

Abstract
The kappa opioid receptor (KOPR) has been identified as a potential drug target to prevent or alter the course of mood, anxiety and addictive disorders or reduce response to stress. In a search for highly potent and selective KOPR partial agonists as pharmacological tools, we have modified 12-epi-salvinorin A, a compound which we have previously observed to be a KOPR partial agonist. Five analogues of 12-epi-salvinorin A were synthesized and their effects on G protein activation as well as β-arrestin2 recruitment were evaluated. Only 12-epi-salvinorin A (1) partially activated signaling through G proteins, yet acted as a full agonist in the β-arrestin 2 DiscoveRx assay. Other salvinorin analogues tested in these functional assays were full agonists in both assays of KOPR activation. By comparison, the non-selective opioid ligand nalbuphine, known to be a partial agonist for G-protein activation, was also a partial agonist for the β-arrestin mediated signaling pathway activated through KOPR.
Keywords: Kappa opioid receptor, Partial agonist, Mood, Anxiety, Drug addiction, Salvinorin
Kappa opioid receptors (KOPR) and modulation of activity through these receptors might be a part of brain circuitry regulating changes in mood, anxiety and drug taking behaviors.1–5 Specifically, preclinical studies suggest that selective KOPR antagonists may possess antidepressant and anxiolytic effects,1,6 while an open-label clinical trial indicates that selective KOPR full agonists may reduce symptoms of mania.7 KOPR ligands may also have therapeutic utility for drug addiction, with selective KOPR antagonists preventing stress-induced reinstatement of cocaine taking.8 While selective KOPR agonists may prevent cocaine-induced reinstatement of drug seeking and reduce drug self-administration,9 they can also potentiate stress-induced reinstatement of drug seeking.10 Most behavioral studies designed to evaluate the effects of KOPR ligands on mood, anxiety and addictive disorders were performed with full agonists and antagonists, drugs which completely activate KOPR or block activation of KOPR by agonists. Little is known about the effects of selective KOPR partial agonists, which might produce a stable signal, because of the lack of availability of such agents. It is possible that, in some cases, a selective partial agonist might have a greater potential for the treatment or prevention of specific symptoms of mood, anxiety or other stress-related disorders.
We have previously identified 12-epi-salvinorin A (12-epi-sal-vA) as a selective KOPR partial agonist with moderate potency in vitro.11 In this study, we designed and synthesized analogues of 12-epi-salvA modified at C-2, C-15, and C-16, with the goal of improving potency and exploring if configuration at C-12 is responsible for partial efficacy.
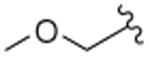
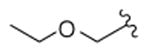
Based on previous SAR findings with salvinorin A (salvA),12 we hypothesized that replacement of the C-2 acetate unit of 12-epi-salvA with methoxymethyl (MOM) or ethoxymethyl (EtOMe) ethers might improve potency, and prepared compounds 2 and 3. 12-epi-salvinorin B (12-epi-salvB, 4) was prepared as an intermediate. To further investigate whether (R) configuration at C-12 is responsible for partial agonism in the [35S]GTPγS assay, we synthesized the C-16 brominated analogue of 12-epi-salvA (5) since C-16 bromination of salvA had very little effect on in vitro pharmacological profile.11,13 Dibromated 12-epi-salvA (6) was isolated during the optimization of the synthetic conditions to 5.
The synthetic routes to 2–6 are depicted in Scheme 1.
Scheme 1.
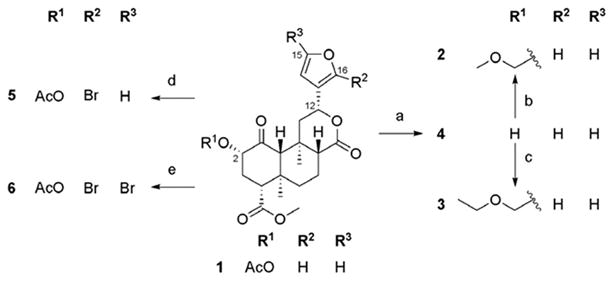
(a) NaHCO3, H2O2, THF, 58%; (b) MOMCl, NaI, DIPEA, DMA, 80 °C, 60%; (c) EtOMeCl, NaI, DIPEA, DMA, 80 °C, 65%; (d) NBS (1.1 equiv), CHCl3, 28%; (e) NBS (2.2 equiv), CHCl3, 31%.
