

Abstract
Although acute lymphocytic leukemia (ALL) is the most common childhood cancer, genetic predisposition to ALL remains poorly understood. Whole-exome sequencing was performed in an extended kindred in which five individuals had been diagnosed with leukemia. Analysis revealed a nonsense variant of TP53 which has been previously reported in families with sarcomas and other typical Li Fraumeni syndrome-associated cancers but never in a familial leukemia kindred. This unexpected finding enabled identification of an appropriate sibling bone marrow donor and illustrates that exome sequencing will reveal atypical clinical presentations of even well-studied genes.
Keywords: exome sequencing, acute lymphocytic leukemia, genetic predisposition to disease, genetic testing
INTRODUCTION
Molecular diagnosis for cancer susceptibility typically proceeds in a sequential process starting with gene or genes most consistent with the clinical presentation. This process is effective if there is a small set of implicated genes, but is expensive and time consuming for families with atypical presentation or a large number of possible causative genes. The cost of genome-scale sequencing has decreased so that it can be effectively used in clinical practice and potentially obviate these problems.
A Hispanic kindred (pedigree in Fig. 1) was first referred for genetic evaluation when patient 1 (III-7) was diagnosed with B cell ALL as a teenager. Family history included a prior sibling who died from childhood ALL (individual III-11) and her deceased father with leukemia diagnosed at age 51. Patient 1’s older brother had recently been diagnosed with a thoracic mass subsequently reported to be an esophageal carcinoma. Patient 1’s leukemia was refractory to chemotherapy, and she died from recurrent disease 51 days after peripheral blood stem cell transplant (PBSCT). Eight years later, patient 2 (individual IV-2), a 13 year old paternal cousin once removed of patient 1, presented with a diagnosis of pre-B cell ALL. Bone marrow cytogenetics revealed multiple hyperdiploid clones apparently secondary to doubling of a hypodiploid clone which is highly associated with early treatment failure [1]. PBSCT was recommended. The family contains five individuals with leukemia (three documented to be childhood ALL) with only one solid tumor reported, consistent with autosomal dominant familial leukemia. Thus, it was recommended not to use a sibling donor unless testing for the familial ALL mutation was possible.
Figure 1.
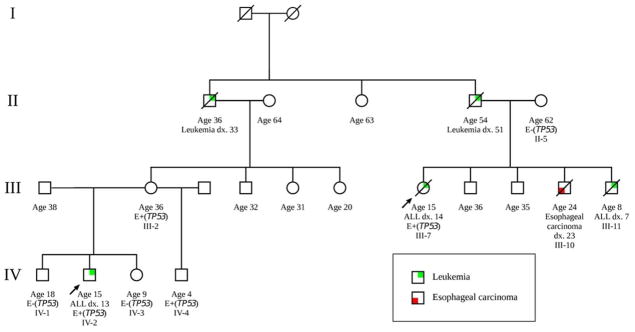
Clinical pedigree of the individuals involved in this study. Arrows indicate consultands who presented for genetic evaluation and “dx.” indicates age at cancer diagnosis. This kindred includes five individuals who had developed leukemia with only one other individual being affected with esophageal carcinoma. Exome sequence analysis was performed the affected individuals III-7 (patient 1) and IV-2 (patient 2) as well as the familial control (mother of patient 1; individual II-5) who did not transmit the cancer risk. Targeted analysis for the TP53 familial mutation by Sanger sequencing subsequently revealed that individual IV-4 carries the mutation (E+(TP53)) but individuals IV-1 and IV-3 do not (E−(TP53)).
Li Fraumeni syndrome (OMIM: 151623) was considered, but TP53 testing was not pursued because of the absence of LFS associated cancers (sarcomas, breast cancer, brain tumors or adrenal tumors) as well as prior studies suggesting that TP53 mutations did not cause familial ALL or lymphoma [2–4]. A search for an unrelated donor was unsuccessful, making identification of the specific familial mutation critical to further therapy.
METHODS
All subjects were enrolled under a protocol approved by Institutional Review Board of the Baylor College of Medicine which includes specific language permitting whole exome sequencing and reporting of clinically-relevant results. Whole exome sequencing was performed on (1) DNA from remission blood or lymphoblastoid cell lines from patient 1 and 2 and Patient 1’s mother (a married-in familial control) and (2) leukemia bone marrow diagnostic sample from patient 2.
Exome sequencing was performed using a capture platform, Vcrome 2.1, comprising 42 megabases of target sequences including coding sequences from VEGA [5], CCDS [6] and RefSeq [7] genesets, followed by sequencing on Illumina HiSeq with 100 bp paired-end reads. Reads were aligned to the hg19 human reference genome using BWA 0.5.9 [8] with realignment around indel sites using the GATK 1.0.5336 [9]. Variants were called using Atlas2 [10]. Analysis focused on rare or novel sequence variants (with population minor allele frequency undefined or less than or equal to 1% in both Thousand Genomes whole-genome phase 1 data and the NIEHS Environmental Genome Project).
