

Abstract
HIV integrase (IN) catalyzes the insertion into the genome of the infected human cell of viral DNA produced by the retrotranscription process. The discovery of raltegravir validated the existence of the IN, which is a new target in the field of anti-HIV drug research. The mechanism of catalysis of IN is depicted, and the characteristics of the inhibitors of the catalytic site of this viral enzyme are reported. The role played by the resistance is elucidated, as well as the possibility of bypassing this problem. New approaches to block the integration process are depicted as future perspectives, such as development of allosteric IN inhibitors, dual inhibitors targeting both IN and other enzymes, inhibitors of enzymes that activate IN, activators of IN activity, as well as a gene therapy approach.
Introduction
Acquired immunodeficiency syndrome (AIDS) was reported for the first time in 1981 in a small number of patients1−3 but has developed into a major epidemic. There were more than 34 million people living with human immunodeficiency virus (HIV) at the end of 2011. The worldwide distribution is not uniform. Sub-Saharan Africa is the most severely affected area, with nearly 5% of the entire population infected with HIV; 69% of the worldwide HIV-infected population lives in this region (www.unaids.org). Therefore, AIDS and HIV infection are global health hazards with huge social, economic, and ethical consequences. Since the clinical identification of AIDS, there have been extraordinary scientific efforts to find a good therapeutic approach to combat this disease, and the first results appeared rapidly. Six years after the identification of HIV as the pathogenic virus that caused AIDS, a sensitive test was developed to detect infected people during the latency period, and AZT was introduced as a clinically effective drug, which was rationally designed to reduce the progress of AIDS. The prognosis of AIDS patients with full access to current therapies has dramatically changed since the first cases of AIDS were reported. The life expectancy for AIDS patients was less than 1 year before AZT was introduced in 1987; today, HIV infection is often treated as a chronic infection rather than a lethal disease.4 The ability to detect HIV-positive individuals early and the development of several drugs, which effectively block the virus cycle, have caused this dramatic change in the prognosis of HIV-positive patients. In fact, the efforts to understand the mechanisms of resistance displayed by the virus have led to the rational development of new drugs and to the understanding that combination therapy could overcome resistance. However, AIDS remains a major worldwide health problem, especially in developing countries where combating the epidemic must overcome societal issues.
Highly active antiretroviral therapy (HAART) utilizes cocktails of different drug classes to target various steps in the HIV replication cycle: entry, fusion, reverse transcription, integration, and protein maturation. HAART, however, is not often well-tolerated by patients because of the harsh side effects; this regimen also requires a high degree of compliance, incurs significant expense, and leads to multidrug resistance.5 Therefore, additional efforts to improve the current therapeutic approaches are needed.
From the approval of AZT in 1987 until late 2007, four different drug classes have been approved by Food and Drug Administration (FDA) for the treatment of AIDS: (i) the nucleoside reverse transcriptase inhibitors (NRTI),(ii) the non-nucleoside transcriptase inhibitors (NNRTI), (iii) the protease inhibitors (PIs), and (iv) the fusion inhibitors.6,7 These drugs successfully control the HIV infection, but their adverse effects and the emergence of resistant strains drive the need for new therapies,8,9 which may focus on novel targets. Consequently, new research has led to the development of maraviroc, which was approved in 2007 as an entry inhibitor that acts as a CCR5 antagonist,10 and raltegravir (RAL), the first integrase (IN) inhibitor. The discovery of RAL validated the existence of the IN, which is a new target in the field of anti-HIV drug research.11−13 Although the clinical armamentarium available for the treatment of HIV infection has grown to include approximately 30 drugs, RAL remains the sole IN inhibitor used in clinical practice as stand-alone drug. More recently, two compounds have been studied: elvitegravir14 (EVG), which was approved by FDA in late 2012 and in EU, while this paper was under submission, and dolutegravir15 (DTG), which is in advanced clinical trials. These agents are integrase strand transfer inhibitors (INSTIs) and represent the latest class of antiretroviral drugs approved for the clinical treatment of HIV infections.
Integrase Function and Structure
IN catalyzes the insertion of viral DNA (vDNA) into the genome of infected cells, although it can act as a cofactor for reverse transcription as well.16 Integration is required for viral replication because the transcription of the viral genome and the production of viral proteins require that the vDNA is integrated into the host chromosome.17 Following reverse transcription, vDNA is primed for integration in the cytoplasm by the IN-mediated trimming of two nucleosides from its 3′-ends. After this cleavage, which is referred to as 3′-processing (3′-P), the IN remains bound to the vDNA as a multimeric complex, which is referred to as a preintegration complex (PIC), that bridges both ends of the vDNA.
The in vivo process that requires PIC is operationally similar to the in vitro process that integrates vDNA into a heterologous DNA target;18 however, the in vitro integration reaction only requires vDNA and IN.19 The estimated size of the PIC20 suggests that it has a complicated composition, which includes a variety of viral and cellular factors; the structure of this complex may change as it travels through the cytoplasm to the nuclear membrane and beyond. Some studies report inconsistent recoveries of viral proteins from PICs, which are likely due to differences in the purification method used and to the dynamic, yet delicate nature of these complexes. In addition to IN,21 matrix (MA),20,22 reverse transcriptase (RT),20,22 and viral protein R (Vpr)23 were observed to be associated with PICs. Nucleocapsid (NC) has also been demonstrated to support the processing and function of the PIC.24 These viral proteins aid the transport of the PIC through the nuclear envelope. IN acts as a karyophilic element that contributes to, but is not essential for, the transport of the PIC and processed DNA within the nucleus.25,26
Some cellular proteins, which are packaged alongside IN within PICs,27 have been identified. These proteins stimulate IN enzymatic activities, regulate integration by binding to DNA, stimulate intermolecular integration, or suppress autointegration (for more information, see the following sections). A number of additional proteins have been identified, but their roles have yet to be elucidated.28
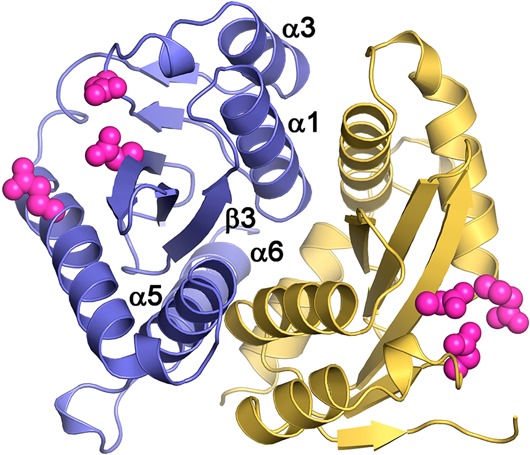
IN is a 32 kDa protein comprising three structural domains: an N-terminal domain (NTD) (residues 1–50) that contains a zinc-binding HHCC motif,29 a catalytic core domain (CCD) (residues 51–212) that contains the enzymatic active site and the catalytic triad (D64, D116, and E152),30 and a C-terminal nonspecific DNA-binding domain (CTD) (residues 213–288) (Figure 1).31 The structures of the three individual IN domains have been determined,29,31,32 in addition to the structures of the CCD when bound to the NTD33 and CTD.34 All of these structures show that IN exists as a dimer or in higher oligomeric states. The interactions between IN subunits are highly dynamic, a property that is essential for the biological function of IN.35 The dimerization interface is composed of four α-helices (α1, residues 95–109; α3, residues 123–133; α5, residues 171–187; and α6, residues 188–208) and one β-strand (β3, residues 248–252) from each monomer of the CCD. The dimer is stabilized through additional interactions between the monomers in the NTD (residues 29–35).34,36 In the CCD, the helix-to-helix contacts between α1 and α5′ and between α1′ and α5 contribute to dimer stabilization by strong hydrophobic and electrostatic interactions.30 Several models of DNA complexed with full-length IN have been proposed,37 but the structure of this complex has not yet been solved.
Figure 1.

Structure of HIV-1 IN. (A) IN domains, where the catalytic triad is shown in pink. (B–D) Structures of single IN domains: (B) NTD (PDB code 1wje); (C) CCD (PDB code 1bis); (D) CTD (PDB code 1ihv). (E–F) IN two-domain structures: (E) NTD + CCD (PDB code 1k6y); (F) CTD + CCD (PDB code 1ex4). Each structure consists of two IN monomers, shown in yellow and blue. The zinc ions in the NTD are shown in green, and the catalytic triad (D64, D116, and E152) in the CCD is shown in pink.
Recently, the crystal structure of the DNA-bound full-length prototype foamy virus (PFV) was determined.38 This structure was used to establish the model for full-length HIV-1 IN when bound to DNA.39 In this model, a tetramer of IN is attached to a pair of vDNA molecules. The CCD retains the known dimeric interface,30,40 but only two of the four active sites in the tetramer, one from each CCD dimer, associate with the vDNA. Both the NTD and the CTD contribute to the stabilization of the tetramer (Figure 2).
Figure 2.

Architecture of the PFV intasome. (A) Views along (left) and perpendicular (right) to the crystallographic 2-fold axis. The subunits of the IN tetramer, which are in blue and green, are engaged with viral DNA. The external IN chains are in yellow. The DNA strands are orange and magenta, and the last one is the most reactive. D128, D185, E221, or the catalytic triad is in red. Gray spheres are Zn atoms. (B) Focus on IN chains with domains and linkers indicated.
Integrase Mechanism of Catalysis
Because the structure of the catalytic domain of IN revealed that the enzyme belongs to a superfamily of polynucleotidyl transferases,41 which includes RNase H, transposases, and polymerases, these enzymes are thought to share a similar mechanism during catalysis. Therefore, catalysis using two metal ions, which is a property that the above enzymes share, is actually the accepted mechanism for IN reactions. This mechanism was first proposed for the 3′-5′ exonuclease reaction of Escherichia coli DNA polymerase I42 and then applied to other polynucleotide polymerases.43
For HlV-l IN, several experiments have established that the integration process consists of two catalytic steps: the first step is 3′-P, a hydrolysis step, followed by strand transfer (ST), a transesterification step (Figure 3),44,45 in which two highly cooperative divalent cations catalyze these two reactions centered on a phosphodiester bond. A long debate regarding the nature and the number of divalent cations that are involved as cofactors has persisted. It is not clear whether both metals are present within CCD or if the second metal is brought by the incoming vDNA, even though the two metal ions cooperate in the catalysis of IN. However, the IN catalytic DDE triad coordinates metal ions such as Mn2+ or Mg2+, with the latter assumed to be the physiologically relevant species.46
Figure 3.

Outline of the in vivo integration process.
The 3′-P reaction is performed in the cytoplasm. The 3′ end of the DNA synthesized by RT consists of the terminal tetranucleotide CAGT; the GT unit is not present in the integrated DNA. In fact, during this step, both of the vDNA termini are endonucleolytically cleaved by IN to remove the GT dinucleotides. The resulting vDNA is recessed at each 3′ end47 and displays a CA dinucleotide that is exposed by 3′-P and is highly conserved among diverse mobile genetic elements. The alterations to this sequence prevent the IN from catalyzing 3′-P. After 3′-P, the HIV-1 PIC enters the nucleus, where IN catalyzes the insertion of the vDNA ends into the host chromosome (Figure 3).
The exposed CA-3′-OH DNA ends are then ready for the second catalytic step, ST. This reaction occurs via divalent metal-mediated phosphodiester transesterification, which utilizes the two 3′-OH groups on the vDNA as nucleophiles.44 According to this mechanism, the two viral 3′-OH groups exposed by the 3′-P step attack phosphodiester bonds on complementary strands of the host DNA. The metal ion cofactors play a dual role during catalysis; they help stabilize the enzyme–DNA complex and facilitate the charge flow from the viral 3′-OH to the departing 3′-OH of the host DNA.48 In this mechanism, MA and MB are capable of interacting with the scissile phosphate in a highly structured SN2-like mechanism. Particularly, metal ion A acts as a Lewis acid to facilitate the function of the nucleophile, which is properly oriented with the help of the protein side chain, and metal B activates the 3′-oxoanion leaving group and stabilizes the pentacoordinate transition state (Figure 4).42,43a,49,50 Therefore, the 5′ end of the host DNA acts as the leaving group of the intermediate, completing the ST reaction. After this ST step, the resulting DNA gaps between the proviral and host DNA are filled in by host cell’s DNA polymerases (Figure 3).51
Figure 4.
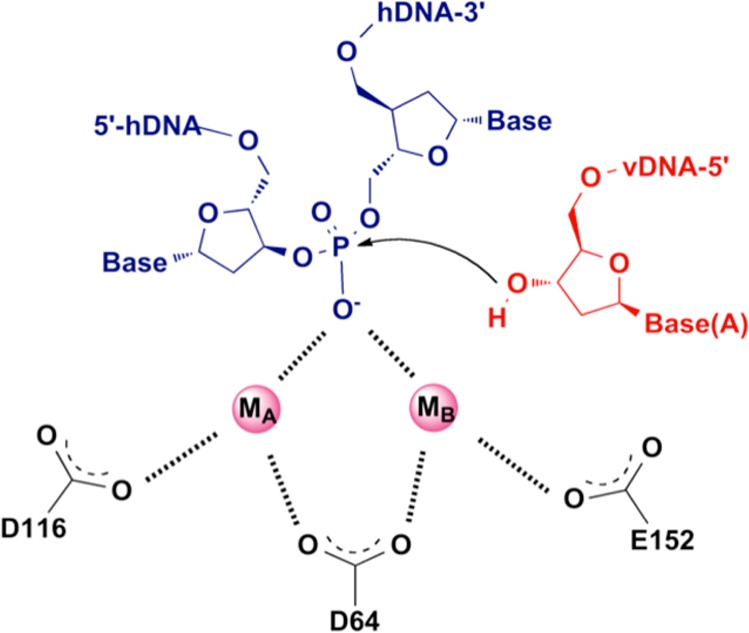
ST step. The attack of the 3′ ends of vDNA on the phosphodiester bonds of host DNA is coordinated by metal ions.
A recent report regarding the 3′-P and ST reactions catalyzed by PFV in crystal furnished an interesting mechanistic confirmation about the mechanism that can be transposed to IN activities.52 Snapshots of the complexes were obtained during different steps: (i) the IN/vDNA complex in the absence of metal, (ii) the complex after metal addition and before the 3′-P reaction, (iii) the complex of IN/processed vDNA/target DNA before the ST reaction, and (iv) the complex after the ST reaction. Analyzing the complexes elucidated the different roles of the metal ions. Metal A is responsible for activating the attacking nucleophile in 3′-P, while metal B activates the attacking 3′-OH of the IN/processed vDNA/target DNA complex. Additionally, the analysis of the final complex suggested that the second metal ion dissociates slowly. The apparent loss of the binding affinity of metal B (second ion) after ST could be caused by the ejection of the newly formed phosphodiester from the active site.
The sites of attack for each vDNA 3′ end, which are on the two target DNA strands, are five nucleotides apart. Both of the attacks take place across the chromosomal DNA major groove (Figure 3). The 3′ ends of the processed vDNA are joined to the 5′ ends of the target DNA, which results in the integration intermediate. In this intermediate, the 5′ ends of the vDNA and the 3′ ends of the target DNA are not joined, and the two protruding 5′ CA dinucleotides are left out. Ligation completes the integration process by filling in the single-strand gaps between the viral and target DNA by removing the two extra nucleotides at the 5′ ends of the vDNA. The viral IN is responsible for catalyzing 3′ end processing and joining, but once the integration intermediate is formed, the cellular machinery carries out the remaining step and completes the integration process (Figure 3).53,54 The site selection for HIV integration shows minimal sequence selectivity toward the target DNA.55,56 However, HIV integrates preferentially inside transcribed genes.56 It is plausible that cellular factors such as transcription complexes, including those bound to the IN within the PICs, as well as chromatin remodeling are implicated in the selection of the HIV integration sites within the transcribing genes.
Inhibitors of Integrase Catalytic Site
Early IN inhibitors (INIs) included peptides,36,57 nucleotides,58 DNA complexes,59 and polyhydroxylated aromatic compounds, which are a class of small molecules derived either from natural products60 or by drug design strategies.61 However, none of these compounds were able to be developed into an effective anti-HIV agent. Although potent IN inhibitors were discovered in in vitro assays against a recombinant IN, the majority of these compounds did not exhibit antiviral activity, and the remaining compounds generally exhibited high cytotoxicity. However, within the limited number of structures with proven antiviral efficacy and low cytotoxicity, the compounds with a catechol moiety were demonstrated to inhibit viral entry as their primary mechanism of action, which was determined in cell-based assays.62
The first major breakthrough for the development of IN inhibitors for clinical use was the development of virologically relevant assay by a Merck team and their subsequent discovery of the aryl diketo acids (DKA) such as pyrrole derivative 1 (L-731,988)63 and its derivatives as effective IN inhibitors in their antiviral assay. These compounds were identified from a focus screen of a collection of flu endonuclease inhibitors discovered earlier by the Merck group.63 Later, Shionogi patented 1-(5-chloroindol-3-yl)-3-hydroxy-3-(2H-tetrazol-5-yl)propenone (2, 5-ClTEP) and related derivatives.64 The chemical structures of these agents featured an α,γ-diketo acid function linked to an aromatic portion (Chart 1).
Chart 1. DKAs and Bioisosteres as IN Selective ST Inhibitors.

A distinctive feature of DKAs is that these compounds selectively inhibit the ST reaction. In general, DKAs are effective inhibitors at nanomolar concentrations and can block integration without interfering with vDNA synthesis in cells.63 The generation of drug-resistant mutant viruses, which bear single or multiple mutations in their IN coding sequence, validated the HIV-1 IN as a molecular target. The identified mutations (T66I, L74M, E92Q, G140G/A, Y143R/C, Q148R/H/K, S153Y, M154I, and N155H) are located around the DDE catalytic triad of amino acids (D64, D116, and E152).63,65 The inhibition was found to be metal-dependent,66 and it also interfered with the binding of the flexible loop on the LTR vDNA end.67 This was consistent with the proposed binding mode of the drug within the IN catalytic site. The increased formation of 2-LTR viral circles, which were not competent for integration, further supported the proposed selective inhibition of the IN in HIV infected cells.63 Binding studies revealed that the DKAs bind to the catalytic site of the HIV-1 IN only in the presence of LTR vDNA68 and that they also interact with the 5′-end of vDNA.69,70 Mutating one of the catalytic residues in the HIV IN core is sufficient to abrogate drug binding in the presence of a vDNA substrate.68 The DKAs act as competitive inhibitors with human DNA; the binding of the target DNA substrate to the IN–vDNA complex prevents the binding of DKAs and the inhibition of integration by DKAs in a concentration-dependent manner.68,69,71
The importance of the work on DKAs prompted an extensive search for antiretroviral agents that could better define the structure–activity relationship (SAR) within this class of inhibitors and elucidate their mechanism of action.70,72 However, the real breakthrough was to reach both clinical trials and practice. The designs of DKAs isosters that include the diketo acid pharmacophore within a heterocyclic ring have achieved this goal.
8-Hydroxyquinoline, 8-hydroxy-1,6-naphthyridine, and related derivatives such as their carboxamides were identified as potent INSTIs. 8-Hydroxy-1,6-naphthyridine 3 (Chart 1) showed excellent potency against ST and HIV replication.73 The napthyridinecarboxamide 4 (L-870,812)74 strongly inhibited ST and HIV replication and had only a moderate affinity for serum proteins. This compound also showed efficacy against the Simian immunodeficiency virus, with an IC95 of 350 nM, and was the first IN inhibitor to demonstrate activity against this virus in the rhesus model.74,75 The analogue naphthyridine 5 (L-870,810)76 exhibited better enzyme inhibition activity than 4, showed very good pharmacokinetic properties, and reached phase II clinical trials.76 The 4-hydroxy-2-oxo-1,2-dihydro-1,5-naphthyridines 6 and 7 (GSK-364735)77 showed excellent potency, and the last one reached phase II clinical trials.
GSK used a heterocyclic azole isoster to replace the carboxamide group present in 5 and related analogues. Oxadiazole and triazole substituted naphthyridines were also patented as IN inhibitors (e.g., 8), as they had impressive biological and toxicological activities.78 Gilead also reported a tricyclic scaffold, which contained an 8-hydroxyquinoline moiety, as a class of IN inhibitors.79 Of those compounds, only 9 (GS-9160)80 entered phase I clinical trials but was not pursued further because of unfavorable bioavailability.80
DKA bioisosters were also proven to bind divalent cations,76 and several studies have been performed to better define the mechanism of action of DKA bioisosters, which included the study that reported 4 and 5. This study demonstrated that the IN–vDNA complex was “trapped” by these ST inhibitors via a transient intermediate within the PIC.81 The most important class of DKA bioisoster has been designed and reported by a Merck team in IRBM in Rome. IRBM had been studying the inhibitors of HCV polymerase and described compounds with a dihydroxypyrimidinecarboxylic moiety that were also potent INSTIs (Figure 5).82
Figure 5.

Design of early RAL-like inhibitors: from HCV polymerase to HIV IN inhibitors.
The dihydroxypyrimidinecarboxamides were described as selective HIV INSTIs; they demonstrated low nanomolar activity in a cellular HIV spread assay in the presence of 50% normal human serum and demonstrated very good pharmacokinetics in preclinical studies.83 The further study of the dihydroxypyrimidinecarboxamides included an optimization for potency, physicochemical properties, and pharmacokinetic profiles. This work led to the discovery of RAL (MK-0518).13,84
First Generation of Integrase Inhibitors Approved for Clinical Trials
Raltegravir (RAL)
RAL is the first commercially available antiretroviral agent to target HIV IN. It was approved by the FDA for the treatment of HIV-1 infections in late 2007.13,85 RAL, which is orally administered at doses of 400 mg twice daily, proved to be efficient in combination therapy; viral loads were reduced in both naive and highly experienced antiretroviral patients (Chart 2).86 RAL has been coadministered with etravirine (NNRTI) and darunavir or ritonavir (PIs) as a salvage therapy for treatment experienced patients who were facing virological failure due to extensive multidrug resistances. This treatment achieved virological suppression similar to that observed in HIV treatment naive patients.87 After approval, the clinical efficacy and tolerability of RAL gave a second chance to patients who, after the failure of HAART, were left with almost no treatment alternatives. RAL has also been recently reported to be a potential alternative treatment for enfuvirtide treated patients with a stable suppressed viral load.88,86a
Chart 2. First and Second Generation of the INSTIs Approved for Clinical Trials.

