

Abstract
The vital process of transcription by RNA polymerase II (Pol II) occurs in chromatin environment in eukaryotic cells; in fact, moderately transcribed genes retain nucleosomal structure. Recent studies suggest that chromatin structure presents a strong barrier for transcribing Pol II in vitro, and that DNA-histone interactions are only partially and transiently disrupted during transcript elongation on moderately active genes. Furthermore, elongating Pol II complex is one of the major targets during gene regulation. Below we describe a highly purified, defined experimental system that recapitulates many important properties of transcribed chromatin in vitro and allows detailed analysis of the underlying mechanisms.
Keywords: transcript elongation, nucleosome, histone octamer, DNA uncoiling, structures
1. Introduction
Recent studies have established that transcript elongation is a major checkpoint during regulation of expression of thousands of human and Drosophila genes (Guenther et al., 2007; Zeitlinger et al., 2007). Strong, regulated stalling of RNA polymerase II (Pol II) occurs as the enzyme proceeds from initiation into transcript elongation, about 50 bp downstream of transcription start (Core and Lis, 2008; Gilmour, 2009; Nechaev and Adelman, 2008; Price, 2008), when Pol II enters into the first (+1) nucleosome on the gene (Churchman and Weissman, 2011; Koerber et al., 2009; Mavrich et al., 2008; Schones et al., 2008; Seila et al., 2008). Once Pol II overcomes the +1 nucleosomal barrier, it can continue transcript elongation over hundreds of kb of predominantly nucleosomal template at a high rate (3–4 kb/min (Singh and Padgett, 2009)), similar with the rate observed on histone-free DNA in vitro (Cheng and Price, 2007; Izban and Luse, 1992). Nucleosomes are not lost from thousands of moderately active genes (Jiang and Pugh, 2009; Kristjuhan and Svejstrup, 2004; Lee et al., 2004; Nacheva et al., 1989; Schwabish and Struhl, 2004). Furthermore, on these genes, fast and extensive transcription-dependent displacement/exchange of only H2A/H2B, but not H3/H4, histones was observed (Dion et al., 2007; Jamai et al., 2007; Rufiange et al., 2007; Schwartz and Ahmad, 2005; Thiriet and Hayes, 2005; Wirbelauer et al., 2005). Accordingly, our studies suggested that only one H2A/H2B dimer is displaced during Pol II transcription in vitro (Belotserkovskaya et al., 2003; Kireeva et al., 2002). The lack of H3/H4 displacement or exchange indicates that these histones most likely do not leave transcribed DNA even transiently ((Kulaeva et al., 2007), confirmed in our in vitro studies (Kulaeva and Studitsky, 2010)). Since Pol II has to disrupt some DNA-histone interactions during transcription, this disruption is transient and does not involve all H3/H4 tetramer-DNA interactions at any given time (Kulaeva et al., 2009). At the same time, during intense transcription, there is partial and transient loss of all core histones at the transcribed regions (Kristjuhan and Svejstrup, 2004; Lee et al., 2004; Petesch and Lis, 2008; Schwabish and Struhl, 2004; Zhao et al., 2005) and exchange (Dion et al., 2007; Jamai et al., 2007; Rufiange et al., 2007; Schwartz and Ahmad, 2005; Thiriet and Hayes, 2005; Wirbelauer et al., 2005). Efficient maintenance of chromatin structure during and after passage of Pol II is essential for gene regulation, cell survival (Martens et al., 2005) and aging (Feser et al., 2010). The Pol II-type mechanism of transcription through chromatin is conserved from yeast to human (Bondarenko et al., 2006) and shared by Pol II and E. coli RNA polymerase (RNAP), but not by other RNA polymerases (Kulaeva et al., 2009; Studitsky, 1999; Studitsky et al., 1994; Studitsky et al., 1997).
Several types of Pol II-based experimental in vitro systems are available for analysis of the mechanism of transcription through chromatin (Dedrick and Chamberlin, 1985; Izban and Luse, 1991; Orphanides et al., 1998; Walter et al., 2003b). Systems that support promoter-dependent transcription initiation in crude extracts (Izban and Luse, 1991) or with highly purified proteins (Orphanides et al., 1998) are characterized by only a small fraction of transcribed templates (Knezetic et al., 1988). This low efficiency of template utilization makes analysis of the fate of nucleosomes after transcription and the structures of transcribed complexes nearly impossible. A different type of DNA templates containing a single-stranded, 3′-extending DNA “tail” support efficient end-initiation by Pol II in vitro (Dedrick and Chamberlin, 1985). However, in this system, stable DNA-Pol II complexes are formed at the end of DNA (Liu et al., 2003). Moreover, end-initiated and promoter-initiated elongation complexes are functionally distinct and most likely have different structures (Liu et al., 2003).
More recently, a method for assembly of “authentic” elongation complexes (ECs) using histidine-tagged yeast Pol II and synthetic RNA and DNA oligonucleotides (Kireeva et al., 2002; Sidorenkov et al., 1998) has been applied to analysis of the mechanism of transcription through chromatin (Kireeva et al., 2002). This experimental system faithfully recapitulates many important properties of chromatin transcribed in vivo (Hsieh et al., 2010; Kireeva et al., 2002; Kulaeva et al., 2009; Kulaeva et al., 2010). However the fraction of functionally active ECs is relatively low, making their direct structural analysis very difficult. Since E. coli RNA polymerase (RNAP) and Pol II use very similar mechanisms for transcription through nucleosomes, the bacterial experimental model remains useful for analysis of general aspects of the mechanism (Hsieh et al., 2010; Kulaeva et al., 2009; Kulaeva et al., 2010; Walter et al., 2003a).
Below we describe experimental approaches developed for analysis of the structures of the ECs formed during transcription through chromatin by E. coli RNAP and Pol II. Currently the first system allows footprinting of the ECs stalled in different positions within a nucleosome. The latter system allows mapping of sites accessible to restriction endonucleases within the stalled ECs (Hsieh et al., 2010; Kulaeva et al., 2009).
2. Materials and methods
The ECs can be assembled by two ways (Fig. 1). ECs can be formed after initiation of transcription on a strong E. coli T7A1 promoter using the σ70-containing holoenzyme. Alternatively, authentic ECs are assembled on synthetic oligonucleotides using the core enzyme (Sidorenkov et al., 1998). Both approaches produce ECs having very similar properties during transcription of DNA (Sidorenkov et al., 1998) or nucleosomal templates (Walter et al., 2003a; Walter et al., 2003b). The first approach can be used only with E. coli RNAP due to the low efficiency of promoter-dependent transcription initiation in purified Pol II-dependent in vitro systems. The ECs can be immobilized on Ni-2+NTA beads through a hexahistidine tag positioned on the C-terminus of one of the large subunits of the enzymes. Below we describe detailed protocols for promoter-dependent formation of ECs for E. coli RNAP and the protocol for assembly of authentic ECs for Pol II. The low adhesion microcentrifuge tubes (USA Scientific, Ocala) are used in all experiments.
