

Description
A 5-month-old male infant was referred to our department for evaluation of abdominal distension and failure to thrive. Clinical examination revealed hepatosplenomegaly. High cholesterol and triglyceride levels, pancytopenia and abnormal liver function were found on blood examination. Bone marrow aspirate demonstrated vacuolated macrophages.
Plain radiographs of the abdomen showed symmetrically enlarged adrenal glands with maintained triangular shape and extensive calcifications (figures 1 and 2). Ultrasound of the abdomen revealed hepatomegaly with fatty infiltration, splenomegaly and enlarged adrenal glands with calcification (figures 3–5). A contrast-enhanced CT scan of the abdomen was also performed which additionally showed low-attenuation abdominal lymphadenopathy (figures 6–10).
Figure 1.
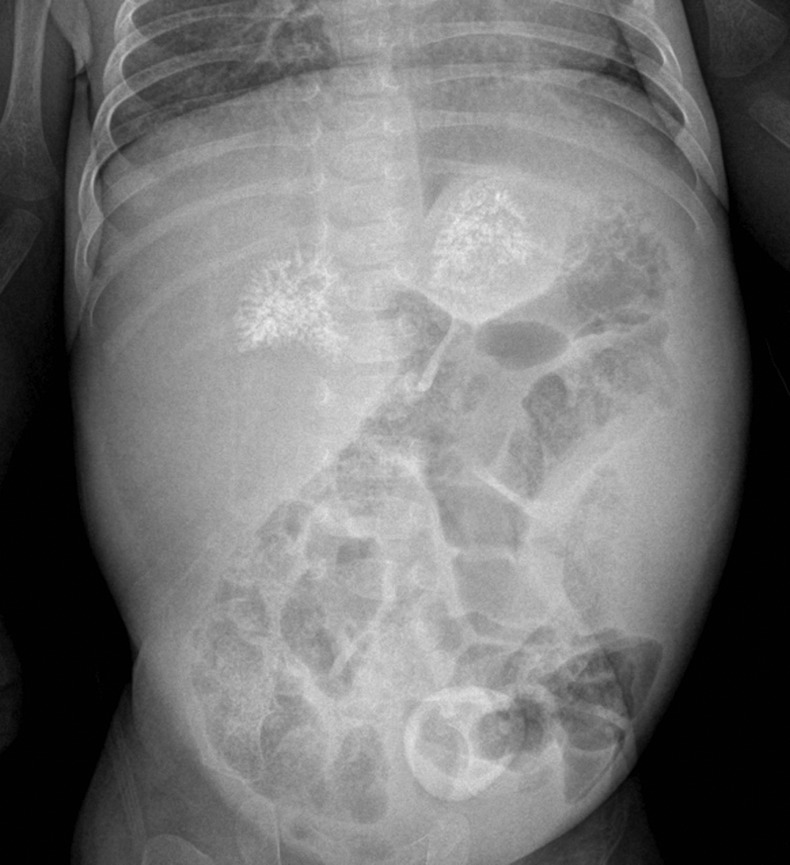
Frontal radiograph of the abdomen showing bilateral symmetrical calcification in the suprarenal region.
Figure 2.
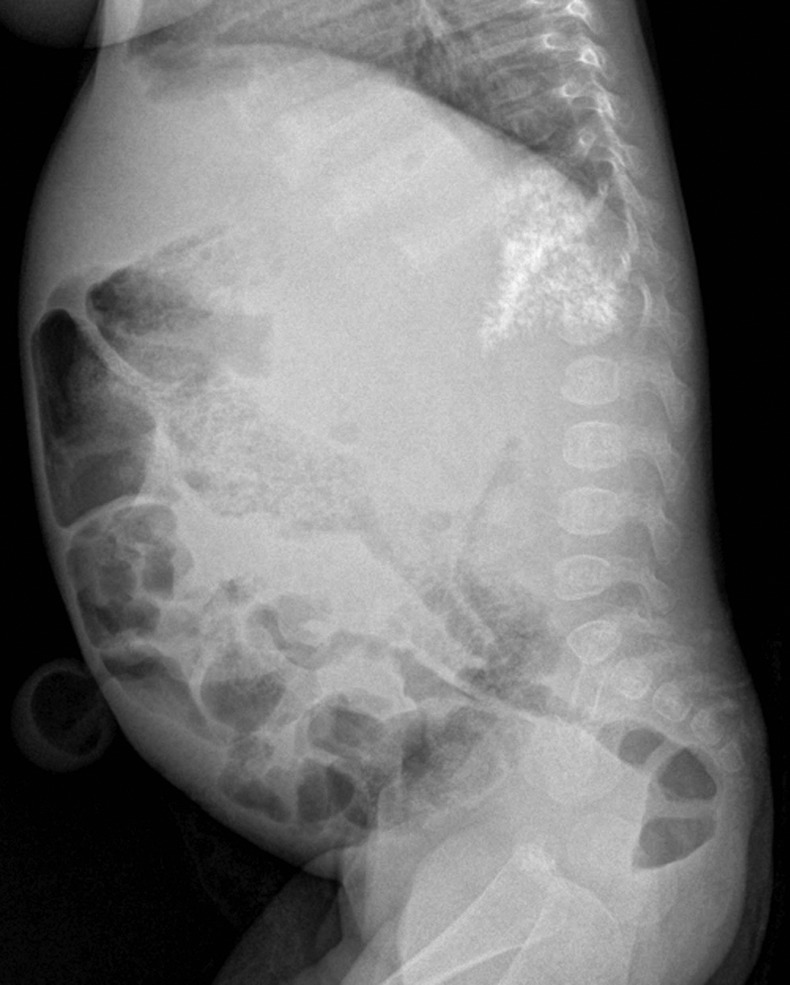
Lateral radiograph of the abdomen confirming the triangular shape of calcification in the suprarenal region. Umbilical hernia containing mesenteric fat is also seen.
