop from MvNei1-WT (blue) and MvNei1-G86D (burgundy) highlights the differences caused by the G86D mutation. (C) Electrostatic surface representation of G86D bound to DNA. The dashed box highlights the negatively charged region introduced by the G86D mutation. The surface was created in PyMOL (The PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC.) and is colored according to electrostatic potential, where blue and red signify positive and negative charges, respectively. The scale is set to ± 65 kT/e.
The G83D mutation introduces a negative charge in an otherwise hydrophobic pocket and was therefore predicted to affect the conformation of the β4/5 loop [35]. The crystal structure of G86D indeed shows that the Asp86 side chain is inserted in a pocket in MvNei1 lined by hydrophobic residues Phe64, Phe82, Trp88, Phe110, and Phe119. The corresponding residues in human NEIL1 are Leu55, Phe79, Phe85, Phe114, and Trp123.
Asp86 causes the β4/5 loop and residues on either side of the loop (residues 80–87) to shift by an r.m.s.d. of 1.3 Å, with a maximum deviation of 3.9 Å (Fig. 7B). The main-chain oxygen of Phe82 is flipped ~180° and moves within van der Waals distance (3.8 Å) of the sugar moiety of the guanine adjacent to the lesion (Fig. 7B). The rearrangement of the β4/5 loop introduced by the G86D mutation causes the void-filling Leu84 residue to adopt a different rotamer conformation and shift away from the furan moiety by about 1.5 Å. The electrostatic surface potential of the solvent accessible residues of MvNei1 reveals a positively charged binding pocket suitable for binding DNA that runs orthogonal to the protein. A region of unfavorable negative charge is created by mutating Gly86 to Asp (Fig. 7C) in the vicinity of the β4/5 loop. Thus, the displacement of residues of the β4/5 loop along with an unfavorable charge together yield an enzyme with severely reduced glycosylase activity.
3.6. G83D Does Not Rescue the Spontaneous Mutation Frequency in E. coli Mutant Strains
In order to test the ability of NEIL1 and its variant to complement spontaneous mutation frequencies in vivo, we used a Rifampin resistance (RifR) assay and expressed WT NEIL1 or NEIL1-G83D in an E. coli fpg−mutY−nei− triple mutant strain [16, 43, 64]. The fpg−mutY−nei− triple mutant strain exhibits a high spontaneous mutation rate, which is reduced by the expression of WT NEIL1 in the mutant cells (Fig. 8) [44]. The triple mutant strain accumulates G to T transversions due to buildup of FapyG, 8-oxoG, and its further oxidation products including Gh and Sp1. WT NEIL1 can compensate for the spontaneous mutations generated through excision of Sp1, Gh, and FapyG from DNA. In contrast, the G83D variant displays a statistically significant, greater build-up of RifR resistant mutants due to inefficient cleavage of the guanine-derived lesions (Fig. 8). This finding is consistent with our in vitro data showing that G83D cleaves Sp1 to a lesser extent than WT, and has little to no activity with FapyG substrates. We note that when NEIL1-G83D is expressed the mutation rate is higher than in the non-complemented triple mutant strain. One possible explanation is that the G83D variant binds to lesions but does not cleave them and thus prevents Nth from excising oxidized DNA lesions.
Figure 8.