Figure 1.

The experimental approach for stalling of yeast Pol II (A) or E. coli RNAP (B) elongation complexes (ECs) at unique positions on the 603-42 templates. The sequences of the 603-42 templates allow stalling of Pol II or E. coli RNAP ECs at the −5 or +41 positions (relative to the promoter-proximal nucleosome boundary) upon addition of different partial combinations of NTPs. In case of E. coli RNAP, the initiation complex (IC) is formed first (small arrow). Then corresponding stalled ECs are characterized and analyzed using restriction enzyme mapping or DNase I footprinting, respectively (see text for details).
3. DNaseI footprinting of elongation complexes formed by E. coli RNAP during transcription through a nucleosome
3.1. Design and preparation of mononucleosomal DNA templates for transcription by E. coli RNAP
The “minimal” nucleosomal templates for transcription by E. coli RNAP contain a strong 110-bp T7A1 promoter region with the start site localized 50 bp upstream of promoter-proximal boundary of one of the strong 147-bp nucleosome-positioning sequences (Bondarenko et al., 2006). Nucleosome positions on these templates are unique and were mapped with high resolution (Morozov et al., 2009). The sequences of the templates are designed to allow stalling of RNAP at different locations along the templates (Bondarenko et al., 2006; Hsieh et al., 2010; Kulaeva et al., 2009; Kulaeva et al., 2010). The template 603-42 is described here as an example; it allows stalling at the positions −39, −5 and +41 (the numbers indicate positions of the active center of RNAP relative to promoter-proximal nucleosome boundary) along the templates using different combinations of NTPs (Fig. 1 (Hsieh et al., 2010; Kulaeva et al., 2009)).
To obtain the templates, DNA fragments containing the nucleosome-positioning sequences and the T7A1 promoter are amplified separately using different pairs of primers, one of primers in each pair contains TspRI sequence. After the amplification and digestion with TspRI, the promoter fragment is ligated to the 147-bp 603-42 template through complementary TspRI sticky ends (Bondarenko et al., 2006; Kulaeva et al., 2009), and the entire template is amplified by PCR again.
T7A1 promoter-containing DNA fragment is prepared as described in (Walter et al., 2004). The upper oligo (5′-AAAGGATCCAGATCCCGAAAATTTATCAAAAAGAGTATT GACTTAAAGTCTAACCTATAGGATACTTACAGCCATCGAGAGGG-3′) and lower oligo (5′-GTTTCCTGTGTGTGCCCAGTGCCGGTGTCGCTTGGGTTGGCTTTTCGC CGTGTCCCTCTCGATGGCTGTAAGTATCCTATAGG-3′, Invitrogen Corporation, Carlsbad, CA) were mixed in equimolar quantities (200 pmole each) in annealing buffer (10 mM Tris-HCl, pH 7.5; 100 mM NaCl; 1 mM EDTA) in a final volume of 50 μl.
The oligos were annealed by heating them to 85°C in a H2O bath for 10 min and allowing them to cool to RT slowly. The annealed oligos were filled in by incubating at 72°C for 20 min in the presence of 1× Vent DNA polymerase buffer (10 mM KCl, 20 mM Tris-HCl (pH 8.8), 10 mM (NH4)2SO4, 2 mM MgSO4, 0.1% Triton X-100), 200 μM dNTPs (Amersham Pharmacia Biotech, Piscataway, NJ), 0.1 mg/ml BSA, and 1.5 units of Vent (exo-) DNA polymerase (New England Biolabs, Beverly, MA). 10 μl of annealed oligo was used to conduct subsequent fill-in reaction.
The 603-42 fragment was amplified from pGEM-3Z/603 plasmid with forward primer (containing TspRI restriction site): 5′-ACACCGGCACTGGGGCCCGGTTCGCGCGCCCG CCTTCCGTGTGTTGTCGTCTCTCGAGCTTC-3′ and reverse primer: 5′-TACCCCAGGGACTTGAAGTAATAAGGAC-3′.
Both PCR products were extracted with ethanol, re-suspended in 1× NEBuffer4 (NEB, New England Biolabs, Beverly, MA), and digested with TspRI (NEB) at 65°C for 4 hr or overnight.
Both fragments were separately purified in 1.5% agarose gel containing 4M Urea. The 113- and 147-bp DNA fragment were excised on trans-illuminator (Fisher Scientific Inc.) using long UV wavelength (312 nm).
DNA fragments were further purified using QIAquick Gel Extraction kit (Qiagen Chatsworth, CA) and the manufacturer’s protocol, and eluted with 30–50 μl of TE buffer (TE buffer: 10 mM Tris-HCl, pH 8.0, and 1 mM EDTA). DNA concentrations were determined by measuring the A260; purified DNA fragments were stored at −20°C.
The 113-bp T7A1 DNA fragment was ligated to the 147-bp 603-42 DNA fragment using T4 DNA ligase (NEB) in 20 μl of ligation buffer (NEB) at 16°C overnight to obtain the 269-bp DNA fragment.
The following PCR amplification step was used for DNA end-labeling with 32P isotope. Either promoter or nucleosome end of the template can be labeled. Only one DNA end was labeled to allow straightforward interpretation of the footprinting results.
T7A1 forward or 603-42 reverse primers were end-labeled with 10–30 μCi γ-[32P] ATP (6000 Ci/mmol, PerkinElmer Life Sciences, Boston, MA) using T4 polynucleotide kinase (NEB, as per NEB recommendations) prior to the PCR. Reaction was continued for 1 hr at 30°C, followed by enzyme inactivation for 20 min at 65°C. Radiolabeled primer was directly used during subsequent PCR amplification without additional purification.
The 269-bp ligated DNA fragment was used as a template for PCR amplification with T7A1 forward and 603 reverse primers (one of the primers was radiolabeled). Amplification was done in preparative amount (200–500 μl volume) using Taq DNA polymerase (NEB).
The PCR product was extracted once with one volume of 1:1 (v/v) phenol:chloroform, ethanol-precipitated, washed with 80% ethanol, dried and dissolved in 30–50 μl of ddH2O.