Deacetylation of 12-epi-salvA 1 provided 12-epi-salvB 4. MOM ether 2 and EtOMe ether 3 were prepared from 4 using the alkyl chlorides MOMCl and EtOMeCl, respectively, along with NaI and Hunig's base in warm DMA.12 Monobrominated 12-epi-salvA 5 was synthesized in modest yield from 1 using 1.1 equiv of NBS in chloroform.11 However, when 2.2 equiv of NBS were used, dibrominated 12-epi-salvA 6 was obtained. Spectral data consistent with the proposed structures were obtained for all the compounds prepared in this study. Compounds purity was >95% as evaluated by 1H NMR and HPLC.
The activities of analogues 1–6 are shown in Table 1. In some assays, data for the full agonists salvA and U50,488H and for the non-selective KOPR partial agonist nalbuphine are provided for comparison. The affinities for the human KOPR, the rat mu opioid receptors (MOPR) and the mouse delta opioid receptors (DOPR) were determined by competitive inhibition of [3H]diprenorphine binding to membranes prepared from Chinese hamster ovary (CHO) cells stably transfected with the KOPR, MOPR and DOPR, respectively.14 None of the test compounds inhibited [3H]diprenorphine binding to the DOPR at 3 μM.
Table 1. In vitro activities of 12-epi-salvinorin A and its analogues.
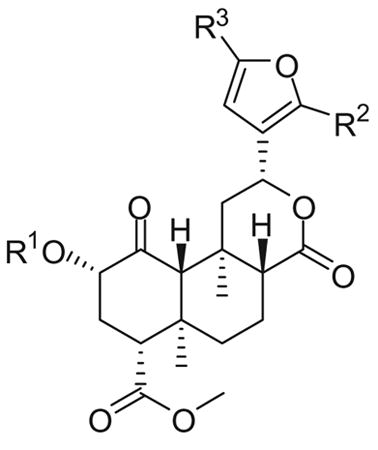
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | hKOPR binding Ki± SEMa,b (nM) | rMOPR binding Ki± SEMb,c (nM) | hKOPR [35S]GTPγS | hKOPR β-arrestin 2 | ||
|
|
|
||||||||
| EC50 ± SEMb,d (nM) | Emax ± SEMe | EC50f (nM) | Emaxg | ||||||
| 1, 12-epi-salvA | Ac | H | H | 41 ± 5h | _h,i | 41 ± 6 | 73 ± 6 | 382 ±85 | 126 ± 10 |
| 2 |
|
H | H | 4.4 ± 0.4 | _i | _j | ∼100%k | _j | _j |
| 3 |
|
H | H | 2.5 ± 0.3 | _i | 9.5 ± 1.2 | 103 ± 3 | 123 ±38 | 127 ±9 |
| 4 | H | H | H | 63 ±5 | _i | _j | ∼100%k | _j | _j |
| 5 | Ac | Br | H | 18 ± 1 | _i | _j | ∼100%k | _j | _j |
| 6 | Ac | Br | Br | 40 ± 1 | 318 ±39 | _j | ∼100%k | _j | _j |
| SalvA | 3.0 ± 0.1 | _i | 6.9 ± 0.5 | 98±3 | 31 ± 14 | 100 | |||
| U50,488H | 4.5 ± 0.2 | _i | 5.7 ± 0.5 | 100 | _j | _j | |||
| Nalbuphine | _j | _j | 65 ± 10 | 61 ± 3 | 250 ±152 | 13 ±9 | |||
Ki values in inhibiting [3H]diprenorphine binding to human KOPR (hKOPR).
Each value represents the mean ± SEMs of at least three independent experiments performed in duplicate.
Ki values in inhibiting [3H]diprenorphine binding to rat MOPR (rMOPR).
EC50 values in activating the hKOPR to enhance [35S]GTPγS binding.
Efficacy determined as the % of maximal response ± SEMs produced by U50,488H run in parallel experiments.
EC50 values in promoting the hKOPR to couple to β-arrestin 2.
Efficacy determined as the % of maximal response ± SEMs produced by salvA run in parallel experiments.