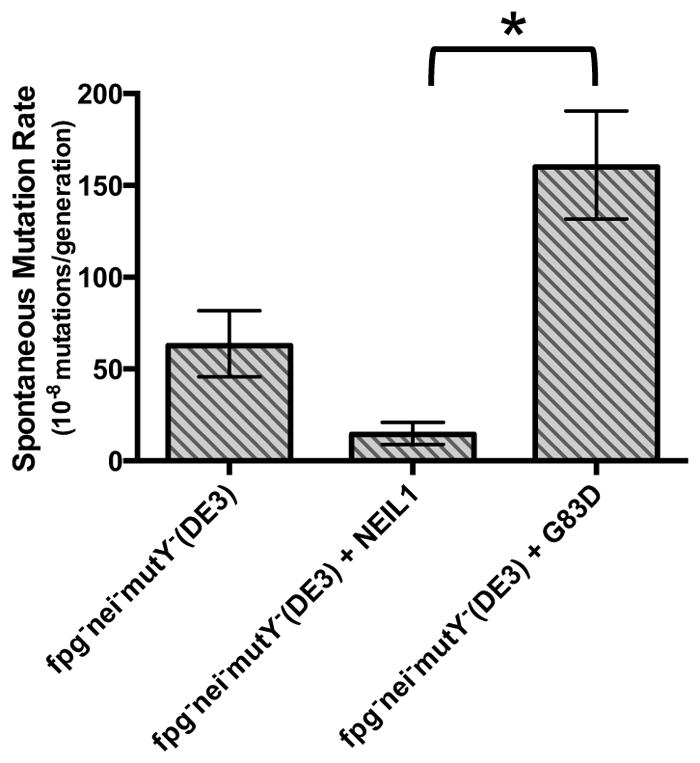
Spontaneous mutation rates to rifampin resistance in fpg−nei−mutY− E. coli. Mutants per 108 cells are shown. Data are expressed as mean of three independent measurements. Uncertainties are displayed at a 95% confidence interval. Statistical significance was determined by the student’s t test and p values were calculated relative to the fpg−nei−mutY− (DE3)-WT NEIL1 cells; *, p < 0.01.
4. DISCUSSION
The accumulation of oxidative DNA damage from both exogenous and endogenous sources have been implicated in the development and progression of numerous disease states such as cancer, obesity, diabetes, Alzheimer’s disease, and others [65–67]. Over 500 SNPs have been identified within DNA repair genes [68] and over 100 germline SNPs were found in enzymes of the BER pathway [30, 69]. SNPs which result in non-synonymous amino acid substitutions in the proteins they encode could result in inactive enzymes and thereby contribute to genomic instability [68].
The current investigation focused on four genome and cancer polymorphic variants of human NEIL1. Although S82C, G83D, and ΔE28 have been studied previously, the studies were conducted with a limited number of lesions. In this report we further characterize these variants using a broader array of damaged bases. NEIL1 ΔE28, which is observed as a heterozygous mutation in gastric cancer patients, exhibited a reduced ability to cleave a double-stranded oligonucleotide containing Tg:A [33]. In our current analysis, ΔE28 was able to efficiently cleave Sp1 and Gh, and Tg:A, albeit to a lesser extent. NEIL1-P208S was reported in patients presenting with colorectal adenomas [34]. We show here that NEIL1-P208S has equivalent to WT or better than WT activity with the substrates used in this study and this represents the first comprehensive biochemical analysis of the substrate preferences for this variant enzyme. As mentioned previously P208S precedes a helix extension region that is unique to NEIL1 and whose function is unknown.
Forsbring et al. reported the presence of the G83D variant in patients with cholangiocarcinoma [35]. Furthermore, their results showed that the glycosylase activity for this variant does not cleave ds 8-oxoG, DHT, and Tg, but is as active as WT enzyme using lesions present in ssDNA substrates. Roy et al. reported that G83D lacked DNA glycosylase activity toward dsTg and Fapy lesions but has intact lyase activity [32]. In the current study, we evaluated NEIL1-G83D activity in the context of dsDNA and showed that the variant was unable to cleave most lesions. Moreover, G83D displayed weak glycosylase activity regardless of the edited state of NEIL1. The crystal structure of the analogous MvNei1 variant revealed that the aspartate mutation introduces changes in the β4/5 loop and in the rotamer of the void-filling residue Leu84, which moves further away from the furan moiety than in the WT structure. This void-filling, or helix-invading, residue plays a crucial role in stabilizing the everted state of the lesion [60]. Given the importance of this void-filing residue it is not surprising that the G83D variant exhibits very weak glycosylase activity, both in vitro and in vivo.