DNA fragments were resolved in 1.5 % (w/v) agarose gel containing 0.5 mg/ml ethidium bromide in TAE buffer at 4–6 V/cm for 1–2 h.
The 269-bp DNA fragment was excised on trans-illuminator using long UV wavelength.
DNA fragment was further purified using QIAquick Gel Extraction kit (Qiagen, Chatsworth, CA as per kit protocol) and was eluted with 30–50 μl of TE buffer.
DNA concentration was determined by measuring the A260. Purified DNA fragments were stored at −20°C.
3.2. Reconstitution of uniquely positioned mononucleosomes
The use of the 600-series nucleosome positioning sequences allows efficient nucleosome assembly at single unique positions on DNA fragments as long as 1500 bp (Gaykalova et al., 2009). Nucleosomes can be reconstituted using H1-depleted (–H1) donor chromatin or core histones purified after overexpression in bacteria (Gaykalova et al., 2009). Only a minimal molar excess of donor chromatin or purified histones is required for complete nucleosome assembly on the radiolabeled DNA fragment. The properties of mononucleosomes assembled using either donor chromatin or purified core histones are indistinguishable. The nucleosomes obtained by these methods do not require additional purification and can be directly used in various transcriptional assays described below.
250 ml of each of CRB1 to CRB6 buffers were cooled to 4°C. CRB 1–6 (core reconstitution buffers): all six buffers contain 10 mM Tris-HCl, pH 8.0, 0.2 mM EDTA, 5 mM β-mercaptoethanol, 0.1% NP-40, and NaCl at the following concentrations: buffer 1 – 2 M, 2 – 1.5 M, 3 – 1 M, 4 – 0.75 M, 5 – 0.5 M, and 6 – 0.01 M.
0.5–3 μg of single end-labeled DNA was dissolved in the CRB1 buffer (for assembly from purified histones) or in CRB3 buffer (for assembly by histone octamer transfer).
For nucleosome assembly by histone octamer transfer, donor chromatin (purified as described in (Gaykalova et al., 2009)) was taken in the amount of three-fold weight excess (quantified by DNA content) relative to the amount the radiolabeled DNA fragment.
In case of nucleosome assembly from purified histones, H3 and H4 histones were added in two-fold molar excess relative to the amount of radiolabeled DNA, while H2A and H2B were added in three-fold molar excess. Additionally, salmon sperm dsDNA (Sigma-Aldrich, St. Louis, MO) was added to achieve two-fold weight excess over the amount of the DNA fragment.
The reaction volume in all cases was adjusted to 40–100 μl to achieve 10–100 ng/μl total concentration of DNA.
In case of nucleosome assembly from purified histones, the samples were subsequently dialyzed against CRB1, CRB2, CRB3 and CRB4 each for 1hr, CRB5 for 2.5hr and CRB6 overnight at 4°C. Only CRB3-6 buffers were used for assembly by histone octamer transfer.
The resulting nucleosomes were stored at 4°C. Freezing of the samples may result in nucleosome disassembly.
The quality of nucleosome reconstitution was evaluated by analysis of the samples by 4.5% (39:1 acrylamide:bis) native PAGE in 0.5× TBE buffer for 3 hr at 110 V (Gaykalova et al., 2009).
The gel was transferred to Whatman 3MM paper, covered with a plastic wrap, vacuum-dried and exposed with a PhosphorImager screen at room temperature for 2–3 h or overnight. Assembled nucleosomes migrate as a single band that has a lower mobility in the gel as compared with histone-free DNA (Fig. 2).
Figure 2.

Characterization of nucleosomes formed on the 603-42 templates for transcription by Pol II or E. coli RNAP. Nucleosomes were reconstituted on DNA-end-labeled 603-42 templates (269 and 147 bp, for transcription by E. coli RNAP and yeast Pol II, respectively). The templates were analyzed by 4.5% native PAGE. Mononucleosomes (mono-N), histone-free DNA and transcriptionally inactive close-packed dinucleosomes (di-N) are indicated. M – end-labeled pBR322-MspI digest.
3.3. Preparation of the complexes containing E. coli RNAP stalled at the unique +41 intranucleosomal position
In order to perform high-resolution structural analysis of the elongation complexes (ECs) formed on the positioned nucleosomes, the RNAP should also be uniquely positioned. To obtain uniquely positioned ECs, the sequences of the DNA templates were modified to allow stalling of E. coli RNAP at various unique positions (e.g. Fig. 1 (Kulaeva et al., 2009)). The resulting sequences usually contain long DNA regions that are missing specific DNA letter(s) in one of the two DNA strands (e.g. the 603-42 DNA template does not contain Ts in the upper DNA strand between the transcription start-site and the position −5, Fig. 1). If transcription is conducted in the presence of partial combination of NTP(s) (e.g. in the absence of UTP, Fig. 1), RNAP continues transcription until it reaches the position where the missing NTP has to be incorporated (e.g. position −5 on the 603-42 template) and then stalls. In most cases, the stalled complexes are reasonably stable (for up to an hour at 150 mM KCl and room temperature) and maintain unique positions on DNA. To evaluate the positions of RNAP on the template, RNA in the ECs is pulse-labeled and the resulting radiolabeled transcripts are analyzed by denaturing PAGE.
All proteins and DNA-protein complexes (E. coli RNAP, Pol II, core histones proteins and –H1 donor chromatin) were purified as described (Gaykalova et al., 2009; Walter et al., 2003b).
To form active initiation complexes, 200 ng of nucleosomal templates or histone-free DNA were incubated with 5-fold molar excess of E. coli RNAP in 20 μl of the transcription buffer containing 40 mM KCl (TB40). Transcription Buffer (TB) contains 20 mM Tris-HCl, pH 8.0, 5 mM MgCl2, 2 mM β-mercaptoethanol, and various concentrations of KCl (e.g. TB40 contains 40mM KCl). The complexes were incubated at 37°C for 10 min.
ApUpC RNA primer (Oligos, Etc., Wilsonville, OR) was added to the active initiation complexes to final concentration of 20 μM, together with 1 μM ATP (Amersham Pharmacia Biotech, Piscataway, NJ) and 1 μM α-[32P] GTP (6000 Ci/mmol, PerkinElmer) to allow formation of 11-nt labeled RNA transcript (EC-39 complex where the active center of the enzyme is positioned 39 bp upstream of promoter-proximal nucleosomal boundary) at 37°C for 10 min.
Unlabeled GTP was added to the reaction to final concentration of 20 μM to allow formation of the 11-mer (EC-39) on the majority of the templates at 37°C for 5 min.
