

Abstract
Recently, many therapeutic agents for prostate cancer (PCa) have been approved that target the androgen receptor and/or the prostate tumor microenvironment. Each of these therapies has modestly increased patient survival. However, if a better understanding as to when in the course of PCa progression specific therapies should be applied, and what biomarkers would indicate when resistance arises, survival due to these therapies would almost certainly improve. Thus, applying the armamentarium of therapeutic agents in the right sequences in the right combination at the right time is a major goal in prostate cancer treatment. For this to occur, an understanding of prostate cancer evolution during progression is required. In this review, we discuss the current understanding of PCa progression, but challenge the prevailing view by proposing a new model of PCa progression, with the goal of improving biologic classification and treatment strategies. We use this model to discuss how integrating clinical and basic understanding of PCa will lead to better implementation of molecularly-targeted therapeutics and improve patient survival.
Significance
Rapid development of drugs with efficacy against prostate cancer now makes it possible to consider applying these agents with curative intent in men with currently incurable cancers. However, when to apply these new drugs as well as those under development to obtain the best outcomes is a challenge that must be addressed. To meet this challenge, better classification of the disease based on the underlying molecular mechanisms of progression will facilitate the implementation of current and emerging therapies.
Current Clinical Model of PCa Progression
The pathological classification of PCa is defined by Gleason sum score (Gleason, UICC), which is based on morphologic criteria. The Gleason score is the major method for prostate cancer tissue grading (1) and the most important prognostic factor in prostate cancer (2). High Gleason score predicts more rapid progression and suggests aggressive treatments are needed. However, the Gleason score does not provide information on therapy selection. As a result, patients are currently grouped by clinical stage or treatment status (e.g. with or without bone metastasis, resistance to androgen ablation therapy or not, with or without chemotherapy). This framework categorizes patients with similar prognoses (3, 4). Thus, these factors currently dictate clinical trial design. However, this approach does not have a mechanistic foundation that can guide the proper sequences or combinations of molecularly targeted agents. The current model of PCa progression also fails to account for the observation that the state of cancer progression determines drug-specific efficacy. For example, androgen ablation, but not chemotherapy, is more efficacious when given at early stage PCa progression (5). Paradoxically, chemotherapy is more effective at the later stages of PCa progression (6–8). This stage-dependent response to treatments indicates that PCa undergoes an evolution into different states during disease progression. Furthermore, prostate cancer shows site-specific preference of progression in that prostate and bone are two preferred areas of persistent or recurrent cancer. Although PCa also metastasizes to lymph nodes, these metastases are usually not resistant to therapy. These observations suggest that prostate cancer has a unique relationship with the microenvironment in the prostate and bone (9, 10). Each of these features is therapeutically relevant, but they do not provide a guideline for therapy selection.
Why a New Classification is Needed
The above problems argue that a new classification system will be needed to guide therapy selection. Rather than using tumor morphology as a criterion for therapy selection, molecular markers that define a specific stage of progression would be preferable in choosing therapy, independent of the tumor stage. Our recent understanding of molecular mechanisms of PCa progression has identified that the androgen receptor (AR), oncogenes/tumor suppressors, and microenvironment are the major mechanisms that lead to PCa progression and will provide such biomarkers to guide therapy approaches. Therefore, these mechanisms need to be incorporated into a classification system designed to guide therapy.
Multiple lines of evidence demonstrate that AR function plays a central role throughout the entire process of PCa progression. The prostate is an androgen-dependent organ and androgen ablation is commonly used in the treatment of advanced PCa. However, nearly all patients with advanced cancers will develop progressive disease despite castrate levels of androgens (testosterone level<50 ng/mL), i.e. castration-resistant prostate cancer (CRPC). Recent studies have suggested multiple “escape mechanisms” that lead to AR signaling under “castration levels of androgens” (11–13), which will be discussed below. Thus, the complex alterations in AR signaling need to be considered and potentially targeted nearly throughout the entirety of PCa progression (14, 15).
Loss of tumor suppressor genes is also critical in PCa progression. The loss of PTEN, p53, and RB (16–19) is a common occurrence in PCa progression. Loss of PTEN, observed in some cancers at diagnosis, is linked to a shorter progression free and overall survival (20). However, to date, PTEN loss has not been predictive of response to specific therapies. Similarly, the loss of p53 and RB, while linked to more advanced stages of the cancer, are also not predictive of response to specific therapies.
Aberrant activation or expression of oncogenes, e.g. Src, MET, Axl and FGFR, are prevalent events in the late stage of PCa progression. Several inhibitors of tyrosine kinases have been used in clinical trials. Each appears to have some efficacy on subsets of patients, supporting their roles as drivers of PCa progression in a limited subset of tumors. Although activation of oncogenes may play an important role in PCa progression, AR signaling and oncogene activation are not independent mediators of this process. Decreased androgen levels have been shown to induce MET oncogene expression (21) and the aberrant expression of oncogenes also affects AR function through AR phosphorylation and/or direct association of AR with oncogenes (22–25). Therefore, a biologic classification scheme needs to consider both the AR status and the expression and activation of oncogenes.
The salient feature of advanced prostate cancer progression is the bone-forming and bone-homing nature of metastases. These findings led investigators to hypothesize that the prostate tumor microenvironment in bone plays a role in PCa progression. This hypothesis is supported by the clinical trial in which Sr89, a radionuclide that targets bone environment following chemotherapy, exhibited an increase in survival, which concurs with a more recent report with Radium 223, a bone homing alpha-emitter with a favorable therapeutic index (26–28). Though one cannot attribute the effects of Sr89 and Radium 223 exclusively to treatment effects on the tumor microenvironment, the bone targeting nature of these radiopharmaceuticals implicates bone in the lethal progression of prostate cancer and serves as an impetus to focus mechanistic and clinical studies to understand growth of bone metastases. These factors should be put in a disease progression context that can be exploited therapeutically.