More recently, the FDA approved the use of RAL for the treatment of HIV/AIDS in treatment-naive and pediatric patients. The replacement of efavirenz by RAL has led to higher efficiency in the optimized background Truvada regimen, which is composed of the NRTIs emtricitabine and tenofovir disoproxil fumarate.86a It is very likely that RAL will become the keystone of future multidrug cocktails, which may lead to an oral, once-daily, highly active antiretroviral therapy.89
Despite the effectiveness of RAL for both first-line and salvage therapy, resistance mutations can still reduce this inhibitor’s activity against HIV. The resistance pathways of RAL involve mutations of HIV-1 IN Q148 or N155.90 Mutations at these positions interfere with the coordination of metal cofactors near the active site carboxylate groups, as proposed recently.91 RAL has a modest genetic barrier to resistance development because the occurrence of single point mutations confers high-level resistance (fold change (FC) of >5). To date, three major resistance pathways, which involve nonpolymorphic residues, have been extensively described and characterized for RAL: E92QV/N155H, T97A/Y143CHR, and G140CS/Q148HKR.92 Although these three pathways have been shown to arise separately, some recent reports suggest that they may be linked. The G140S/C and E92Q/V mutations alone impart greater than 5- to 10-fold resistance to RAL93 but usually appear only after the N155H and Q148HKR mutations,94 which leads to FC > 100 for the combined mutations. In addition to these major resistance mutations, several polymorphic and nonpolymorphic residues that impart a greater than 5-fold resistance to RAL have been identified. Some of these mutations, such as T66I/L, act synergistically with pre-existing major resistance mutations (Table 1).95
Table 1. Main Drug Resistance Mutations That Confer Resistance to INSTIsa.
| fold
change (FC) |
||||
|---|---|---|---|---|
| mutation | RAL | EVG | DTG | 10 |
| T66R | + | ++++ | _ | _ |
| E92Q | + | +++ | + | _ |
| T97A | + | + | _ | NDb |
| G118R | + | _ | + | + |
| E138K | + | + | + | + |
| G140S | + | + | _ | + |
| Y143C | + | _ | _ | + |
| Y143R | +++ | _ | _ | + |
| Q148H | ++ | + | _ | + |
| Q148K | ++++ | ++++ | _ | _ |
| Q148R | +++ | ++++ | _ | + |
| N155H | +++ | +++ | _ | + |
| R263K | _ | + | ++ | _ |
| T66I/R263K | _ | +++++ | NDb | NDb |
| E92Q/N155H | ++++ | +++++ | + | + |
| E138K/Q148H | _ | ++ | + | NDb |
| E138K/Q148K | ++ | +++++ | +++++ | NDb |
| G140S/Q148H | +++++ | +++++ | _ | ++ |
| G140S/Q148K | +++++ | +++++ | _ | + |
| T97A/Y143C | +++++ | _ | _ | NDb |
| T97A/Y143R | +++++ | _ | _ | NDb |
Susceptibilities of common drug resistance mutations to first and second generation INSTIs are expressed as FC in the susceptibility of mutant virus or IN enzyme relative to wild-type. FC scores are designated as follows: <2, −; 2–10, +; 10–20, ++; 20–50, +++; 50–100, ++++; >100, +++++.
ND, not done.
Like other INSTIs, RAL has been proven to inhibit IN selectively by blocking the ST reaction. The cocrystals of RAL, HIV IN, and the vDNA ternary complex have never been solved. However, the structure of the full-length IN from PFV complexed with RAL has been recently solved, and this helped to define the structural details of retroviral DNA integration. These studies also facilitated the modeling of the HIV-1 intasome, which has aided in the development of antiretroviral drugs.38 The crystal structure confirmed that the three coplanar oxygen or nitrogen atoms of the metal binding motif coordinate the two Mg2+. The p-F-benzyl group penetrates somewhat deeply into the hydrophobic cleft between the residues P145 and Q146 created by the opening of the conserved 5′C4pA33′ nucleotide at the end of the processed strand. This opening is caused by the displacement of 3′A operated by the p-F-benzyl group, which forms a well-oriented π-stacking interaction with the penultimate C residue of the processed DNA strand. The halobenzyl group induced fit, which is caused by displacement of the 3′ A, explains why the deletion of this base dramatically increased INSTI on and off rates for binding to the HIV-1 IN–DNA complexes.96 RAL also places its terminal oxadiazole ring in a cleft to engage in π-stacking with the Y143 residue of IN. This π–π stacking is unique for RAL and does not appear to be vital for inhibitory activity; it can be replaced by other interactions within the 140 amino acid residues of the loop. The resolution of this structure was crucial to the rationalization of RAL’s mechanism of action and resistance profile. Although Q148H and N155H do not make direct contact with the INSTIs, mutations of these two strategically located residues are thought to trigger conformational changes within the catalytic pocket, which result in a decrease in the binding efficacy of INSTIs to mutated IN. Y143 is involved in a π-stacking interaction with the oxadiazole group of RAL, which explains the Y143 mutation resistance.97
Elvitegravir (EVG)
After the discovery of RAL, further research into new INSTIs, which are DKA bioisosteres, also led to the investigation of a molecular simplification of the DKA group. These studies led to the discovery of EVG (GS-9137) (Chart 2). EVG is a monoketo acid that resulted from early modifications of the DKA motif.14a,14b The initial approach utilized 4-quinolone-3-glyoxylic acids, and these were replaced by 4-quinolone-3-carboxylic acids, which retained three chelating groups, including the carbonyl of the quinolinone ring. While the glyoxylic derivative was inactive, the quinolonylcarboxylic acids showed high specificity and efficacy against the ST reaction, which was similar to the DKA compounds. EVG has demonstrated an in vitro IC50 of 7 nM against IN and an EC90 of 1.7 nM in cell based assays, which were performed in the presence of normal human serum. EVG displayed an approximately 30% bioavailability in dogs and rats, with maximal plasma concentrations being achieved 0.5–1 h after administration, and was found to be well tolerated and efficacious in clinical trials.98 However, EVG shares a moderate genetic barrier to IN resistance with RAL, which includes extensive cross-resistance between the two compounds. This factor is a major drawback for clinical use, along with the need to be co-dosed with a CYP3A inhibitor for QD dosing (150 mg). The mutations in N155H, Q148H/R/K, and G140A/C/S were selective for EVG both in culture and in patients.99 Because these are mutations typical of RAL, the use of EVG is precluded for the treatment of most RAL-resistant viruses. The only major RAL-associated mutations not selective for EVG were Y143C/R/H. Subsequent studies showed that viruses containing Y143C/R/H remained susceptible to EVG.100 This has been rationalized by X-ray structural studies, which indicate that the π-stacking interaction with RAL’s oxadiazole ring with Y143 is completely absent from the complex between IN and ELV.38 In addition to the RAL-associated resistance mutations, EVG developed other mutational pathways: T66I did not confer a high-level of resistance to RAL99a but gave FC > 10 for EVG, while a T66R mutation showed FC > 10 for RAL and FC > 80 for EVG.101 The T66I mutation is associated with a series of accessory mutations, which include F121Y, S153Y, and R263K; the second two have not been associated with RAL resistance (Table 1).102
Second Generation Integrase Inhibitors (INSTI)
Dolutegravir (DTG)
First generation IN inhibitors were remarkably efficient at reducing viral load in both treated and naive patients, but the moderate genetic barrier for resistance demonstrated the pressing need for second-generation INSTIs. These compounds should be active against RAL- and EVG-resistant viral strains.
DTG (S/GSK1349572) is a promising HIV INI. DTG specifically inhibits the ST reaction with recombinant IN in enzyme assays (IC50 value is 2.7 nM) and is highly active against HIV replication in infected PBMC cells (EC50 value is 0.5 nM) (Chart 2). DTG also demonstrated efficacy against most viral clones that were resistant to RAL and EVG, as well as against clinical isolates of HIV-1 and HIV-2; however, some viruses containing E138K, G140S, or Q148H mutations were not as susceptible to DTG. Double mutants, which contained combinations of E138K, G140S, and R148H, had FC of >10 for DTG, but this was favorable in comparison to RAL, which yielded an FC of >330 and >140, respectively (Table 1). In vitro combination antiviral studies demonstrated that DTG did not increase the cytotoxicity of the combination but instead exhibited a synergistic effect with EFV, nevirapine, stavudine, abacavir, lopinavir, amprenavir, and enfuvirtide, as well as an additive effect in combination with maraviroc.103
New drug application of DTG was filed in late 2012 with the FDA. The good pharmacokinetic profile, anti-HIV effect, and potency of DTG are well documented,67,68 without evidence of serious adverse effects.104 Primary INI resistance mutations have not yet been reported for DTG in either culture or the clinic. In vitro resistance selection studies over 112 weeks identified, in order of appearance, viruses harboring T124S/S153F, T124A/S153Y, L101I/T124A/S153F, and S153Y by week 84. Although these mutations persisted throughout serial passaging, they did not confer a high-level resistance to DTG.103 Position 124 of IN is modestly polymorphic, and S153F/Y has previously been described in EVG selection studies. Despite an apparently high genetic barrier for resistance selection, recent tissue culture and biochemical studies report that a R263K mutation in IN may confer modest resistance to DTG (Table 1).105
Recently, the use of DTG in RAL-treated patients, who were infected with subtype-B viruses that harbored mutations at position Y143 and N155, was provided as a proof-of-principle.106 To model the effects of DTG in RAL-treated patients, several serial passaging studies have been carried out. These studies demonstrate that the presence of N155H and Y143CHR resistances did not lead to additional resistance mutations because of DTG pressure nor did it cause decreased DTG susceptibility.107 In contrast, the presence of Q148HRK mutations did lead to further mutations; a > 100 FC was found for DTG susceptibility relative to the wild-type in subtype B viruses.103 Y143 is thought to connect with RAL via π-stacking between the oxadiazole group and the tyrosine phenol ring.97 DTG, 10 (MK-2048, see below),108 and EVG do not have this oxadiazole group and are largely resistant to mutations at position Y143.100 The mutation for DTG-associated resistance, R263K, is not well characterized but may be linked to decreased vDNA binding.105
It has been suggested that DTG shows a high barrier for resistance because it binds more tightly to IN when compared to RAL and EVG.109 Assays also confirmed that DTG exhibited tight binding and, therefore, had a longer dissociative half-life from IN than either RAL or EVG.110 In this model, the half-life of binding was directly related to the inhibitory potential of INIs when the binding half-life (t1/2) was below 4 h. FC > 3 for drug resistance, relative to the wild type, was observed when t1/2 dropped below 1 h.109 In assays with wild-type enzymes, t1/2 values of DTG, RAL, and EVG were 71, 8.8, and 2.7 h, respectively. RAL and EVG have a shorter t1/2 than DTG, which suggests that resistance mutations that affect the binding of RAL and EVG might compromise antiviral potency. For example, the Y143CHR mutations compromise interactions between IN and RAL but do not do so between IN and DTG or between IN and EVG.111 This is further supported by data for mutations E92Q/N155H, E138K/Q148R, and G140S/Q148R, which have been shown to significantly reduce both the t1/2 and antiviral potency.109 This hypothesis had also been suggested for another second generation INSTI, 10 (see below). This compound has a relatively high barrier for resistance because it also has a slower dissociation rate (t1/2 = 32 h) for IN when compared to RAL (t1/2 ≥ 7.3 h).110
MK-2048 (10)
The optimization of the tricyclic 10-hydroxy-7,8-dihydropyrazinopyrrolopyrazine-1,9-dione compounds led to the development of 10, which is a potent inhibitor (EC95 < 50 nM) in the presence of 50% human serum (Chart 2).108 Compound 10 was effective against both RAL- and EVG-resistant viruses in tissue culture, as well as rIN mutated enzymes. Only slightly diminished effectiveness against viruses containing at least two of the following mutations was found: E138K, G140S, and Q148R.112 However, selection studies in culture selected a novel substitution at position G118R that, in concert with E138K, conferred an approximately 8-fold resistance to 10 (Table 1).113 G118R may cause changes in the geometry of the catalytic triad, which decreases the ability of second-generation INSTIs to chelate the divalent ions within the catalytic site.114 This substitution also decreases drug binding via steric hindrance.115 Despite its favorable resistance profile, clinical development of this inhibitor has been arrested because of a poor pharmacokinetic profile.
S/GSK-1265744 (11)
Compound 11 (S/GSK-1265744)116 is another second generation INSTI that has been tested in double-blind randomized placebo-controlled trials. It has shown a promising short-term efficacy and an excellent pharmacokinetic profile and was well tolerated in patients with HIV (Chart 2).116 The development of 11 is in progress as a weekly suspension concentrate formulation.
Toward Third Generation Integrase Inhibitors (INSTIs)
Although all of the anti-HIV drugs targeted to IN have been prone to drug resistance, a second generation of IN inhibitors may be relatively resilient to this problem and therefore retain efficacy over long periods. Several newly identified resistance mutations, such as G118R, R263K, and S153Y, have been identified in cell assays with the second generation INSTIs. These new mutations add to our understanding of the three identified resistance pathways discovered with the first generation INSTIs, which involve mutations at positions Y143, N155, and Q148. Although resistance mutations against DTG have not yet emerged in clinical trials,117 viruses that contain Q148 mutations combined with several secondary mutations may require an increased clinical dose.118 Therefore, in contrast with the N155 and Y143 pathways, mutations that emerge from the Q148 pathway can confer cross-resistance to second generation INSTIs.
In summary, second-generation inhibitors have been developed to avoid cross-resistance with first-generation drugs but bind to the same catalytic site and, therefore, possess similar chemical properties as the first-generation INSTIs. This shared binding site creates the potential for multiple cross-resistances for the first-generation drugs RAL and EVG.119 The main pathways of resistance to INSTIs act by reducing the association time of the drugs with the IN enzyme; a tentative association between a slow dissociation rate and reduced resistance has been demonstrated.108,120 Therefore, to successfully design a third generation of INSTI, researchers should treat the dissociation rate as an important metric for the quality of a drug.
Dual Inhibitors of Integrase and Ribonuclease H Function of Reverse Transcriptase
Although HAART offers reduced single drug doses, which mitigate treatment toxicity, chronic treatment still has several drawbacks, which include long-term toxicity (i.e., lipodystrophy, dyslipidemia, high cardiovascular risk, etc.), emergence of drug resistant strains, drug–drug interactions, and the financial strain caused by the use of multiple drugs. Additionally, patient compliance with combinational treatments is an issue because noncompliance can lead to the rapid emergence of viral resistance.121 Therefore, new anti-HIV agents are still urgently needed, particularly against novel viral targets and/or that demonstrate activity against existing drug resistant virus strains. However, while the introduction of an additional drug increases the armamentarium of antivirals against HIV, this addition would not reduce the problems intrinsic to HAART; it would simply strengthen the “polypharmacy” approach to fighting HIV/AIDS.
To overcome the problems associated with polypharmacy, various strategies such as sustained-release drugs and fixed-dose combination regimens (polypills) have been developed. The use of dual-action drugs, which are compounds that combine two different desired pharmacological actions at a similarly effective dose, is a novel and highly promising approach. In these systems, a single compound possesses dual mechanistic action due to targeting of different effector mechanisms. The use of these drugs leads to efficacious outcomes because they would reduce pill burden, improve medication compliance, and decrease the adverse effects or drug–drug interactions. A single molecule with dual activity is superior to combination therapy from both a developmental and a clinical perspective. This approach can be assessed by conventional toxicology studies and avoids the pharmacokinetic disadvantages of using two separate agents with different absorption and distribution properties. Interest in a class of compounds known as dual inhibitors (a single molecule possessing dual inhibitory activity) is experiencing a surge, both in the scientific and clinical fields. Dual inhibitors possess two different biological inhibitory activities, target two different receptors or effector mechanisms, target two different enzymes, or target two separate functions of a single enzyme. This is currently an active area of drug research across a wide range of fields122 and has been validated in the oncology arena. Dual inhibitors of tyrosine and phosphoinositide kinases show a very promising physiological activity.123 More recently, this approach has also been used for antimalarial treatments,124 but it has not yet reached the HIV field. Therefore, a polypharmacological strategy, which is based on the use of dual-action drugs, is an innovative approach to rational drug development against HIV, particularly in the area of dual inhibitors.
The design of dual inhibitors that are active against HIV RT and IN is an active area of research.125 Compounds endowed with a unique pharmacophore, which could inhibit two different targets, could act on the catalytic sites of the IN enzyme and the ribonuclease H (RNase H) domain of the HIV RT.
The rationale behind this approach is that the structure of the HIV-1 RNase H CCD has a fold that is similar to that of HIV-1 IN; consequently, their catalytic sites share a similar geometry. Both enzymes also have the same DDE motif, which is required for catalytic activity. Other similar structural characteristics, including three aspartate residues and two magnesium ions at a distance of 3.57 Å from each other, were found in the active site of RT complexed with a DNA primer-template and an incoming nucleotide.126
Dual inhibitors of the IN and RNase H function of RT have been recently described. Both DKA derivatives127 and bioisosteres128 have been reported to inhibit the above functions (Chart 3).
Chart 3. Dual Inhibitors of IN Enzyme and RNase H Function of RT.

Some pyrrolyl (12) and quinolonyl (13) DKA derivatives, which were previously developed as IN inhibitors,72 have been found to be active against the RNase H function of RT. The best IC50 value was 2 μM in the enzyme assays. These compounds were also active against HIV replication in cell based assays.127,129 Similar results have been reported for benzoylaminothienyl DKA 14 that inhibited both the IN activity and RNase H function of RT at similar concentrations (1.9 and 3.2 μM, respectively). Unfortunately, 14 showed no activity against HIV infected cells (Chart 3).130
A series of 2-hydroxyisoquinoline-1,3(2H,4H)-diones (i.e., 15) was proven to be active against both targets at submicromolar concentrations, but the authors suggested that the compounds’ high cytotoxicity limits their development (Chart 3).128a,128b
Madurahydroxylactone derivatives (i.e., 16) also exhibited similar potencies for both enzymes at micromolar concentrations. The authors suggested that a systematic screening for both IN and RNase H should be utilized when developing novel inhibitors (Chart 3).128c
In general, the dual inhibitor research field seems very interesting. The main goal in this area is to obtain compounds with a similar potency against both enzymes.
DNA Binders
The structure of PFV IN cocrystallized with RAL demonstrates extensive contact between the inhibitor and the vDNA.38 This finding clarifies why the INSTIs preferentially interact with and inhibit the DNA-bound form of the HIV-1 IN. An intriguing hypothesis has been recently reported, which treats vDNA as the primary target of RAL and INSTIs.131 In this study, RAL was shown to bind specifically to both the unprocessed and processed LTR ends. RAL supposedly binds directly and selectively to DNA, similar to other small organic molecules with chemotherapeutic activities. However, the authors emphasized that, unlike the anticancer agents, RAL occupies selective binding sites on DNA strands. RAL binds to the unprocessed LTR end, which could be the cause of the observed 3′-P inhibition, and causes a small damping of motion (fraying) in the terminal base pairs. Initial studies44,132 have shown that any restriction caused by fraying in the terminal base pairs, by either the extension of the duplex44,132a or a chemical linkage of the duplex ends,132e impairs the 3′-P reaction. Remarkably, RAL keeps the same conformation within the complex with both unprocessed and processed DNAs. Its halogenated ring forms a face-to-face contact with the cytosine base of the conserved 5′C4pA33′ step and the adenine A3, which bears a recessed 3′-OH group moved from its operative position. RAL binds to the LTR end prior to the 3′-P reaction, but the impact on this reaction is small. The selectivity of RAL for the processed LTR end is attributed to the nucleotides C4, A3, and G.24 This finding causes speculation that new drugs that interact more fully with these highly conserved bases, expense of interactions with amino acid side chains of the protein active site, will be better INSTIs and less prone to inducing resistance.
Allosteric Integrase Inhibitors
Over the past 2 decades, IN drug design and discovery has mainly focused on the direct inhibition of enzyme catalytic activities, leading to clinically approved INI-like RAL, EVG, and DTG. These compounds share a similar mode of action at the IN active site; they chelate the metals coordinated by the three catalytic residues and interact with vDNA in the complex. Because of the limited chemical space available for inhibitor design specific to IN, an overlap in resistance for the future generation INSTIs is inevitable. Therefore, although the successful design of future-generation INSTIs is attainable, this development will most likely result in only a temporary abatement in drug resistance. Therefore, future efforts should be directed to obtain compounds that block the integration process using different mechanisms of action.
Recently, research interests have moved toward the design of inhibitors with an allosteric mechanism of action or inhibitors of the interactions between cellular cofactors that are essential for integration. The first group of inhibitors would bind IN at a different region from the substrate-binding active site while still inhibiting its enzymatic activity. The second group of inhibitors would inhibit the protein–protein interactions between IN and its cofactor. Inhibiting cofactor binding leads to allosteric modification. Therefore, both groups can be described as IN allosteric inhibitors (ALLINIs). Additionally, the nonactive-site-binding IN inhibitors could display synergy with the current generation of INSTIs and other antiretroviral agents in clinical use.
The first efforts to find ALLINIs were targeted toward peptides that interfered with the integration process without binding to the catalytic site; however, it is clear that the final drug leads will be small molecules, which have high bioavailabilities and low molecular masses. To learn more about the potential pharmacophores and targets, peptides are useful tools to conduct the basic research because they can mimic the binding interface of the different protein–protein interactions that are necessary for successful integration.
Host Proteins Associated with the Retroviral Integration Complex
The PIC is the key nucleoprotein complex responsible for integration. The PIC is formed in the cytoplasm after the reverse transcription of vDNA from the RNA genome. Although the exact composition of the PIC has yet to be fully determined, the proteins that comprise this complex can be classified as (i) the viral proteins derived from the core of the infecting virion, such as IN itself, RT, MA, CA, and some HIV-1 accessory proteins,133 and (ii) the cellular components (Figure 6).134
Figure 6.

Representative list of IN- and PIC-associated viral (blue) and cellular (red) proteins in retroviral replication.
Several host proteins are involved with the interaction and constitution of the PIC, as well as the activation of enzymatic activities (Figure 6). Intriguingly, the host protein, Gemin2, may be important for the reverse transcription process in HIV-1 because it is associated with IN. Gemin2 may serve as a cofactor that stimulates and/or stabilizes the formation of the reverse transcription complex, which initiates DNA synthesis through its interaction with IN.135
After the 3′-P step takes place in the cytoplasm, the PIC needs to be shuttled into the nucleus to allow the integration process to take place. The PIC easily crosses the nuclear envelope because of its karyophilic properties.133 To date, importin 7 and TNPO3, which are both members of the importin β family, have been identified as IN-interacting importins that direct the HIV-1 PICs to the nucleus.136 NUP153, which is another cellular protein that regulates nucleocytoplasmic trafficking, was also shown to have a role in the nuclear transport of the PIC (Figure 6).137
PIC components include high-mobility group (HMG) proteins, barrier-to-autointegration factor (BAF), Ku, and LEM proteins. These components may help stabilize the nucleoprotein complex, promote the nuclear retention of the PIC, or protect host cells from vDNA termini-induced apoptosis. Any of these actions indirectly involve these components in the integration process.27,133,138 HMGA1 and BAF regulate integration by binding to DNA directly. HMGA1 stimulates IN activity,139,140 and BAF stimulates intermolecular integration while suppressing autointegration. Other proteins packaged in the PIC include integrase interactor 1 (INI1),141 lens epithelium-derived growth factor (LEDGF),142 and embryonic ectoderm-development protein.143 In addition to the host proteins listed above, more extensive discussions regarding the other cellular cofactors’ (Figure 6) interactions with the IN/PIC and their roles in retroviral replication are presented in comprehensive reviews.27,144
INI1 was the first binding partner of HIV-1 IN that was identified.141 The function of INI1 in HIV-1 integration was demonstrated in an in vitro integration assay where it stimulated the ST activity of recombinant IN.141 Subsequent studies showed that INI1 was specifically incorporated into HIV-1 virions during virus production,145 which suggests a possible role of INI1 in the late stage of HIV-1 replication rather than in the integration step.