20 μl of Ni2+-NTA agarose (50% suspension in alcohol, Qiagen) was washed 3 times with 0.5 ml of TB40, incubated in the presence of 0.5 mg/ml of acetylated BSA (Sigma-Aldrich, St. Louis, MO) for 10 min, and washed 2 times with 0.5 ml TB40. The volume was adjusted to 25 μl.
EC-39 was incubated with 1 μM CTP, 20 ug/ml rifampicin, 0.5 mg/ml acetylated BSA, 150 mM KCl at 37°C for 10min to advance EC-39 to EC-5 at the end of the –T DNA track (upper DNA strand).
Efficient formation and DNase I footprinting of the EC+41 can be achieved in solution without additional washing steps. However extra wash steps after immobilization of the complexes on Ni2+-NTA beads remove the unlabeled long chromatin or salmon sperm DNA used in nucleosome reconstitution and excess amount of ATP and other nucleotides, improving the site-specific pausing and decreasing the background signals.
The EC-5 was immobilized on the Ni-NTA-agarose beads in TB40 and washed once with TB40, 2 times with TB300, and 2 times with TB200. The volume was adjusted to 25 μl.
EC-5 was incubated for 5 min in the presence of 100 mM imidazole to elute the complex from Ni2+-NTA beads.
Eluted EC-5 was diluted twice with TB200 to decrease imidazole concentration to 50 mM. Higher concentrations of imidazole inhibit activity of DNase I during footprinting.
EC+41 was formed by incubating EC-5 in the presence of 300 μM CTP, UTP, GTP and 150 μM 3′-dATP (Trilink Biotechnologies, Inc., San Diego, CA) in TB200 for 4 min at room temperature.
The reaction was terminated by adding EDTA to 20 mM, the reaction volume was adjusted to 100 μl and the samples were extracted once with one volume of 1:1 (v/v) phenol:chloroform.
Radiolabeled RNA was precipitated with ethanol, washed with 80% ethanol, dried, and dissolved in 5 μl of RLB buffer (RNA Loading Buffer: 95% formamide, 10 mM EDTA, 0.1% SDS, and 0.01% of each bromophenol blue and xylene cyanol dyes).
Samples were boiled for 3 min and analyzed by denaturing PAGE in 8% (19:1 acrylamide:bis) PAG in 0.5× TBE buffer for 1–1.5 hr at 2000 V.
Gel was dried on Whatman 3MM paper and exposed to a Phosphor-Imager screen at room temperature overnight. An example of RNA analysis is shown in Fig. 3.
Figure 3.

Characterization of RNA of the EC-5 and EC+41 formed by Pol II and E. coli RNAP on the 603-42 templates. EC-5 and EC+41 containing pulse-labeled RNA were formed as described in Fig. 1, RNA was purified and analyzed by denaturing PAGE (see text for detail). Arrows indicate positions of the transcripts and labeled DNA templates. M – end-labeled pBR322-MspI digest.
3.4. Footprinting of stalled EC-5 and EC+41 using DNase I
After characterizing the nucleosomes and the ECs containing stalled RNAP, the footprinting experiments using DNA endonuclease DNase I are performed. In this case, the complexes are obtained using single DNA-end-labeled templates, and the transcription reactions are conducted in the presence of unlabeled NTPs. Usually DNaseI is added immediately after formation of the stalled ECs and the footprinting reaction is performed for a short time (30 sec) to avoid decomposition of the ECs. The concentration of DNaseI used during footprinting has to be determined in preliminary experiments (using histone-free DNA) to allow introduction of single-strand breaks in less then 30% of all templates present in the reaction (to guarantee mostly single-hit reaction conditions). The positions of DNA-protein complexes (RNAP and/or nucleosome) on DNA are determined by analysis of the digested end-labeled DNA by denaturing PAGE.
The EC+41 formed in TB200 is diluted by equal volume of TB0 immediately after formation, to achieve final KCl concentration of 100 mM. Higher concentrations of KCl inhibit activity of DNase I.
At this point, concentration of DNA template in reaction mix is approximately 2.5 μg/ml. Unlabeled –H1 chromatin is added to achieve total DNA concentration of 25 μg/ml to increase stability of the EC and prevent overdigestion by DNaseI.
0.2–0.3 units of DNase I per 100 ng of total DNA is added to the reaction and samples are incubated for 30 sec at 37°C.
The reaction is terminated by adding EDTA to final concentration of 20 mM.
The samples are supplemented with one third of reaction volume of 4× CLB (chromatin loading buffer: 40% sucrose (w/v), 40 mM EDTA, 1 mg/ml ssDNA, 0.4 mg/ml-H1 chromatin and 0.5× TBE buffer) before loading into 4.5% (39:1 acrylamide:bis) native PAGE containing 0.5× TBE buffer.
ECs, nucleosomes and histone-free DNA are resolved by native PAGE for 3 hr at 110 V.
To localize the ECs, the wet gel is exposed to a PhosphorImager screen for 1 hr at room temperature. The bands containing different complexes (Fig. 4) are cut out of the gel.
The gel slices are crashed, and the DNA is extracted overnight at 4°C in 3–5 volumes of NEB buffer (nucleic acid extraction buffer: 10 mM Tris-HCl, pH 8.0, 0.2 mM EDTA, 0.1% SDS).
Eluates are purified from gel pieces with spin-x centrifugation tube filter (0.45 uM cellulose acetate filter, Corning-Costar, Lowell, MA) by centrifugation at 13,000 rpm for 30 min at room temperature on a microcentrifuge.
The sample volume is adjusted to 0.2 ml by extracting with 100% buthanol. Then the samples are extracted once with one volume of 1:1 (v/v) phenol:chloroform.
DNA is precipitated with ethanol, washed with 80% ethanol, dried, and dissolved in 5 μl of RLB buffer.
Samples are boiled for 3 min and analyzed by denaturing PAGE in 8% (19:1 acrylamide:bis) PAG in 0.5× TBE buffer for 1–1.5 hr at 2000 V.
Gel was dried on Whatman 3MM paper and exposed to a Phosphor-Imager screen at room temperature overnight. An example of DNase I footprinting of stalled ECs is shown in Fig. 5.
Figure 4.

Analysis of the EC-5 and EC+41 formed by E. coli RNAP on the 603-42 DNA and nucleosomal templates. DNA-labeled ECs were incubated in the presence of DNase I and separated by native PAGE. Different ECs have distinct electrophoretic mobilities indicating their structural homogeneity.
Figure 5.