Although G83D has little to no activity on oxidized pyrimidines and FapyG, it still cleaves Sp1 and Gh (Fig. 1). This finding is reminiscent of what we observed with Neil3, where this glycosylase does not cleave dsDNA substrates, with the notable exception of these two hydantoin lesions [16]. A combination of NMR data, microcalorimetry, and DNA melting curves showed that Sp introduces severe distortions in the DNA double helix that destabilize dsDNA and affect the Watson-Crick base pairs flanking the base lesion [70]. A crystal structure of a DNA polymerase bound to Gh showed that the lesion is extrahelical and rotated towards the major groove [71]. The distortions introduced in the DNA double helix by the hydantoin lesions may be such that these lesions are particularly apt at being extruded.
In contrast to the drastic effect of the G83D mutation, mutating the adjacent serine 82 into cysteine results in a variant, which possesses near WT glycosylase activity on most lesions [32]. Even though the cysteine variant is nearly isosteric with serine and is thus not expected to cause severe distortions, we still fully characterized this variant with a panel of substrates, surmising that there may be subtle differences between the two enzymes. The glycosylase activity panel revealed that although S82C exhibits near WT level activity on most lesions, it has reduced activity on Tg. The hydroxyl of Ser82 engages in a H-bond with the main-chain amide of the void-filling Arg118 of the β7/8 loop [47]. The most likely cysteine rotamer would place the sulfur ~4.5 Å away from the Arg118 amide, too far for a H-bond. This mutation might partially destabilize the β7/8 loop and lead to a decrease in glycosylase activity. Why this affects mostly the Tg lesion is not fully clear at this point.
With all the NEIL1 variants used in this report the major product after cleavage is the β,δ-elimination product. Others have reported formation of both β-elimination and the β,δ-elimination products [35]. We attribute some of this discrepancy to the temperature at which the reactions were performed. We collected circular dichroism spectra at various temperatures and noticed that the truncated, crystallizable form of NEIL1, i.e. NEIL1Δ56, is unstable at 37°C. This finding correlates with dynamic light scattering data that showed that full-length NEIL1 was polydisperse at 35°C. Thus all our assays were performed at room temperature. Other factors that could contribute to the discrepancy in the elimination products include DNA sequence context, DNA length, and differences in assay buffer conditions.
In summary, we present here a comprehensive analysis of four variants of human NEIL1, ΔE28, G82C, G83D, and P208S. Our structural, biochemical, and in vivo data highlight the importance of the NEIL1-G83D variant, which was found as a heterozygous mutation in patients with cholangiocarcinoma. Gly83 is highly conserved among members of the Fpg/Nei family and is present in close proximity to the void-filling Met81 in the β4/5 loop. The analogous Gly to Asp mutation in MvNei1 alters the conformation of the β4/5 loop where the void-filling leucine moves away from an AP site analog, resulting in an enzyme with severely diminished glycosylase activity, both in vitro and in vivo.
Supplementary Material
Highlights.
We present a comprehensive analysis of four variants of the human NEIL1 glycosylase
The G83D variant shows a decreased activity on all DNA lesions tested in this study
RNA editing of NEIL1 does not affect the activity of the G83D variant
The structure of MvNei1-G86D reveals a rearrangement in the void-filling β4/5 loop
Acknowledgments
We would like to thank Dr. Cynthia J. Burrows, University of Utah, for providing oligodeoxyribonucleotides containing spiroiminodihydantoin, and guanidinohydantoin; Dr. Carmelo Rizzo (Vanderbilt) for the oligodeoxynucleotide containing methyl-FapyG; Drs. Mark Rould, and Brian E. Eckenroth for helpful crystallographic suggestions; Drs. Jeff Bond and Julie Dragon for help with identifying the variants. This work was supported by National Institutes of Health Grant P01CA098993 awarded by the National Cancer Institute. AP was also supported by a J. Walter Juckett post-doctoral fellowship awarded by the Lake Champlain Cancer Research Organization and the Vermont Cancer Center. Support from the Vermont Cancer Center is acknowledged.