Biologic Classification of Prostate Cancer
Based on the above considerations, the purpose of this review is to propose a new molecular classification of prostate cancer, which incorporates AR, oncogenes/tumor suppressors, and the tumor/bone microenvironment in the disease model. Because of the heterogeneity of PCa, responses to therapies have given us better insights into which of these players are drivers of specific stages of progression. This “responses-to-therapies” concept forms the basis of a new molecular classification of prostate cancer and should help to determine at which stage during progression specific inhibitors can be most effectively applied.
In the alternative model, PCa progression is grouped into three categories: endocrine driven, microenvironment dependent, and tumor cell autonomous. Androgen signaling plays a central role in this model of tumor progression. In the early phase of prostate cancer, androgen signaling responds to DHT depletion (endocrine-driven) (Figure 1). A portion of low grade and low stage cancers are in this “DHT-dependent” stage. However, upon “escaping” DHT-dependence, cancers are characterized by paracrine-driven progression where androgen signaling remains important, albeit by different mechanisms (detailed below). The transition from an endocrine to a paracrine-driven PCa is a milestone that signals the potential lethal progression of the cancer. In this phase of PCa progression, the cancer enters into what we term a “progression spiral”, in which numerous changes in androgen signaling are accompanied with altered microenvironment/tumor interactions. In the last phase of the disease, the cancer cells lose AR dependence, exit the “paracrine progression spiral” and become tumor cell autonomous. The model serves as a framework to group prostate cancers into therapeutically relevant subsets.
Figure 1. DHT-dependent phase of PCa.

In this phase of PCa, some tumors rely mainly on DHT. The source of testosterone (T) is mainly from the testis and adrenal glands provide dehydroepiandrosterone (DHEA). Testosterone and DHEA are converted to DHT by 5-alpha reductases (SRD5A) present in the prostate. AR has higher affinity for DHT than testosterone. For this reason, finasteride and dutasteride, which inhibit type 1 or both type 1 and type 2 5-alpha reductase activity, respectively, can suppress tumor development or growth, accounting for suppression of low-grade cancers, but not high-grade cancers, in a proportion of men with PCa.
Endocrine-driven Phase
The endocrine-driven phase of prostate cancer depends on the presence of 5α dihydrotestosterone (DHT), occurring by 5-alpha reduction of testosterone (see Figure 1). Although testosterone and DHT both bind to AR, and activate AR and its downstream-targeted genes, DHT has a lower dissociation rate and therefore a greater effect on AR signaling than does testosterone (29, 30). Thus, inhibition of DHT formation should reduce AR activity. Indeed, in the Endocrine-driven Phase of prostate cancer, androgen signaling responds to DHT depletion. The 5-alpha-reductase inhibitors finasteride and dutasteride are used to prevent the conversion of testosterone to DHT to interrupt DHT’s growth-promoting signaling. However, the PCPT (31) and REDUCE (32) trial showed that finasteride and dutasteride reduced the rate of low-grade cancers but did not have an effect on high-grade cancers. Additionally, the REDEEM trial (33) demonstrated that a significant number of patients with low-grade cancers at initial diagnosis did not have detectable cancer on subsequent repeat biopsies after dutasteride treatment compared with placebo. These findings support the hypothesis that some low-grade cancers are dependent on dihydrotestosterone (34). The grade-dependent effect of finasteride or dutasteride point to therapeutically relevant heterogeneity of androgen signaling networks that are different between low-grade and higher-grade cancers.
Molecular Mechanisms of Endocrine-driven Phase
Differential effects of DHT and testosterone on AR signaling were observed by several groups. In characterizing the different roles of testosterone and DHT on signaling, Lin and Chang (35) found that the expression of TDD5, an androgen target gene, was differentially repressed by testosterone as compared to DHT. In animal studies, Dadras et al. (36) reported that castrated rats treated with testosterone and finasteride expressed higher levels of genes responsible for prostate growth inhibition and differentiation than in those treated with testosterone alone. Because finasteride treatment led to high intraprostatic testosterone levels, the studies concluded that the effects of DHT on proliferation and differentiation were not the same as those of testosterone (36).
In another set of experiments, Li et al. (37) found that finasteride or dutasteride caused an overall reduction in intraprostatic DHT levels. However, the central axis of androgen signaling is subject to multiple regulatory pathways. Their results indicated that despite the AR status, 5-alpha-reductase genotypic variants, and varying expression levels of 5-alpha-reductase in prostate cancer cells are all responsible for modulating AR signaling. They found that 5-alpha-reductase expression varied among prostate cell lines, and further, the expression of each of the three 5-alpha-reductase enzymes in response to androgen was also cell type specific. Recent studies suggest that the type-1 5-alpha reductase is increased in most prostate tumors (relative to type 2 that is more predominant in the normal prostate), leading the authors to argue that the efficacy of dutasteride, a dual 5-alpha reductase inhibitor, would be superior to finasteride, a type 2 5-alpha reductase inhibitor (38).
More recently, laboratory studies have been undertaken in an attempt to explain the clinical trial results with 5-alpha-reductase inhibitors. Using samples from men with prostate cancer who had undergone treatment with one of two dutasteride dosages for four months before prostatectomy, Mostaghel et al. (39) microdissected normal prostate epithelial cells and analyzed the expression of ninety androgen-regulated genes. The results showed that treated samples could be grouped into high- or low-AR gene activity groups based on their gene expression profiles. The relationship between AR gene activity and treatment response to dutasteride has not yet been clarified.
Marker and therapy for endocrine-driven phase
The grade- and stage-dependent differential responses of prostate epithelial cells to DHT depletion by 5 alpha-reductase inhibitors support the conclusion that a portion of early prostate cancers can be identified by the development of markers that point to DHT dependence. The differences between the DHT-dependent and -independent group are most likely due to the changes in AR sensitivity to DHT. However, the biochemical basis for these changes remains unclear. Understanding the androgen signaling networks, including the expression of the 5-alpha reductase enzymes, during early stage of PCa progression will be important in determining whether the cancer is in the endocrine-driven stage and if the patient will respond to treatment with 5-alpha reductase inhibitors.