LEDGF is a transcriptional regulatory protein that is strongly associated with chromatin throughout the cell cycle. It is expressed as two spliced variants: the LEDGF/p52 and LEDGF/p75 proteins.146 LEDGF comprises several functional domains implicated in the integration process. The N-terminus contains the PWWP (proline–tryptophan–tryptophan–proline) domain, three charged domains, the nuclear localization signal (NLS), and dual copies of the AT-hook DNA-binding motif.147 The C-terminus is different for the splicing variants: LEDGF/p75 shows a more extended domain that includes the integrase-binding domain (IBD), which was crucial for specific interaction with HIV-1 IN (Figure 7A).147 This protein was identified as an interaction partner of HIV-1 IN in human cells148 and has been shown to stimulate the in vitro integration activity of IN (Figure 6).149
Figure 7.

(A) LEDGF/p75 domains: N-terminal PWWP motif and the charged regions (CR1–3) critical for chromatin recognition and the central DNA binding domain (blue) and the C-terminal IBD (magenta) essential for binding to IN and cellular proteins. (B) Cocrystallized structure of LEDGF/p75–IBD (magenta) and the CCD dimer of integrase (green and blue). The catalytic triad is represented in orange (PDB code 2B4J). (C) Cartoon focused on CCD–IBD binding (PDB code 2B4J). IN CCDs are shown in green and blue, whereas the LEDGF/p75 IBD is in magenta. Residues of IN (dark green) and IBD (magenta) critical for the interaction are highlighted.
LEDGF is the first cellular protein demonstrated to be a bona fide cofactor for HIV-1 integration.147 It plays a critical, but not strictly essential, role. A significant reduction of HIV-1 replication in human CD4+ T cells with a knockdown of endogenous LEDGF was demonstrated.150 Additionally, a knockout study performed in mouse embryonic fibroblasts cell lines reported a 90% reduction in HIV-1 infectivity upon the depletion of LEDGF/p75, which was recovered upon the re-expression of LEDGF.151 The blockage of HIV-1 infection was shown to occur specifically at the integration step, and both the PWWP and IBD domains were proven to be of critical importance for HIV-1 integration and replication.150,151 On the basis of these findings, LEDGF is proposed to be a molecular adaptor that tethers HIV-1 IN to the target DNA. Because LEDGF is a transcriptional coactivator, this tethering activity might be responsible for targeting the integration site of HIV-1 toward transcriptionally active regions (Figure 8).147
Figure 8.

Schematic representation of the LEDGF support of the HIV-1 integration process. On the right, the different mechanisms of inhibition by LEDGINs (up) and INSTIs (down) are described. Inhibitors are represented in red.
LEDGF/p75–Integrase Interaction as a Drug Target for Anti-HIV Therapy
A crystal structure of a dimer of the IN CCD bound to IBD was recently reported (Figure 7B).40 These new data identified the amino acids K364, I365, D366, F406, and V408 from LEDGF/p75 as relevant for the mediation of the interactions with IN (Figure 7C). The IBD structure is composed of four α-helices linked by interhelical loops, which are responsible for binding to IN. The IN amino acids involved in the interaction with LEDGF/p75 are W131, W132, and the region extending from I161 to E170.40,152,153 The interface is located in a pocket that is formed by the two subunits of the IN-core dimer (α1 and α3 of one monomer and the six residues from the α4/5 connector of the other monomer). Residues located in the α4/5 connector and a hydrophobic pocket, which is formed by the other subunit, engage tightly with the two interhelical loops of LEDGF/p75–IBD. The I365 residue of LEDGF/p75 contacts the hydrophobic pocket formed by L102, A128, A129, and W132 of one IN subunit and T174 and M178 from the other subunit. Furthermore, I365 establishes a hydrogen bond with the backbone carbonyl group of IN E168, whereas D366 of LEDGF/p75 forms a hydrogen bond with E170.40 The importance of the IN amino acids A128, H170, T174, W131, W132, Q168, and E170, as well as the LEDGF/p75 residues I365, D366, F406, and V408, has been confirmed by mutagenesis studies. Mutation of these residues decreased or eliminated the binding of LEDGF/p75 to IN.40,152−155 Therefore, the protein–protein interaction surface of LEDGF/p75 and IN provides a well-defined pocket with multiple hydrophobic and hydrogen bond interactions. Rationally, this pocket is a good target for the design of small molecules for the purpose of inhibiting LEDGF/IN protein–protein interactions (Figure 7C).
Biological evidence highlighted the fact that an effective LEDGF/IN interaction is relevant for viral replication. In fact, (i) the depletion of LEDGF/p75 from cells by RNAi or knockout techniques significantly reduced the infectivity of HIV in those cells;150,151,156,157 (ii) the overexpression of the IBD of LEDGF/p75 in human cells inhibits HIV replication;158 and (iii) the serial passaging of HIV cells overexpressing this LEDGF/p75 fragment selects for a virus strain resistant to this phenotype.154 Interestingly, two mutations in IN were required to render IN resistant: A128T and E170G. These results confirmed the mutagenesis data and the validity of the IBD/IN CCD structure.40,152 These findings provided the proof-of-concept that the LEDGF/p75–IN interaction might be a feasible and druggable target for anti-HIV therapy.
The peptide corresponding to IBD efficiently competed with the endogenous LEDGF, which inhibited HIV replication and integration by more than 100-fold.158 Further validation was granted by reports that shorter peptides derived from LEDGF/p75 blocked the interaction between LEDGF/p75 and IN.159,160 Although peptides are not ideal compounds for drug development, these reports provided further support for the identification of the LEDGF/p75–IN interaction as a good target for anti-HIV drug development.
Inhibitors of LEDGF/p75–Integrase Interaction
Peptides
Usually, the protein–protein interface is so flat that it is difficult to identify small molecules that can effectively block the protein–protein interaction. However, the interaction between LEDGF and IN shows a peculiarity, a narrow part of IBD located in a deep pocket that is formed by the two subunits of the IN dimer.40 A first approach is to find compounds that interfere with the interaction between IN and its cofactor, LEDGF/p75. In fact, although the stability and/or bioavailability of peptides is always an issue, the design of small synthetic peptides that interact with one of the binding partners of a protein–protein interaction is a valid starting point to facilitate the development of peptidomimetic derivatives or small molecule inhibitors.
The initial work on the peptides derived from the LEDGF/p75 sequence has been focused on the design and synthesis of the three peptides: LEDGF/p75 353–378, 361–370, and 402–411.160 The authors described these peptides as inhibitors of DNA–IN binding. They shift the IN oligomerization equilibrium from the active dimer toward the inactive tetramer, which is unable to catalyze the 3′-P step. The LEDGF/p75-derived peptides inhibited the enzymatic activity of IN in vitro and blocked HIV-1 replication in cells by completely stopping integration. In this report, the inhibition of the LEDGF/p75–IN interaction by the described peptides was not presented, and the authors attributed the observed antiviral effect solely to the inhibition of the IN catalytic activity. Later it was reported that the peptide LEDGF/p75 355–377 was capable of competing with LEDGF/p75 for binding to IN; therefore, it inhibited the cofactor–IN interaction with an IC50 of 25 μM. The inhibition of the catalytic activity (both 3′-P and ST) was less pronounced for this peptide and was lost when the IN–DNA complex was assembled before the addition of the peptide. This finding led to the hypothesis that the peptide might disrupt the initial DNA-binding of IN, therefore exerting its effect on the catalytic activity.169 LEDGF/p75 361–370 was identified later as the smallest peptide that demonstrated inhibitory activity.161 In a more recent study, this peptide was cyclized, which increased its IN inhibitory activity.162 Additional LEDGF/p75 derived cyclic peptides have been synthesized, which has helped to identify new interactions in the LEDGF/p75–IN interface. Particularly, a hydrogen bonding interaction with IN–E168 gave valuable input for structure-based design efforts toward novel small molecules that inhibit the LEDGF/p75–IN interaction.163
Recently an approach has been developed to design peptides that can block the LEDGF–IN interaction. This is the approach of “reciprocal peptides” that is designed to bind to LEDGF/p75 and, as a consequence, to inhibit the LEDGF/p75–IN interaction from the side of the cellular cofactor. Because the LEDGF/p75 binding pocket in IN is not linear and, therefore, cannot guide the development of IN-derived inhibitory peptides, a phage display strategy was employed to select for peptides with an affinity for the IN interaction side of IBD. As peptides are notoriously difficult to deliver to cells, the authors chose a stable lentiviral expression of the selected active and mutant peptides. Expression of the active peptides led to the potent inhibition of HIV replication. Biochemical and biophysical studies, as well as antiviral profiling, demonstrated that the selected peptides inhibit HIV replication through their binding to the cellular cofactor, LEDGF/p75.164 Notably, although the peptides bind to the IBD of LEDGF/p75, no cellular toxicity was observed. This finding can be rationalized because the IBD of LEDGF/p75 interacts with IN as well as cellular partners, such as PogZ and JPO2 (Figure 7A), by a distinct type of interaction.165 Viral protein resistance selection failed because the interaction occurred with a cellular cofactor instead of viral fragments. The above studies provide the proof-of-concept that intracellular cofactors, such as LEDGF/p75, are competent drug targets for antiviral therapy and might have a higher barrier toward resistance selection.
Small Molecules
The effective interaction between LEDGF/p75 and IN for the integration process, the HIV replication cycle, and the findings regarding peptides that effectively block this protein–protein interaction led to the discovery of small molecules as LEDGF/IN interaction inhibitors (Chart 4).
Chart 4. Small Molecule Inhibitors of the LEDGF/p75–IN Interaction.

The benzoic acid derivative 17 (D77),166 discovered in a library containing approximately 300 compounds, disrupts the interaction between IN and the LEDGF/p75 IBD (Chart 4). Compound 17 was observed to inhibit LEDGF/IN binding in a dose-dependent manner and demonstrated antiretroviral activity, with an EC50 value of 23.8 μg mL–1 in cell assays. A molecular docking analysis using the IN CCD revealed that 17 makes significant contact with T174 from monomer A as well as Q95, T125, and W131 from monomer B. Site-directed mutagenesis experiments demonstrated that an alanine substitution at position T125 significantly decreased the 17–IN interaction, whereas the Q95A, W131A, and T174A substitutions practically eliminated the binding of the inhibitor to IN.166
DKA derivatives have been studied as LEDGF/IN interaction inhibitors as well. The indole derivative 18 (CHIBA-3002)167 is a small molecule that was previously described as INSTI and is able to inhibit both ST and LEDGF/p75–IN protein–protein interactions with micromolar activity (Chart 4). Molecular modeling (MM) studies of the congeners of these benzylindole derivatives highlighted that the CHIBA compounds form hydrogen bonds with the main chains of E170 and H171 of IN, while the diketo acid moiety creates a hydrogen bond with Q95 of the other IN–CCD subunit.167 Follow-up studies explored the chemical space of the LEDGF–F406 contact with W131 and generated 19 (CHIBA-3053),168 which was a more potent congener, with an IC50 in the lower micromolar range (Chart 4). Further optimization is needed to reach potent antiviral activity for this class of small molecules.168
A scaffold hopping approach was used to design another set of small molecules capable of inhibiting the catalytic site as well as the LEDGF/p75–IN interaction.169 By merging of the pharmacophores of salicylate and catechol, the 2,3-dihydroxybenzamide was identified as a new scaffold to efficiently inhibit the ST reaction. This active scaffold dramatically inhibited the interaction between IN and the LEDGF/p75 cofactor. The prototype example, N-(cyclohexylmethyl)-2,3-dihydroxy-5-(piperidin-1-ylsulfonyl)benzamide (20), inhibited the IN–LEDGF/p75 interaction with an IC50 value of 8 μM (Chart 4). MM studies on the mechanism of action led to the proposed involvement of the chelation of the divalent metal ions inside the IN active site. However, this compound developed strong interactions with IN residues in the LEDGF/p75 binding site. The compound did not induce any cytotoxicity in H630 cells, but no antiviral activity was reported. Further optimization of the described structures is required to reach higher in vitro activities as well as antiviral activity.
A fragment based screen to identify small molecules has been performed that took into account the relevant hydrogen bonding interactions between LEDGF and IN–Q168, which was identified, but not yet exploited, against the LEDGF–IN interaction.163 Interestingly, this contact might lead to the development of compounds with a higher affinity and/or a higher barrier to resistance. Initially, 500 fragments were screened using surface plasmon resonance (SPR), NMR, and crystallography to identify hits with good density in the LEDGF/p75 binding site of IN.170 Synthesis of small molecules based on these hits led to the best compound, a N-bis(4-methoxyphenyl)methylbenzamide (21)170 that showed an IC50 of 8.1 μM in the AlphaScreen based LEDGF/p75–IN interaction assay (Chart 4). Crystallography demonstrated that 21 penetrates deep into the hydrophobic pocket on the IN-dimer interface by directly interacting with the IN–Q168 and, therefore, targeting the 167–173 interaction site for LEDGF/p75 in IN. Compound 21 has moderate antiviral activity, with an EC50 of 29 μM, and no apparent toxicity. Compound 21 is also not cross-resistant with RAL resistance mutants such as IN Q148H/G140S and N155H/E92Q.
A repositioning approach with old drugs led to the identification of eight inhibitors via a MM approach, which was based on the flexible docking of the ligands. First, 1467 clinically approved drugs from a public drug bank were screened.171 The hits were capable of inhibiting the LEDGF/p75–IN interaction with moderate IC50 values, which ranged from 6.54 μM for carbidopa (22) (Chart 4) to 36.85 μM for eprosartan. Note that one of the selected compounds, atorvastatin (23) (IC50 = 8.9 μM), has been used in HIV patients to reduce cholesterol levels, and an effect on HIV replication has already been reported (Chart 4). Whether the in vitro and antiviral activities are linked with each other is still hypothetical, however, and requires further investigation.
A series of 2-(quinolin-3-yl)acetic acids were discovered to be inhibitors of LEDGF–IN interactions by a rational drug design approach. These inhibitors were called “LEDGINs” and represent the first class of authentic small-molecule allosteric inhibitors to display antiretroviral activity tied to a specific disruption within the IN–LEDGF/p75 interaction.172 A set of 200 000 commercially available compounds was filtered based on chemoinformatic parameters that incorporated the known chemical properties of small-molecule inhibitors of protein–protein interactions. Following this initial step, the remaining 160 000 compounds were screened with a pharmacophore model, which used the known crystal structures of IN and the cocrystal structure of the IN–LEDGF/p75 binding domains.40 The first LEDGIN hit displayed modest activity against the IN–LEDGF/p75 interaction, with a 36% inhibition at 100 μm. Starting from this compound, multiple rounds of SAR studies led to molecules with increased potency, which eventually resulted in the synthesis of the highly potent 2-(quinolin-3-yl)acetic acid derivative 24 (CX0516)154 (Chart 4). The lead LEDGIN exhibited an IC50 value of 1.37 μm against the IN–LEDGF/p75 interaction in vitro and exhibited an EC50 value of 2.35 μm against HIV replication in a cell assay. 24 is highly specific for the IN–LEDGF/p75 interaction; it did not show any activity against the interactions of LEDGF/p75 with other cellular partners. Importantly, it is active against multiple clinically relevant drug-resistant viral strains, including viral strains resistant to NRTIs, NNRTIs, CXCR4 chemokine receptor agonists, and most significantly RAL. The efficacy of 24 against RAL resistant viral strains is an indirect validation of its allosteric inhibition of IN. Additionally, this finding is particularly relevant because it provides an opportunity to produce an ALLINI to treat viral strains resistant to the INSTIs currently in clinical use. Compound 24 was inactive against a mutant viral strain containing IN substitutions (A128T/E170G) previously selected to be resistant to the transdominant inhibition of overexpressed LEDGF/p75 IBD.154 This is an additional support of the LEDGIN inhibition of viral replication through a direct disruption of the IN–LEDGF/IN interaction. Additionally, the selective pressure of the LEDGIN on the virus selected an A128T IN substitution located at the protein–protein interface, which clearly indicated that 24 was an effective allosteric LEDGF/p75–IN interaction inhibitor.172 A cocrystal of IN CCD with 24 was also reported. The LEDGIN carboxyl moiety forms hydrogen bonds with both main-chain nitrogen atoms of IN E170 and H171, which mimics the IN protein contacts of the LEDGF/p75 residue D366. The IN residue A128 occupies a space adjacent to the chlorine atom between the phenyl and conjugated ring system of the 2-(quinolin-3-yl)acetic acid derivative.
The above structural information helped to design and synthesize more potent LEDGINs with improved biological activities, such as 25 (CX14442),173 and facilitated a complete antiviral profiling of this compound class (Chart 4). Compound 25 is the first LEDGIN reported to display antiviral activity in the low nanomolar range, with an EC50 of 69 nM and a selectivity index of 1391.173 To date, the 2-(tert-butoxy)-2-substituted acetic acid derivatives are the most heavily studied LEDGINs; congeners of these compounds are in advanced preclinical development.
A series of 2-(quinolin-3-yl)acetic acid derivatives, including the prototype 26 (BI-1001),174 have also been disclosed in an international patent application by Boehringer Ingelheim Pharmaceuticals Inc. (Chart 4).174 These compounds bind to the allosteric region of IN, disrupt the IN–LEDGF/p75 protein–protein interaction, but also inhibit the LEDGF-indipendent IN catalytic function.175
LEDGINs inhibit the ST and 3′-P reactions to the same extent, and this complete inhibition can only be achieved when LEDGINs are added to IN before the DNA substrate.173,175,176 This mode of inhibition is different when compared to the uncompetitive one utilized by INSTIs, which require prior binding and 3′-P of vDNA ends (Figure 8).38,68 The inhibition of both the 3′-P and ST reactions by LEDGIN suggests that the binding to IN influences the active site of the enzyme.
LEDGF/p75 can be considered an allosteric effector of IN activity. In fact, this cofactor is supposed to modulate the IN multimerization required for enzymatic activity.177 Evidence has been provided for this modulation.173,175,176 In biochemical studies, LEDGINs are proven to bind to the interface of the IN dimer, which leads to its stabilization, restricts IN’s oligomeric flexibility, and consequently decreases the formation of the effective intasome. Therefore, in addition to their function as small molecule protein–protein interaction inhibitors, LEDGINs can be considered allosteric enzymatic inhibitors.
Recently, it was demonstrated that LEDGINs induce a significant decrease of deletions at the 2-LTR junctions in the 2-LTR circles produced during HIV replication in cell culture. This is consistent with an antiviral mechanism involving the inhibition of 3′-P and therefore is also consistent with the biochemical characterizations.176 Both the protein–protein interaction inhibition and the allosteric mechanism are relevant for their biological activity, cannot be uncoupled, and lead to the inhibition of the integration reaction. In the discussion of whether one mechanism should be considered more important than the other, one should keep in mind that in vivo LEDGINs will always encounter LEDGF/p75 bound to the dimer interface of IN and therefore are required to displace LEDGF/p75, which is essential for HIV replication.156
LEDGINs were proven to inhibit the integration step of HIV replication by quantitative PCR (Q-PCR) and time-of-addition (TOA) experiments.172,173 This is similar to the studies reported for INSTIs. Most importantly, LEDGINs are not cross resistant with INSTIs.
The first report regarding resistance selection with 24 demonstrated that a single point mutation was sufficient to render HIV resistance to the action of LEDGINs.172 However, the more potent derivative 25 was found to be active against A128T, and at least one additional resistance mutation is required to render IN resistant against this LEDGIN action.173 This finding could be very important for the further clinical development of this compound class because it proves that the expansion of the chemical space raises the barrier toward resistance development. Combination experiments demonstrate that LEDGINs and INSTIs do not antagonize each other but instead act in an additive or even slightly synergistic way. These studies suggest the implementation of a possible design of LEDGIN/INSTI combination therapy within the HAART treatment.173
As described above, cyclic peptides can bind to LEDGF/p75 and inhibit HIV replication in cell culture, which produced an impaired infectivity of viral particles.164 The most recent report on the mechanism of action of LEDGINs confirms this multimodal inhibition pathway.173 Presence of LEDGINs during virus production not only blocks provirus integration but also affects the infectivity of the residual virus progeny. This observation is unique for LEDGINs when compared with INSTIs or other early replication inhibitors, such as entry blockers, NRTIs, and NNRTIs. The finding that LEDGINs not only block the integration of the viral genome but additionally impair the infectivity of viral particles when present during production makes them highly interesting candidates for further clinical development.
Several LEDGF–IN interaction inhibitors were reported to have a dual mechanism of action. They inhibited both the protein–protein interaction and the activity of IN binding in the catalytic site.167,169,178 Whether the strategy of designing compounds that bind both the catalytic site of IN and the LEDGF/p75 binding site, and therefore inhibit both of the functionalities of HIV-IN simultaneously, will be a valid strategy to potently inhibit integration with a reduced risk of resistance selection or will lead to undesired side effects due to unequal affinity for both inhibitory sites still remains to be investigated.
Toward the Inhibition of Integrase–Viral Cofactors Interaction
The excellent results leading to the discovery of effective inhibitors of the LEDGF/p75–IN protein–protein interaction could be very helpful in the near future to obtain compounds useful in clinical practice that also circumvent the problem of resistance associated with the use of INSTIs. A similar approach to the one used for the discovery of LEDGINs could be applied to obtain molecules that inhibit other protein–protein interactions that IN maintains with its cofactors. IN interacts with numerous viral proteins and cellular cofactors in addition to LEDGF. Despite the excellent results obtained for the inhibition of the IN–host cofactor LEDGF interaction, which could be a unique case, in general, targeting host proteins for therapeutic intervention is a risky strategy because many host proteins are essential for cell viability, and interfering with their natural function may have undesired toxic side effects. Therefore, a better strategy to inhibit the integration process could be to target the interaction of IN with viral proteins. Inhibiting the interaction between two viral proteins is a better target for antiviral intervention because no cellular function would be interrupted by the inhibition of either protein. The in vivo integration process depends on multiple interactions of IN with various proteins.27,133,144a If binding to any of these cofactors is required for optimal IN activity, then disrupting these interactions would result in possible therapeutic compounds with great potential to complement existing HIV-1 treatments.