Data from Ref. 11.
Test compound at 3 μM inhibited <50% radioligand binding.
Not determined.
Test compound at 3 μM induced maximum activation of the KOPR.
In a previous study,12 substitution of the C-2 acetate of salvA with MOM and EtOMe ethers improved KOPR binding affinity by 4- and 7.5-fold, respectively. Similar substitutions to 12-epi-salvA leads to an even greater increase in KOPR binding affinity (MOM ether 2: 9-fold and EtOMe ether 3: 16-fold). 12-epi-salvB (4) and 12-epi-salvA (1) have similar binding affinities, whereas previous reports indicated that salvinorin B was approximately 100-fold less active than salvA.15–18 C-16 bromination of 12-epi-salvA increases binding affinity by two fold. In comparison, when salvA was brominated at the same position, a modest decrease (1.1 to 1.5-fold) in binding affinity was observed.11,13 While bromination at the C-15 and C-16 positions of 12-epi-salvA had no effect on binding to KOPR, it increased binding affinity to MOPR, resulting in a decrease in KOPR over MOPR selectivity.
The potencies and efficacies of compounds 1–6 on hKOPR were determined by their abilities to regulate [35S]GTPγS binding to membranes of CHO-hKOPR cells, an assay of receptor activation and G-protein coupling.19 Each test compound was initially screened at 3 μM and concentration-response curves were generated for 1, 3, salvA and nalbuphine. The selective KOPR full agonist U50,488H was used as a reference, with its efficacy designated as 100%. Among the salvinorin analogues, only 12-epi-salvA produced partial stimulation of [35S]GTPγS binding at 3 μM. Detailed evaluation of the effects of 1, 3, salvA and nalbuphine confirmed that 12-epi-salvA and nalbuphine were each partial agonists, with efficacies of 73 and 61%, respectively, and with comparable potencies (41 and 65 nM, respectively). Consistent with the SAR trends observed in the binding assay, introduction of an EtOMe ether at C-2 provided a KOPR agonist with enhanced potency (3: EC50 = 9.5 nM) in the G protein coupling assay.
Finally, to determine functional activity in another KOPR stimulated pathway, the ability of ligands 1, 3, salvA and nalbuphine to induce β-arrestin2 binding to the KOPR was assessed using a β-arrestin PathHunter® assay. The β-arrestin assay EC50 values were higher than the ones obtained in the [35S]GTPγS assay (3- to 12-fold), which may reflect differences in the degree of stimulation needed to promote the different cellular signaling events.20 In the β-arrestin2 interaction studies, nalbuphine acted as a weak partial agonist (13% efficacy), while the other three test compounds, including 12-epi-salvA (1), had full agonist activity (100– 112% efficacy). Again, introduction of an EtOMe ether at the C-2 position of 12-epi-salvA improved potency by threefold. These findings were mostly consistent with results observed in a visual β-arrestin-2 translocation assay (Fig. 1). Qualitatively speaking, nalbuphine appeared to induce a stronger β-arrestin2 response in the visual assay, with weaker albeit easily visible and numerous puncta. In contrast, nalbuphine had a very weak effect in the DiscoveRx assay (13% max). The difference between the results in these two assays may reflect differences in the treatment paradigm which is on a faster time scale (10–20 min) for the visual assay and a slower time scale for the DiscoveRx (90 min). Thus, if nalbuphine induced a transient β-arrestin2 recruitment, the signal could be lost over the longer time scale of the DiscoveRx assay. Alternatively, the difference could be due to differences in cellular context, receptor expression, or assay sensitivity which could all alter nalbuphine's response. Regardless, it is clear that nalbuphine is less effective at β-arrestin2 recruitment than the other compounds.
Figure 1.