Figure 3.
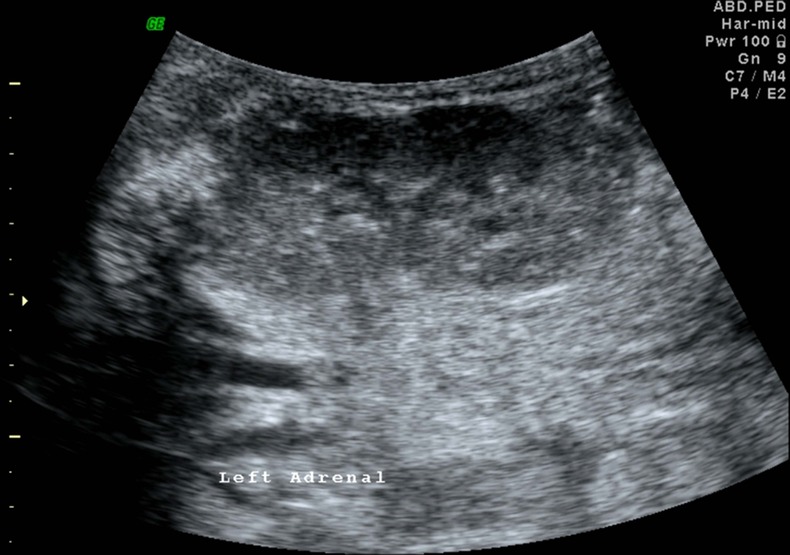
Ultrasound image of the left kidney with multiple calcifications showing posterior acoustic shadowing in the enlarged left adrenal gland.
Figure 4.
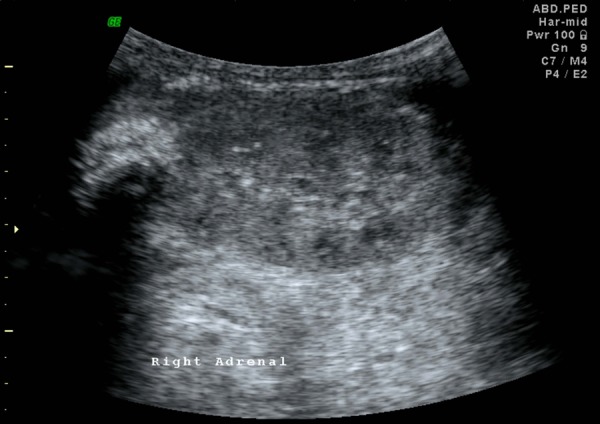
Ultrasound image of the right kidney with similar findings in the enlarged right adrenal gland.
Figure 5.
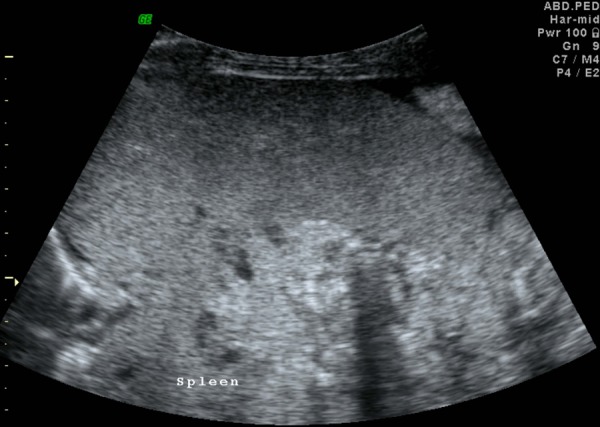
Ultrasound image showing splenomegaly.