Microenvironment-dependent phase (Endocrine-to-paracrine transition)
The transition from endocrine to paracrine-driven PCa signals potential lethal progression of the cancer. For patients with high-grade prostate cancer, androgen ablation (depletion of gonadal androgen), e.g. by Lupron, is more effective than 5-alpha reductase in inhibiting high-grade or metastatic prostate cancer (40). However, the response to androgen ablation is heterogeneous in that some patients have a sustained suppressive effect while others are refractory to the treatment within a few years. When the disease advances to the metastatic stage, only a minority of men with PCa will have sustained control of cancer with androgen ablation. This heterogeneity in response to androgen ablation indicates a clinically and biologically meaningful difference in the role of AR signaling among different stages of prostate cancers. These clinical observations suggest that different mechanisms of AR signaling need to be considered in categorizing patients (Figure 2).
Figure 2. Proposed Spiral Model for PCa progression.

The model proposes three main phases in PCa progression. The first phase is the DHT-dependent phase, during which the tumor is responsive to 5-alpha reductase inhibitor treatments, as indicated by the yellow arrow on the left-hand side of the figure. When the tumor is no longer responsive to inhibitors of 5-alpha reductase, it enters the progression spiral, as marked by a broad up arrow, where multiple factors, including AR signaling changes, oncogene activation, tumor suppressor gene downregulation (not shown), and microenvironment changes, affect tumor progression. Each “turn” is defined by a predictive marker(s) that can be targeted. The pitch in each spiral reflects the duration the tumors that remain responsive to a specific therapy. The adaptive changes in tumors in response to therapy accounts for resistance, leading the tumor to progress to the next turn of the progression spiral, which signals additional alterations in the tumor and its microenvironment. Tumors in this new “turn” will require different therapeutics that specifically target the altered properties that define this turn. Markers that reflect the biology that drive each turn can be used to guide timely therapy application in anticipation of progression. Exit from the spiral occurs when a series of mutations arise, including the loss of AR, RB or p53, and upregulation of PLK1, AURKA and amplification of MYCN. At this stage, the PCa cells are no longer regulated by the microenvironment and become tumor cell autonomous, as indicated by the red arrow on the right-hand side of the figure. Targeted therapies that may affect candidates that drive turns in the spiral are indicated in the figure. Possible disease stages corresponding to the spiral are also indicated.
Associated with AR changes is the activation of oncogenes and loss of tumor suppressor genes, which has been discussed in numerous reviews (41, 42). Loss of the PTEN tumor suppressor gene, leading to the activation of the PI3-kinase/Akt pathway, is one of the earliest genetic changes detected during PCa progression (43). Several oncogenes, as described above, are upregulated in advanced PCa (42). Interestingly, androgen depletion leads to upregulation of genes associated with epithelial-to-mesenchymal transition (44), as observed with the expression of mesenchymal cadherins, e.g. N-cadherin (45) or cadherin-11 (46), in castrate-resistant prostate cancer. The combination of these changes leads to PCa progression and metastasis (see Figure 2).
Acquired resistance to androgen-deprivation coincides with progression of cancer in bone, the preferred area of recurrent cancer, pointing to the presence of a specific bone – epithelial interaction that drives the striking organ-specific progression (see Figure 2).
Together, these observations indicate that under the selective pressure of androgen ablation, prostate cancers evolve from an endocrine-driven (by the gonadal steroid hormones) to a paracrine-driven (by the factors present in the tumor microenvironment) cancer. The endocrine-to-paracrine transition of prostate cancer often signals the presence of disseminated cancer that is incurable with current standard therapy.
During the microenvironment-dependent phase, cancer progression is dominated by tumor adaptation over time. These adaptations comprise continuous vicious cycles, in which the microenvironment alters the tumors and the tumors in turn alter the microenvironment. We propose the term “progression spiral” to illustrate these serial changes over time (See Figure 2). The serial molecular mechanisms that drive vicious cycles are reflected in the “turns” of the spiral. The interval between each turn, the “pitch”, reflects the rate of adaptation of the tumor. While there are many changes that may occur in a “turn”, we refer to the changes that are most critical in driving the tumor progression as the key player in each turn.
Because PCa is heterogeneous, with varied “drivers” for different tumors at different time of tumor progression, the “turns” may be identified through the response to specific therapy. For example, a tumor that is responsive to abiraterone, a Cyp17 (a steroid synthesis enzyme) inhibitor, may comprise one such turn (Figure 2). The duration during which the tumor remains responsive to abiraterone therapy is the “pitch”. When the tumor is no longer responsive to androgen depletion therapy, it indicates that another turn has occurred and the tumor has progressed into another phase of the progression spiral, which signals that additional alterations in the tumor and its microenvironment have occurred. Tumors in this new “spiral” will require different therapeutics that specifically target the altered properties that define this phase of the spiral. At present, the early “turns”, which are detected by responses to specific targeted therapy, have been identified (Figure 2). Because novel antiandrogens targeting AR signaling as initial therapy of PCa are more effective in early rather than in later stages of progression, altered androgen biosynthesis and changes in the androgen receptor are the drivers of the initial turns of the progression spiral. Candidate drivers of the later turns in the spiral have been identified, but their sequence remains unclear (shaded area, Figure 2). Examination of the molecular changes before and after challenge with targeted agents will clarify the pathways to help guide targeted therapy to specific turns in the spiral.