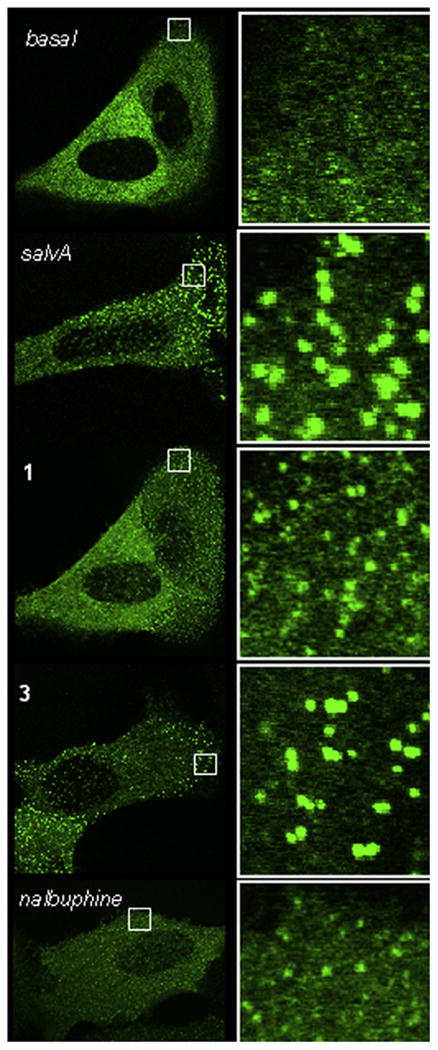
Agonist-induced recruitment of βarr2-GFP to KOPR in U2OS cells. Live cell confocal imaging of KOPR-βarr2eGFP-U2OS cells reveals robust recruitment of β-arrestin 2 to the KOPR by salvA and its derivatives as evidenced by the formation of intense green punctae after 10 min of drug exposure. A 5× magnification of the region outlined by the white square in each image is shown on the right. SalvA and the two derivatives (1 and 3) (10 μM) robustly recruit β-arrestin 2 to the KOPR (bright punctae), while nalbuphine (10 μM), a partial agonist, recruits less βarr2-GFP. Similar results obtained from three independent experiments performed on separate days, representative images shown.
Nalbuphine has been characterized as a non-selective opioid ligand for G-protein mediated signaling pathways, with the most pronounced effects consisting of partial agonism at KOPR and antagonism at MOPR.21 We have not found any previous reports of the effect of nalbuphine on β-arrestin2 recruitment. Our data indicate that nalbuphine is also a partial agonist on KOPR signaling through β-arrestin2. In comparison, 12-epi-salvA produced a partial response in the [35S]GTPγS assay system and a full response in the β-arrestin2 recruitment assay. This differential effect of one drug on separate signaling pathways through the same receptor may constitute an example of drug functional selectivity by 12-epi-salvA.22–24
In conclusion, introduction of MOM and EtOMe ethers at the C-2 position of 12-epi-salvA (2, 3) enhances potency while furan ring bromination (5, 6) has a minimal effect. All analogues of 12-epi-salvA were full agonists in the [35S]GTPγS assay indicating that there is no clear correlation between configuration at C-12 and maximum efficacy. Our data indicate that under our testing conditions, only 12-epi-salvA is a biased ligand, being more efficacious at activating β-arrestin2 mediated signaling pathways. In view of its unique signaling profile in vitro, 12-epi-salvA might serve as a pharmacological tool to investigate how bias towards β-arrestin 2 recruitment would affect behavior in animal models used to study drug abuse and mood and anxiety disorders.
Supplementary Material
Acknowledgments
This work was supported by the Stanley Medical Research Institute and NIH grants DA 17302 and P30 DA013429 (to L.-Y.L.-C.).
Footnotes
Supplementary data: Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bmcl.2011.11.128.