Figure 6.
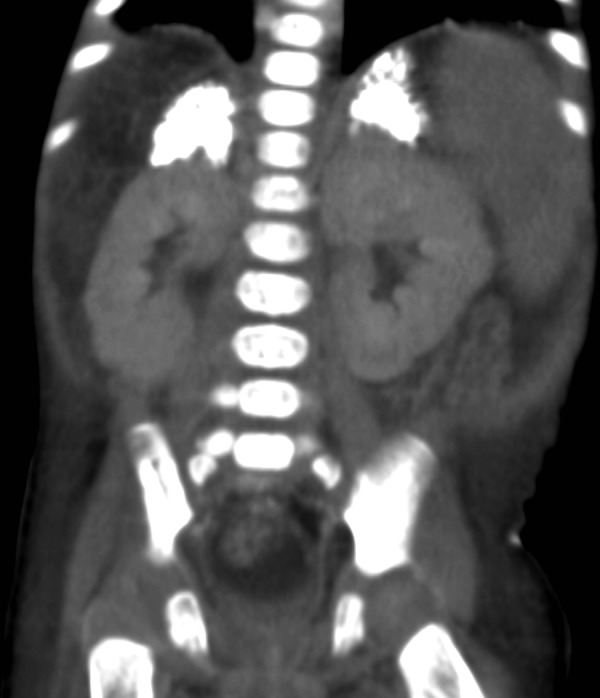
Coronal CT section of abdomen showing maintained triangular shape of the enlarged adrenal glands with dense calcification.
Figure 7.
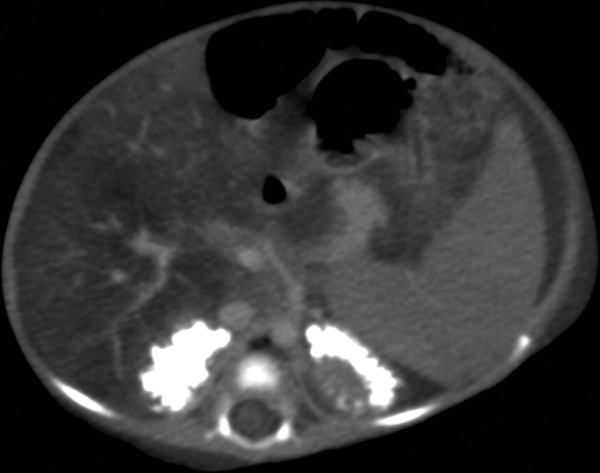
Axial CT section of abdomen showing bilateral adrenal gland calcification and fatty infiltration of liver.
Figure 8.
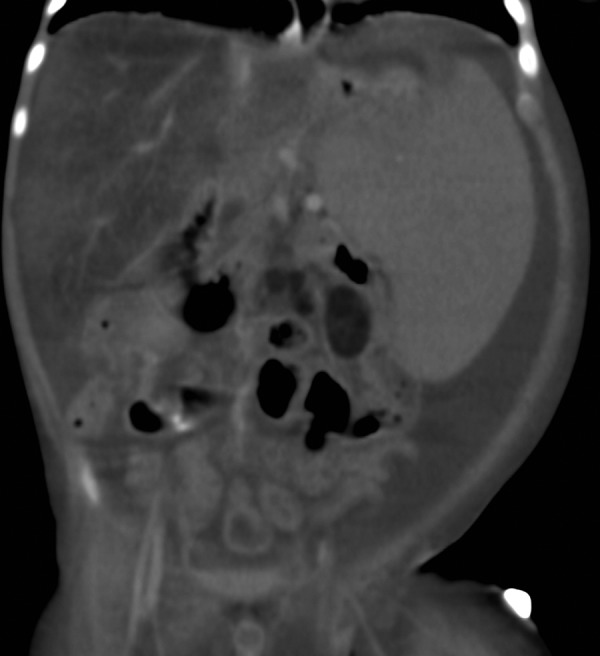
Coronal CT section of abdomen showing fatty liver, splenomegaly, low attenuation mesenteric lymph nodes and ascites.
Figure 9.