Footnotes
Conflict of Interest
None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References Cited
- 1.Duclos S, Doublie S, Wallace SS. Consequences and Repair of Oxidative DNA Damage. In: Greim H, Albertini R, editors. The Cellular Response to the Genotoxic Insult: The Question of Threshold for Genotoxic Carcinogens. The Royal Society of Chemistry; 2012. pp. 115–159. [Google Scholar]
- 2.Friedberg EC, Walker GC, Seide W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. 2. ASM Press; Washington, D.C: 2006. [Google Scholar]
- 3.Maynard S, Schurman SH, Harboe C, de Souza-Pinto NC, Bohr VA. Base excision repair of oxidative DNA damage and association with cancer and aging. Carcinogenesis. 2009;30:2–10. doi: 10.1093/carcin/bgn250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wallace SS, Murphy DL, Sweasy JB. Base excision repair and cancer. Cancer Lett. 2012;327:73–89. doi: 10.1016/j.canlet.2011.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fromme JC, Verdine GL. Base excision repair. Adv Protein Chem. 2004;69:1–41. doi: 10.1016/S0065-3233(04)69001-2. [DOI] [PubMed] [Google Scholar]
- 6.Zharkov DO. Base excision DNA repair. Cellular and molecular life sciences : CMLS. 2008;65:1544–1565. doi: 10.1007/s00018-008-7543-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wallace SS. Enzymatic processing of radiation-induced free radical damage in DNA. Radiat Res. 1998;150:S60–79. [PubMed] [Google Scholar]
- 8.Fortini P, Pascucci B, Parlanti E, D’Errico M, Simonelli V, Dogliotti E. The base excision repair: mechanisms and its relevance for cancer susceptibility. Biochimie. 2003;85:1053–1071. doi: 10.1016/j.biochi.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 9.McCullough AK, Dodson ML, Lloyd RS. Initiation of base excision repair: glycosylase mechanisms and structures. Annual review of biochemistry. 1999;68:255–285. doi: 10.1146/annurev.biochem.68.1.255. [DOI] [PubMed] [Google Scholar]
- 10.Hegde ML, Hazra TK, Mitra S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell research. 2008;18:27–47. doi: 10.1038/cr.2008.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Katafuchi A, Nakano T, Masaoka A, Terato H, Iwai S, Hanaoka F, Ide H. Differential specificity of human and Escherichia coli endonuclease III and VIII homologues for oxidative base lesions. J Biol Chem. 2004;279:14464–14471. doi: 10.1074/jbc.M400393200. [DOI] [PubMed] [Google Scholar]
- 12.Burgess S, Jaruga P, Dodson ML, Dizdaroglu M, Lloyd RS. Determination of active site residues in Escherichia coli endonuclease VIII. J Biol Chem. 2002;277:2938–2944. doi: 10.1074/jbc.M110499200. [DOI] [PubMed] [Google Scholar]
- 13.Bandaru V, Sunkara S, Wallace SS, Bond JP. A novel human DNA glycosylase that removes oxidative DNA damage and is homologous to Escherichia coli endonuclease VIII. DNA Repair (Amst) 2002;1:517–529. doi: 10.1016/s1568-7864(02)00036-8. [DOI] [PubMed] [Google Scholar]
- 14.Prakash A, Doublie S, Wallace SS. The Fpg/Nei family of DNA glycosylases: substrates, structures, and search for damage. Prog Mol Biol Transl Sci. 2012;110:71–91. doi: 10.1016/B978-0-12-387665-2.00004-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brooks SC, Adhikary S, Rubinson EH, Eichman BF. Recent advances in the structural mechanisms of DNA glycosylases. Biochim Biophys Acta. 2013;1834:247–271. doi: 10.1016/j.bbapap.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu M, Bandaru V, Bond JP, Jaruga P, Zhao X, Christov PP, Burrows CJ, Rizzo CJ, Dizdaroglu M, Wallace SS. The mouse ortholog of NEIL3 is a functional DNA glycosylase in vitro and in vivo. Proc Natl Acad Sci U S A. 2010;107:4925–4930. doi: 10.1073/pnas.0908307107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dou H, Mitra S, Hazra TK. Repair of oxidized bases in DNA bubble structures by human DNA glycosylases NEIL1 and NEIL2. J Biol Chem. 2003;278:49679–49684. doi: 10.1074/jbc.M308658200. [DOI] [PubMed] [Google Scholar]