Viral RT
IN interacts with the HIV-1 RT.16,179,180 RT was proven to inhibit IN activity in vitro and is believed to act as an inhibitor of integration in infected cells.181 A library of RT-derived peptides was synthesized and screened in vitro for binding and inhibiting IN. Within this library, two peptides inhibited the 3′-P and ST activities in the low micromolar range. These peptides are located on the surface of RT, and their docking with the IN CCD structure suggested that the IN activities could be inhibited by steric hindrance.182 In another study, a library of peptides derived from the HIV-1 Pol sequence was evaluated for their IN-inhibitory activity, and five peptides derived from the RT sequence were found to be potential inhibitors.183
Vpr Protein
Vpr is a small protein in HIV-1 that is critical for efficient viral infection and the impairment of anti-HIV-1 immunity.184 It plays an important role in the nuclear localization of the PIC through direct interaction with IN and stimulates the binding of IN to vDNA through the interacting sequence, Vpr 52–96.185
A library of peptides covering the full Vpr sequence was screened in vitro for binding to IN and RT and the inhibition of their enzymatic activities.186 The two partly overlapping peptides, Vpr 57–71 and Vpr 61–75, were proven to inhibit both IN activities at micromolar concentrations. These peptides are exposed on the surface of Vpr and thus are accessible for protein–protein interactions in the context of the full protein.187 The docking of Vpr 33–47 and Vpr 61–75 with the IN CCD revealed that these peptides bind to the dimerization interface between the two IN subunits.186
In a further study regarding Vpr-derived peptides, three partly overlapping Vpr-derived peptides were found to inhibit IN activity in a dose-dependent manner. The sequence of the overlapping domain was Vpr 64–69, which is located in one of the helices of Vpr. Therefore, these inhibiting peptides were hypothesized to have an α-helical conformation and to interact with the cleft between the NTD and the CCD of IN. This region is distinct from the nucleic acid interacting surfaces of IN, which suggests that the Vpr-derived peptides inhibit IN in an allosteric manner.188
In a SAR study of these peptides, a rational design step was performed to obtain new compounds in which the helix formation of the inhibitory peptide was promoted. Not all of these peptides retained IN-inhibitory properties. Modification via the addition of eight arginines to the C-terminus of the peptides made them cell-penetrating and allowed for the examination of their IN-inhibitory activity and anti-HIV-1 activity in infected cells. One of these peptides (Vpr15) inhibited both 3′-P and ST with IC50 of 40 and 90 nm, respectively, and showed anti-HIV-1 activity at 1–2 μM.189
Rev Protein
IN was found to bind the HIV-1 Rev protein both in vitro and in vivo.190,191 Rev inhibited IN-mediated integration during the early phase of the virus replication cycle.192 Recently, a structural docking model of the Rev–IN complex was published supporting two possible mechanisms of IN inhibition by Rev: (i) the Rev-binding sites on IN overlap with the LEDGF/p75-binding sites, and its mode of action could simply be a displacement of LEDGF/p75; (ii) Rev could also act by shifting the oligomerization equilibrium of IN from the active dimer to the inactive tetramer.193
IN- and Rev-derived peptides have been synthesized and have been proven to inhibit the IN–Rev interaction. Rev-derived peptides, Rev 13–23 and Rev 53–67, bound to IN and were found to inhibit its enzymatic activities as well as the HIV-1 infectivity of cell cultures.190 In vitro assays revealed that Rev and the Rev-derived peptides inhibited the enzymatic activity of IN in a dose dependent manner, which indicated that the inhibition was specific.190,194 Conversely, two IN-derived peptides, IN 66–80 and IN 118–128, were reported to block the inhibitory effect of Rev, disrupting the Rev–IN interaction by binding to Rev.195
Modulators of Integrase Multimerization
Blocking the IN oligomerization process represents a very attainable allosteric inhibitory approach toward the design of a new class of IN inhibitors. Peptides and small molecules that inhibit the LEDGF/p75 protein–protein interaction were proven to influence the oligomerization of IN. LEDGINs bind to a site distinct from the catalytic site and show no cross-resistance to any known IN inhibitor. As IN requires a dynamic equilibrium between at least dimers and tetramers for its activity, the oligomerization of the enzyme seems to be a good candidate target for the design of ALLINIs.160
First, researchers were focused on peptides derived from the IN CCD dimer interface that were proven to effectively compete with IN dimer formation.196,197 Some of these peptides, which mimicked α-helices 1–5 of the CCD, were able to shift the IN oligomerization equilibrium entirely to the monomer.196 Recently, a focused library of potential HIV IN dimerization inhibitors was designed based on cross-linking peptides α5 and α6 to mimic a larger interfacial region of IN. The best cross-linked inhibitors are approximately 5-fold more potent against HIV-1 IN than the individual peptides alone or in combination. The most active inhibitors have an inhibitory concentration in the mid-nanomolar range and were proven to function via a dissociative mechanism of inhibition.198
Small molecules have been identified that bind across the IN dimer interface, which allows them to establish contact with both monomers (Chart 5). Consequently, these compounds stimulate dimerization and shift the oligomerization equilibrium to certain species, resulting in a loss of catalytic activity.199
Chart 5. Small Molecule Inhibitors of IN Multimerization.

The acetylated bis-caffeoyl derivative (2E)-3-[3,4-bis(acetoxy)phenyl]-2-propenoate-N-[(2E)-3-[3,4-bis(acetyloxy)phenyl]-1-oxo-2-propenyl]-l-serine methyl ester (27) was an early inhibitor of IN catalytic activities, which exhibited an IC50 value of 3 μM (Chart 5). This compound was later confirmed to acetylate the K173 residue. MM studies suggested that T174 and M178 could serve as additional IN residues involved in INI binding.200 This acetylated bis-caffeoyl inhibitor directly disrupts the dynamic exchange of IN monomeric forms between IN multimers. The compound preferentially binds and stabilizes a multimeric IN complex, thereby disrupting the dynamics of the free IN subunit exchanges required for IN–vDNA nucleoprotein complex formation.201
N,N′-(Methylene-di-4,1-phenylene)bis-1-pyrrolidineacetamide (12) was also shown to bind the IN dimeric interface and to specifically interact with residues K173 and T174 (Chart 5). MM studies suggested that IN residue K173 engages inhibitor 12 through cation−π interactions, whereas IN residue T174 forms hydrogen bond interactions. Compound 12 was proven to inhibit IN–vDNA binding with an IC50 value of 7.29 μM via an SPR-based competitive assay. This compound also exhibited antiretroviral activity, with an EC50 value of 17.05 μg mL–1.202
The coumarin derivative 29 was identified as an inhibitor of both 3′-P and ST IN catalytic activities, with IC50 values of 27 and 18 μM, respectively (Chart 5). This compound was shown to bind the IN dimeric interface region by using a benzophenone photoactivatable affinity labeling approach. Moreover, molecular docking studies revealed that 29 binds a pocket at the IN dimeric interface, suggesting it may disrupt the IN multimerization process. Compound 29 was much less effective at inhibiting preassembled IN–DNA complexes, with IC50 values that increased for both 3′-P and ST. The decrease in potency against IN–DNA preassembled complexes supports an inhibitory mechanism of action, in which the compound binds the allosteric site at the dimeric interface and disrupts IN multimerization.199a
A successful approach based on virtual screening recently led to the identification of new small molecule inhibitors of IN dimerization.203 A novel AlphaScreen-based IN dimerization assay was used to evaluate the inhibitory activities of the compounds.204 The most interesting hit was the hydrazone 30, which inhibited 83% of IN dimerization at 100 μM (Chart 5). For this compound, an IC50 value of 38.4 μM was determined.
A recent study that detailed the binding region of the ALLINIs at the dimeric interface at a high resolution was reported. The structure contains a sucrose molecule, which was bound in a symmetrical binding site at the dimer interface comprising residues E87, V88, I89, P90, E96, Y99, F100, K103, and K173 from both monomer chains, and characterizes an inhibitory binding pocket, which was previously reported for other inhibitors, such as compounds 27, 28, and 29. This site is one of a number of clefts on the surface of IN and may represent a functional binding pocket for protein–protein recognition. The large number of interactions between sucrose and IN could be used to develop compounds with better binding affinities. Although sucrose does not exhibit an IN inhibitory activity, the structural elucidation of the allosteric site it occupies represents a remarkably useful platform for the rational design of multimeric disrupting compounds with improved affinities and inhibitory potentials.205
Integrase Enzyme Indirect Inhibition
Post-translational modifications (PTMs), which include phosphorylation, ubiquitination, and acetylation, are the chemical alteration of the primary structure of a protein after its translation. This process has a demonstrable impact on various protein functions, such as enzymatic activity, protein–protein interaction, and subcellular localization.206 The histone acetyltransferase (HAT) p300 protein is a transcriptional coactivator that is important for cell proliferation and differentiation, as was demonstrated with in vitro assays through the use of HAT p300 synthetic inhibitors, which showed an anti-acetylase activity in mammalian cells.207
Although the association between PTMs and the retroviral integration process has not been fully addressed, it was revealed that HAT p300 regulates the viral expression of both DNA viruses and lentiviruses;208 the enzymatic activity of HIV-1 IN is positively regulated by both p300 and GCN5 HATs (Figure 9).209 A specific interaction between HAT p300 and IN both in vitro and in vivo has been described.209a The authors demonstrated that endogenous p300 co-immuno-precipitated with IN when using a codon-optimized flag-tagged IN. HAT p300 was found to acetylate IN at residues K264, K266, and K273 within the IN C-terminus both in vitro and in vivo. The acetylation of IN increased its affinity for DNA and enhanced its ST activity without influencing the 3′-P catalytic activity in vitro. HAT GCN5 was found to mediate the acetylation of HIV-1 IN at the same C-terminal lysines as p300, and HIV-1 integration was shown to be impaired in GCN5 knockdown cells.209b
Figure 9.
Regulation of HIV-1 integration by the acetylation and deacetylation of IN.
Although casting doubt on the viral importance of HAT p300-mediated IN acetylation, a different report confirmed that the IN C-terminal lysine residues were efficiently acetylated by p300 in vitro.210 The reactivity between the IN C-terminal lysine residues and p300 was also found to be specific because two other HATs were unable to acetylate this specific IN region. In vivo acetylation of the IN C-terminal triple-arginine mutant suggests that different IN lysine residues at positions other than the CTD are also targets for acetylation by different host-cell acetyltransferases. Additionally, IN might be subject to other PTMs by as-yet-unidentified cellular cofactors during the integration process.
In general, it is believed that HAT is responsible for the acetylation of IN that appears to enhance the DNA-binding and ST activity of IN. The restriction of HIV-1 integration by KAP1 is also effective. In a proposed model, KAP1 specifically recognizes the acetylated form of IN and induces a protein complex formation with histone deacetylase (HDAC), which results in the deacetylation of IN and the reduced integration activity (Figure 9).211
Although further studies are needed to better understand the influence of the acetylation levels of IN on the enzyme activity, the inhibition of IN by inhibiting its acetylating human cofactors seems to be an attractive approach with great potential. In fact, the variability of a human enzyme (HAT in this case) is lower than that of viral enzymes, and this can help to overcome resistance problems. Targeting HAT or HDAC is an effective and safe therapeutic approach that stretches across several therapeutic fields.212
Toward Gene Therapy?
Since the discovery of the self-cleavage and ligation activity of the group I intron for the manipulation of biomolecules, research interest in catalytic nucleic acids has increased dramatically. Within the group I intron, deoxyribozymes, which are also known as DNA enzymes or DNAzymes, are single strands of DNA that are not found in nature but are endowed with catalytic activity. A wide variety of DNAzymes have been discovered via in vitro evolution to catalyze a number of chemical reactions. The major advantages of using DNAzymes instead of other antisense oligonucleotides are the simplicity of design, target-specific cleavage, serum stability, and catalytic activity of these molecules. These findings led DNAzymes to become promising candidates for drug therapy in a wide range of diseases, such as cancer and cardiovascular disorders.213
Some DNAzymes can also cleave RNA phosphodiester linkages.214 Particularly, the 10–23 DNAzyme is able to cleave various combinations of RNA substrates, on which cleavage occurs at the 3′ side of a single unpaired purine followed by a paired pyrimidine. Although its cleavage site is ubiquitous, it is not always possible for a DNAzyme to cleave every target with the same efficiency because of the intensive self-folding of the single-stranded RNA molecule in the cellular milieu.215 Therefore, the cleavage reactions operated by DNAzymes will occur at superficial target junctions.
A potential tool for HIV IN gene therapy has been recently proposed. This was based on the design and synthesis of 10–23 DNAzymes that were effective against HIV-1 IN and were designed to interfere with the replication of HIV-1 in a sequence-specific manner.216 A computational approach was used to predict the HIV-1 IN RNA secondary structure, to select the most accessible target sites, and to design and synthesize the 10–23 DNAzymes tailored to the predicted target site. The full-length HIV-IN mRNA structure was evaluated, and three DNAzymes, DIN54, DIN116, and DIN152, were considered to be ideal. The target sites demonstrate a high degree of conservation among the different strains of HIV-1217 and are less compactly folded, thus, allowing ample space for the binding and activity of the DNAzymes. DIN116 showed a significant target-specific cleavage of full-length RNA in the experiments, whereas the other two DNAzymes did not cleave the target. This was most likely because a different target site was accessible to the three DNAzymes. However, the activity, specificity, and inhibitory effects of the DNAzymes have been demonstrated. These results suggest that 10–23 DNAzymes could inhibit HIV IN gene expression in vitro, which could be a useful strategy for gene therapy.
Activating the Integrase Activity: Integrase Activators
Within each virus particle are packed approximately 30–100 molecules of IN protein. Despite this large availability of IN molecules, as well as numerous vDNA, a low number of integration events occur in each infected cell, leading to the integration of only 1–10% of the reverse-transcribed DNA into the genome of virus-infected cells.218,219 In most cases, only one to two copies of integrated vDNA molecules are present within the host DNA.218 Therefore, the majority of vDNA remains unintegrated, and the integration process for most of the free DNA is blocked.219 This has been attributed to the viral Rev protein, which was verified to inhibit IN enzymatic activity in vitro, as well as the integration process in virus-infected cells.195,220−222 In fact, Rev was demonstrated to bind IN, forming a complex that was not competent for integration in infected cells. Rev decreased integration by binding IN and inhibiting its activity. This inhibition could be ascribed to a displacement of LEDGF or a modulation of IN multimerization toward inactive tetramers.
The activity of IN-derived and Rev-derived peptides that were able to inhibit the IN–Rev interaction has been described, and some Rev-derived peptides (Rev 13–23 and Rev 53–67) were shown to bind to IN, such as Rev does, and to inhibit its enzymatic activities, as well as the HIV-1 infectivity in cells cultures.190 However, the disruption of the Rev–IN complex can activate IN enzymatic activity and, consequently, the integration process.195,220−222 The dissociation of the Rev–IN complex has been achieved by IN-derived peptides designated as INrs (IN-derived Rev-interacting peptides), which have been selected based on their specific interaction with Rev.195 The disruption of the Rev–IN complex resulted in the activation of the inhibited IN.220 More recently, a new cell-permeable IN-derived peptide was reported that, in addition to its ability to remove Rev from the inhibitory Rev–IN complex, directly activates IN enzymatic activity in vitro.223 This peptide, INS (IN stimulatory), enhances vDNA integration in virus-infected cells. Its role in integration enhancement is supported by its ability to stimulate integration events in cells infected with the ΔRev virus. The stimulatory effect of INS has been attributed to the presence of a C-terminal lysine, which has a replacement by glutamic acid that converts it to an inhibitory peptide. The multi-integration of vDNA in AIDS patients may lead to host genome instability.224 A correlation between the promotion of multi-integration and an increase in cell death was recently demonstrated.225 On the basis of these observations, a novel approach has been proposed to eradicate HIV-1 infected cells as well as to eliminate infectious virions from cultured cells by using INS or INr peptides in combination with PI.226 In fact, as expected, the addition of INS, INr peptides, or a combination of these peptides with HIV-1 infected cells significantly increased the appearance of new virions during the first 6–8 days after infection. However, after this time, a decrease in virus production was observed. Almost complete eradication of the virions was obtained when cells were infected, in the presence of the INS and the INr peptides, by a relatively high titer of the virus. This eradication is most likely due to the promotion of cell death. By addition of a PI with either the INS or the INrs peptides or with both, the increase in virus production and in viral vDNA integration was observed only during the first 2–4 days after infection. After the fourth day postinfection, a drastic reduction in both virus production and vDNA integration could be observed, reaching values below the detection levels in the presence of the mixture of inhibitors. This combination did not have an effect on noninfected cells. Therefore, it appears that cell death is promoted only in infected cells. This interesting result suggests a new and general antiviral therapy based on a novel approach, in which the death of HIV-1 infected cells is specifically promoted.
Conclusions and Perspectives
Since AIDS was first clinically identified, the scientific efforts to find good therapeutic treatments have been extraordinary. Rapidly, the first results were addressed, and AZT was introduced into clinical practice as the first rationally designed drug that was able to reduce the progress of AIDS. Further drugs have been discovered and have contributed to an increase in the anti-HIV armamentarium. HAART is based on drug cocktails that contain drugs that belong to different classes of compounds and target various steps of the HIV replication cycle. However, HAART is not a definitive cure and suffers from the problem of multidrug resistance. Therefore, additional efforts to improve therapeutic approaches are still needed.
Research to obtain compounds with new mechanisms of action led to the discovery of RAL, the first IN inhibitor for treatment of HIV infection. Recently, two IN inhibitors have been studied and are now in advanced clinical trials: EVG and DTG. These inhibitors belong to the INSTI class. DTG is considered a second generation inhibitor because it is active against RAL-resistant and EVG-resistant viral strains. There is hope that second generation IN inhibitors may prove to be relatively robust toward the problem of resistance while retaining efficacy over long periods. Despite second generation inhibitors having been developed to avoid cross-resistance with first generation drugs, second generation inhibitors bind to the same catalytic site and possess similar chemical properties as the first generation of INSTIs, leading to the potential of multiple cross-resistances. The design of a third generation of INSTIs should take into account that the main pathways of resistance toward INSTIs can act by reducing the residence time of the drugs within the IN enzyme because a possible correlation between a slow dissociation rate and the development of resistance has been shown. Therefore, the dissociation rate is an important factor that determines the quality of a drug and should be considered in the design and development of the next generation of IN inhibitors. However, the structural studies that described the structure of IN in complex with inhibitors also elucidated the mechanism of binding of these inhibitors to IN. These studies showed that there is a limited amount of chemical space available to design inhibitors against the catalytic activity of IN. These findings note that an overlap of resistance for future generation INSTIs is likely inevitable. Therefore, although a successful design of future-generation INSTIs is attainable, this could most likely result only in a temporary abatement of drug resistance. Therefore, future efforts should be focused on obtaining compounds able to block the integration process by different mechanisms of action.
An intriguing research study has recently reported using vDNA as the primary target of RAL and INSTIs. In this study, RAL was shown to bind specifically to both the unprocessed and processed LTR ends. As with other small organic molecules with chemotherapeutic activities, RAL binds directly and selectively to DNA but more specifically. This result allows for the speculation that new inhibitors, which exhibit an increased number of interactions with these highly conserved bases at the expense of interactions with the amino acid side chains of the protein active site, will have higher genetic barriers to emergence of resistance mutants.
Because patient compliance may be an issue with combinational treatments, such as HAART, and noncompliance can lead to the more rapid emergence of viral resistance, new anti-HIV agents are still urgently needed. However, the introduction of an additional drug, while increasing the armamentarium of antivirals against HIV, would not reduce the problems related to HAART: it would simply strengthen the “polypharmacy” approach to fighting HIV/AIDS. A novel and promising approach is the use of dual-action drugs, which are compounds that combine two different pharmacological actions in one similarly effective dose. A single molecule with dual activity could be better than combination therapy from both a developmental and a clinical perspective. It would also be an innovative approach in the HIV field to use a polypharmacological strategy via the use of dual inhibitors. An interesting approach that could lead toward this polypharmacological approach was recently tested by designing compounds characterized by a unique pharmacophore that are able to bind both the catalytic sites of the IN enzyme and the RNase H domain of the HIV RT.
Strategies should be pursued that target integration and are not based on binding to the catalytic triad, as they may lead to compounds that retain full activity against INSTI resistant viruses. Therefore, the development of ALLINIs should have priority. During the past few years, great efforts have moved toward the design of inhibitors with an allosteric mechanism of action. A relevant issue within this topic is that nonactive-site-binding IN inhibitors would also display synergy with current INSTIs and other antiretroviral agents in clinical use. To date, the best results in this field have been obtained through the identification of the LEDGF/p75–IN interaction as a good target for anti-HIV drug development. Inhibitors of the LEDGF–IN protein–protein interaction, such as LEDGINs, are designed by a strictly rational approach and are potential drugs that could reach clinical trials in the near future. The excellent results that led to the discovery of effective inhibitors of LEDGF/p75–IN protein–protein interactions could be very helpful to obtain compounds useful in clinical practice that circumvent the problem of resistance associated with the use of INSTIs.
An approach similar to that used for the discovery of LEDGINs could be applied to obtain molecules that inhibit the other protein–protein interactions between IN and its cofactors. IN interacts with numerous viral proteins and cellular cofactors in addition to LEDGF. Despite the excellent results obtained for the inhibition of the IN–host cofactor LEDGF interaction, which could be a unique case, targeting host proteins for therapeutic intervention could be a risky strategy because many host proteins are essential for cell viability and interfering with their natural function may have undesired toxic side effects. Therefore, a better strategy to inhibit the integration process could be to target the interaction of IN with viral proteins. Inhibiting the interaction between two viral proteins is a better strategy for antiviral intervention because no cellular function would be interrupted by inhibiting either protein. The in vivo integration process depends on a multitude of interactions of IN with various proteins. If binding to these cofactors is required for optimal IN activity, then disrupting these interactions would result in possible therapeutics with the potential to complement existing HIV-1 treatments. Relevant results have been obtained with peptides that inhibit IN–RT, −Vpr, and −Rev protein–protein interactions. Although peptides are not ideal drugs because of stability and/or bioavailability problems, the design of small synthetic peptides to interact with one of the binding partners of a protein–protein interaction is a valid starting point to facilitate the development of peptidomimetic derivatives or small molecule inhibitors. Thus far, the studies regarding a number of transport and other cofactors are still in the preliminary stages; it is too soon to consider the rapid development of other protein–protein inhibitors. However, the prospects of this field could be interesting.
Inhibitors of the LEDGF–IN interaction may possibly influence the oligomerization state of IN. Compounds that specifically influence IN oligomerization have been discovered and represent a very promising class of ALLINIs. Several independent studies have identified an inhibitor binding region at the IN dimeric interface of the CCD using chemically diverse compounds. Additionally, X-ray structural determinations of the IN allosteric region have been solved using two separate ligands. The identified inhibitor binding region at the dimeric interface is highly useful as a rational drug design platform to develop allosteric inhibitors of multimerization that may possess improved target affinity and inhibitory potency.
An indirect enzymatic inhibition can also be considered a potential future method to block the HIV integration process. In fact, IN needs to be acetylated to optimally perform its catalytic activity. Human enzymes belonging the HAT family are responsible for this acetylation. In particular the p300 and GCN5 HATs seem to be involved in the acetylation of lysine residues, which results in the activation of the IN enzyme. Small molecule HAT inhibitors could be indirect IN inhibitors. Although further studies are needed to better understand the influence of acetylation levels of IN on the enzyme activity, the inhibition of IN by inhibiting its acetylating human cofactors seems to be a promising new approach. In fact, the variability of human enzymes is lower than viral enzymes, and this reduced variability can help to overcome resistance problems. It is worthy to note that targeting HAT or HDAC is a good and safe therapeutic approach that stretches across several therapeutic fields.
Gene therapy has also been suggested, for the first time, to inhibit HIV IN activity. The activity, specificity, and inhibitory effects caused by DNAzymes, which were designed to specifically target the HIV-IN mRNA structure, have been demonstrated. Thus, the development of DNAzymes that inhibit HIV IN gene expression in vitro could be useful for a gene therapy approach.
Finally, a novel approach has been proposed to eradicate HIV-1 infected cells and to eliminate infectious virions from cultured cells by using INS or INr peptides in combination with PI. These peptides inhibit the Rev–IN interaction (INr), which results in an increase of IN activity, or directly stimulate the enzyme activity (INS). The treatment of HIV-1 infected cells with INS and INr peptides (and in combination with or without a PI) significantly increased the appearance of new virions 2–8 days after infection. However, after this time, a decrease in virus production was observed, and it was possible to obtain an almost complete eradication of virions, most likely due to the promotion of cell death. The combination of INr, INS, and PI did not have an effect on noninfected cells. Therefore, it appears that cell death is promoted only in infected cells. This result suggests that a new and general antiviral therapy could be based on this novel approach, in which the death of HIV-1 infected cells is specifically promoted.