References and notes
- 1.Knoll AT, Muschamp JW, Sillivan SE, Ferguson D, Dietz DM, Meloni EG, Carroll FI, Nestler EJ, Konradi C, Carlezon WA., Jr Biol Psychiatry. 2011;70:425. doi: 10.1016/j.biopsych.2011.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carlezon WA, Jr, Béguin C, Knoll AT, Cohen BM. Pharmacol Ther. 2009;123:334. doi: 10.1016/j.pharmthera.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Knoll AT, Carlezon WA., Jr Brain Res. 2010;1314:56. doi: 10.1016/j.brainres.2009.09.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bruchas MR, Chavkin C. Psychopharmacology (Berl) 2010;210:137. doi: 10.1007/s00213-010-1806-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bruchas MR, Land BB, Chavkin C. Brain Res. 2010;1314:44. doi: 10.1016/j.brainres.2009.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mague SD, Pliakas AM, Todtenkopf MS, Tomasiewicz HC, Zhang Y, Stevens WC, Jr, Jones RM, Portoghese PS, Carlezon WA., Jr J Pharmacol Exp Ther. 2003;305:323. doi: 10.1124/jpet.102.046433. [DOI] [PubMed] [Google Scholar]
- 7.Cohen BM, Murphy B. Int J Neuropsychopharmacol. 2008;11:243. doi: 10.1017/S1461145707008073. [DOI] [PubMed] [Google Scholar]
- 8.Beardsley PM, Pollard GT, Howard JL, Carroll FI. Psychopharmacology (Berl) 2010;210:189. doi: 10.1007/s00213-010-1846-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schenk S, Partridge B, Shippenberg TS. Psychopharmacology (Berl) 1999;144:339. doi: 10.1007/s002130051016. [DOI] [PubMed] [Google Scholar]
- 10.McLaughlin JP, Land BB, Li S, Pintar JE, Chavkin C. Neuropsychopharmacology. 2006;31:787. doi: 10.1038/sj.npp.1300860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Béguin C, Duncan KK, Munro TA, Ho DM, Xu W, Liu-Chen LY, Carlezon WA, Jr, Cohen BM. Bioorg Med Chem. 2009;17:1370. doi: 10.1016/j.bmc.2008.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Munro TA, Duncan KK, Xu W, Wangn Y, Liu-Chen LY, Carlezon WA, Jr, Cohen BM, Béguin C. Bioorg Med Chem. 2008;16:1279. doi: 10.1016/j.bmc.2007.10.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simpson DS, Katavic PL, Lozama A, Harding WW, Parrish D, Deschamps JR, Dersch CM, Partilla JS, Rothman RB, Navarro H, Prisinzano TE. J Med Chem. 2007;50:3596. doi: 10.1021/jm070393d. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Tang K, Inan S, Siebert D, Holzgrabe U, Lee DY, Huang P, Li JG, Cowan A, Liu-Chen LY. J Pharmacol Exp Ther. 2005;312:220. doi: 10.1124/jpet.104.073668. [DOI] [PubMed] [Google Scholar]
- 15.Munro TA, Rizzacasa MA, Roth BL, Toth BA, Yan F. J Med Chem. 2005;48:345. doi: 10.1021/jm049438q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Béguin C, Richards MR, Wang Y, Chen Y, Liu-Chen LY, Ma Z, Lee DY, Carlezon WA, Jr, Cohen BM. Bioorg Med Chem Lett. 2005;15:2761. doi: 10.1016/j.bmcl.2005.03.113. [DOI] [PubMed] [Google Scholar]
- 17.Lee DY, Karnati VV, He M, Liu-Chen LY, Kondaveti L, Ma Z, Wang Y, Chen Y, Béguin C, Carlezon WA, Jr, Cohen B. Bioorg Med Chem Lett. 2005;15:3744. doi: 10.1016/j.bmcl.2005.05.048. [DOI] [PubMed] [Google Scholar]
- 18.Tidgewell K, Harding WW, Lozama A, Cobb H, Shah K, Kannan P, Dersch CM, Parrish D, Deschamps JR, Rothman RB, Prisinzano TE. J Nat Prod. 2006;69:914. doi: 10.1021/np060094b. [DOI] [PubMed] [Google Scholar]
- 19.Zhu J, Luo LY, Li JG, Chen C, Liu-Chen LY. J Pharmacol Exp Ther. 1997;282:676. [PubMed] [Google Scholar]
- 20.Clark RB, Knoll BJ, Barber R. Trends Pharmacol Sci. 1999;20:279. doi: 10.1016/s0165-6147(99)01351-6. [DOI] [PubMed] [Google Scholar]
- 21.Pick CG, Paul D, Pasternak GW. J Pharmacol Exp Ther. 1992;262:1044. [PubMed] [Google Scholar]
- 22.Kenakin T. J Pharmacol Exp Ther. 2011;336:296. doi: 10.1124/jpet.110.173948. [DOI] [PubMed] [Google Scholar]
- 23.Mailman RB. Trends Pharmacol Sci. 2007;28:390. doi: 10.1016/j.tips.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mailman RB, Murthy V. Neuropsychopharmacology. 2010;35:345. doi: 10.1038/npp.2009.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.