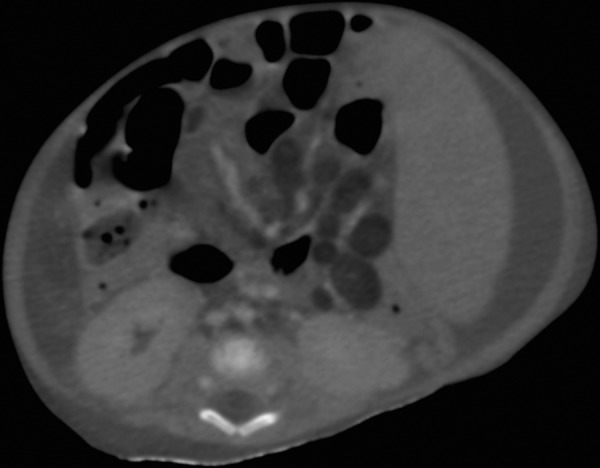
Axial CT section of abdomen showing low attenuation fatty lymph nodes, splenomegaly and ascites.
Figure 10.
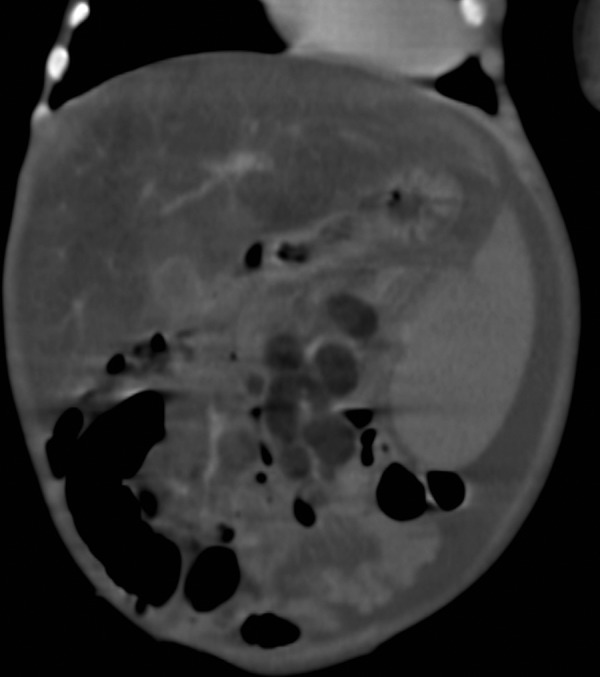
Coronal CT section of abdomen showing fatty liver, low attenuation mesenteric lymph nodes and ascites.
Wolman disease is a rare autosomal recessive lysosomal storage disorder presenting in infancy. It occurs due to a deficiency in lysosomal acid lipase which is an enzyme essential for the metabolism of triglycerides and cholesterol esters. A deficiency in this enzyme causes elevated levels of triglycerides and cholesterol which leads to deposition of excessive amount of lipid esters in the viscera such as liver, spleen, adrenals, lymph nodes and bowel. Infants generally present during the second to third months of life with features of abdominal distension, malnutrition, diarrhoea and failure to thrive.1
Diagnosis of this condition is based on both laboratory parameters and its unique imaging features. Blood tests typically show high cholesterol and triglyceride levels. Peripheral blood smear shows an increase in lymphocytes while bone marrow aspirate shows vacuolated macrophages. Various imaging modalities such as plain radiography, ultrasonography and CT are used to detect this condition of which contrast-enhanced CT is the most important. The characteristic feature of this disease is symmetrically enlarged adrenal glands with maintained triangular shape and extensive calcifications. This occurs due to saponification of the excessive fat deposited in the adrenals. Other imaging features include organomegaly, fatty infiltration of liver and low-attenuation abdominal lymphadenopathy.2
This condition comes with a poor prognosis and is invariably fatal with death occurring within a few months. Affected infants are generally treated conservatively with parenteral nutrition, steroids, dietary supplements etc. Bone marrow transplant has been tried in a few cases and has shown some success.3 Enzyme replacement therapy is, however, still under development.
Learning points.
Wolman disease is a rare autosomal recessive lysosomal storage disorder which presents with a constellation of typical laboratory and imaging findings.
A definite diagnosis of this disease can be made in the presence of hepatosplenomegaly, bilateral adrenal calcification and foamy macrophages in the bone marrow aspirate.
Footnotes
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Majumder N, Ayubi AW, More SS, et al. Wolman's disease. J Case Rep 2013;3:85–8 [Google Scholar]
- 2.Al Essa M, Nounou R, Sakati N, et al. Wolman's disease: the King Faisal Specialist Hospital and research centre experience. Ann Saudi Med 1998;18:2. [DOI] [PubMed] [Google Scholar]
- 3.Krivit W, Peters C, Dusenbery K, et al. Wolman disease successfully treated by bone marrow transplantation. Bone Marrow Transplant 2000;26:567–70 [DOI] [PubMed] [Google Scholar]