In conclusion, a few new approaches seem to be on the horizon that could lead to the inhibition of IN activity, both through the design of more specific and effective catalytic site inhibitors that can delay the emergence of resistant strains and through ALLINIs that target the protein–protein interaction. Inhibitors of interactions of the IN–human cofactor LEDGF are in the pipeline, but the inhibitors of interactions with viral proteins can also be developed into effective drugs, including the inhibitors of IN oligomerization. Other more original approaches, such as indirect IN inhibition (targeting HAT), gene therapy, or IN stimulation to obtain the selective apoptosis of infected cells, can also produce relevant perspectives.
Acknowledgments
Thanks are due to Sandro Cosconati and Francesco Saverio Di Leva for their help in designing the figures adapted from PBD files. This work was supported by the CHAARM Project from the FP7 European Commission.
Glossary
Abbreviations Used
- AIDS
acquired immunodeficiency syndrome
- HIV
human immunodeficiency virus
- FDA
Food and Drug Administration
- NRTI
nucleoside reverse transcriptase inhibitor
- NNRTI
non-nucleoside reverse transcriptase inhibitor
- PI
protease inhibitor
- RAL
raltegravir
- IN
integrase
- INI
integrase inhibitor
- EVG
elvitegravir
- DTG
dolutegravir
- INSTI
integrase strand transfer inhibitor
- vDNA
viral DNA
- 3′-P
3′-processing
- PIC
preintegration complex
- MA
matrix
- RT
reverse transcriptase
- Vpr
viral protein R
- NC
nucleocapsid
- INI1
integrase interactor 1
- LEDGF
lens epithelium-derived growth factor
- HSP60
heat-shock protein 60
- PML
promyelocytic leukemia
- HMG
high mobility group protein
- BAF
barrier to autointegration factor
- Nup62
nucleoporin 62
- NTD
N-terminal domain
- CCD
catalytic core domain
- CTD
C-terminal domain
- PFV
prototype foamy virus
- ST
strand transfer
- FC
fold change
- RNase H
ribonuclease H
- ALLINI
allosteric integrase inhibitors
- IBD
integrase binding domain
- PTM
post-translational modification
- SAR
structure–activity relationship
- MM
molecular modeling
- SPR
surface plasmon resonance
- HAT
histone acetyltransferase
- HDAC
histone deacetylase
- INr
integrase-derived Rev-interacting peptide
- INS
integrase stimulatory
Biography
Roberto Di Santo obtained his Master’s degree in Chemistry and then in Pharmacy, both with distinction, at “Sapienza” University of Rome, Italy. He was a Fellow at Pasteur Institute of Rome, where he gained valuable expertise in heterocyclic chemistry. He became researcher and then Associate Professor of Medicinal Chemistry at “Sapienza” University of Rome. He obtained his Full Professor position at Torino University, Italy, and then was recruited to “Sapienza” University, where he established a group working on drug design and development, with particular focus on chemotherapeutic agents for treating cancer and infectious diseases. His group was one of the first to publish synthetic HIV integrase inhibitors as analogues of the polyhydroxylated natural products discovered by the Y. Pommier group at NIH.
The authors declare no competing financial interest.
References
- Gottlieb M. S.; Schroff R.; Schanker H. M; Weisman J. D.; Fan P. T; Wolf R. A.; Saxon A. Pneumocystis carinii pneumonia and mucosal candidiasis in previously healthy homosexual men: evidence of a new acquired cellular immunodeficiency. N. Engl. J. Med. 1981, 305, 1425–1431. [DOI] [PubMed] [Google Scholar]
- Masur H.; Michelis M. A.; Greene J. B.; Onorato I.; Stouwe R. A.; Holzman R. S.; Wormser G.; Brettman L.; Lange M.; Murray H. W.; Cunningham-Rundles S. An outbreak of community-acquired Pneumocystis carinii pneumonia: initial manifestation of cellular immune dysfunction. N. Engl. J. Med. 1981, 305, 1431–1438. [DOI] [PubMed] [Google Scholar]
- Siegal F. P.; Lopez C.; Hammer G. S.; Brown A. E.; Kornfeld S. J.; Gold J.; Hassett J.; Hirschman S. Z.; Cunningham-Rundles C.; Adelsberg B. R.; Parham D. M.; Siegal M.; Cunningham-Rundles S.; Armstrong D. Severe acquired immunodeficiency in male homosexuals, manifested by chronic perianal ulcerative herpes simplex lesions. N. Engl. J. Med. 1981, 305, 1439–1444. [DOI] [PubMed] [Google Scholar]
- Barton K. M.; Burch B. D.; Soriano-Sarabia N.; Margolis D. M. Prospects for treatment of latent HIV. Clin. Pharmacol. Ther. 2013, 93, 46–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen J. Therapies. Confronting the limits of success. Science 2002, 296, 2320–2324. [DOI] [PubMed] [Google Scholar]
- Meadows D. C.; Gervay-Hague J. Current developments in HIV chemotherapy. ChemMedChem 2006, 1, 16–29. [DOI] [PubMed] [Google Scholar]
- De Clercq E. New approaches toward anti-HIV chemotherapy. J. Med. Chem. 2005, 48, 1297–1313. [DOI] [PubMed] [Google Scholar]
- Yin P.; Das D.; Mitsuya H. Overcoming HIV drug resistance through rational drug design based on molecular, biochemical, and structural profiles of HIV resistance. Cell. Mol. Life Sci. 2006, 63, 1706–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamichi T. Action of anti-HIV drugs and resistance: reverse transcriptase inhibitors and protease inhibitors. Curr. Pharm. Des. 2004, 10, 4039–4053. [DOI] [PubMed] [Google Scholar]
- Meanwell N. A.; Kadow J. F. Maraviroc, a chemokine CCR5 receptor antagonist for the treatment of HIV infection and AIDS. Curr. Opin. Invest. Drugs 2007, 8, 669–681. [PubMed] [Google Scholar]
- Dayam R.; Gundla R.; Al-Mawsawi L. Q.; Neamati N. HIV-1 integrase inhibitors: 2005–2006 update. Med. Res. Rev. 2008, 28, 118–154. [DOI] [PubMed] [Google Scholar]
- Evering T. H.; Markowitz M. Raltegravir (MK-0518): an integrase inhibitor for the treatment of HIV-1. Drugs Today 2007, 43, 865–877. [DOI] [PubMed] [Google Scholar]
- Summa V.; Petrocchi A.; Bonelli F.; Crescenzi B.; Donghi M.; Ferrara M.; Fiore F.; Gardelli C.; Paz O. G.; Hazuda D. J.; Jones P.; Kinzel O.; Laufer R.; Monteagudo E.; Muraglia E.; Nizi E.; Orvieto F.; Pace P.; Pescatore G.; Scarpelli R.; Stillmock K.; Witmer M. V.; Rowley M. Discovery of raltegravir, a potent, selective orally bioavailable HIV-integrase inhibitor for the treatment of HIV-AIDS infection. J. Med. Chem. 2008, 51, 5843–5855. [DOI] [PubMed] [Google Scholar]
- a Shimura K.; Kodama E.; Sakagami Y.; Matsuzaki Y.; Watanabe W.; Yamataka K.; Watanabe Y.; Ohata Y.; Doi S.; Sato M.; Kano M.; Ikeda S.; Matsuoka M. Broad antiretroviral activity and resistance profile of the novel human immunodeficiency virus integrase inhibitor elvitegravir (JTK-303/GS-9137). J. Virol. 2008, 82, 764–774. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sato M.; Motomura T.; Aramaki H.; Matsuda T.; Yamashita M.; Ito Y.; Kawakami H.; Matsuzaki Y.; Watanabe W.; Yamataka K.; Ikeda S.; Kodama E.; Matsuoka M.; Shinkai H. Novel HIV-1 integrase inhibitors derived from quinolone antibiotics. J. Med. Chem. 2006, 49, 1506–1508. [DOI] [PubMed] [Google Scholar]; c Sato M.; Kawakami H.; Motomura T.; Aramaki H.; Matsuda T.; Yamashita M.; Ito Y.; Matsuzaki Y.; Yamataka K.; Ikeda S.; Shinkai H. Quinolone carboxylic acids as a novel monoketo acid class of human immunodeficiency virus type 1 integrase inhibitors. J. Med. Chem. 2009, 52, 4869–4882. [DOI] [PubMed] [Google Scholar]
- Katlama C.; Murphy R. Dolutegravir for the treatment of HIV. Expert Opin. Invest. Drugs 2012, 21, 523–530. [DOI] [PubMed] [Google Scholar]
- Zhu K.; Dobard C.; Chow S. A. Requirement for integrase during reverse transcription of human immunodeficiency virus type 1 and the effect of cysteine mutations of integrase on its interactions with reverse transcriptase. J. Virol. 2004, 78, 5045–5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman A.; Englund G.; Orenstein J. M.; Martin M. A.; Craigie R. Multiple effects of mutations in human immunodeficiency virus type 1 integrase on viral replication. J. Virol. 1995, 69, 2729–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison V.; Abrams H.; Roe T.; Lifson J.; Brown P. Human immunodeficiency virus integration in a cell-free system. J. Virol. 1990, 64, 2711–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bushman F. D.; Fujiwara T.; Craigie R. Retroviral DNA integration directed by HIV integration protein in vitro. Science 1990, 249, 1555–1558. [DOI] [PubMed] [Google Scholar]; b Craigie R.; Mizuuchi K.; Bushman F. D.; Engelman A. A rapid in vitro assay for HIV DNA integration. Nucleic Acids Res. 1991, 19, 2729–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M. D.; Farnet C. M.; Bushman F. D. Human immunodeficiency virus type 1 preintegration complexes: studies of organization and composition. J. Virol. 1997, 71, 5382–5390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnet C. M.; Haseltine W. A. Determination of viral proteins present in the human immunodeficiency virus type 1 preintegration complex. J. Virol. 1991, 65, 1910–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukrinsky M. I.; Sharova N.; McDonald T. L.; Pushkarskaya T.; Tarpley W. G.; Stevenson M. Association of integrase, matrix, and reverse transcriptase antigens of human immunodeficiency virus type 1 with viral nucleic acids following acute infection. Proc. Natl. Acad. Sci. U.S.A. 1993, 90, 6125–6129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzinger N. K.; Bukrinsky M. I.; Haggerty S. A.; Ragland A. M.; Kewalramani V.; Lee M.; Gendelman H. E.; Ratner L.; Stevenson M.; Emerman M. The vpr protein of human immunodeficiency virus type 1 influences nuclear localization of viral nucleic acids in nondividing host cells. Proc. Natl. Acad. Sci. U.S.A. 1994, 91, 7311–7315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Buckman J. S.; Bosche W. J.; Gorelick R. J. Human immunodeficiency virus type 1 nucleocapsid Zn(2+) fingers are required for efficient reverse transcription, initial integration processes, and protection of newly synthesized viral DNA. J. Virol. 2003, 77, 1469–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lapadat-Tapolsky M.; De Rocquigny H.; Van Gent D.; Roques B.; Plasterk R.; Darlix J. L. Interactions between HIV-1 nucleocapsid protein and viral DNA may have important functions in the viral life cycle. Nucleic Acids Res. 1993, 21, 831–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita M.; Emerman M. The cell cycle independence of HIV infections is not determined by known karyophilic viral elements. PLoS Pathog. 2005, 1, e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivière L.; Darlix J.-L.; Cimarelli A. Analysis of the viral elements required in the nuclear import of HIV-1 DNA. J. Virol. 2010, 84, 729–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turlure F.; Devroe E.; Silver P.; Engelman A. Human cell proteins and human immunodeficiency virus DNA integration. Front. Biosci. 2004, 9, 3187–3208. [DOI] [PubMed] [Google Scholar]
- Schweitzer C. J.; Jagadish T.; Haverland N.; Ciborowski P.; Belshan M. Proteomic analysis of early HIV-1 nucleoprotein complexes. J. Proteome Res. 2013, 12, 559–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai M.; Huang Y.; Caffrey M.; Zheng R.; Craigie R.; Clore G. M.; Gronenborn A. M. Solution structure of the His12→Cys mutant of the N-terminal zinc binding domain of HIV-1 integrase complexed to cadmium. Protein Sci. 1998, 7, 2669–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldgur Y.; Dyda F.; Hickman A. B.; Jenkins T. M.; Craigie R.; Davies D. R. Three new structures of the core domain of HIV-1 integrase: an active site that binds magnesium. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 9150–9154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodi P. J.; Ernst J. A.; Kuszewski J.; Hickman A. B.; Engelman A.; Craigie R.; Clore G. M.; Gronenborn A. M. Solution structure of the DNA binding domain of HIV-1 integrase. Biochemistry 1995, 34, 9826–9833. [DOI] [PubMed] [Google Scholar]
- Goldgur Y.; Craigie R.; Cohen G. H.; Fujiwara T.; Yoshinaga T.; Fujishita T.; Sugimoto H.; Endo T.; Murai H.; Davies D. R. Structure of the HIV-1 integrase catalytic domain complexed with an inhibitor: a platform for antiviral drug design. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 13040–13043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. Y.; Ling H.; Yang W.; Craigie R. Structure of a two-domain fragment of HIV-1 integrase: implications for domain organization in the intact protein. EMBO J. 2001, 20, 7333–7343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. C.; Krucinski J.; Miercke L. J.; Finer-Moore J. S.; Tang A. H.; Leavitt A. D.; Stroud R. M. Crystal structure of the HIV-1 integrase catalytic core and C-terminal domains: a model for viral DNA binding. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 8233–8238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee C. J.; Kessl J. J.; Shkriabai N.; Dar M. J.; Engelman A.; Kvaratskhelia M. Dynamic modulation of HIV-1 integrase structure and function by cellular LEDGF protein. J. Biol. Chem. 2008, 283, 31802–31812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluis-Cremer N.; Tachedjian G. Modulation of the oligomeric structures of HIV-1 retroviral enzymes by synthetic peptides and small molecules. Eur. J. Biochem. 2002, 269, 5103–5111. [DOI] [PubMed] [Google Scholar]
- Karki R. G.; Tang Y.; Burke T. R. Jr.; Nicklaus M. C. Model of full-length HIV-1 integrase complexed with viral DNA as template for anti-HIV drug design. J. Comput.-Aided Mol. Des. 2004, 18, 739–760. [DOI] [PubMed] [Google Scholar]
- Hare S.; Gupta S. S.; Valkov E.; Engelman A.; Cherepanov P. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature 2010, 464, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan L.; Li X.; Naraharisetty H. L.; Hare S.; Cherepanov P.; Engelman A. Structure-based modeling of the functional HIV-1 intasome and its inhibition. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 15910–15915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherepanov P.; Ambrosio A. L.; Rahman S.; Ellenberger T.; Engelman A. Structural basis for the recognition between HIV-1 integrase and transcriptional coactivator p75. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 17308–17313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Dyda F.; Hickman A. B.; Jenkins T. M.; Engelman A.; Craigie R.; Davies D. R. Crystal structure of the catalytic domain of HIV-1 integrase: similarity to other polynucleotidyl transferases. Science 1994, 266, 1981–1986. [DOI] [PubMed] [Google Scholar]; b Rice P. A.; Baker T. A. Comparative architecture of transposase and integrase complexes. Nat. Struct. Biol. 2001, 8, 302–307. [DOI] [PubMed] [Google Scholar]
- Beese L. S.; Steitz T. A. Structural basis for the 3′-5′ exonuclease activity of Escherichia coli DNA polymerase I: a two metal ion mechanism. EMBO J. 1991, 10, 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Steitz T. A.; Steitz J. A. A general two-metal-ion mechanism for catalytic RNA. Proc. Natl. Acad. Sci. U.S.A. 1993, 90, 6498–6502. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Steitz T. A. DNA polymerases: structural diversity and common mechanisms. J. Biol. Chem. 1999, 274, 17395–17398. [DOI] [PubMed] [Google Scholar]; c Steitz T. A. A mechanism for all polymerases. Nature 1998, 391, 231–232. [DOI] [PubMed] [Google Scholar]; d Horton N. C.; Perona J. J. Making the most of metal ions. Nat. Struct. Biol. 2001, 8, 290–293. [DOI] [PubMed] [Google Scholar]
- Engelman A.; Mizuuchi K.; Craigie R. HIV-1 DNA integration: mechanism of viral DNA cleavage and DNA strand transfer. Cell 1991, 67, 1211–1221. [DOI] [PubMed] [Google Scholar]
- a Gerton J. L.; Herschlag D.; Brown P. O. Stereospecificity of reactions catalyzed by HIV-1 integrase. J. Biol. Chem. 1999, 274, 33480–33487. [DOI] [PubMed] [Google Scholar]; b Mazumder A.; Neamati N.; Pilon A. A.; Sunder S.; Pommier Y. Chemical trapping of ternary complexes of human immunodeficiency virus type 1 integrase, divalent metal, and DNA substrates containing an abasic site. Implications for the role of lysine 136 in DNA binding. J. Biol. Chem. 1996, 271, 27330–27338. [DOI] [PubMed] [Google Scholar]
- a Marchand C.; Johnson A. A.; Karki R. G.; Pais G. C.; Zhang X.; Cowansage K.; Patel T. A.; Nicklaus M. C.; Burke T. R. Jr.; Pommier Y. Metal-dependent inhibition of HIV-1 integrase by beta-diketo acids and resistance of the soluble double-mutant (F185K/C280S). Mol. Pharmacol. 2003, 64, 600–609. [DOI] [PubMed] [Google Scholar]; b Marchand C.; Neamati N.; Pommier Y. In vitro human immunodeficiency virus type 1 integrase assays. Methods Enzymol. 2001, 340, 624–633. [DOI] [PubMed] [Google Scholar]
- Craigie R. HIV integrase, a brief overview from chemistry to therapeutics. J. Biol. Chem. 2001, 276, 23213–23216. [DOI] [PubMed] [Google Scholar]
- Pemberton I. K.; Buckle M.; Buc H. The metal ion-induced cooperative binding of HIV-1 integrase to DNA exhibits a marked preference for Mn(II) rather than Mg(II). J. Biol. Chem. 1996, 271, 1498–1506. [DOI] [PubMed] [Google Scholar]
- Nowotny M.; Yang W. Stepwise analyses of metal ions in RNase H catalysis from substrate destabilization to product release. EMBO J. 2006, 25, 1924–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W.; Lee J. Y.; Nowotny M. Making and breaking nucleic acids: two-Mg2+-ion catalysis and substrate specificity. Mol. Cell 2006, 22, 5–13. [DOI] [PubMed] [Google Scholar]
- Craigie R. HIV integrase, a brief overview from chemistry to therapeutics. J. Biol. Chem. 2001, 276, 23213–23216. [DOI] [PubMed] [Google Scholar]
- Hare S.; Maertens G. N.; Cherepanov P. 3′-Processing and strand transfer catalysed by retroviral integrase in crystallo. EMBO J. 2012, 31, 3020–3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder K. E.; Bushman F. D. Repair of gaps in retroviral DNA integration intermediates. J. Virol. 2000, 74, 11191–11200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel R.; Greger J. G.; Katz R. A.; Taganov K. D.; Wu X.; Kappes J. C.; Skalka A. M. Evidence that stable retroviral transduction and cell survival following DNA integration depend on components of the nonhomologous end joining repair pathway. J. Virol. 2004, 78, 8573–8581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carteau S.; Hoffmann C.; Bushman F. Chromosome structure and human immunodeficiency virus type 1 cDNA integration: centromeric alphoid repeats are a disfavored target. J. Virol. 1998, 72, 4005–4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder A. R.; Shinn P.; Chen H.; Berry C.; Ecker J. R.; Bushman F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell 2002, 110, 521–529. [DOI] [PubMed] [Google Scholar]
- Maurin C.; Bailly F.; Cotelle P. Structure–activity relationships of HIV-1 integrase inhibitors—enzyme–ligand interactions. Curr. Med. Chem. 2003, 10, 1795–1810. [DOI] [PubMed] [Google Scholar]
- Singh S. B.; Jayasuriya H.; Salituro G. M.; Zink D. L.; Shafiee A.; Heimbuch B.; Silverman K. C.; Lingham R. B.; Genilloud O.; Teran A.; Vilella D.; Felock P.; Hazuda D. The complestatins as HIV-1 integrase inhibitors. Efficient isolation, structure elucidation, and inhibitory activities of isocomplestatin, chloropeptin I, new complestatins, A and B, and acid-hydrolysis products of chloropeptin I. J. Nat. Prod. 2001, 64, 874–882. [DOI] [PubMed] [Google Scholar]
- Jing N.; Xu X. Rational drug design of DNA oligonucleotides as HIV inhibitors. Curr. Drug Targets: Infect. Disord. 2001, 1, 79–90. [DOI] [PubMed] [Google Scholar]
- a Fesen M. R.; Kohn K. W.; Leteurtre F.; Pommier Y. Inhibitors of human immunodeficiency virus integrase. Proc. Natl. Acad. Sci. U.S.A. 1993, 90, 2399–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Mazumder A.; Gazit A.; Levitzki A.; Nicklaus M.; Yung J.; Kohlhagen G.; Pommier Y. Effects of tyrphostins, protein kinase inhibitors, on human immunodeficiency virus type 1 integrase. Biochemistry 1995, 34, 15111–15122. [DOI] [PubMed] [Google Scholar]; c Fesen M. R.; Pommier Y.; Leteurtre F.; Hiroguchi S.; Yung J.; Kohn K. W. Inhibition of HIV-1 integrase by flavones, caffeic acid phenethyl ester (CAPE) and related compounds. Biochem. Pharmacol. 1994, 48, 595–608. [DOI] [PubMed] [Google Scholar]; d Lin Z.; Neamati N.; Zhao H.; Kiryu Y.; Turpin J. A.; Aberham C.; Strebel K.; Kohn K.; Witvrouw M.; Pannecouque C.; Debyser Z.; De Clercq E.; Rice W. G.; Pommier Y.; Burke T. R. Jr. Chicoric acid analogues as HIV-1 integrase inhibitors. J. Med. Chem. 1999, 42, 1401–1414. [DOI] [PubMed] [Google Scholar]
- a Artico M.; Di Santo R.; Costi R.; Novellino E.; Greco G.; Massa S.; Tramontano E.; Marongiu M. E.; De Montis A.; La Colla P. Geometrically and conformationally restrained cinnamoyl-compounds as inhibitors of HIV-1 integrase: synthesis, biological evaluation and molecular modeling. J. Med. Chem. 1998, 41, 3948–3960. [DOI] [PubMed] [Google Scholar]; b Di Santo R.; Costi R.; Artico M.; Tramontano E.; La Colla P.; Pani A. HIV-1 integrase inhibitors that block HIV-1 replication in infected cells. Planning synthetic derivatives from natural products. Pure Appl. Chem. 2003, 75, 195–206. [Google Scholar]; c Costi R.; Di Santo R.; Artico M.; Massa S.; Ragno R.; Loddo R.; La Colla M.; Tramomtano E.; La Colla P.; Pani A. 2,6-Bis(3,4,5-trihydroxybenzylidene) derivatives of cyclohexanone: novel potent HIV-1 integrase inhibitors that prevent HIV-1 multiplication in cell-based assays. Bioorg. Med. Chem. 2004, 12, 199–215. [DOI] [PubMed] [Google Scholar]
- Pluymers W.; Neamati N.; Pannecouque C.; Fikkert V.; Marchand C.; Burke T. R. Jr.; Pommier Y.; Schols D.; De Clercq E.; Debyser Z.; Witvrouw M. Viral entry as the primary target for the anti-HIV activity of chicoric acid and its tetra-acetyl esters. Mol. Pharmacol. 2000, 58, 641–648. [DOI] [PubMed] [Google Scholar]
- Hazuda D. J.; Felock P.; Witmer M.; Wolfe A.; Stillmock K.; Grobler J. A.; Espeseth A.; Gabryelski L.; Schleif W.; Blau C.; Miller M. D. Inhibitors of strand transfer that prevent integration and inhibit HIV-1 replication in cells. Science 2000, 287, 646–650. [DOI] [PubMed] [Google Scholar]
- Fujishita T.; Yoshinaga T.; Sato A.. Aromatic Heterocycle Compounds Having HIV Integrase Inhibiting Activities. WO2000039086, June 6, 2000.