RESULTS
Analysis of each individual demonstrated approximately 2400 rare variant alleles of which 700 were predicted to affect protein sequence through missense, frameshift, nonsense, or codon insertion/deletion events (Fig. 2A). The union of rare variants affecting protein sequence in the two affected individuals comprised 201 variants. Subtraction of the rare variants from the unaffected familial control resulted in a final set of 72 heterozygous rare variants for further consideration. Strikingly, this process identified (Fig. 2B) a nonsense variant of TP53 (c.916C>T; p.R306X) which was confirmed in patient 2 from an independent remission blood sample by Sanger sequencing in a clinical molecular diagnostic laboratory. This information was then used to determine that his HLA matched brother was negative for the TP53 mutation. A bone marrow transplant with his brother as donor was initiated. Another brother, age 5, was positive for the mutation and, along with his mother who is an obligate carrier, is undergoing cancer surveillance [11]. Exome sequencing of Patient 2’s leukemic bone marrow revealed the p.R306X germline mutation and a second somatic p.T118A TP53 mutation which is a rare variant in a conserved DNA binding domain consistent with a second hit in this tumor suppressor gene [12].
Figure 2.

The heterozygous TP53 mutation c.916C>T was present in the two affected individuals but not in the nontransmitting parent. (A) Germline variants were filtered by familial relationships, rarity (allele frequency <= 0.01 in 1000 Genomes Project data and NIEHS Environmental Genome Project) and potential for functional change to protein sequence (missense, nonsense, frameshift, codon insertion/deletion or splice-altering). 231 rare variants (72 predicted to change protein sequence) were present in both affected individuals but not in the familial control. Further details regarding these variants are given in Supplementary Table 1. (B) The variant c.916C>T was present in two patients but not in the familial control through whole exome sequencing. Sequence reads near this mutation are displayed using the Integrative Genomics Viewer, with reads colored by strand (red and blue for positive and negative strand, respectively). The view is centered at cDNA position 916 of TP53 and variant (non-reference) base calls are highlighted for each subject.
DISCUSSION
The highly aneuploid nature of the leukemias seen in this family as well as the high mortality in this kindred (four of the five individuals have died and patient 2 has an extremely high-risk form of childhood ALL) is consistent with that reported for leukemia containing somatic TP53 mutations [13]. TP53 mutations may be a rare cause of familial leukemia. In addition to prior negative TP53 studies in familial leukemia/lymphoma [2–4], our own exome and deletion analysis of five other small familial ALL kindreds did not demonstrate other TP53 constitutional mutations.
Conversely, the family’s pattern of cancer does not include any of the characteristic LFS tumors (sarcomas, brain tumors, breast cancers and adrenocortical carcinomas) and does not meet the Chompret or other criteria used to decide on TP53 testing [14, 15]. Review of a large cohort of TP53 mutation positive extended kindreds (n=107) revealed only a single family with multiple cases of ALL (two sisters from a kindred with other typical LFS tumors [16]), further demonstrating that familial ALL is not typically associated with TP53 mutations. The predisposition to leukemia does not appear to be related to this specific p.R306X TP53 mutation. In the International Agency for Research on Cancer’s locus-specific database of TP53 mutations (version R15, November 2010 [17]), this mutation has been described in both germline and somatic samples. Among the five p.R306X germline families with 11 affected individuals, there is the typical LFS pattern with breast (4), soft-tissue (3), nasopharynx (1), brain (1), unknown site (1) malignancies and only one hematopoetic cancer. There are 150 somatic p.R306X mutation reports, again with a typical tumor pattern and only one somatic mutation each of acute myelogenous leukemia, precursor cell leukemia NOS and mantle cell lymphoma. Therefore, in addition to the known MDM2 [18] promoter SNP309T>G (rs2279744) and TP53 R72P (rs1042522) polymorphisms [19] associated with age of tumor onset in TP53 mutation carriers, the atypical pattern of leukemia in this family suggests the possibility of a rare leukemia modifier cosegregating with the TP53 mutation. Supplementary Table 1 provides the other rare sequence variants that cosegregate with leukemia, although none obviously participate in the p53 pathway. However, verification of a leukemia modifier will require analysis of other rare TP53 mutation kindreds with this unusual ALL predisposition. We also cannot exclude that the leukemia phenotype could result from interactions with epigenetic or environmental exposures. In summary, identification of the TP53 mutation in this kindred through exome sequencing provided clinically actionable information including identification of (1) the appropriate sibling donor for transplant, (2) a young sibling at risk for cancer requiring cancer surveillance, (3) the mother of patient 2, an obligate TP53 mutation carrier who requires early, intensive breast cancer surveillance and (4) two other mutation negative siblings not at increased cancer risk. As exome analysis enters the clinical arena we can anticipate similar unexpected findings to broaden our understanding of the clinical spectrum of mutations in many previously well-studied genes.
Supplementary Material
Heterozygous rare variants present in the two affected individuals and not present in the familial control. Chromosome positions are in hg19 coordinates.