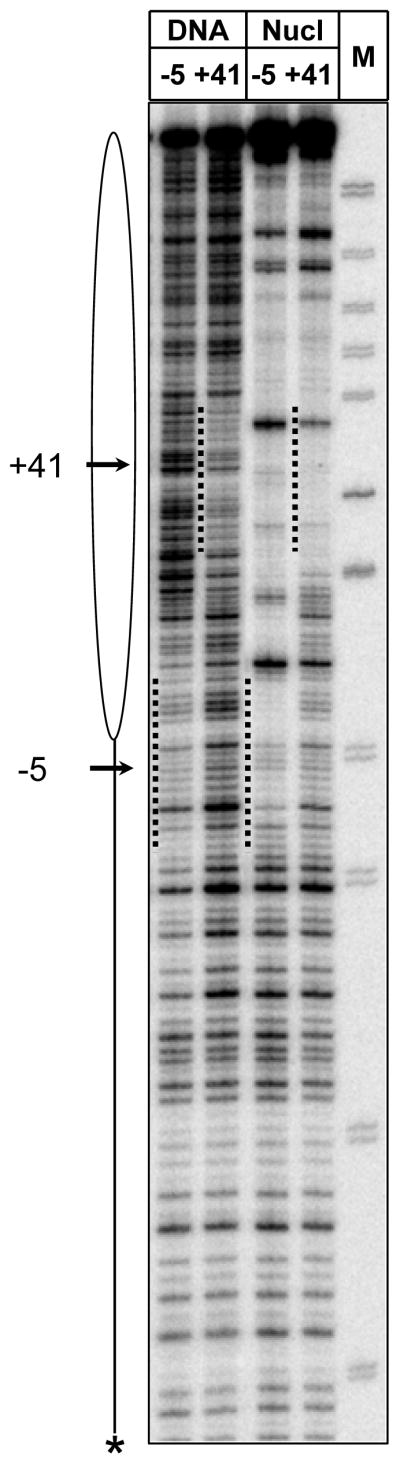
DNase I footprinting of the EC-5 and EC+41 formed by E. coli RNAP on the 603-42 DNA and nucleosomal templates. The promoter end-labeled DNA and nucleosomal templates were incubated in the presence of DNase I and separated by native PAGE (Fig. 4). DNA from different bands was gel-purified and separated by denaturing PAGE. The regions occupied by RNAP in the EC-5 and EC+41 are indicated by dashed lines. The position of the labeled DNA end is indicated by asterisk. M – end-labeled pBR322-MspI digest.
4. Mapping of elongation complexes formed by Pol II during transcription through a nucleosome using restriction endonucleases
In the case of Pol II, authentic ECs are assembled using synthetic oligonucleotides and the core enzyme (Sidorenkov et al., 1998) and then ligated to pre-assembled nucleosomes.
4.1. Design and preparation of mononucleosomal DNA templates for transcription by Pol II
To obtain the nucleosomes, DNA fragments containing the nucleosome-positioning sequences are PCR-amplified separately (one of the primers contains TspRI sequence). After the amplification and digestion with TspRI, the fragment is purified, nucleosome is reconstituted and ligated to the pre-assembled Pol II EC through the TspRI sticky end (Bondarenko et al., 2006; Kulaeva et al., 2009). In order to evaluate the quality of nucleosomes, the promoter-distal end of nucleosomal DNA is weakly labeled with γ-[32P] ATP. This weak labeling does not interfere with subsequent, more intense DNA end-labeling and mapping using restriction endonucleases (part 4.3).
603-42 reverse primer was labeled with 0.1 – 0.3 μCi of γ-[32P] ATP (6000 Ci/mmol, PerkinElmer) using T4 polynucleotide kinase (NEB) in the TB40 buffer for 20 min at 37°C with subsequent enzyme inactivation for 20 min at 65°C.
The radiolabeled primer was used for fragment amplification from pGEM-3Z/603 vector with non-labeled forward 603-42 primer, as described in part 3.1.
The PCR product was extracted with ethanol, re-suspended in 1× NEBuffer 4 (New England Biolabs, Beverly, MA), and digested with TspRI at 65°C for 4 hr or overnight.
The 147-bp product of digestion was purified in 1.5% agarose gel containing 4 M Urea. The 147-bp DNA fragment was excised on the trans-illuminator using long UV wavelength.
The 147-bp DNA fragment was further purified using QIAquick Gel Extraction kit columns (Qiagen Chatsworth, CA) using the manufacturer protocol and eluted with 30–50 μl of TE buffer (10 mM Tris-HCl, pH 8.0, and 1 mM EDTA). DNA concentrations were determined by measuring the A260; purified DNA fragments were stored at −20°C.
Nucleosome reconstitution on the 147-bp DNA fragment was performed as described in part 3.2. The quality of nucleosome assembly was evaluated by native PAGE (part 3.2, Fig. 2).
4.2. Preparation and identification of stalled EC
Yeast Pol II EC are assembled from two single-stranded DNA oligonucleotides (template and non-template strands), 9-nt RNA primer and the core enzyme that was purified as described in (Walter et al., 2003b). The template oligonucleotide is end-labeled, annealed with the RNA primer and this RNA:DNA duplex is incubated with Pol II. Formation of EC is completed by addition of an excess amount of non-template DNA oligo. The DNA oligonucleotides are designed to generate the TspRI sticky end after annealing. Then the assembled Pol II EC is immobilized through the His-tag positioned on Pol II complex (Walter et al., 2003b), extensively washed, ligated to the pre-assembled nucleosomal template through the TspRI sticky end, eluted into solution with imidazole, and transcription is continued in the presence of partial combination of NTPs to stall Pol II at the desired intranucleosomal locations.
After the ligation, the resulting sequence contains DNA regions that are missing specific DNA letter(s) in one of the two DNA strands. Thus the 603-42 DNA template does not contain Ts in the upper DNA strand between the transcription start-site and the position −5, Fig. 1). If transcription is conducted in the absence of UTP, RNAP proceeds until it reaches the position where the missing UTP has to be incorporated (position −5 on the 603-42 template) and stalls, forming EC-5.
-
1
200 pmoles of template ssDNA oligonucleotide, 50T: 5′-GGTGTCGCTTGGGTTGGCTTTTCGGGCTGTCCCTCTCGATGGCTGTAAGT-3′ was end-labeled with 10–40 μCi of γ-[32P] ATP (6000 Ci/mmol, PerkinElmer) using T4 polynucleotide kinase (NEB) in TB40 buffer for 20 min at 37°C with subsequent enzyme inactivation for 20 min at 65°C.
-
2
RNA:DNA duplex was obtained by addition of 200 pmoles of 9-nt RNA primer: 5′-AUCGAGAGG-3′ to 200 pmoles of radiolabeled 50T DNA oligo in 20 μl of TB40. Reaction was incubated for 10 min at 45°C; then the temperature was decreased by 2°C every 2 min to the room temperature.