- Fikkert V.; Van Maele B.; Vercammen J.; Hantson A.; Van Remoortel B.; Michiels M.; Gurnari C.; Pannecouque C.; De Maeyer M.; Engelborghs Y.; De Clercq E.; Debyser Z.; Witvrouw M. Development of resistance against diketo derivatives of human immunodeficiency virus type 1 by progressive accumulation of integrase mutations. J. Virol. 2003, 77, 11459–11470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karki R. G.; Pais G. C.; Zhang X.; Cowansage K.; Patel T. A.; Nicklaus M. C.; Burke T. R. Jr.; Pommier Y. Metal-dependent inhibition of HIV-1 integrase by {beta}-diketo acids and resistance of the soluble double-mutant (F185K/C280S). Mol. Pharmacol. 2003, 64, 600–609. [DOI] [PubMed] [Google Scholar]
- Johnson A. A.; Marchand C.; Patil S. S.; Costi R.; Di Santo R.; Burke T. R. Jr.; Pommier Y. Probing HIV-1 integrase inhibitor binding sites with position-specific integrase-DNA cross-linking assays. Mol. Pharmacol. 2007, 71, 893–901. [DOI] [PubMed] [Google Scholar]
- Espeseth A. S.; Felock P.; Wolfe A.; Witmer M.; Grobler J.; Anthony N.; Egbertson M.; Melamed J. Y.; Young S.; Hamill T.; Cole J. L.; Hazuda D. J. HIV-1 integrase inhibitors that compete with the target DNA substrate define a unique strand transfer conformation for integrase. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 11244–11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchand C.; Zhang X.; Pais G. C.; Cowansage K.; Neamati N.; Burke T. R. Jr.; Pommier Y. Structural determinants for HIV-1 integrase inhibition by beta-diketo acids. J. Biol. Chem. 2002, 277, 12596–12603. [DOI] [PubMed] [Google Scholar]
- Pais G. C.; Zhang X.; Marchand C.; Neamati N.; Cowansage K.; Svarovskaia E. S.; Pathak V. K.; Tang Y.; Nicklaus M. C.; Pommier Y.; Burke T. R. Jr. Structure activity of 3-aryl-1,3-diketo-containing compounds as HIV-1 integrase inhibitors. J. Med. Chem. 2002, 45, 3184–3194. [DOI] [PubMed] [Google Scholar]
- Grobler J. A.; Stillmock K. A.; Hazuda D. J. Scintillation proximity assays for mechanistic and pharmacological analyses of HIV-1 integration. Methods 2009, 47, 249–253. [DOI] [PubMed] [Google Scholar]
- a Costi R.; Di Santo R.; Artico M.; Roux A.; Ragno R.; Massa S.; Tramontano E.; La Colla M.; Loddo R.; Marongiu M. E.; Pani A.; La Colla P. 6-Aryl-2,4-dioxo-5-hexenoic acids, novel integrase inhibitors active against HIV-1 multiplication in cell-based assays. Bioorg. Med. Chem. Lett. 2004, 14, 1745–1749. [DOI] [PubMed] [Google Scholar]; b Di Santo R.; Costi R.; Artico M.; Roux A.; Ragno R.; Greco G.; Novellino E.; Marchand C.; Pommier Y. Design, synthesis and biological evaluation of heteroaryl diketohexenoic and diketobutanoic acids as HIV-1 integrase inhibitors endowed with antiretroviral activity. Farmaco 2005, 60, 409–417. [DOI] [PubMed] [Google Scholar]; c Di Santo R.; Costi R.; Roux A.; Artico M.; Lavecchia A.; Marinelli L.; Novellino E.; Palmisano L.; Andreotti M.; Amici R.; Galluzzo C. M.; Nencioni L.; Palamara A. T.; Pommier Y.; Marchand C. Novel bifunctional quinolonyl diketo acid derivatives as HIV-1 integrase inhibitors: design, synthesis, biological activities and mechanism of action. J. Med. Chem. 2006, 49, 1939–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Di Santo R.; Costi R.; Roux A.; Miele G.; Cuzzucoli Crucitti G.; Iacovo A.; Rosi F.; Lavecchia A.; Marinelli L.; Di Giovanni C.; Novellino E.; Palmisano L.; Andreotti M.; Amici R.; Galluzzo C. M.; Nencioni L.; Palamara A. T.; Pommier Y.; Marchand C. Novel quinolinonyl diketo acid derivatives as HIV-1 integrase inhibitors: design, synthesis and biological activities. J. Med. Chem. 2008, 51, 4744–4750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang L.; Wai J. S.; Embrey M. W.; Fisher T. E.; Egbertson M. S.; Payne L. S.; Guare J. P. Jr.; Vacca J. P.; Hazuda D. J.; Felock P. J.; Wolfe A. L.; Stillmock K. A.; Witmer M. V.; Moyer G.; Schleif W. A.; Gabryelski L. J.; Leonard Y. M.; Lynch J. J. Jr.; Michelson S. R.; Young S. D. Design and synthesis of 8-hydroxy-[1,6]naphthyridines as novel inhibitors of HIV-1 integrase in vitro and in infected cells. J. Med. Chem. 2003, 46, 453–456. [DOI] [PubMed] [Google Scholar]
- Guare J. P.; Wai J. S.; Gomez R. P.; Anthony N. J.; Jolly S. M.; Cortes A. R.; Vacca J. P.; Felock P. J.; Stillmock K. A.; Schleif W. A.; Moyer G.; Gabryelski L. J.; Jin L.; Chen I. W.; Hazuda D. J.; Young S. D. A series of 5-aminosubstituted 4-fluorobenzyl-8-hydroxy-[1,6]naphthyridine-7-carboxamide HIV-1 integrase inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 2900–2904. [DOI] [PubMed] [Google Scholar]
- Hazuda D. J.; Young S. D.; Guare J. P.; Anthony N. J.; Gomez R. P.; Wai J. S.; Vacca J. P.; Handt L.; Motzel S. L.; Klein H. G.; Dornadula G.; Danovich R. M.; Witmer M. V.; Wilson K. A.; Tussey L.; Schleif W. A.; Gabryelski L. S.; Jin L.; Miller M. D.; Casimiro D. R.; Emini E. A.; Shiver J. W. Integrase inhibitors and cellular immunity suppress retroviral replication in rhesus macaques. Science 2004, 305, 528–532. [DOI] [PubMed] [Google Scholar]
- Hazuda D. J.; Anthony N. J.; Gomez R. P.; Jolly S. M.; Wai J. S.; Zhuang L.; Fisher T. E.; Embrey M.; Guare J. P. Jr.; Egbertson M. S.; Vacca J. P.; Huff J. R.; Felock P. J.; Witmer M. V.; Stillmock K. A.; Danovich R.; Grobler J.; Miller M. D.; Espeseth A. S.; Jin L.; Chen I. W.; Lin J. H.; Kassahun K.; Ellis J. D.; Wong B. K.; Xu W.; Pearson P. G.; Schleif W. A.; Cortese R.; Emini E.; Summa V.; Holloway M. K.; Young S. D. A naphthyridine carboxamide provides evidence for discordant resistance between mechanistically identical inhibitors of HIV-1 integrase. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 11233–11238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garvey E. P.; Johns B. A.; Gartland M. J.; Foster S. A.; Miller W. H.; Ferris R. G.; Hazen R. J.; Underwood M. R.; Boros E. E.; Thompson J. B.; Weatherhead J. G.; Koble C. S.; Allen S. H.; Schaller L. T.; Sherrill R. G.; Yoshinaga T.; Kobayashi M.; Wakasa-Morimoto C.; Miki S.; Nakahara K.; Noshi T.; Sato A.; Fujiwara T. The naphthyridinone GSK364735 is a novel, potent human immunodeficiency virus type 1 integrase inhibitor and antiretroviral. Antimicrob. Agents Chemother. 2008, 52, 901–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Johns B. A.; Weatherhead J. G.; Allen S. H.; Thompson J. B.; Garvey E. P.; Foster S. A.; Jeffrey J. L.; Miller W. H. The use of oxadiazole and triazole substituted naphthyridines as HIV-1 integrase inhibitors. Part 1: establishing the pharmacophore. Bioorg. Med. Chem. Lett. 2009, 19, 1802–1806. [DOI] [PubMed] [Google Scholar]; b Johns B. A.; Weatherhead J. G.; Allen S. H.; Thompson J. B.; Garvey E. P.; Foster S. A.; Jeffrey J. L.; Miller W. H. 1,3,4-Oxadiazole substituted naphthyridines as HIV-1 integrase inhibitors. Part 2: SAR of the C5 position. Bioorg. Med. Chem. Lett. 2009, 19, 1807–1810. [DOI] [PubMed] [Google Scholar]
- Metobo S. E.; Jin H.; Tsiang M.; Kim C. U. Design, synthesis, and biological evaluation of novel tricyclic HIV-1 integrase inhibitors by modification of its pyridine ring. Bioorg. Med. Chem. Lett. 2006, 16, 3985–3988. [DOI] [PubMed] [Google Scholar]
- Jones G. S.; Yu F.; Zeynalzadegan A.; Hesselgesser J.; Chen X.; Chen J.; Jin H.; Kim C. U.; Wright M.; Geleziunas R.; Tsiang M. Preclinical evaluation of GS-9160, a novel inhibitor of human immunodeficiency virus type 1 integrase. Antimicrob. Agents Chemother. 2009, 53, 1194–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey K. K.; Bera S.; Zahm J.; Vora A.; Stillmock K.; Hazuda D.; Grandgenett D. P. Inhibition of human immunodeficiency virus type 1 concerted integration by strand transfer inhibitors which recognize a transient structural intermediate. J. Virol. 2007, 81, 12189–12199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrocchi A.; Koch U.; Matassa V. G.; Pacini B.; Stillmock K. A.; Summa V. From dihydroxypyrimidine carboxylic acids to carboxamide HIV-1 integrase inhibitors: SAR around the amide moiety. Bioorg. Med. Chem. Lett. 2007, 17, 350–353. [DOI] [PubMed] [Google Scholar]
- Summa V.; Petrocchi A.; Matassa V. G.; Gardelli C.; Muraglia E.; Rowley M.; Paz O. G.; Laufer R.; Monteagudo E.; Pace P. 4,5-Dihydroxypyrimidine carboxamides and N-alkyl-5-hydroxypyrimidinone carboxamides are potent, selective HIV integrase inhibitors with good pharmacokinetic profiles in preclinical species. J. Med. Chem. 2006, 49, 6646–6649. [DOI] [PubMed] [Google Scholar]
- Anker M.; Corales R. B. Raltegravir (MK-0518): a novel integrase inhibitor for the treatment of HIV infection. Expert Opin. Invest. Drugs 2008, 17, 97–103. [DOI] [PubMed] [Google Scholar]
- Grinsztejn B.; Nguyen B. Y.; Katlama C.; Gatell J. M.; Lazzarin A.; Vittecoq D.; Gonzalez C. J.; Chen J.; Harvey C. M.; Isaacs R. D. Safety and efficacy of the HIV-1 integrase inhibitor raltegravir (MK-0518) in treatment-experienced patients with multidrug-resistant virus: a phase II randomised controlled trial. Lancet 2007, 369, 1261–1269. [DOI] [PubMed] [Google Scholar]
- a Lennox J. L.; DeJesus E.; Lazzarin A.; Pollard R. B.; Madruga J. V.; Berger D. S.; Zhao J.; Xu X.; Williams-Diaz A.; Rodgers A. J.; Barnard R. J.; Miller M. D.; DiNubile M. J.; Nguyen B. Y.; Leavitt R.; Sklar P. Safety and efficacy of raltegravir-based versus efavirenz-based combination therapy in treatment-naive patients with HIV-1 infection: a multicentre, double-blind randomised controlled trial. Lancet 2009, 374, 796–806. [DOI] [PubMed] [Google Scholar]; b Markowitz M.; Nguyen B. Y.; Gotuzzo E.; Mendo F.; Ratanasuwan W.; Kovacs C.; Prada G.; Morales-Ramirez J. O.; Crumpacker C. S.; Isaacs R. D.; Gilde L. R.; Wan H.; Miller M. D.; Wenning L. A.; Teppler H. Rapid and durable antiretroviral effect of the HIV-1 integrase inhibitor raltegravir as part of combination therapy in treatment-naive patients with HIV-1 infection: results of a 48-week controlled study. J. Acquired Immune Defic. Syndr. 2007, 46, 125–133. [DOI] [PubMed] [Google Scholar]; c Steigbigel R. T.; Cooper D. A.; Kumar P. N.; Eron J. E.; Schechter M.; Markowitz M.; Loutfy M. R.; Lennox J. L.; Gatell J. M.; Rockstroh J. K.; Katlama C.; Yeni P.; Lazzarin A.; Clotet B.; Zhao J.; Chen J.; Ryan D. M.; Rhodes R. R.; Killar J. A.; Gilde L. R.; Strohmaier K. M.; Meibohm A. R.; Miller M. D.; Hazuda D. J.; Nessly M. L.; DiNubile M. J.; Isaacs R. D.; Nguyen B. Y.; Teppler H. Raltegravir with optimized background therapy for resistant HIV-1 infection. N. Eng. J. Med. 2008, 359, 339–354. [DOI] [PubMed] [Google Scholar]
- a Yazdanpanah Y.; Fagard C.; Descamps D.; Taburet A. M.; Colin C.; Roquebert B.; Katlama C.; Pialoux G.; Jacomet C.; Piketty C.; Bollens D.; Molina J. M.; Chêne G. High rate of virologic suppression with raltegravir plus etravirine and darunavir/ritonavir among treatment-experienced patients infected with multidrug-resistant HIV: results of the ANRS 139 TRIO trial. Clin. Infect. Dis. 2009, 49, 1441–1449. [DOI] [PubMed] [Google Scholar]; b Thuret I.; Chaix M. L.; Tamalet C.; Reliquet V.; Firtion G.; Tricoire J.; Rabaud C.; Frange P.; Aumaitre H.; Blanche S. Raltegravir, etravirine and darunavir combination in adolescents with multidrug-resistant virus. AIDS 2009, 23, 2364–2366. [DOI] [PubMed] [Google Scholar]; c McKinnell J. A.; Lin H. Y.; Nevin C. N.; Willig J. H.; McFarland G.; Genz M.; Raper J. L.; DeLaitsch L. L.; Mrus J. M.; Klaskala W.; Mugavero M. J.; Saag M. S. Early virologic suppression with three-class experienced patients: 24-week effectiveness in the darunavir outcomes study. AIDS 2009, 23, 1539–1546. [DOI] [PubMed] [Google Scholar]; d Imaz A.; del Saz S. V.; Ribas M. A.; Curran A.; Caballero E.; Falcó V.; Crespo M.; Ocaña I.; Diaz M.; de Gopegui E. R.; Riera M.; Ribera E. Raltegravir, etravirine, and ritonavir-boosted darunavir: a safe and successful rescue regimen for multidrug-resistant HIV-1 infection. J. Acquired Immune Defic. Syndr. 2009, 52, 382–386. [DOI] [PubMed] [Google Scholar]
- a Towner W.; Klein D.; Kerrigan H. L.; Follansbee S.; Yu K.; Horberg M. Virologic outcomes of changing enfuvirtide to raltegravir in HIV-1 patients well controlled on an enfuvirtide based regimen: 24-week results of the CHEER study. J. Acquired Immune Defic. Syndr. 2009, 51, 367–373. [DOI] [PubMed] [Google Scholar]; b De Castro N.; Braun J.; Charreau I.; Pialoux G.; Cotte L.; Katlama C.; Raffi F.; Weiss L.; Meynard J. L.; Yazdanpanah Y.; Delaugerre C.; Madelaine-Chambrin I.; Aboulker J. P.; Molina J. M. Switch from enfuvirtide to raltegravir in virologically suppressed multidrug-resistant HIV-1-infected patients: a randomized open-label trial. Clin. Infect. Dis. 2009, 49, 1259–1267. [DOI] [PubMed] [Google Scholar]; c Grant P. M.; Palmer S.; Bendavid E.; Talbot A.; Slamowitz D. C.; Cain P.; Kobayashi S. S.; Balamane M.; Zolopa A. R. Switch from enfuvirtide to raltegravir in virologically suppressed HIV-1 infected patients: effects on level of residual viremia and quality of life. J. Clin. Virol. 2009, 46, 305–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clercq E. A new drug combination therapy for treatment-naive patients with HIV-1 infection, consisting of raltegravir, emtricitabine and tenofovir disoproxil fumarate. Expert Opin. Pharmacother. 2009, 10, 2935–2937. [DOI] [PubMed] [Google Scholar]
- Buzon M. J.; Dalmau J.; Puertas M. C.; Puig J.; Clotet B.; Martinez-Picado J. The HIV-1 integrase genotype strongly predicts raltegravir susceptibility but not viral fitness of primary virus isolates. AIDS 2010, 24, 17–25. [DOI] [PubMed] [Google Scholar]
- Grobler J. A.; Stillmock K.; Miller M. D.; Hazuda D. J. Mechanism by which the HIV integrase activesite mutation N155H confers resistance to raltegravir. Antiviral Ther. 2008, 13, A41. [Google Scholar]
- a Malet I.; Delelis O.; Valantin M. A.; Montes B.; Soulie C.; Wirden M.; Tchertanov L.; Peytavin G.; Reynes J.; Mouscadet J. F.; Katlama C.; Calvez V.; Marcelin A. G. Mutations associated with failure of raltegravir treatment affect integrase sensitivity to the inhibitor in vitro. Antimicrob. Agents Chemother. 2008, 52, 1351–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Garrido C.; de Mendoza C.; Soriano V. Resistance to integrase inhibitors. Enferm. Infecc. Microbiol. Clin. 2008, 26, 40–46. [DOI] [PubMed] [Google Scholar]
- Bar-Magen T.; Donahue D. A.; McDonough E. I.; Kuhl B. D.; Faltenbacher V. H.; Xu H.; Michaud V.; Sloan R. D.; Wainberg M. A. HIV-1 subtype B and C integrase enzymes exhibit differential patterns of resistance to integrase inhibitors in biochemical assays. AIDS 2010, 24, 2171–2179. [DOI] [PubMed] [Google Scholar]
- Delelis O.; Malet I.; Na L.; Tchertanov L.; Calvez V.; Marcelin A. G.; Subra F.; Deprez E.; Mouscadet J. F. The G140S mutation in HIV integrases from raltegravir-resistant patients rescues catalytic defect due to the resistance Q148H mutation. Nucleic Acids Res. 2009, 37, 1193–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson V. A.; Brun-Vézinet F.; Clotet B.; Günthard H. F.; Kuritzkes D. R.; Pillay D.; Schapiro J. M.; Richman D. D. Update of the drug resistance mutations in HIV-1: December 2010. Top. HIV Med. 2010, 18, 156–163. [PubMed] [Google Scholar]
- Langley D. R.; Samanta H. K.; Lin Z.; Walker M. A.; Krystal M. R.; Dicker I. B. The terminal (catalytic) adenosine of the HIV LTR controls the kinetics of binding and dissociation of HIV integrase strand transfer inhibitors. Biochemistry 2008, 47, 13481–13488. [DOI] [PubMed] [Google Scholar]
- Hare S.; Vos A. M.; Clayton R. F.; Thuring J. W.; Cummings M. D.; Cherepanov P. Molecular mechanisms of retroviral integrase inhibition and the evolution of viral resistance. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 20057–20062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klibanov O. M. Elvitegravir, an oral HIV integrase inhibitor, for the potential treatment of HIV infection. Curr. Opin. Invest. Drugs 2009, 10, 190–200. [PubMed] [Google Scholar]
- a Goethals O.; Clayton R.; Van Ginderen M.; Vereycken I.; Wagemans E.; Geluykens P.; Dockx K.; Strijbos R.; Smits V.; Vos A.; Meersseman G.; Jochmans D.; Vermeire K.; Schols D.; Hallenberger S.; Hertogs K. Resistance mutations in human immunodeficiency virus type 1 integrase selected with elvitegravir confer reduced susceptibility to a wide range of integrase inhibitors. J. Virol. 2008, 82, 10366–10374. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Taiwo B.; Zheng L.; Gallien S.; Matining R. M.; Kuritzkes D. R.; Wilson C. C.; Berzins B. I.; Acosta E. P.; Bastow B.; Kim P. S.; Eron J. J. Jr. ACTG A5262 team: efficacy of a nucleoside-sparing regimen of darunavir/ritonavir plus raltegravir in treatment-naive HIV-1-infected patients (ACTG A5262). AIDS 2011, 25, 2113–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Métifiot M.; Vandegraaff N.; Maddali K.; Naumova A.; Zhang X.; Rhodes D.; Marchand C.; Pommier Y. Elvitegravir overcomes resistance to raltegravir induced by integrase mutation Y143. AIDS 2011, 25, 1175–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Shimura K.; Kodama E.; Sakagami Y.; Matsuzaki Y.; Watanabe W.; Yamataka K.; Watanabe Y.; Ohata Y.; Doi S.; Sato M.; Kano M.; Ikeda S.; Matsuoka M. Broad antiretroviral activity and resistance profile of the novel human immunodeficiency virus integrase inhibitor elvitegravir (JTK-303/GS-9137). J. Virol. 2008, 82, 764–774. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Dicker I. B.; Terry B.; Lin Z.; Li Z.; Bollini S.; Samanta H. K.; Gali V.; Walker M. A.; Krystal M. R. Biochemical analysis of HIV-1 integrase variants resistant to strand transfer inhibitors. J. Biol. Chem. 2008, 283, 23599–23609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McColl D. J.; Chen X. Strand transfer inhibitors of HIV-1 integrase: bringing in a new era of antiretroviral therapy. Antiviral Res. 2010, 85, 101–118. [DOI] [PubMed] [Google Scholar]
- Kobayashi M.; Yoshinaga T.; Seki T.; Wakasa-Morimoto C.; Brown K. W.; Ferris R.; Foster S. A.; Hazen R. J.; Miki S.; Suyama-Kagitani A.; Kawauchi-Miki S.; Taishi T.; Kawasuji T.; Johns B. A.; Underwood M. R.; Garvey E. P.; Sato A.; Fujiwara T. In vitro antiretroviral properties of S/GSK1349572, a next-generation HIV integrase inhibitor. Antimicrob. Agents Chemother. 2011, 55, 813–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Min S.; Song I.; Borland J.; Chen S.; Lou Y.; Fujiwara T.; Piscitelli S. C. Pharmacokinetics and safety of S/GSK1349572, a next-generation HIV integrase inhibitor, in healthy volunteers. Antimicrob. Agents Chemother. 2010, 54, 254–258. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Min S.; Sloan L.; Dejesus E.; Hawkins T.; McCurdy L.; Song I.; Stroder R.; Chen S.; Underwood M.; Fujiwara T.; Piscitelli S.; Lalezari J. Antiviral activity, safety, and pharmacokinetics/pharmacodynamics of dolutegravir as 10-day monotherapy in HIV-1-infected adults. AIDS 2011, 25, 1737–1745. [DOI] [PubMed] [Google Scholar]
- Quashie P. K.; Mesplède T.; Han Y. S.; Oliveira M.; Singhroy D. N.; Fujiwara T.; Underwood M. R.; Wainberg M. A. Characterization of the R263K mutation in HIV-1 integrase that confers low-level resistance to the second generation integrase strand transfer inhibitor dolutegravir. J. Virol. 2012, 86, 2696–2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eron J.; Livrozet J. M.; Morlat P.; Lazzarin A.; Katlama C.; Hawkins T.; Fujiwara T.; Cuffe R.; Vavro C.; Santiago J.; Ait-Khaled M.; Min S.; Yeo J. M. Activity of integrase inhibitor S/GSK9572 in subjects with HIV exhibiting raltegravir resistance: week 24 results of VIKING study. J. Int. AIDS Soc. 2010, 13Suppl. 4O51. [Google Scholar]
- a Sloan R. D.; Wainberg M. A. The role of unintegrated DNA in HIV infection. Retrovirology 2011, 8, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Underwood M.; Johns B.; Sato A.; Fujiwara T.; Spreen W.. S/GSK1349572: A Next Generation Integrase Inhibitor with Activity against Integrase Inhibitor-Resistant Clinical Isolates from Patients Experiencing Virologic Failure while on Raltegravir Therapy. Presented at the 5th International AIDS Society’s Conference on HIV Pathogenesis, Cape Town, South Africa, 2009. Levin, J. Conference Reports for NATAP, 2009; http://www.natap.org/2009/IAS/IAS_06.htm.