Molecular Mechanisms of Microenvironment-dependent Phase
The progression spiral is characterized by a combination of signaling through AR and the tumor microenvironment. With respect to AR signaling, recent studies have suggested multiple “escape mechanisms” by which AR can sustain signaling under “castrate level” of androgen (11, 14). These escape mechanisms are important in driving tumor progression. Intracrine production of androgen by upregulation of the steroid synthesis enzymes, e.g. cyp17, induces the transition from endocrine to intracrine androgen dependence (11). AR copy number consistently increases after prolonged castration (47). The involvement of these mechanisms in PCa progression is demonstrated by a recent report that therapeutic agents, e.g. abiraterone (12) or MDV3100 (48), that deplete tumor- produced androgens or inhibit the function of the androgen receptor, respectively, lead to improved clinical outcomes (Figure 2). It was also shown that mutations of AR broaden AR ligand specificity such that glucocorticoids besides androgen are able to activate AR (49). Another mechanism that accounts for castration resistance and occurs frequently after therapy is the expression of AR isoforms (AR-Vs) that lack the AR ligand-binding domain but nevertheless can transduce signals (50). Importantly, an increasing number of studies demonstrate that these AR-Vs induce the transcription of additional genes (51–53). The AR-V signature is comprised of numerous genes involved in mitosis, including UBE2C, whose expression correlates with AR-V7 expression in clinical specimens (52). Several groups demonstrated that in androgen-independent PCa cells, androgen receptor regulates a distinct transcriptional program that is different from AR-dependent PCa cells, notably the upregulation of mitotic cell cycle genes (13, 54). Androgen depletion has also been reported to lead to an increase in the expression of mesenchymal cell adhesion molecules, including N-cadherin (45, 55) and cadherin-11 (46), which increase the migratory property of tumor cells as well as contribute to the interaction of tumor cells with osteoblasts in bone microenvironment. Thus, as androgen becomes further and further depleted, AR signaling becomes altered, favoring metastasis and growth-promoting functions over differentiation functions (54). These alterations may ultimately signal the final turn in the progression spiral, i.e., poorly differentiated neuroendocrine carcinoma, which is devoid of androgen receptor.
The changes in “turns” in the “progression spiral” are determined not only by AR alterations but also by activation of oncogenes and changes in the tumor microenvironment. As a result of these interactions, numerous oncogenes are activated. As this topic has been discussed in Gallick et al. (42), only a few illustrative examples are given here. When and how these oncogenes are activated is complex and not well understood. Examples of oncogenes that may play a role in turns of the spiral include Src, IGF-1R, FGF-R, MET, Axl and ACK. At later stages of the spiral, Plk1, AURKA, UBEC2, and MYCN are upregulated due to androgen depletion (Figure 2). These may drive late stages of disease progression.
Activation of oncogenes and androgen depletion are not independent functions. Emerging evidence suggests that there is bi-directional cross talk between oncogenes and AR. Resistance to AR blockade has been shown to be associated with an increase in Src activity, as evidenced by pSFK expression (Figure 3), thus activation of Src family proteins (SPK) may be used as markers, but the mechanism is not known. Androgen depletion has been shown to increase the expression of MET (21). Hence, this may contribute to the response seen by cabozantinib in castration-resistant PCa (56). Oncogenes, e.g. Src, Her2, AKT, ACK, have been shown to phosphorylate AR, which may change the functions of AR as well as affect oncogene activation (57). Each of these changes may represent an individual “turn” in the spiral, or may work in conjunction or sequence to lead to progressive turns. Because we do not have detailed knowledge as to how many of the identified AR changes affect PCa progression, nor do we know the time and sequence of oncogene activation, it is not yet possible to assign the specific “AR signaling changes” and “oncogene changes” to specific turns in the spiral progression at present (hence, the ambiguity represented by the shaded box in Figure 2). The combination of AR signaling blockers and molecular targeting agents will allow us to define these “turns”, which represent critical changes in disease status. It is likely that the factors that determine the “turns” may be different among individuals, as the specific oncogene and sequence of oncogene activation may vary among tumors. Nevertheless, if targeted therapy against a given oncogene is successful, it will prolong the time a patient remains in a specific turn as dictated by the activation of that oncogene (Figure 4). Subsequent identification of markers for patients entering a specific “turn” in the spiral will likely determine future therapy selection.
Figure 3. Src family kinase phosphorylation correlates with therapy resistance to androgen signaling inhibitors.
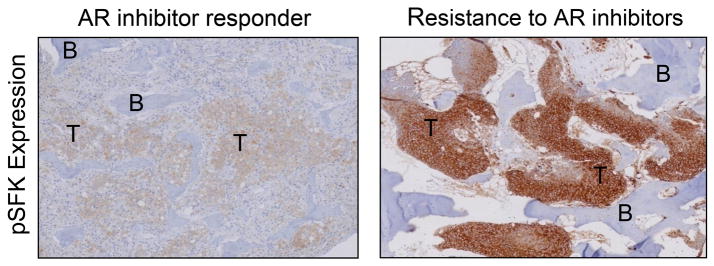
By immunohistochemical staining, Src family kinase phosphorylation is higher in bone marrow biopsies of men who lack response to therapy with androgen signaling inhibitors.
Figure 4. Possible outcomes of therapeutic treatments using the Spiral model.

A turn in the spiral is defined through the expression or alteration of a predictive marker. Three possible outcomes of therapeutic treatment may occur based on a predictive marker that defines a turn in the spiral. Abiraterone is used as an example. After application of abiraterone, (A) No further “turns” in the spiral occur, suggesting that the therapy may be curative. (B) The turn may be elongated, suggesting that abiraterone is effective in reducing androgen generated from Cyp17. Following the treatment, tumor adaptation may occur, leading to the next turn in the spiral. Successful application of the correct therapeutic agent at this stage of the spiral will elongate the next turn. (C) Application of targeted therapy does not have a positive therapeutic effect. This result would suggest that the tumor is in a more complex phase of the disease.