-
3
The RNA:DNA duplex was incubated with 3-fold molar excess of yeast core Pol II to form an active EC for 10 min at room temperature.
-
4
Formation of EC was completed by adding 50-fold molar excess of unlabeled non-template DNA strand, 59NT: (5′-ACTTACAGCCATCGAGAGGGACACGGCGAAAAGCCAACCCAAGCGACACCGGC ACTGGG-3′) for 10 min at room temperature.
-
5
(In some experiments, in order to identify Pol II positions on the template, the RNA transcript was pulse-labeled in the presence of 1 μM α-[32P] GTP (6000 Ci/mmol, PerkinElmer) for 10 min at room temperature. RNA labeling was done only in reactions that were conducted in parallel with the restriction mapping. For the restriction enzyme mapping experiments, the reactions were conducted with unlabeled RNA.)
-
6
20 μl of Ni2+-NTA agarose (50% suspension in alcohol, Qiagen) was washed 3 times with 0.5 ml of TB40, incubated in the presence of 0.5 mg/ml of acetylated BSA (Sigma-Aldrich, St. Louis, MO) for 10 min, and washed 2 times with 0.5 ml TB40. The volume was adjusted to 25 μl.
-
7
Pol II EC was incubated with 25 μl of Ni2+-NTA suspension for 15 min at room temperature.
-
8
Immobilized EC was washed once with TB40, twice with TB300, and twice with TB40. The volume was adjusted to 25 μl.
-
9
The EC was ligated with 100–200 ng of the nucleosomal template in the presence of 100 μM ATP, 1% PEG-8000, and 50 units of T4 DNA ligase (NEB) in a volume of 50 μl at 16°C for 1–2 hours.
-
10
After ligation, immobilized ECs were washed with TB40, incubated for 10 min with TB300, and washed twice with TB300. These washing steps remove excess of unbound DNA templates.
-
11
To extend the Pol II transcript, the product of ligation is incubated in the presence of 200 μM CTP, ATP and GTP in TB40 for 15 min at room temperature to form EC-5.
-
12
EC-5 is extensively washed with TB40, incubated for 10 min with TB300, and washed twice with TB300.
-
13
EC-5 is eluted from Ni2+-NTA beads in the presence of 100 mM imidazole after incubating for 5 min at room temperature.
-
14
Eluted EC-5 was diluted twice with TB200 to decrease final imidazole concentration to 50 mM.
-
15
EC+41 was formed by incubating EC-5 in the presence of 200 μM CTP, UTP, GTP and 150 μM 3′dATP in TB300 for 4 min at room temperature.
-
16
For the analysis of the complexes using restriction enzymes, after formation of the EC+41 the reaction mix was diluted 3 times by TB0 to final KCl concentration to 100 mM and incubated in the presence of restriction enzymes (see below).
-
16
To characterize pulse-labeled RNA, the reaction was terminated by adding EDTA to final concentration of 20 mM. Radiolabeled RNA was precipitated with ethanol, washed with 80%
-
17
Samples were boiled for 3 min and analyzed by denaturing PAGE in 8% (19:1 acrylamide:bis) PAG in 0.5× TBE buffer for 1–1.5 hr at 2000 V.
-
18
Gel was dried on Whatman 3MM paper and exposed to a Phosphor-Imager screen at room temperature overnight. An example of RNA analysis is shown in Fig. 3.
4.3. Mapping of Pol II elongation complexes using restriction endonucleases
Although DNase I footprinting provides detailed information on the structures of analyzed complexes, it could only be applied to highly homogeneous samples (the desired, structurally homogeneous complex should be formed on more than 60% of the templates). At the same time, during Pol II transcription large amounts of non-productive elongation complexes arrested early during transcription of the templates is formed (Kireeva et al., 2002). Therefore in case of Pol II DNA accessibility within the complexes was analyzed using restriction enzymes. Nucleosomes strongly protect DNA from digestion with restriction enzymes (Polach and Widom, 1999); thus DNA regions that are uncoiled from the surface of the histone octamer would become sensitive to corresponding restriction enzymes (Kulaeva et al., 2009). Sensitivity of DNA in various (productive and non-productive) complexes can be easily evaluated after separation of the complexes by native PAGE.
After 3-fold dilution of EC+41 by TB0 to final KCl concentration of 100 mM, reactions were equally divided to three tubes and incubated with 10 units of Cac81 (NEB) that recognizes promoter-proximal end of nucleosomal DNA, 10 units of StyI (NEB) that recognizes promoter-distal end of nucleosomal DNA, or in the absence of restriction endonucleases (control reaction), respectively. Reactions were performed at room temperature for 15 min.
Reactions were stopped by adding EDTA to final concentration of 20 mM.
4× CLB (one third volume of each reaction) was added to the reaction before loading on 4.5% (39:1 acrylamide:bis) native PAG containing 0.5× TBE buffer.
ECs, nucleosomes and histone-free DNA were resolved by native PAGE for 3 hr at 110 V.
Gel was dried on Whatman 3MM paper and exposed to a Phosphor-Imager screen at room temperature overnight (Fig. 6). The bands were quantified using a PhosphorImager software.
Both the Cac8I and StyI intranucleosomal sites are protected from digestion in EC-5 (Fig. 6) and in intact nucleosomes. As expected, in EC+41, the DNA behind Pol II (the Cac8I site) is sensitive to digestion and the DNA in front of the enzyme (the StyI site) is resistant. These data are consistent with the results obtained using DNase I footprinting (E. coli RNAP, Fig. 5) and suggest that yeast Pol II and E. coli RNAP induce similar structural rearrangements of DNA/histone contacts during transcription through nucleosomes (Kulaeva et al., 2009).
Figure 6.

Analysis of the EC-5 and EC+41 formed by Pol II by the restriction enzyme sensitivity assay. DNA-end-labeled elongation complexes were incubated in the presence of either Cac81 or StyI restriction enzymes and analyzed by native PAGE. The positions of the ECs, nucleosomes, DNA fragments and sites for the restriction endonucleases are indicated.
Acknowledgments
We thank Dr. J. Widom for providing pGEM-3Z/603 plasmid. This work was supported by NIH GM58650, NSF (MCB-1050470) and Government of the Russian Federation (order #220) grants to V.M.S.