- Wiscount C. M.; Williams P. D.; Tran L. O.; Embrey M. W.; Fisher T. E.; Sherman V.; Homnick C. F.; Donnette Staas D.; Lyle T. A.; Wai J. S.; Vacca J. P.; Wang Z.; Felock P. J.; Stillmock K. A.; Witmer M. V.; Miller M. D.; Hazuda D. J.; Day A. M.; Gabryelski L. J.; Ecto L. T.; Schleif W. A.; Di Stefano D. J.; Kochansky C. J.; Anari M. R. 10-Hydroxy-7,8-dihydropyrazino[1′,2′:1,5]pyrrolo[2,3-d]pyridazine-1,9(2H,6H)-diones: potent, orally bioavailable HIV-1 integrase strand-transfer inhibitors with activity against integrase mutants. Bioorg. Med. Chem. Lett. 2008, 18, 4581–4583. [DOI] [PubMed] [Google Scholar]
- Hightower K. E.; Wang R.; Deanda F.; Johns B. A.; Weaver K.; Shen Y.; Tomberlin G. H.; Carter H. L.; Broderick T.; Sigethy S.; Seki T.; Kobayashi M.; Underwood M. R. Dolutegravir (S/GSK1349572) exhibits significantly slower dissociation than raltegravir and elvitegravir from wild-type and integrase inhibitor-resistant HIV-1 integrase-DNA complexes. Antimicrob. Agents Chemother. 2011, 55, 4552–4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascolini M. Merck offers unique perspective on second-generation integrase inhibitor. http://www.natap.org/2009/PK/PK_10.htm.
- Hare S.; Smith S. J.; Métifiot M.; Jaxa-Chamiec A.; Pommier Y.; Hughes S. H.; Cherepanov P. Structural and functional analyses of the second generation integrase strand transfer inhibitor dolutegravir (S/GSK1349572). Mol. Pharmacol. 2011, 80, 565–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Al-Mawsawi L. Q.; Al-Safi R. I.; Neamati N. Anti-infectives: clinical progress of HIV-1 integrase inhibitors. Expert Opin. Emerging Drugs 2008, 13, 213–225. [DOI] [PubMed] [Google Scholar]; b Van Wesenbeeck L.; Rondelez E.; Feyaerts M.; Verheyen A.; Van der Borght K.; Smits V.; Cleybergh C.; De Wolf H.; Van Baelen K.; Stuyver L. J. Cross-resistance profile determination of two second-generation HIV-1 integrase inhibitors using a panel of recombinant viruses derived from raltegravir treated clinical isolates. Antimicrob. Agents Chemother. 2011, 55, 321–325. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Pandey K. K.; Bera S.; Vora A. C.; Grandgenett D. P. Physical trapping of HIV-1 synaptic complex by different structural classes of integrase strand transfer inhibitors. Biochemistry 2010, 49, 8376–8387. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Goethals O.; van Ginderen M.; Vos A.; Cummings M. D.; van Der Borght K.; van Wesenbeeck L.; Feyaerts M.; Verheyen A.; Smits V.; van Loock M.; Hertogs K.; Schols D.; Clayton R. F. Resistance to raltegravir highlights integrase mutations at codon 148 in conferring cross-resistance to a second generation HIV-1 integrase inhibitor. Antiviral Res. 2011, 91, 167–176. [DOI] [PubMed] [Google Scholar]
- Bar-Magen T.; Sloan R. D.; Donahue D. A.; Kuhl B. D.; Zabeida A.; Xu H.; Oliveira M.; Hazuda D. J.; Wainberg M. A. Identification of novel mutations responsible for resistance to MK-2048, a second-generation HIV-1 integrase inhibitor. J. Virol. 2010, 84, 9210–9216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Magen T.; Sloan R. D.; Faltenbacher V. H.; Donahue D. A.; Kuhl B. D.; Oliveira M.; Xu H.; Wainberg M. A. Comparative biochemical analysis of HIV-1 subtype B and C integrase enzymes. Retrovirology 2009, 6, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delelis O.; Carayon K.; Saib A.; Deprez E.; Mouscadet J. F. Integrase and integration: biochemical activities of HIV-1 integrase. Retrovirology 2008, 5, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Highleyman L.Early studies demonstrate potent activity and safety of experimental integrase inhibitor S/GSK1265744. http://www.hivandhepatitis.com/2009icr/icaac/docs/092209_a.html.
- van Lunzen J.; Maggiolo F.; Arribas J. R.; Rakhmanova A.; Yeni P.; Young B.; Rockstroh J. K.; Almond S.; Song I.; Brothers C.; Min S. Once daily dolutegravir (S/GSK1349572) in combination therapy in antiretroviral-naive adults with HIV: planned interim 48 week results from SPRING-1, a dose-ranging, randomised, phase 2b trial. Lancet Infect. Dis. 2012, 12, 111–118. [DOI] [PubMed] [Google Scholar]
- a Enron J.; Kumar P.; Lazzarin A.; Richmond G.; Soriano V.; Huang J.; Vavro C.; Ait-Khaled M.; Min S.; Yeo J.. DTG in Subjects with HIV Exhibiting RAL Resistance: Functional Monotherapy Results of VIKING Study Cohort II. Presented at the 18th CROI, Conference on Retroviruses and Opportunistic Infections, Boston, MA, 2011; Abstract 151LB.; b Kobayashi M.; Seki T.; Yoshinaga T.; Sato A.; Fujiwara T.; Underwood M.; Johns B.. Antiviral Activity in Vitro of the INI, Dolutegravir, against Raltegravir-Resistant HIV-2 Mutants. Presented at the 19th CROI, Conference on Retroviruses and Opportunistic Infections, Seattle, WA, 2012; Abstract 691.
- a Mouscadet J. F.; Delelis O.; Marcelin A. G.; Tchertanov L. Resistance to HIV-1 integrase inhibitors: a structural perspective. Drug Resist. Updates 2010, 13, 139–150. [DOI] [PubMed] [Google Scholar]; b Blanco J. L.; Varghese V.; Rhee S. Y.; Gatell J. M.; Shafer R. W. HIV-1 integrase inhibitor resistance and its clinical implications. J. Infect. Dis. 2011, 203, 1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland R. A.; Pompliano D. L.; Meek T. D. Drug–target residence time and its implications for lead optimization. Nat. Rev. Drug Discovery 2006, 5, 730–739. [DOI] [PubMed] [Google Scholar]
- Mehellou Y.; De Clercq E. Twenty-six years of anti-HIV drug discovery: Where do we stand and where do we go?. J. Med. Chem. 2010, 53, 521–538. [DOI] [PubMed] [Google Scholar]
- Patyar S.; Prakash A.; Medhi B. Dual inhibition: a novel promising pharmacological approach for different disease conditions. J. Pharm. Pharmacol. 2011, 63, 459–471. [DOI] [PubMed] [Google Scholar]
- Apsel B.; Blair J. A.; Gonzalez B. Z.; Nazif T. M.; Feldman M. E.; Aizenstein B.; Hoffman R.; Williams R. L.; Shokat K. M.; Knight Z. A. Targeted polypharmacology: discovery of dual inhibitors of tyrosine and phosphoinositide kinases. Nat. Chem. Biol. 2008, 4, 691–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muregi F. W.; Kirira P. G.; Ishih A. Novel rational drug design strategies with potential to revolutionize malaria chemotherapy. Curr. Med. Chem. 2011, 18, 113–143. [DOI] [PubMed] [Google Scholar]
- a Wang Z.; Bennett E. M.; Wilson D. J.; Salomon C.; Vince R. Rationally designed dual inhibitors of HIV reverse transcriptase and integrase. J. Med. Chem. 2007, 50, 3416–3419. [DOI] [PubMed] [Google Scholar]; b Wang Z.; Vince R. Synthesis of pyrimidine and quinolone conjugates as a scaffold for dual inhibitors of HIV reverse transcriptase and integrase. Bioorg. Med. Chem. Lett. 2008, 18, 1293–1296. [DOI] [PubMed] [Google Scholar]; c Wang Z.; Vince R. Design and synthesis of dual inhibitors of HIV reverse transcriptase and integrase: introducing a diketoacid functionality into delavirdine. Bioorg. Med. Chem. 2008, 16, 3587–3595. [DOI] [PubMed] [Google Scholar]
- a Kaushik N.; Rege N.; Yadav P. N.; Sarafianos S. G.; Modak M. J.; Pandey V. N. Biochemical analysis of catalytically crucial aspartate mutants of human immunodeficiency virus type 1 reverse transcriptase. Biochemistry 1996, 35, 11536–11546. [DOI] [PubMed] [Google Scholar]; b Patel H. P.; Jacobo-Molina A.; Ding J.; Tantillo C.; Clark A. D.; Raag R.; Nanni R. G.; Hughes S. H.; Arnold E. Insights into DNA polymerization mechanisms from structure and function analysis of HIV-1 reverse transcriptase. Biochemistry 1995, 34, 5351–5363. [DOI] [PubMed] [Google Scholar]
- Di Santo R. Diketo acids derivatives as dual inhibitors of human immunodeficiency virus type 1 integrase and the reverse transcriptase RNase H domain. Curr. Med. Chem. 2011, 18, 3335–3342. [DOI] [PubMed] [Google Scholar]
- a Billamboz M.; Bailly F.; Barreca M. L.; De Luca L.; Mouscadet J. F.; Calmels C.; Andréola M. L.; Witvrouw M.; Christ F.; Debyser Z.; Cotelle P. Design, synthesis, and biological evaluation of a series of 2-hydroxyisoquinoline-1,3(2H,4H)-diones as dual inhibitors of human immunodeficiency virus type 1 integrase and the reverse transcriptase RNase H domain. J. Med. Chem. 2008, 51, 7717–7730. [DOI] [PubMed] [Google Scholar]; b Billamboz M.; Bailly F.; Lion C.; Calmels C.; Andréola M. L.; Witvrouw M.; Christ F.; Debyser Z.; De Luca L.; Chimirri A.; Cotelle P. 2-Hydroxyisoquinoline-1,3(2H,4H)-diones as inhibitors of HIV-1 integrase and reverse transcriptase RNase H domain: influence of the alkylation of position 4. Eur. J. Med. Chem. 2011, 46, 535–546. [DOI] [PubMed] [Google Scholar]; c Marchand C.; Beutler J. A.; Wamiru A.; Budihas S.; Möllmann U.; Heinisch L.; Mellors J. W.; Le Grice S. F.; Pommier Y. Madurahydroxylactone derivatives as dual inhibitors of human immunodeficiency virus type 1 integrase and RNase H. Antimicrob. Agents Chemother. 2008, 52, 361–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tramontano E.; Esposito F.; Badas R.; Di Santo R.; Costi R.; La Colla P. 6-[1-(4-Fluorophenyl)methyl-1H-pyrrol-2-yl)]-2,4-dioxo-5-hexenoic acid ethyl ester a novel diketo acid derivative which selectively inhibits the HIV-1 viral replication in cell culture and the ribonuclease H activity in vitro. Antiviral Res. 2005, 65, 117–124. [DOI] [PubMed] [Google Scholar]
- Shaw-Reid C. A.; Munshi V.; Graham P.; Wolfe A.; Witmer M.; Danzeisen R.; Olsen D. B.; Carroll S. S.; Embrey M.; Wai J. S.; Miller M. D.; Cole J. L.; Hazuda D. J. Inhibition of HIV-1 ribonuclease H by a novel diketo acid, 4-[5-(benzoylamino)thien-2-yl]-2,4-dioxobutanoic acid. J. Biol. Chem. 2003, 278, 2777–2780. [DOI] [PubMed] [Google Scholar]
- Ammar F. F.; Abdel-Azeim S.; Zargarian L.; Hobaika Z.; Maroun R. G.; Fermandjian S. Unprocessed viral DNA could be the primary target of the HIV-1 integrase inhibitor raltegravir. PLoS One 2012, 7, e40223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Scottoline B. P.; Chow S.; Ellison V.; Brown P. O. Disruption of the terminal base pairs of retroviral DNA during integration. Genes Dev. 1997, 11, 371–382. [DOI] [PubMed] [Google Scholar]; b Katz R. A.; Merkel G.; Andrake M. D.; Roder H.; Skalka A. M. Retroviral integrases promote fraying of viral DNA ends. J. Biol. Chem. 2011, 286, 25710–25718. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Vink C.; van Gent D. C.; Elgersma Y.; Plasterk R. H. Human immunodeficiency virus integrase protein requires a subterminal position of its viral DNA recognition sequence for efficient cleavage. J. Virol. 1991, 65, 4636–4644. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Vink C.; Yeheskiely E.; van der Marel G. A.; van Boom J. H.; Plasterk R. H. Site-specific hydrolysis and alcoholysis of human immunodeficiency virus DNA termini mediated by the viral integrase protein. Nucleic Acids Res. 1991, 19, 6691–6698. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Agapkina J.; Smolov M.; Zubin E.; Mouscadet J. F.; Gottikh M. HIV-1 integrase can process a 39-end crosslinked substrate. Eur. J. Biochem. 2004, 271, 205–211. [DOI] [PubMed] [Google Scholar]
- Suzuki Y.; Craigie R. The road to chromatin—nuclear entry of retroviruses. Nat. Rev. Microbiol. 2007, 5, 187–196. [DOI] [PubMed] [Google Scholar]
- Turlure F.; Devroe E.; Silver P. A.; Engelman A. Human cell proteins and human immunodeficiency virus DNA integration. Front. Biosci. 2004, 9, 3187–3208. [DOI] [PubMed] [Google Scholar]
- Masuda T. Non-enzymatic functions of retroviral integrase: the next target for novel anti-HIV drug development. Front. Microbiol. 2011, 2, 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Fassati A.; Gorlich D.; Harrison I.; Zaytseva L.; Mingot J. M. Nuclear import of HIV-1 intracellular reverse transcription complexes is mediated by importin 7. EMBO J. 2003, 22, 3675–3685. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ao Z.; Huang G.; Yao H.; Xu Z.; Labine M.; Cochrane A. W.; Yao X. Interaction of human immunodeficiency virus type 1 integrase with cellular nuclear import receptor importin 7 and its impact on viral replication. J. Biol. Chem. 2007, 282, 13456–13467. [DOI] [PubMed] [Google Scholar]; c Christ F.; Thys W.; De Rijck J.; Gijsbers R.; Albanese A.; Arosio D.; Emiliani S.; Rain J. C.; Benarous R.; Cereseto A.; Debyser Z. Transportin-SR2 imports HIV into the nucleus. Curr. Biol. 2008, 18, 1192–1202. [DOI] [PubMed] [Google Scholar]
- Woodward C. L.; Prakobwanakit S.; Mosessian S.; Chow S. A. Integrase interacts with nucleoporin NUP153 to mediate the nuclear import of human immunodeficiency virus type 1. J. Virol. 2009, 83, 6522–6533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Maele B.; Busschots K.; Vandekerckhove L.; Christ F.; Debyser Z. Cellular co-factors of HIV-1 integration. Trends Biochem. Sci. 2006, 31, 98–105. [DOI] [PubMed] [Google Scholar]
- Hindmarsh P.; Ridky T.; Reeves R.; Andrake M.; Skalka A. M.; Leis J. HMG protein family members stimulate human immunodeficiency virus type 1 and avian sarcoma virus concerted DNA integration in vitro. J. Virol. 1999, 73, 2994–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnet C.; Bushman F. D. HIV-1 cDNA integration: requirement of HMG I (Y) protein for function of preintegration complexes in vitro. Cell 1997, 88, 483–492. [DOI] [PubMed] [Google Scholar]
- Kalpana G. V.; Marmon S.; Wang W.; Crabtree G. R.; Goff S. P. Binding and stimulation of HIV-1 integrase by a human homolog of yeast transcription factor SNF5. Science 1994, 266, 2002–2006. [DOI] [PubMed] [Google Scholar]
- Cherepanov P.; Maertens G.; Proost P.; Devreese B.; Van Beeumen J.; Engelborghs Y.; De Clercq E.; Debyser Z. HIV-1 integrase forms stable tetramers and associates with LEDGF/p75 protein in human cells. J. Biol. Chem. 2002, 278, 372–381. [DOI] [PubMed] [Google Scholar]
- Violot S.; Hong S. S.; Rakotobe D.; Petit C.; Gay B.; Moreau K.; Billaud G.; Priet S.; Sire J.; Schwartz O.; Mouscadet J. F.; Boulanger P. The human polycomb group EED protein interacts with the integrase of human immunodeficiency virus type 1. J. Virol. 2003, 77, 12507–12522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Al-Mawsawi L. Q.; Neamati N. Blocking interactions between HIV-1 integrase and cellular cofactors: an emerging anti-retroviral strategy. Trends Pharmacol. Sci. 2007, 28, 526–535. [DOI] [PubMed] [Google Scholar]; b Goff S. P. Host factors exploited by retroviruses. Nat. Rev. Microbiol. 2007, 5, 253–263. [DOI] [PubMed] [Google Scholar]; c Studamire B.; Goff S. P. Interactions of host proteins with the murine leukemia virus integrase. Viruses 2010, 2, 1110–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung E.; Sorin M.; Wang E. J.; Perumal S.; Ott D.; Kalpana G. V. Specificity of interaction of INI1/hSNF5 with retroviral integrases and its functional significance. J. Virol. 2004, 78, 2222–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh D. P.; Ohguro N.; Kikuchi T.; Sueno T.; Reddy V. N.; Yuge K.; Chylack L. T. Jr.; Shinohara T. Lens epithelium-derived growth factor: effects on growth and survival of lens epithelial cells, keratinocytes, and fibroblasts. Biochem. Biophys. Res. Commun. 2000, 267, 373–381. [DOI] [PubMed] [Google Scholar]
- Engelman A.; Cherepanov P. The lentiviral integrase binding protein LEDGF/p75 and HIV-1 replication. PLoS Pathog. 2008, 4, e1000046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherepanov P.; Maertens G.; Proost P.; Devreese B.; Van Beeumen J.; Engelborghs Y.; DeClercq E.; Debyser Z. HIV-1 integrase forms stable tetramers and associates with LEDGF/p75 protein in human cells. J. Biol. Chem. 2003, 278, 372–381. [DOI] [PubMed] [Google Scholar]
- Cherepanov P.; Devroe E.; Silver P. A.; Engelman A. Identification of an evolutionarily conserved domain in human lens epithelium-derived growth factor/transcriptional co-activator p75 (LEDGF/p75) that binds HIV-1integrase. J. Biol. Chem. 2004, 279, 48883–48892. [DOI] [PubMed] [Google Scholar]
- Llano M.; Saenz D. T.; Meehan A.; Wongthida P.; Peretz M.; Walker W. H.; Teo W.; Poeschla E. M. An essential role for LEDGF/p75 in HIV integration. Science 2006, 314, 461–464. [DOI] [PubMed] [Google Scholar]
- Shun M. C.; Raghavendra N. K.; Vandegraaff N.; Daigle J. E.; Hughes S.; Kellam P.; Cherepanov P.; Engelman A. LEDGF/p75 functions downstream from preintegration complex formation to effect gene-specific HIV-1 integration. Genes Dev. 2007, 21, 1767–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busschots K.; Voet A.; De Maeyer M.; Rain J. C.; Emiliani S.; Benarous R.; Desender L.; Debyser Z.; Christ F. Identification of the LEDGF/p75 binding site in HIV-1 integrase. J. Mol. Biol. 2007, 365, 1480–1492. [DOI] [PubMed] [Google Scholar]
- Emiliani S.; Mousnier A.; Busschots K.; Maroun M.; Van Maele B.; Tempe D.; Vandekerckhove L.; Moisant F.; Ben-Slama L.; Witvrouw M.; Christ F.; Rain J. C.; Dargemont C.; Debyser Z.; Benarous R. Integrase mutants defective for interaction with LEDGF/p75 are impaired in chromosome tethering and HIV-1 replication. J. Biol. Chem. 2005, 280, 25517–25523. [DOI] [PubMed] [Google Scholar]
- Hombrouck A.; De Rijck J.; Hendrix J.; Vandekerckhove L.; Voet A.; De Maeyer M.; Witvrouw M.; Engelborghs Y.; Christ F.; Gijsbers R.; Debyser Z. Virus evolution reveals an exclusive role for LEDGF/p75 in chromosomal tethering of HIV. PLoS Pathog. 2007, 3, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherepanov P.; Sun Z. Y.; Rahman S.; Maertens G.; Wagner G.; Engelman A. Solution structure of the HIV-1 integrase-binding domain in LEDGF/ p75. Nat. Struct. Mol. Biol. 2005, 12, 526–532. [DOI] [PubMed] [Google Scholar]
- Schrijvers R.; De Rijck J.; Demeulemeester J.; Adachi N.; Vets S.; Ronen K.; Christ F.; Bushman F. D.; Debyser Z.; Gijsbers R. LEDGF/p75-independent HIV- 1 replication demonstrates a role for HRP-2 and remains sensitive to inhibition by LEDGINs. PLoS Pathog. 2012, 8, e1002558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandekerckhove L.; Christ F.; Van Maele B.; De Rijck J.; Gijsbers R.; Van den Haute C.; Witvrouw M.; Debyser Z. Transient and stable knockdown of the integrase cofactor LEDGF/p75 reveals its role in the replication cycle of human immunodeficiency virus. J. Virol. 2006, 80, 1886–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rijck J.; Vandekerckhove L.; Gijsbers R.; Hombrouck A.; Hendrix J.; Vercammen J.; Engelborghs Y.; Christ F.; Debyser Z. Overexpression of the lens epithelium-derived growth factor/p75 integrase binding domain inhibits human immunodeficiency virus replication. J. Virol. 2006, 80, 11498–11509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Mawsawi L. Q.; Christ F.; Dayam R.; Debyser Z.; Neamati N. Inhibitory profile of a LEDGF/p75 peptide against HIV-1 integrase: insight into integrase-DNA complex formation and catalysis. FEBS Lett. 2008, 582, 1425–1430. [DOI] [PubMed] [Google Scholar]
- Hayouka Z.; Rosenbluh J.; Levin A.; Loya S.; Lebendiker M.; Veprintsev D.; Kotler M.; Hizi A.; Loyter A.; Friedler A. Inhibiting HIV-1 integrase by shifting its oligomerization equilibrium. Proc. Nat. Acad. Sci. U.S.A. 2007, 104, 8316–8321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayouka Z.; Levin A.; Maes M.; Hadas E.; Shalev D. E.; Volsky D. J.; Loyter A.; Friedler A. Mechanism of action of the HIV-1 integrase inhibitory peptide LEDGF 361–370. Biochem. Biophys. Res. Commun. 2010, 394, 260–265. [DOI] [PubMed] [Google Scholar]
- Hayouka Z.; Hurevich M.; Levin A.; Benyamini H.; Iosub A.; Maes M.; Shalev D. E.; Loyter A.; Gilon C.; Friedler A. Cyclic peptide inhibitors of HIV-1 integrase derived from the LEDGF/p75 protein. Bioorg. Med. Chem. 2010, 18, 8388–8395. [DOI] [PubMed] [Google Scholar]
- Rhodes D. I.; Peat T. S.; Vandegraaff N.; Jeevarajah D.; Newman J.; Martyn J.; Coates J. A.; Ede N. J.; Rea P.; Deadman J. J. Crystal structures of novel allosteric peptide inhibitors of HIV integrase identify new interactions at the LEDGF binding site. ChemBioChem 2011, 12, 2311–2315. [DOI] [PubMed] [Google Scholar]
- Desimmie B. A.; Humbert M.; Lescrinier E.; Hendrix J.; Vets S.; Gijsbers R.; Ruprecht R. M.; Dietrich U.; Debyser Z.; Christ F. Phage display-directed discovery of LEDGF/p75 binding cyclic peptide inhibitors of HIV replication. Mol. Ther. 2012, 20, 2064–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bartholomeeusen K.; De Rijck J.; Busschots K.; Desender L.; Gijsbers R.; Emiliani S.; Benarous R.; Debyser Z.; Christ F. Differential interaction of HIV-1 integrase and JPO2 with the C terminus of LEDGF/p75. J. Mol. Biol. 2007, 372, 407–421. [DOI] [PubMed] [Google Scholar]; b Bartholomeeusen K.; Gijsbers R.; Christ F.; Hendrix J.; Rain J. C.; Emiliani S.; Benarous R.; Debyser Z.; De Rijck J. Lens epithelium derived growth factor/p75 interacts with the transposase derived DDE domain of PogZ. J. Biol. Chem. 2009, 284, 11467–11477. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Maertens G. N.; Cherepanov P.; Engelman A. Transcriptional co-activator p75 binds and tethers the Myc-interacting protein JPO2 to chromatin. J. Cell Sci. 2006, 119, 2563–2571. [DOI] [PubMed] [Google Scholar]
- Du L.; Zhao Y.; Chen J.; Yang L.; Zheng Y.; Tang Y.; Shen X.; Jiang H. D77, one benzoic acid derivative, functions as a novel anti-HIV-1 inhibitor targeting the interaction between integrase and cellular LEDGF/p75. Biochem. Biophys. Res. Commun. 2008, 375, 139–144. [DOI] [PubMed] [Google Scholar]
- De Luca L.; Barreca M. L.; Ferro S.; Christ F.; Iraci N.; Gitto R.; Monforte A. M.; Debyser Z.; Chimirri A. Pharmacophore-based discovery of small-molecule inhibitors of protein–protein interactions between HIV-1 integrase and cellular cofactor LEDGF/p75. Chem MedChem 2009, 4, 1311–1316. [DOI] [PubMed] [Google Scholar]
- De Luca L.; Ferro S.; Gitto R.; Barreca M. L.; Agnello S.; Christ F.; Debyser Z.; Chimirri A. Small molecules targeting the interaction between HIV-1 integrase and LEDGF/p75 cofactor. Bioorg. Med. Chem. 2010, 18, 7515–7521. [DOI] [PubMed] [Google Scholar]
- Fan X.; Zhang F. H.; Al-Safi R. I.; Zeng L. F.; Shabaik Y.; Debnath B.; Sanchez T. W.; Odde S.; Neamati N.; Long Y. Q. Design of HIV-1 integrase inhibitors targeting the catalytic domain as well as its interaction with LEDGF/p75: a scaffold hopping approach using salicylate and catechol groups. Bioorg. Med. Chem. 2011, 19, 4935–4952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peat T. S.; Rhodes D. I.; Vandegraaff N.; Le G.; Smith J. A.; Clark L. J.; Jones E. D.; Coates J. A.; Thienthong N.; Newman J.; Dolezal O.; Mulder R.; Ryan J. H.; Savage G. P.; Francis C. L.; Deadman J. J. Small molecule inhibitors of the LEDGF site of human immunodeficiency virus integrase identified by fragment screening and structure based design. PLoS One 2012, 7, e40147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G.; Li X.; Sun X.; Lu W.; Liu G.; Huang J.; Shen X.; Tang Y. Identification of old drugs as potential inhibitors of HIV-1 integrase—human LEDGF/p75 interaction via molecular docking. J. Mol. Model. 2012, 18, 4995–5003. [DOI] [PubMed] [Google Scholar]
- Christ F.; Voet A.; Marchand A.; Nicolet S.; Desimmie B. A.; Marchand D.; Bardiot D.; Van der Veken N. J.; Van Remoortel B.; Strelkov S. V.; De Maeyer M.; Chaltin P.; Debyser Z. Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nat. Chem. Biol. 2010, 6, 442–448. [DOI] [PubMed] [Google Scholar]
- Christ F.; Shaw S.; Demeulemeester J.; Desimmie B. A.; Marchand A.; Butler S.; Smets W.; Chaltin P.; Westby M.; Debyser Z.; Pickford C. Small-molecule inhibitors of the LEDGF/p75 binding site of integrase block HIV replication and modulate integrase multimerization. Antimicrob. Agents Chemother. 2012, 56, 4365–4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsantrizos Y. S.; Boes M.; Brochu C.; Fenwick C.; Malenfant E.; Mason S.; Pesant M.. Inhibitors of Human Immunodeficiency Virus Replication. PCT Int. Appl. WO 131350, 2007.