Castration-resistant PCa progression (CRPC) is also dependent on paracrine factors from the tumor environment. Chung’s group showed that by co-inoculating the low tumorigenic androgen-responsive LNCaP cells with human bone fibroblast, a castration-resistant cell line C4-2 was generated, indicating that stromal cells are able to modify the tumor cell properties and render LNCaP cells castrate resistant (58–60). The tumor cell-environment interactions are mostly driven by paracrine factors. It has been shown that PCa cells secrete BMP (61–63), VEGF (64), or FGF9 (65), which affect the proliferation of stromal cells and increase tumor angiogenesis. In turn, tumor-educated stromal cells provide factors, such as osteonectin (66, 67), that increase tumor cell invasiveness, survival, or proliferation. In addition to bone formation, proliferation of tumor cells in the bone marrow frequently induces an osteolytic response, as reflected in the increase in RANKL (68) (69) and N-telopeptide (70). Increased bone resorption leads to the release of TGF beta from the bone matrix (71), further altering tumor cell properties. These vicious cycles are also important in driving “turns” in the spiral. In addition, both the tumor and the microenvironment adapt under selection pressure of therapies, and such adaptations can lead to turns into different phases of the spiral. A combination of all these factors amplifies the bi-directional interactions to multidimensional, multi-level communication representing another example of a vicious cycle. Thus, the combination of alterations in AR signaling, oncogene activation, and environmental factors all contribute to the spiral model presented in Figure 2.
Markers and Treatment Strategies for Microenvironment-driven Phase
The spiral model suggests that therapy should be applied before complex interactions between tumor and its microenvironment occur. Some patients with primary PCa have a sustained suppressive effect in response to androgen ablation, suggesting that there is a stage of PCa that is sensitive to gonadal androgen. These patients eventually become refractory to treatment within a few years and this state of PCa defines an initial “turn” in the spiral. The markers that point to the entry into the initial turn may be an increase in Cyp17 expression in PCa cells, an indication of intracrine androgen biosynthesis. If this were the case, application of abiraterone may lead to prolongation of the “Cyp17 turn” (see Figure 4). In support of this possibility, recent studies from our group showed that application of abiraterone at the early stage of PCa significantly improves clinical outcome (unpublished). AR amplification and/or mutations may lead to a lack of response to abiraterone treatment. Increased Src family kinase gene activity may be developed as a marker for predicting abiraterone resistance (Figure 3) and entry into the next phase of the spiral. Once an increase in Src family kinases is detected, this would suggest that an inhibitor such as dasatinib may be applied in preventing the disease from advancing into the next turn of the spiral. These are potential examples of how the proposed model may be used to predict disease progression and implement targeted therapy at the right time with the right sequence. However, some men with prostate cancer may have very short pitches, indicating very rapid disease progression. These short pitches indicate multiple alterations occur in a very short period of time. In these instances, a single therapy may be unable to elicit a response. As of yet, no predictive markers exist that can guide treatment for this subset of men with prostate cancer. In this case, a combination therapy based on the emergence of multiple markers may be considered.
As shown in the model in Figure 4, it is possible that no further “turns” in the spiral will occur, suggesting that the therapy may be curative (scenario A). This possibility is likely to occur by earlier application of therapy to a known predictive marker. Alternatively, the turn may be elongated, suggesting that the targeted therapy has been effective against the predictive marker (scenario B). Following the treatment, tumor adaptation may occur, leading to the next turn in the spiral. Successful application of the correct therapeutic agent at this stage of the spiral will also elongate the next turn (scenario B). It is possible that application of targeted therapy will not have a positive therapeutic effect (scenario C). This result would suggest that tumor is in a more complex phase of the disease.
Tumor cell autonomous phase
The exit from the microenvironment-dependent progression spiral is heralded by a distinct clinical manifestation characterized by a large tumor mass in the prostate or lymph nodes, and visceral metastases without a commensurate increase in serum PSA. If bone metastases occur, they are predominately lytic bone metastases. These findings reflect the emergence of a rapidly proliferating androgen-independent cancer that will require different treatment. In contrast to the microenvironment-dependent phase, PCa responds to chemotherapeutics including docetaxel by reducing the tumor volume, an indication that the tumor is no longer regulated by its microenvironment, a stage we term the “tumor cell autonomous” phase.
This form of PCa can be distinguished from other castrate-resistant PCa because it does not express the androgen receptor and/or secrete PSA, thus its growth is truly androgen-independent. The clinical features of the “tumor cell autonomous” phase of PCa mimic small cell carcinomas. Pathologically, most of these cancers share some neuroendocrine features and in their pure form are termed neuroendocrine prostate cancer (NEPC) or small cell prostate cancer (SCPC) (72–74). NEPC is rare at de novo diagnosis as less than 1% of PCa at initial diagnosis comprise this histologic/morphology subtype (75). However, NEPC appears to occur most commonly after failure of hormone therapy and is thus almost certain to increase in frequency as better androgen deprivation therapies are used clinically. It was noted that the tumor showed the presence of a morphologic continuum between acinar adenocarcinoma of the prostate to a cancer with small cell carcinoma features (19, 75, 76). Rearrangement of TMPRSS2-ERG is found in NEPC (77, 78), providing additional support that NEPC progresses from PCa adenocarcinoma. These findings are in line with clinical observations that most NEPC are detected upon sudden and rapid progression in the setting of advanced adenocarcinoma of the prostate. NEPC is therefore predicted to be responsible for an increasing percentage of lethality from PCa estimated to be as high as 30% (19, 76, 79, 80).
Molecular Mechanisms of Cell Autonomous Phase
The mechanism by which the “tumor cell autonomous” phase arises is under intense study but is yet unclear, including how the tumor becomes enriched with AR-negative cells. The hallmark of this transition is loss of RB and loss or mutation of p53. Loss of tumor suppressor genes leads to chromosomal instability, resulting in many genomic changes including loss of AR. The presence of AR-negative cells in the tumor may also be due to enrichment of prostate cancer stem cells, reported to be low in AR expression (81). Beltran et al. (80) found that MYCN amplification occurs in these tumors and this may, in part, explain the neuroendocrine properties of the tumor.