- 18.Morland I, Rolseth V, Luna L, Rognes T, Bjoras M, Seeberg E. Human DNA glycosylases of the bacterial Fpg/MutM superfamily: an alternative pathway for the repair of 8-oxoguanine and other oxidation products in DNA. Nucleic Acids Res. 2002;30:4926–4936. doi: 10.1093/nar/gkf618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Das A, Rajagopalan L, Mathura VS, Rigby SJ, Mitra S, Hazra TK. Identification of a zinc finger domain in the human NEIL2 (Nei-like-2) protein. J Biol Chem. 2004;279:47132–47138. doi: 10.1074/jbc.M406224200. [DOI] [PubMed] [Google Scholar]
- 20.Grin IR, Zharkov DO. Eukaryotic endonuclease VIII-like proteins: new components of the base excision DNA repair system. Biochemistry Biokhimiia. 2011;76:80–93. doi: 10.1134/s000629791101010x. [DOI] [PubMed] [Google Scholar]
- 21.Krishnamurthy N, Zhao X, Burrows CJ, David SS. Superior removal of hydantoin lesions relative to other oxidized bases by the human DNA glycosylase hNEIL1. Biochemistry. 2008;47:7137–7146. doi: 10.1021/bi800160s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parsons JL, Kavli B, Slupphaug G, Dianov GL. NEIL1 is the major DNA glycosylase that processes 5-hydroxyuracil in the proximity of a DNA single-strand break. Biochemistry. 2007;46:4158–4163. doi: 10.1021/bi0622569. [DOI] [PubMed] [Google Scholar]
- 23.Onizuka K, Yeo J, David SS, Beal PA. NEIL1 binding to DNA containing 2′-fluorothymidine glycol stereoisomers and the effect of editing. Chembiochem. 2012;13:1338–1348. doi: 10.1002/cbic.201200139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao X, Krishnamurthy N, Burrows CJ, David SS. Mutation versus repair: NEIL1 removal of hydantoin lesions in single-stranded, bulge, bubble, and duplex DNA contexts. Biochemistry. 2010;49:1658–1666. doi: 10.1021/bi901852q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hailer MK, Slade PG, Martin BD, Rosenquist TA, Sugden KD. Recognition of the oxidized lesions spiroiminodihydantoin and guanidinohydantoin in DNA by the mammalian base excision repair glycosylases NEIL1 and NEIL2. DNA Repair (Amst) 2005;4:41–50. doi: 10.1016/j.dnarep.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 26.Hu J, de Souza-Pinto NC, Haraguchi K, Hogue BA, Jaruga P, Greenberg MM, Dizdaroglu M, Bohr VA. Repair of formamidopyrimidines in DNA involves different glycosylases: role of the OGG1, NTH1, and NEIL1 enzymes. J Biol Chem. 2005;280:40544–40551. doi: 10.1074/jbc.M508772200. [DOI] [PubMed] [Google Scholar]
- 27.Frosina G. Commentary: DNA base excision repair defects in human pathologies. Free Radic Res. 2004;38:1037–1054. doi: 10.1080/10715760400011445. [DOI] [PubMed] [Google Scholar]
- 28.Hung RJ, Hall J, Brennan P, Boffetta P. Genetic polymorphisms in the base excision repair pathway and cancer risk: a HuGE review. Am J Epidemiol. 2005;162:925–942. doi: 10.1093/aje/kwi318. [DOI] [PubMed] [Google Scholar]