Acknowledgments
This work was supported by research grant RP10189 from the Cancer Prevention and Research Institute of Texas and R01-CA138836 to SEP and training grant T32 GM007526 (BP).
Footnotes
CONFLICT OF INTEREST STATEMENT
Dr. Richard Gibbs receives compensation as a consultant for GE Healthcare. Donna Muzny has been a co-owner of SeqWright, which also held investments in Lasergen and Visigen. She divested from SeqWright and Visigen in 2012. The authors declare no other potential conflict of interest.
References
- 1.Nachman JB, Heerema NA, Sather H, et al. Outcome of treatment in children with hypodiploid acute lymphoblastic leukemia. Blood. 2007;110:1112–5. doi: 10.1182/blood-2006-07-038299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Felix CA, D’Amico D, Mitsudomi T, et al. Absence of hereditary p53 mutations in 10 familial leukemia pedigrees. The Journal of Clinical Investigation. 1992;90:653–8. doi: 10.1172/JCI115907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weintraub M, Lin AY, Franklin J, et al. Absence of germline p53 mutations in familial lymphoma. Oncogene. 1996;12:687–91. [PubMed] [Google Scholar]
- 4.Pötzsch C, Schaefer HE, Lübbert M. Familial and metachronous malignant lymphoma: absence of constitutional p53 mutations. American Journal of Hematology. 1999;62:144–9. doi: 10.1002/(sici)1096-8652(199911)62:3<144::aid-ajh3>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 5.Wilming LG, Gilbert JGR, Howe K, et al. The vertebrate genome annotation (Vega) database. Nucleic Acids Research. 2008;36:D753–60. doi: 10.1093/nar/gkm987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pruitt KD, Harrow J, Harte RA, et al. The consensus coding sequence (CCDS) project: Identifying a common protein-coding gene set for the human and mouse genomes. Genome Research. 2009;19:1316–23. doi: 10.1101/gr.080531.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pruitt KD, Tatusova T, Klimke W, et al. NCBI Reference Sequences: current status, policy and new initiatives. Nucleic Acids Research. 2009;37:D32–6. doi: 10.1093/nar/gkn721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–95. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research. 2010;20:1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Challis D, Yu J, Evani US, et al. An integrative variant analysis suite for whole exome next-generation sequencing data. BMC Bioinformatics. 2012;13:8. doi: 10.1186/1471-2105-13-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Villani A, Tabori U, Schiffman J, et al. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: a prospective observational study. The Lancet Oncology. 2011;12:559–67. doi: 10.1016/S1470-2045(11)70119-X. [DOI] [PubMed] [Google Scholar]
- 12.Fen CX, Coomber DW, Lane DP, et al. Directed evolution of p53 variants with altered DNA-binding specificities by in vitro compartmentalization. Journal of Molecular Biology. 2007;371:1238–48. doi: 10.1016/j.jmb.2007.05.099. [DOI] [PubMed] [Google Scholar]
- 13.Shlien A, Tabori U, Marshall CR, et al. Excessive genomic DNA copy number variation in the Li-Fraumeni cancer predisposition syndrome. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:11264–9. doi: 10.1073/pnas.0802970105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gonzalez K, Noltner K, Buzin C. Beyond Li Fraumeni Syndrome: clinical characteristics of families with p53 germline mutations. Journal of Clinical Oncology. 2009;27:1250–1256. doi: 10.1200/JCO.2008.16.6959. [DOI] [PubMed] [Google Scholar]
- 15.Tinat J, Bougeard G, Baert-Desurmont S, et al. 2009 Version of the Chompret Criteria for Li Fraumeni Syndrome. Journal of Clinical Oncology. 2009;27:e108–e109. doi: 10.1200/JCO.2009.22.7967. [DOI] [PubMed] [Google Scholar]
- 16.Wu C-C, Krahe R, Lozano G, et al. Joint effects of germ-line TP53 mutation, MDM2 SNP309, and gender on cancer risk in family studies of Li-Fraumeni syndrome. Human Genetics. 2011;129:663–73. doi: 10.1007/s00439-011-0957-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Petitjean A, Mathe E, Kato S, et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Human Mutation. 2007;28:622–9. doi: 10.1002/humu.20495. [DOI] [PubMed] [Google Scholar]
- 18.Ruijs MWG, Schmidt MK, Nevanlinna H, et al. The single-nucleotide polymorphism 309 in the MDM2 gene contributes to the Li-Fraumeni syndrome and related phenotypes. European Journal of Human Genetics. 2007;15:110–4. doi: 10.1038/sj.ejhg.5201715. [DOI] [PubMed] [Google Scholar]
- 19.Hrstka R, Coates PJ, Vojtesek B. Polymorphisms in p53 and the p53 pathway: roles in cancer susceptibility and response to treatment. Journal of Cellular and Molecular Medicine. 2009;13:440–53. doi: 10.1111/j.1582-4934.2008.00634.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Heterozygous rare variants present in the two affected individuals and not present in the familial control. Chromosome positions are in hg19 coordinates.