The result of genetic instability leads to additional changes, many of which affect cell cycle genes, especially those related to M-phase transition, including aurora kinase A (AURKA) and Polo-like kinase (PLK1) (82). Wang et al. (13) demonstrated that “cell cycle” (52 transcripts) and “mitotic cell cycle” (24 transcripts) are the two top upregulated transcripts by GO analysis. AURKA, PLK1, UBE2C as well as MYCN are all examples of potential markers. AURKA regulates entry into mitosis, as well as assembly of the mitotic spindle apparatus, thereby affecting chromosome separation (83). MYCN amplification is frequently associated with AURKA amplification. In addition, Otto et al. (84) also demonstrated that AURKA stabilizes MYCN.
Another regulator of M Phase overexpressed in prostate cancer is PLK1. Plk1 mediates entry into mitosis as well as centrosome maturation, spindle checkpoint activity, activation of the Anaphase-Promoting Complex (APC) and eventual exit from the M phase with the initiation of cytokinesis (85). PLK1 is overexpressed in prostate cancer with higher expression in high-grade tumors (86). PTEN loss increases PLK1 (87). Recently, Deeraksa et al (82) demonstrated increases in PLK1 expression in androgen-independent LNCaP cells. Importantly, these cells respond to PLK inhibitor BI2536 by undergoing necroptosis (82). Thus, numerous recent results suggest the possible importance of inhibitors of M phase gene products as therapies for NEPC discussed below.
Markers and Treatment Strategies for Cell autonomous Phase
In the late stage of microenvironment-dependent phase, androgen receptor is either not present or has undergone changes as to regulate a distinct transcriptional program, e.g. mitotic cell cycle genes, that is different from AR-dependent PCa cells (13). Thus, detection of the increase in mitotic markers, e.g. UBE2C, AURKA, PLK1, and proliferation marker, e.g. Ki-67, may indicate the exit from microenvironment-dependent phase. NEPC or small cell prostate cancer (SCPC) markers such as chromogranin A, synaptophysin, and neuron-specific enolase are also characteristic markers of this phase (88), as is MYCN amplification.
In the “tumor cell autonomous” phase of the disease, inhibitors that affect mitotic function may be efficacious as opposed to earlier stages when AR signaling affects more “classic” AR-mediated pathways. Currently, first line treatment for this phase is chemotherapy, but patients become rapidly resistant to this approach. As the molecular basis for SCPC becomes better understood, individualized therapy may be possible. For example, AURKA inhibitors such as PHA-739358 (danusertib) have been tested in clinical trials. However, danusertib failed to achieve the primary endpoint of PSA response (89). Based on our analysis, PSA as an endpoint is unlikely to be suitable for tumors that are in the “tumor cell autonomous” phase. In addition, therapeutic treatment in this trial was not directed specifically to patients with amplified AURKA; hence it is not certain if better response would have been achieved by focusing on NEPC patients with amplified AURKA.
PLK1 is receiving increasing interest as a promising target (90) (91). Preclinical studies in osteosarcoma cells have provided evidence that PLK1 is a promoter of oncogenic transformation (92). LNCaP-AI cells were shown to have increased PLK1 and respond to inhibitor by triggering necroptosis (82). PLK1 inhibitors are now reaching trial for solid tumors. BI 2536 is a PLK1 selective inhibitor that reached Phase II trial in several solid tumors, but not PCa, with little efficacy (93). Volasertib (BI 6727) is a potent and relatively selective inhibitor for PLK1. A phase I study in patients with advanced disease demonstrated a favorable pharmacokinetic profile and limited toxicities in patients with advanced solid tumors (94). Phase II studies are ongoing. Based on our model, PLK1 inhibitors might be effective in “tumor cell autonomous” tumor where PLK1 is overexpressed.
Identification of Markers that Predict Therapy Response
At present, very few markers have been identified that predict each turn in the PCa progression spiral. In our proposed model, each of the turns in the spiral occurs in response to a specific “driver” of PCa progression (Figure 2). These “turns” are identified by the patient’s response to a specific therapy. Identification of specific biomarkers is critical to determining appropriate therapy for a given turn and predicting transit to the next turn. Several targeted therapeutic agents have been approved for PCa treatment based on their proven effectiveness in prolonging patients’ survival and others are in active clinical trial. However, only a fraction of treated patients benefit from these targeted therapies. Patients that do benefit comprise true responders and biopsies collected from this group of patients will be invaluable in identifying markers for each “turn”. For example, expression of cyp17, AR, nuclear localization of AR in the tumor, and detectable levels of plasma testosterone, predict the likelihood of response to abiraterone treatment (14). In contrast, a decrease of AR nuclear localization to a more cytoplasmic distribution during abiraterone therapy may indicate changes in AR function that render abiraterone ineffective. Such a result might predict that an AR inhibitor with a different mechanism of action, such as MVD3100, might be useful in therapy. Markers may also provide clues that a more progressive stage of the disease is occurring or is about to occur. For example, irrespective of Gleason score, PTEN status or TMPRSS-ERG-fusion proteins, if markers of the cell autonomous stage of PCa are increased, e.g. Aurora A, MYCN, PLK1, these results would suggest that patients should be treated with chemotherapy. Markers can also help predict impending resistance to targeted therapy. For example, increase in phosphorylation of Src family proteins was found in specimens from patients who were not responsive to abiraterone treatment (Figure 3). These examples suggest that new therapies or combination therapy could be applied when specific markers allow us to predict that a new target (or driver of progression) will emerge, rather than waiting until resistance to a given inhibitor occurs. Most importantly, identification of markers that predict therapy response will enrich the subset of patients who will benefit from specific inhibitors or combination of inhibitors.