- 29.Karahalil B, Bohr VA, Wilson DM., 3rd Impact of DNA polymorphisms in key DNA base excision repair proteins on cancer risk. Human & experimental toxicology. 2012;31:981–1005. doi: 10.1177/0960327112444476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nemec AA, Wallace SS, Sweasy JB. Variant base excision repair proteins: contributors to genomic instability. Semin Cancer Biol. 2010;20:320–328. doi: 10.1016/j.semcancer.2010.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dey S, Maiti AK, Hegde ML, Hegde PM, Boldogh I, Sarkar PS, Abdel-Rahman SZ, Sarker AH, Hang B, Xie J, Tomkinson AE, Zhou M, Shen B, Wang G, Wu C, Yu D, Lin D, Cardenas V, Hazra TK. Increased risk of lung cancer associated with a functionally impaired polymorphic variant of the human DNA glycosylase NEIL2. DNA Repair (Amst) 2012;11:570–578. doi: 10.1016/j.dnarep.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roy LM, Jaruga P, Wood TG, McCullough AK, Dizdaroglu M, Lloyd RS. Human polymorphic variants of the NEIL1 DNA glycosylase. J Biol Chem. 2007;282:15790–15798. doi: 10.1074/jbc.M610626200. [DOI] [PubMed] [Google Scholar]
- 33.Shinmura K, Tao H, Goto M, Igarashi H, Taniguchi T, Maekawa M, Takezaki T, Sugimura H. Inactivating mutations of the human base excision repair gene NEIL1 in gastric cancer. Carcinogenesis. 2004;25:2311–2317. doi: 10.1093/carcin/bgh267. [DOI] [PubMed] [Google Scholar]
- 34.Dallosso AR, Dolwani S, Jones N, Jones S, Colley J, Maynard J, Idziaszczyk S, Humphreys V, Arnold J, Donaldson A, Eccles D, Ellis A, Evans DG, Frayling IM, Hes FJ, Houlston RS, Maher ER, Nielsen M, Parry S, Tyler E, Moskvina V, Cheadle JP, Sampson JR. Inherited predisposition to colorectal adenomas caused by multiple rare alleles of MUTYH but not OGG1. NUDT1, NTH1 or NEIL 1, 2 or 3. Gut. 2008;57:1252–1255. doi: 10.1136/gut.2007.145748. [DOI] [PubMed] [Google Scholar]
- 35.Forsbring M, Vik ES, Dalhus B, Karlsen TH, Bergquist A, Schrumpf E, Bjoras M, Boberg KM, Alseth I. Catalytically impaired hMYH and NEIL1 mutant proteins identified in patients with primary sclerosing cholangitis and cholangiocarcinoma. Carcinogenesis. 2009;30:1147–1154. doi: 10.1093/carcin/bgp118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yeo J, Goodman RA, Schirle NT, David SS, Beal PA. RNA editing changes the lesion specificity for the DNA repair enzyme NEIL1. Proc Natl Acad Sci U S A. 2010;107:20715–20719. doi: 10.1073/pnas.1009231107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Imamura K, Wallace SS, Doublie S. Structural characterization of a viral NEIL1 ortholog unliganded and bound to abasic site-containing DNA. J Biol Chem. 2009;284:26174–26183. doi: 10.1074/jbc.M109.021907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Imamura K, Averill A, Wallace SS, Doublie S. Structural characterization of viral ortholog of human DNA glycosylase NEIL1 bound to thymine glycol or 5-hydroxyuracil-containing DNA. J Biol Chem. 2012;287:4288–4298. doi: 10.1074/jbc.M111.315309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bandaru V, Cooper W, Wallace SS, Doublie S. Overproduction, crystallization and preliminary crystallographic analysis of a novel human DNA-repair enzyme that recognizes oxidative DNA damage. Acta Crystallogr D Biol Crystallogr. 2004;60:1142–1144. doi: 10.1107/S0907444904007929. [DOI] [PubMed] [Google Scholar]
- 40.Bandaru V, Zhao X, Newton MR, Burrows CJ, Wallace SS. Human endonuclease VIII-like (NEIL) proteins in the giant DNA Mimivirus. DNA Repair (Amst) 2007;6:1629–1641. doi: 10.1016/j.dnarep.2007.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kornyushyna O, Berges AM, Muller JG, Burrows CJ. In vitro nucleotide misinsertion opposite the oxidized guanosine lesions spiroiminodihydantoin and guanidinohydantoin and DNA synthesis past the lesions using Escherichia coli DNA polymerase I (Klenow fragment) Biochemistry. 2002;41:15304–15314. doi: 10.1021/bi0264925. [DOI] [PubMed] [Google Scholar]