References
- Belotserkovskaya R, Oh S, Bondarenko VA, Orphanides G, Studitsky VM, Reinberg D. FACT facilitates transcription-dependent nucleosome alteration. Science. 2003;301:1090–3. doi: 10.1126/science.1085703. [DOI] [PubMed] [Google Scholar]
- Bondarenko VA, Steele LM, Ujvari A, Gaykalova DA, Kulaeva OI, Polikanov YS, Luse DS, Studitsky VM. Nucleosomes can form a polar barrier to transcript elongation by RNA polymerase II. Mol Cell. 2006;24:469–79. doi: 10.1016/j.molcel.2006.09.009. [DOI] [PubMed] [Google Scholar]
- Cheng B, Price DH. Properties of RNA polymerase II elongation complexes before and after the P-TEFb-mediated transition into productive elongation. J Biol Chem. 2007;282:21901–12. doi: 10.1074/jbc.M702936200. [DOI] [PubMed] [Google Scholar]
- Churchman LS, Weissman JS. Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature. 2011;469:368–73. doi: 10.1038/nature09652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core LJ, Lis JT. Transcription regulation through promoter-proximal pausing of RNA polymerase II. Science. 2008;319:1791–2. doi: 10.1126/science.1150843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedrick RL, Chamberlin MJ. Studies on transcription of 3′-extended templates by mammalian RNA polymerase II. Parameters that affect the initiation and elongation reactions. Biochemistry. 1985;24:2245–53. doi: 10.1021/bi00330a019. [DOI] [PubMed] [Google Scholar]
- Dion MF, Kaplan T, Kim M, Buratowski S, Friedman N, Rando OJ. Dynamics of replication-independent histone turnover in budding yeast. Science. 2007;315:1405–8. doi: 10.1126/science.1134053. [DOI] [PubMed] [Google Scholar]
- Feser J, Truong D, Das C, Carson JJ, Kieft J, Harkness T, Tyler JK. Elevated histone expression promotes life span extension. Mol Cell. 2010;39:724–35. doi: 10.1016/j.molcel.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaykalova DA, Kulaeva OI, Bondarenko VA, Studitsky VM. Preparation and analysis of uniquely positioned mononucleosomes. Methods Mol Biol. 2009;523:109–23. doi: 10.1007/978-1-59745-190-1_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmour DS. Promoter proximal pausing on genes in metazoans. Chromosoma. 2009;118:1–10. doi: 10.1007/s00412-008-0182-4. [DOI] [PubMed] [Google Scholar]
- Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. A chromatin landmark and transcription initiation at most promoters in human cells. Cell. 2007;130:77–88. doi: 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh FK, Fisher M, Ujvari A, Studitsky VM, Luse DS. Histone Sin mutations promote nucleosome traversal and histone displacement by RNA polymerase II. EMBO Rep. 2010;11:705–10. doi: 10.1038/embor.2010.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izban MG, Luse DS. Transcription on nucleosomal templates by RNA polymerase II in vitro: inhibition of elongation with enhancement of sequence-specific pausing. Genes Dev. 1991;5:683–96. doi: 10.1101/gad.5.4.683. [DOI] [PubMed] [Google Scholar]
- Izban MG, Luse DS. Factor-stimulated RNA polymerase II transcribes at physiological elongation rates on naked DNA but very poorly on chromatin templates. J Biol Chem. 1992;267:13647–55. [PubMed] [Google Scholar]
- Jamai A, Imoberdorf RM, Strubin M. Continuous histone H2B and transcription-dependent histone H3 exchange in yeast cells outside of replication. Mol Cell. 2007;25:345–55. doi: 10.1016/j.molcel.2007.01.019. [DOI] [PubMed] [Google Scholar]
- Jiang C, Pugh BF. Nucleosome positioning and gene regulation: advances through genomics. Nat Rev Genet. 2009;10:161–72. doi: 10.1038/nrg2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kireeva ML, Walter W, Tchernajenko V, Bondarenko V, Kashlev M, Studitsky VM. Nucleosome remodeling induced by RNA polymerase II: loss of the H2A/H2B dimer during transcription. Mol Cell. 2002;9:541–52. doi: 10.1016/s1097-2765(02)00472-0. [DOI] [PubMed] [Google Scholar]
- Knezetic JA, Jacob GA, Luse DS. Assembly of RNA polymerase II preinitiation complexes before assembly of nucleosomes allows efficient initiation of transcription on nucleosomal templates. Mol Cell Biol. 1988;8:3114–21. doi: 10.1128/mcb.8.8.3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koerber RT, Rhee HS, Jiang C, Pugh BF. Interaction of transcriptional regulators with specific nucleosomes across the Saccharomyces genome. Mol Cell. 2009;35:889–902. doi: 10.1016/j.molcel.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristjuhan A, Svejstrup JQ. Evidence for distinct mechanisms facilitating transcript elongation through chromatin in vivo. EMBO J. 2004;23:4243–52. doi: 10.1038/sj.emboj.7600433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulaeva OI, Gaykalova DA, Pestov NA, Golovastov VV, Vassylyev DG, Artsimovitch I, Studitsky VM. Mechanism of chromatin remodeling and recovery during passage of RNA polymerase II. Nat Struct Mol Biol. 2009;16:1272–8. doi: 10.1038/nsmb.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulaeva OI, Gaykalova DA, Studitsky VM. Transcription through chromatin by RNA polymerase II: Histone displacement and exchange. Mutat Res. 2007;618:116–29. doi: 10.1016/j.mrfmmm.2006.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulaeva OI, Hsieh FK, Studitsky VM. RNA polymerase complexes cooperate to relieve the nucleosomal barrier and evict histones. Proc Natl Acad Sci USA. 2010;107:11325–30. doi: 10.1073/pnas.1001148107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulaeva OI, Studitsky VM. Mechanism of histone survival during transcription by RNA polymerase II. Transcr. 2010;1:85–88. doi: 10.4161/trns.1.2.12519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CK, Shibata Y, Rao B, Strahl BD, Lieb JD. Evidence for nucleosome depletion at active regulatory regions genome-wide. Nat Genet. 2004;36:900–5. doi: 10.1038/ng1400. [DOI] [PubMed] [Google Scholar]
- Liu YV, Clark DJ, Tchernajenko V, Dahmus ME, Studitsky VM. Role of C-terminal domain phosphorylation in RNA polymerase II transcription through the nucleosome. Biopolymers. 2003;68:528–38. doi: 10.1002/bip.10302. [DOI] [PubMed] [Google Scholar]