Our model then requires multiple and repetitive tumor samplings during the course of treatment for determining the “turn” for therapy selection. This may not be practical in the clinical setting. A likely solution to this problem lies in the recent advances in “liquid biopsy”, where the circulating tumor cells (CTCs) provide the information on tumor status (stage of progression). Analysis of CTCs is valuable as multiple samplings of blood for marker determination is possible.
Factors in the microenvironment may also “drive” progression and many of these factors are released into the blood. Therefore, development of antibody arrays that capture these paracrine factors in the circulation will provide information on the changes in tumor microenvironment that predict tumor progression (or therapy failure). A comprehensive secretome analysis of the paracrine factors that mediate the bi-directional interaction between PCa cells and bone microenvironment at various stage of tumor progression in bone and the changes of secretome components in response to targeted therapies will augment the effort in identifying specific markers that predict “turns” in the spiral.
A major contribution to understanding key molecular markers of tumor initiation and progression will almost certainly arise from next generation sequencing (NGS) of tumors, an ongoing endeavor. This approach has already identified several genetic changes that may “drive” tumor progression, as discussed by Beltran and Rubin (95). For example, in ETS negative PCa in which PTEN has not been lost, SPINK (serine peptidase inhibitor, Kalal type 1) is overexpressed in 10% of tumors. Mutations in the speckle-type POZ gene (SPOP) occur in 6–13% of tumors. In a very small fraction (<1%) of tumors, NGS has shown rearrangements in the RAF gene. Predictive and prognostic implications of these genetic alterations are uncertain but under intense investigation, and whether development of inhibitors that target these molecules specifically will be of therapeutic value remains to be determined.
NGS studies suggest that some prostate progression may occur through clonal evolution and this is further supported by recent studies from Balk’s group (96) that show that Gleason 3 to Gleason 4 PCa tumors have identical TMPRSS gene rearrangement. While the initial driver may be clonal, experience with all drugs currently in clinical use demonstrates resistance to them will arise, leading to the prediction that the initiating clone will further evolve to express additional drivers described in our spiral model. This prediction is borne out by the great majority of tumors that express, for example, activated oncogenes and altered AR. Whether the clonal origin of the tumor will help predict the order in which these additional “drivers” occur will require further study.
How Will the New Model Alter Therapy Selection?
Therapy selection that incorporates molecular drivers that promote tumor progression in heterogeneous tumors, as proposed in (Figure 2), should improve clinical outcome. Current prognostic-based models do not include the factors described above, i.e., AR alterations, microenvironment factors and oncogene activation. In our proposed biology-based model, we use predictive markers, some of which have been clearly identified and others that will emerge as targeted therapies continue to be used in the clinic. This model will also lead to more effective sequences or combinations of molecularly-targeted agents. For example, in the prevailing prognostic model in which Gleason grade and extent of disease dictate treatment strategy, chemotherapy is reserved for treatment of hormone refractory patients with impending or symptomatic progression. In our proposed model, we suggest that chemotherapy will benefit those patients whose cancer is in the cell autonomous state regardless of whether they are detected in early or late stage of cancer progression. In current prognostic models, the newly approved inhibitors of intracrine/paracrine androgen signaling are reserved for patients with castrate-resistant progression. In our proposed model, we suggest that these inhibitors will be more effective when applied earlier, i.e. in the initial androgen signaling driven turns of the endocrine-driven stage of progression, rather than in the later microenvironment-driven stage. The rationale for this approach is that when the tumor enters a more microenvironment-driven stage, the diseases are in a complex progression spiral where AR inhibitors will be less effective or insufficient to inhibit tumor progression due to activation of oncogenes and complex tumor/microenvironment interactions. As an example, we know that in the prognostic model, patients respond to androgen-ablation therapy with variable duration. However, in our model of progression, we may be able to better predict the duration of therapy by examination of specific markers. For example, our data predict that androgen ablation therapy will be durable until increase of phosphorylation of Src family kinases (SFKs) occurs. When SFK increases occur, a combination therapy that targets both the androgen receptor axis and tyrosine kinases may improve outcome, regardless of the stage the tumor would be classified by the prognostic model. As an understanding of molecular markers in the increasingly complex turns of the spiral advances, this marker-based approach will provide further guidance as to what therapeutic combinations are likely to yield the greatest success.
Concluding Remarks
Currently, new drug development for treatment of PCa outpaces our understanding of biology of the disease. While several recently approved FDA agents have prolonged patient survival, at present, we lack knowledge on how best to apply these agents. Clinical trials using the crude “grouping by disease stage” method showed each drug results in a modest prolongation of life (approximately three months for most agents) for patients with the metastatic late stage PCa. The three-month gain is an average, and likely an underestimate for many patients due to a large portion that fail to respond to the treatments. Selection of the patients who will be responsive will significantly improve the outcomes of targeted therapy. Furthermore, applying the targeted therapy at the right time before the disease enters the progression spiral we describe, i.e. anticipating the need for therapy before clinically apparent disease progression has occurred, will be critical in prolonging the effectiveness of the therapy. The development of markers that predict the disease progression will not only be prognostic, indicating the impending changes in tumor properties (i.e. a “turn” in the spiral model), but also will suggest the application of additional targeted therapy to counter the specific changes. Our model will also help select combination therapy for an even more effective outcome. Marker-guided individualized cancer therapy will have a tremendous impact on the PCa treatment. We expect that our proposed PCa progression model will help guide the development of markers, resulting in logical therapy selection. We hope that with the many targeted drugs that have been developed as well as those currently under development, we will be able to change the current “modest gain” from each of the targeted agents into the possibility of PCa becoming a manageable chronic disease.