- 42.Christov PP, Brown KL, Kozekov ID, Stone MP, Harris TM, Rizzo CJ. Site-specific synthesis and characterization of oligonucleotides containing an N6-(2-deoxy-D-erythro-pentofuranosyl)-2,6-diamino-3,4-dihydro-4-oxo-5-N-methylfor mamidopyrimidine lesion, the ring-opened product from N7-methylation of deoxyguanosine. Chemical research in toxicology. 2008;21:2324–2333. doi: 10.1021/tx800352a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guo Y, Bandaru V, Jaruga P, Zhao X, Burrows CJ, Iwai S, Dizdaroglu M, Bond JP, Wallace SS. The oxidative DNA glycosylases of Mycobacterium tuberculosis exhibit different substrate preferences from their Escherichia coli counterparts. DNA Repair (Amst) 2010;9:177–190. doi: 10.1016/j.dnarep.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hall BM, Ma CX, Liang P, Singh KK. Fluctuation analysis CalculatOR: a web tool for the determination of mutation rate using Luria-Delbruck fluctuation analysis. Bioinformatics. 2009;25:1564–1565. doi: 10.1093/bioinformatics/btp253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. In: Carter CWJ, Sweet RM, editors. Methods in Enzymology. Academic Press; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 46.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Doublie S, Bandaru V, Bond JP, Wallace SS. The crystal structure of human endonuclease VIII-like 1 (NEIL1) reveals a zincless finger motif required for glycosylase activity. Proc Natl Acad Sci U S A. 2004;101:10284–10289. doi: 10.1073/pnas.0402051101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rould MA. The same but different: isomorphous methods for phasing and high-throughput ligand screening. Methods in molecular biology. 2007;364:159–182. doi: 10.1385/1-59745-266-1:159. [DOI] [PubMed] [Google Scholar]
- 49.Zwart PH, Afonine PV, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, McKee E, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Storoni LC, Terwilliger TC, Adams PD. Automated structure solution with the PHENIX suite. Methods Mol Biol. 2008;426:419–435. doi: 10.1007/978-1-60327-058-8_28. [DOI] [PubMed] [Google Scholar]
- 50.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Terwilliger TC, Grosse-Kunstleve RW, Afonine PV, Moriarty NW, Zwart PH, Hung LW, Read RJ, Adams PD. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr D Biol Crystallogr. 2008;64:61–69. doi: 10.1107/S090744490705024X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Afonine PV, Mustyakimov M, Grosse-Kunstleve RW, Moriarty NW, Langan P, Adams PD. Joint X-ray and neutron refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr. 2010;66:1153–1163. doi: 10.1107/S0907444910026582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA C Genomes Project. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McCain J. The cancer genome atlas: new weapon in old war? Biotechnology healthcare. 2006;3:46–51B. [PMC free article] [PubMed] [Google Scholar]