- Kessl J. J.; Jena N.; Koh Y.; Taskent-Sezgin H.; Slaughter A.; Feng L.; de Silva S.; Wu L.; Le Grice S. F.; Engelman A.; Fuchs J. R.; Kvaratskhelia M. A multimode, cooperative mechanism of action of allosteric HIV-1 integrase inhibitors. J. Biol. Chem. 2012, 287, 16801–16811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsiang M.; Jones G. S.; Niedziela-Majka A.; Kan E.; Lansdon E. B.; Huang W.; Hung M.; Samuel D.; Novikov N.; Xu Y.; Mitchell M.; Guo H.; Babaoglu K.; Liu X.; Geleziunas R.; Sakowicz R. New class of HIV-1 integrase (IN) inhibitors with a dual mode of action. J. Biol. Chem. 2012, 287, 21189–21203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessl J. J.; Li M.; Ignatov M.; Shkriabai N.; Eidahl J. O.; Feng L.; Musier-Forsyth K.; Craigie R.; Kvaratskhelia M. FRET analysis reveals distinct conformations of IN tetramers in the presence of viral DNA or LEDGF/p75. Nucleic Acids Res. 2011, 39, 9009–9022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Luca L.; Gitto R.; Christ F.; Ferro S.; De Grazia S.; Morreale F.; Debyser Z.; Chimirri A. 4-[1-(4-Fluorobenzyl)-4-hydroxy-1H-indol-3-yl]-2-hydroxy-4-oxobut-2-enoic acid as a prototype to develop dual inhibitors of HIV-1 integration process. Antiviral Res. 2011, 92, 102–107. [DOI] [PubMed] [Google Scholar]
- Tasara T.; Maga G.; Hottiger M. O.; Hubscher U. HIV-1 reverse transcriptase and integrase enzymes physically interact and inhibit each other. FEBS Lett. 2001, 507, 39–44. [DOI] [PubMed] [Google Scholar]
- Wilkinson T. A.; Januszyk K.; Phillips M. L.; Tekeste S. S.; Zhang M.; Miller J. T.; Le Grice S. F.; Clubb R. T.; Chow S. A. Identifying and characterizing a functional HIV-1 reverse transcriptase-binding site on integrase. J. Biol. Chem. 2009, 284, 7931–7939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oz I.; Avidan O.; Hizi A. Inhibition of the integrases of human immunodeficiency viruses type 1 and type 2 by reverse transcriptases. Biochem. J. 2002, 361, 557–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oz-Gleenberg I.; Avidan O.; Goldgur Y.; Herschhorn A.; Hizi A. Peptides derived from the reverse transcriptase of human immunodeficiency virus type 1 as novel inhibitors of the viral integrase. J. Biol. Chem. 2005, 280, 21987–21996. [DOI] [PubMed] [Google Scholar]
- Zawahir Z.; Neamati N. Inhibition of HIV-1 integrase activity by synthetic peptides derived from the HIV-1 HXB2 Pol region of the viral genome. Bioorg. Med. Chem. Lett. 2006, 16, 5199–5202. [DOI] [PubMed] [Google Scholar]
- Kogan M.; Rappaport J. HIV-1 accessory protein Vpr: relevance in the pathogenesis of HIV and potential for therapeutic intervention. Retrovirology 2011, 8, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischerour J.; Tauc P.; Leh H.; de Rocquigny H.; Roques B.; Mouscadet J. F. The (52–96) C-terminal domain of Vpr stimulates HIV-1 IN-mediated homologous strand transfer of mini-viral DNA. Nucleic Acids Res. 2003, 31, 2694–2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleenberg I. O.; Herschhorn A.; Hizi A. Inhibition of the activities of reverse transcriptase and integrase of human immunodeficiency virus type-1 by peptides derived from the homologous viral protein R (Vpr). J. Mol. Biol. 2007, 369, 1230–1243. [DOI] [PubMed] [Google Scholar]
- Morellet N.; Bouaziz S.; Petitjean P.; Roques B. P. NMR structure of the HIV-1 regulatory protein VPR. J. Mol. Biol. 2003, 327, 215–227. [DOI] [PubMed] [Google Scholar]
- Suzuki S.; Urano E.; Hashimoto C.; Tsutsumi H.; Nakahara T.; Tanaka T.; Nakanishi Y.; Maddali K.; Han Y.; Hamatake M. Peptide HIV-1 integrase inhibitors from HIV-1 gene products. J. Med. Chem. 2010, 53, 5356–5360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S.; Maddali K.; Hashimoto C.; Urano E.; Ohashi N.; Tanaka T.; Ozaki T.; Arai H.; Tsutsumi H.; Narumi T.; Nomura W.; Yamamoto N.; Pommier Y.; Komano J. A.; Tamamura H. Peptidic HIV integrase inhibitors derived from HIV gene products: structure–activity relationship studies. Bioorg. Med. Chem. 2010, 18, 6771–6775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbluh J.; Hayouka Z.; Loya S.; Levin A.; Armon-Omer A.; Britan E.; Hizi A.; Kotler M.; Friedler A.; Loyter A. Interaction between HIV-1 Rev and integrase proteins: a basis for the development of anti-HIV peptides. J. Biol. Chem. 2007, 282, 15743–15753. [DOI] [PubMed] [Google Scholar]
- Grewe B.; Uberla K. The human immunodeficiency virus type 1 Rev protein: menage a trois during the early phase of the lentiviral replication cycle. J. Gen. Virol. 2010, 91, 1893–1897. [DOI] [PubMed] [Google Scholar]
- Levin A.; Hayouka Z.; Brack-Werner R.; Volsky D. J.; Friedler A.; Loyter A. Novel regulation of HIV-1 replication and pathogenicity: rev inhibition of integration. Protein Eng., Des. Sel. 2009, 22, 753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benyamini H.; Loyter A.; Friedler A. A structural model of the HIV-1 Rev-integrase complex: the molecular basis of integrase regulation by Rev. Biochem. Biophys. Res. Commun. 2011, 416, 252–257. [DOI] [PubMed] [Google Scholar]
- Hayouka Z.; Rosenbluh J.; Levin A.; Maes M.; Loyter A.; Friedler A. Peptides derived from HIV-1 Rev inhibit HIV-1 integrase in a shiftide mechanism. Biopolymers 2008, 90, 481–487. [DOI] [PubMed] [Google Scholar]
- Levin A.; Hayouka Z.; Helfer M.; Brack-Werner R.; Friedler A.; Loyter A. Peptides derived from HIV-1 integrase that bind Rev stimulate viral genome integration. PLoS One 2009, 4, e4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroun R. G.; Gayet S.; Benleulmi M. S.; Porumb H.; Zargarian L.; Merad H.; Leh H.; Mouscadet J. F.; Troalen F.; Fermandjian S. Peptide inhibitors of HIV-1 integrase dissociate the enzyme oligomers. Biochemistry 2001, 40, 13840–13848. [DOI] [PubMed] [Google Scholar]
- Zhao L.; O’Reilly M. K.; Shultz M. D.; Chmielewski J. Interfacial peptide inhibitors of HIV-1 integrase activity and dimerization. Bioorg. Med. Chem. Lett. 2003, 13, 1175–1177. [DOI] [PubMed] [Google Scholar]
- Zhao L.; Chmielewski J. Inhibition of HIV-1 integrase dimerization and activity with crosslinked interfacial peptide. Bioorg. Med. Chem. 2013, 21, 4041–4044. [DOI] [PubMed] [Google Scholar]
- a Al-Mawsawi L. Q.; Fikkert V.; Dayam R.; Witvrouw M.; Burke T. R. Jr.; Borchers C. H.; Neamati N. Discovery of a small-molecule HIV-1 integrase inhibitor-binding site. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 10080–10085. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Tsiang M.; Jones G. S.; Hung M.; Mukund S.; Han B.; Liu X.; Babaoglu K.; Lansdon E.; Chen X.; Todd J.; Cai T.; Pagratis N.; Sakowicz R.; Geleziunas R. Affinities between the binding partners of the HIV-1 integrase dimer-lens epithelium-derived growth factor (IN dimer-LEDGF) complex. J. Biol. Chem. 2009, 284, 33580–33599. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Tsiang M.; Jones G. S.; Hung M.; Samuel D.; Novikov N.; Mukund S.; Brendza K. M.; Niedziela-Majka A.; Jin D.; Liu X.; Mitchell M.; Sakowicz R.; Geleziunas R. Dithiothreitol causes HIV-1 integrase dimer dissociation while agents interacting with the integrase dimer interface promote dimer formation. Biochemistry 2011, 50, 1567–1581. [DOI] [PubMed] [Google Scholar]
- Shkriabai N.; Patil S. S.; Hess S.; Budihas S. R.; Craigie R.; Burke T. R. Jr.; Le Grice S. F.; Kvaratskhelia M. Identification of an inhibitor-binding site to HIV-1 integrase with affinity acetylation and mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 6894–6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessl J. J.; Eidahl J. O.; Shkriabai N.; Zhao Z.; McKee C. J.; Hess S.; Burke T. R. Jr.; Kvaratskhelia M. An allosteric mechanism for inhibiting HIV-1 integrase with a small molecule. Mol. Pharmacol. 2009, 76, 824–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du L.; Zhao Y. X.; Yang L. M.; Zheng Y. T.; Tang Y.; Shen X.; Jiang H. L. Symmetrical 1-pyrrolidineacetamide showing anti-HIV activity through a new binding site on HIV-1 integrase. Acta Pharmacol. Sin. 2008, 29, 1261–1267. [DOI] [PubMed] [Google Scholar]
- Tintori C.; Demeulemeester J.; Franchi L.; Massa S.; Debyser Z.; Christ F.; Botta M. Discovery of small molecule HIV-1 integrase dimerization inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 3109–3114. [DOI] [PubMed] [Google Scholar]
- Demeulemeester J.; Tintori C.; Botta M.; Debyser Z.; Christ F. Development of an AlphaScreen-based HIV-1 integrase dimerization assay for discovery of novel allosteric inhibitors. J. Biomol. Screening 2012, 17, 618–628. [DOI] [PubMed] [Google Scholar]
- Wielens J.; Headey S. J.; Jeevarajah D.; Rhodes D. I.; Deadman J.; Chalmers D. K.; Scanlon M. J.; Parker M. W. Crystal structure of the HIV-1 integrase core domain in complex with sucrose reveals details of an allosteric inhibitory binding site. FEBS Lett. 2010, 584, 1455–1462. [DOI] [PubMed] [Google Scholar]
- Walsh C. T.; Garneau-Tsodikova S.; Gatto G. J. Jr. Protein posttranslational modifications: the chemistry of proteome diversifications. Angew. Chem., Int. Ed. 2005, 44, 7342–7372. [DOI] [PubMed] [Google Scholar]
- a Fukuda H.; Sano N.; Muto S.; Horikoshi M. Simple histone acetylation plays a complex role in the regulation of gene expression. Briefings Funct. Genomics Proteomics 2006, 5, 190–208. [DOI] [PubMed] [Google Scholar]; b Costi R.; Di Santo R.; Artico M.; Miele G.; Valentini P.; Novellino E.; Cereseto A. Cinnamoyl compounds as simple molecules that inhibit p300 histone acetyltransferase. J. Med. Chem. 2007, 50, 1973–1977. [DOI] [PubMed] [Google Scholar]; c Toth M.; Boros I. M.; Balint E. Elevated level of lysine 9-acetylated histone H3 at the MDR1 promoter in multidrug-resistant cells. Cancer Sci. 2012, 103, 659–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hottiger M. O.; Nabel G. J. Viral replication and the coactivators p300 and CBP. Trends Microbiol. 2000, 8, 560–565. [DOI] [PubMed] [Google Scholar]
- a Cereseto A.; Manganaro L.; Gutierrez M. I.; Terreni M.; Fittipaldi A.; Lusic M.; Marcello A.; Giacca M. Acetylation of HIV-1 integrase by p300 regulates viral integration. EMBO J. 2005, 24, 3070–3081. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Terreni M.; Valentini P.; Liverani V.; Gutierrez M. I.; Di Primio C.; Di Fenza A.; Tozzini V.; Allouch A.; Albanese A.; Giacca M.; Cereseto A. GCN5-dependent acetylation of HIV-1 integrase enhances viral integration. Retrovirology 2010, 7, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topper M.; Luo Y.; Zhadina M.; Mohammed K.; Smith L.; Muesing M. A. Post-translational acetylation of the HIV-1 integrase carboxyl-terminal domain is dispensable for viral replication. J. Virol. 2007, 81, 3012–3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allouch A.; Di Primio C.; Alpi E.; Lusic M.; Arosio D.; Giacca M.; Cereseto A. The TRIM family protein KAP1 inhibits HIV-1 integration. Cell Host Microbe 2011, 9, 484–495. [DOI] [PubMed] [Google Scholar]
- a Giannini G.; Cabri W.; Fattorusso C.; Rodriquez M. Histone deacetylase inhibitors in the treatment of cancer: overview and perspectives. Future Med. Chem. 2012, 4, 1439–1460. [DOI] [PubMed] [Google Scholar]; b Wiech N. L.; Fisher J. F.; Helquist P.; Wiest O. Inhibition of histone deacetylases: a pharmacological approach to the treatment of non-cancer disorders. Curr. Top. Med. Chem. 2009, 9, 257–271. [DOI] [PubMed] [Google Scholar]
- Chan C. W.; Khachigian L. M. DNAzymes and their therapeutic possibilities. Intern. Med. J. 2009, 39, 249–251. [DOI] [PubMed] [Google Scholar]
- Breaker R. R.; Joyce G. F. A DNA enzyme that cleaves RNA. Chem. Biol. 1994, 1, 223–229. [DOI] [PubMed] [Google Scholar]
- Chandrasekhar K.; Malathhi R. Non-Watson Crick base pairs might stabilize RNA structural motifs in ribozymes—a comparative study of group-I intron structures. J. Biosci. 2003, 28, 547–555. [DOI] [PubMed] [Google Scholar]
- Singh N.; Ranjan A.; Sur S.; Chandra R.; Tandon V. Inhibition of HIV-1 integrase gene expression by 10-23 DNAzyme. J. Biosci. 2012, 3, 493–502. [DOI] [PubMed] [Google Scholar]
- Unwalla H.; Banerjea A. C. Novel mono- and di-DNAenzymes targeted to cleave TAT or TAT-REV RNA inhibit HIV-1 gene expression. Antiviral Res. 2001, 51, 127–139. [DOI] [PubMed] [Google Scholar]
- Butler S. L.; Hansen M. S.; Bushman F. D. A quantitative assay for HIV DNA integration in vivo. Nat. Med. 2001, 7, 631–634. [DOI] [PubMed] [Google Scholar]
- Chun T. W.; Carruth L.; Finzi D.; Shen X.; DiGiuseppe J. A.; Taylor H.; Hermankova M.; Chadwick K.; Margolick J.; Quinn T. C.; Kuo Y. H.; Brookmeyer R.; Zeiger M. A.; Barditch-Crovo P.; Siliciano R. F. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 1997, 387, 183–188. [DOI] [PubMed] [Google Scholar]
- Levin A.; Hayouka Z.; Brack-Werner R.; Volsky D. J.; Friedler A.; Loyter A. Novel regulation of HIV-1 replication and pathogenicity: Rev inhibition of integration. Protein Eng., Des. Sel. 2009, 22, 753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin A.; Hayouka Z.; Friedler A.; Brack-Werner R.; Volsky D. J.; Loyter A. A novel role for the viral Rev protein in promoting resistance to superinfection by human immunodeficiency virus type 1. J. Gen. Virol. 2010, 91, 1503–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin A.; Rosenbluh J.; Hayouka Z.; Friedler A.; Loyter A. Integration of HIV-1 DNA is regulated by interplay between viral rev and cellular LEDGF/p75 proteins. Mol. Med. 2010, 16, 34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin A.; Hayouka Z.; Helfer M.; Brack-Werner R.; Friedler A.; Loyter A. Stimulation of the HIV-1 integrase enzymatic activity and cDNA integration by a peptide derived from the integrase protein. Biopolymers 2010, 93, 740–751. [DOI] [PubMed] [Google Scholar]
- Jung A.; Maier R.; Vartanian J. P.; Bocharov G.; Jung V.; Fischer U.; Meese E.; Wain-Hobson S.; Meyerhans A. Recombination: multiply infected spleen cells in HIV patients. Nature 2002, 418, 144. [DOI] [PubMed] [Google Scholar]
- Levin A.; Hayouka Z.; Brack-Werner R.; Volsky D. J.; Friedler A.; Loyter A. Novel regulation of HIV-1 replication and pathogenicity: Rev inhibition of integration. Protein Eng., Des. Sel. 2009, 22, 753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin A.; Hayouka Z.; Friedler A.; Loyter A. Specific eradication of HIV-1 from infected cultured cells. AIDS Res. Ther. 2010, 7, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]