- Martens JA, Wu PY, Winston F. Regulation of an intergenic transcript controls adjacent gene transcription in Saccharomyces cerevisiae. Genes Dev. 2005;19:2695–704. doi: 10.1101/gad.1367605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavrich TN, Jiang C, Ioshikhes IP, Li X, Venters BJ, Zanton SJ, Tomsho LP, Qi J, Glaser RL, Schuster SC, Gilmour DS, Albert I, Pugh BF. Nucleosome organization in the Drosophila genome. Nature. 2008;453:358–62. doi: 10.1038/nature06929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morozov AV, Fortney K, Gaykalova DA, Studitsky VM, Widom J, Siggia ED. Using DNA mechanics to predict in vitro nucleosome positions and formation energies. Nucleic Acids Res. 2009;37:4707–22. doi: 10.1093/nar/gkp475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nacheva GA, Guschin DY, Preobrazhenskaya OV, Karpov VL, Ebralidse KK, Mirzabekov AD. Change in the pattern of histone binding to DNA upon transcriptional activation. Cell. 1989;58:27–36. doi: 10.1016/0092-8674(89)90399-1. [DOI] [PubMed] [Google Scholar]
- Nechaev S, Adelman K. Promoter-proximal Pol II: when stalling speeds things up. Cell Cycle. 2008;7:1539–44. doi: 10.4161/cc.7.11.6006. [DOI] [PubMed] [Google Scholar]
- Orphanides G, LeRoy G, Chang CH, Luse DS, Reinberg D. FACT, a factor that facilitates transcript elongation through nucleosomes. Cell. 1998;92:105–16. doi: 10.1016/s0092-8674(00)80903-4. [DOI] [PubMed] [Google Scholar]
- Petesch SJ, Lis JT. Rapid, transcription-independent loss of nucleosomes over a large chromatin domain at Hsp70 loci. Cell. 2008;134:74–84. doi: 10.1016/j.cell.2008.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polach KJ, Widom J. Restriction enzymes as probes of nucleosome stability and dynamics. Methods Enzymol. 1999;304:278–98. doi: 10.1016/s0076-6879(99)04017-3. [DOI] [PubMed] [Google Scholar]
- Price DH. Poised polymerases: on your mark. ..get set...go! Mol Cell. 2008;30:7–10. doi: 10.1016/j.molcel.2008.03.001. [DOI] [PubMed] [Google Scholar]
- Rufiange A, Jacques PE, Bhat W, Robert F, Nourani A. Genome-wide replication-independent histone H3 exchange occurs predominantly at promoters and implicates H3 K56 acetylation and Asf1. Mol Cell. 2007;27:393–405. doi: 10.1016/j.molcel.2007.07.011. [DOI] [PubMed] [Google Scholar]
- Schones DE, Cui K, Cuddapah S, Roh TY, Barski A, Wang Z, Wei G, Zhao K. Dynamic regulation of nucleosome positioning in the human genome. Cell. 2008;132:887–98. doi: 10.1016/j.cell.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwabish MA, Struhl K. Evidence for eviction and rapid deposition of histones upon transcriptional elongation by RNA polymerase II. Mol Cell Biol. 2004;24:10111–7. doi: 10.1128/MCB.24.23.10111-10117.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz BE, Ahmad K. Transcriptional activation triggers deposition and removal of the histone variant H3.3. Genes Dev. 2005;19:804–14. doi: 10.1101/gad.1259805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seila AC, Calabrese JM, Levine SS, Yeo GW, Rahl PB, Flynn RA, Young RA, Sharp PA. Divergent transcription from active promoters. Science. 2008;322:1849–51. doi: 10.1126/science.1162253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidorenkov I, Komissarova N, Kashlev M. Crucial role of the RNA:DNA hybrid in the processivity of transcription. Mol Cell. 1998;2:55–64. doi: 10.1016/s1097-2765(00)80113-6. [DOI] [PubMed] [Google Scholar]
- Singh J, Padgett RA. Rates of in situ transcription and splicing in large human genes. Nat Struct Mol Biol. 2009;16:1128–33. doi: 10.1038/nsmb.1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studitsky VM. Preparation and analysis of positioned nucleosomes. Methods Mol Biol. 1999;119:17–26. doi: 10.1385/1-59259-681-9:17. [DOI] [PubMed] [Google Scholar]
- Studitsky VM, Clark DJ, Felsenfeld G. A histone octamer can step around a transcribing polymerase without leaving the template. Cell. 1994;76:371–82. doi: 10.1016/0092-8674(94)90343-3. [DOI] [PubMed] [Google Scholar]
- Studitsky VM, Kassavetis GA, Geiduschek EP, Felsenfeld G. Mechanism of transcription through the nucleosome by eukaryotic RNA polymerase. Science. 1997;278:1960–3. doi: 10.1126/science.278.5345.1960. [DOI] [PubMed] [Google Scholar]
- Thiriet C, Hayes JJ. Replication-independent core histone dynamics at transcriptionally active loci in vivo. Genes Dev. 2005;19:677–82. doi: 10.1101/gad.1265205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter W, Kashlev M, Studitsky VM. Transcription through the nucleosome by mRNA-producing RNA polymerases. Methods Enzymol. 2004;377:445–60. doi: 10.1016/S0076-6879(03)77029-3. [DOI] [PubMed] [Google Scholar]
- Walter W, Kireeva ML, Studitsky VM, Kashlev M. Bacterial polymerase and yeast polymerase II use similar mechanisms for transcription through nucleosomes. J Biol Chem. 2003a;278:36148–56. doi: 10.1074/jbc.M305647200. [DOI] [PubMed] [Google Scholar]
- Walter W, Kireeva ML, Tchernajenko V, Kashlev M, Studitsky VM. Assay of the fate of the nucleosome during transcription by RNA polymerase II. Methods Enzymol. 2003b;371:564–77. doi: 10.1016/S0076-6879(03)71042-8. [DOI] [PubMed] [Google Scholar]
- Wirbelauer C, Bell O, Schubeler D. Variant histone H3.3 is deposited at sites of nucleosomal displacement throughout transcribed genes while active histone modifications show a promoter-proximal bias. Genes Dev. 2005;19:1761–6. doi: 10.1101/gad.347705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitlinger J, Stark A, Kellis M, Hong JW, Nechaev S, Adelman K, Levine M, Young RA. RNA polymerase stalling at developmental control genes in the Drosophila melanogaster embryo. Nat Genet. 2007;39:1512–6. doi: 10.1038/ng.2007.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Herrera-Diaz J, Gross DS. Domain-wide displacement of histones by activated heat shock factor occurs independently of Swi/Snf and is not correlated with RNA polymerase II density. Mol Cell Biol. 2005;25:8985–99. doi: 10.1128/MCB.25.20.8985-8999.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]