- 57.Robbins DE, Gruneberg A, Deus HF, Tanik MM, Almeida JS. A self-updating road map of The Cancer Genome Atlas. Bioinformatics. 2013;29:1333–1340. doi: 10.1093/bioinformatics/btt141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu M, Imamura K, Averill AM, Wallace SS, Doublie S. Structural characterization of a mouse ortholog of human NEIL3 with a marked preference for single-stranded DNA. Structure. 2013;21:247–256. doi: 10.1016/j.str.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dunn AR, Kad NM, Nelson SR, Warshaw DM, Wallace SS. Single Qdot-labeled glycosylase molecules use a wedge amino acid to probe for lesions while scanning along DNA. Nucleic Acids Res. 2011;39:7487–7498. doi: 10.1093/nar/gkr459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sung RJ, Zhang M, Qi Y, Verdine GL. Structural and biochemical analysis of DNA helix-invasion by the bacterial 8-oxoguanine DNA glycosylase MutM. J Biol Chem. 2013 doi: 10.1074/jbc.M112.415612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li JB, Levanon EY, Yoon JK, Aach J, Xie B, Leproust E, Zhang K, Gao Y, Church GM. Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science. 2009;324:1210–1213. doi: 10.1126/science.1170995. [DOI] [PubMed] [Google Scholar]
- 62.Qi Y, Spong MC, Nam K, Banerjee A, Jiralerspong S, Karplus M, Verdine GL. Encounter and extrusion of an intrahelical lesion by a DNA repair enzyme. Nature. 2009;462:762–766. doi: 10.1038/nature08561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Duclos S, Aller P, Jaruga P, Dizdaroglu M, Wallace SS, Doublie S. Structural and biochemical studies of a plant formamidopyrimidine-DNA glycosylase reveal why eukaryotic Fpg glycosylases do not excise 8-oxoguanine. DNA Repair (Amst) 2012;11:714–725. doi: 10.1016/j.dnarep.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Blaisdell JO, Hatahet Z, Wallace SS. A novel role for Escherichia coli endonuclease VIII in prevention of spontaneous G-->T transversions. J Bacteriol. 1999;181:6396–6402. doi: 10.1128/jb.181.20.6396-6402.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2003;17:1195–1214. doi: 10.1096/fj.02-0752rev. [DOI] [PubMed] [Google Scholar]
- 66.Sampath H, McCullough AK, Lloyd RS. Regulation of DNA glycosylases and their role in limiting disease. Free radical research. 2012;46:460–478. doi: 10.3109/10715762.2012.655730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lillenes MS, Espeseth T, Stoen M, Lundervold AJ, Frye SA, Rootwelt H, Reinvang I, Tonjum T. DNA base excision repair gene polymorphisms modulate human cognitive performance and decline during normal life span. Mechanisms of ageing and development. 2011;132:449–458. doi: 10.1016/j.mad.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 68.Xi T, Jones IM, Mohrenweiser HW. Many amino acid substitution variants identified in DNA repair genes during human population screenings are predicted to impact protein function. Genomics. 2004;83:970–979. doi: 10.1016/j.ygeno.2003.12.016. [DOI] [PubMed] [Google Scholar]
- 69.Jones IM, Thomas CB, Xi T, Mohrenweiser HW, Nelson DO. Exploration of methods to identify polymorphisms associated with variation in DNA repair capacity phenotypes. Mutat Res. 2007;616:213–220. doi: 10.1016/j.mrfmmm.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 70.Khutsishvili I, Zhang N, Marky LA, Crean C, Patel DJ, Geacintov NE, Shafirovich V. Thermodynamic profiles and nuclear magnetic resonance studies of oligonucleotide duplexes containing single diastereomeric spiroiminodihydantoin lesions. Biochemistry. 2013;52:1354–1363. doi: 10.1021/bi301566v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Aller P, Ye Y, Wallace SS, Burrows CJ, Doublie S. Crystal structure of a replicative DNA polymerase bound to the oxidized guanine lesion guanidinohydantoin. Biochemistry. 2010;49:2502–2509. doi: 10.1021/bi902195p. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.