References
- 1.Gleason DF. Classification of prostatic carcinomas. Cancer Chemother Rep. 1966;50:125–8. [PubMed] [Google Scholar]
- 2.Gleason DF, Mellinger GT. Prediction of prognosis for prostatic adenocarcinoma by combined histological grading and clinical staging. The Journal of urology. 1974;111:58–64. doi: 10.1016/s0022-5347(17)59889-4. [DOI] [PubMed] [Google Scholar]
- 3.Ryan CJ, Smith A, Lal P, Satagopan J, Reuter V, Scardino P, et al. Persistent prostate-specific antigen expression after neoadjuvant androgen depletion: an early predictor of relapse or incomplete androgen suppression. Urology. 2006;68:834–9. doi: 10.1016/j.urology.2006.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McKenney JK, Simko J, Bonham M, True LD, Troyer D, Hawley S, et al. The potential impact of reproducibility of Gleason grading in men with early stage prostate cancer managed by active surveillance: a multi-institutional study. The Journal of urology. 2011;186:465–9. doi: 10.1016/j.juro.2011.03.115. [DOI] [PubMed] [Google Scholar]
- 5.Gravis G, Fizazi K, Joly F, Oudard S, Priou F, Esterni B, et al. Androgen-deprivation therapy alone or with docetaxel in non-castrate metastatic prostate cancer (GETUG-AFU 15): a randomised, open-label, phase 3 trial. The lancet oncology. 2013;14:149–58. doi: 10.1016/S1470-2045(12)70560-0. [DOI] [PubMed] [Google Scholar]
- 6.Efstathiou E, Abrahams NA, Tibbs RF, Wang X, Pettaway CA, Pisters LL, et al. Morphologic characterization of preoperatively treated prostate cancer: toward a post-therapy histologic classification. European urology. 2010;57:1030–8. doi: 10.1016/j.eururo.2009.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Efstathiou E, Logothetis CJ. A new therapy paradigm for prostate cancer founded on clinical observations. Clinical cancer research. 2010;16:1100–7. doi: 10.1158/1078-0432.CCR-09-1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Millikan RE, Wen S, Pagliaro LC, Brown MA, Moomey B, Do KA, et al. Phase III trial of androgen ablation with or without three cycles of systemic chemotherapy for advanced prostate cancer. J Clin Oncol. 2008;26:5936–42. doi: 10.1200/JCO.2007.15.9830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loberg RD, Gayed BA, Olson KB, Pienta KJ. A paradigm for the treatment of prostate cancer bone metastases based on an understanding of tumor cell-microenvironment interactions. J Cell Biochem. 2005;96:439–46. doi: 10.1002/jcb.20522. [DOI] [PubMed] [Google Scholar]
- 10.Logothetis CJ, Lin SH. Osteoblasts in prostate cancer metastasis to bone. Nat Rev Cancer. 2005;5:21–8. doi: 10.1038/nrc1528. [DOI] [PubMed] [Google Scholar]
- 11.Attard G, Richards J, de Bono JS. New strategies in metastatic prostate cancer: targeting the androgen receptor signaling pathway. Clinical cancer research. 2011;17:1649–57. doi: 10.1158/1078-0432.CCR-10-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, et al. Abiraterone and increased survival in metastatic prostate cancer. The New England journal of medicine. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J, et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell. 2009;138:245–56. doi: 10.1016/j.cell.2009.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Efstathiou E, Titus M, Tsavachidou D, Tzelepi V, Wen S, Hoang A, et al. Effects of abiraterone acetate on androgen signaling in castrate-resistant prostate cancer in bone. Journal of clinical oncology. 2012;30:637–43. doi: 10.1200/JCO.2010.33.7675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mostaghel EA, Marck BT, Plymate SR, Vessella RL, Balk S, Matsumoto AM, et al. Resistance to CYP17A1 inhibition with abiraterone in castration-resistant prostate cancer: induction of steroidogenesis and androgen receptor splice variants. Clinical cancer research. 2011;17:5913–25. doi: 10.1158/1078-0432.CCR-11-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–43. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aparicio A, Den RB, Knudsen KE. Time to stratify? The retinoblastoma protein in castrate-resistant prostate cancer. Nat Rev Urol. 2011;8:562–8. doi: 10.1038/nrurol.2011.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Udayakumar T, Shareef MM, Diaz DA, Ahmed MM, Pollack A. The E2F1/Rb and p53/MDM2 pathways in DNA repair and apoptosis: understanding the crosstalk to develop novel strategies for prostate cancer radiotherapy. Semin Radiat Oncol. 2010;20:258–66. doi: 10.1016/j.semradonc.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 19.Tzelepi V, Zhang J, Lu JF, Kleb B, Wu G, Wan X, et al. Modeling a lethal prostate cancer variant with small-cell carcinoma features. Clinical cancer research. 2012;18:666–77. doi: 10.1158/1078-0432.CCR-11-1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lotan TL, Gurel B, Sutcliffe S, Esopi D, Liu W, Xu J, et al. PTEN protein loss by immunostaining: analytic validation and prognostic indicator for a high risk surgical cohort of prostate cancer patients. Clinical cancer research. 2011;17:6563–73. doi: 10.1158/1078-0432.CCR-11-1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Verras M, Lee J, Xue H, Li TH, Wang Y, Sun Z. The androgen receptor negatively regulates the expression of c-Met: implications for a novel mechanism of prostate cancer progression. Cancer research. 2007;67:967–75. doi: 10.1158/0008-5472.CAN-06-3552. [DOI] [PubMed] [Google Scholar]
- 22.Yu J, Akishita M, Eto M, Koizumi H, Hashimoto R, Ogawa S, et al. Src kinase-mediates androgen receptor-dependent non-genomic activation of signaling cascade leading to endothelial nitric oxide synthase. Biochem Biophys Res Commun. 2012;424:538–43. doi: 10.1016/j.bbrc.2012.06.151. [DOI] [PubMed] [Google Scholar]
- 23.Liu Y, Karaca M, Zhang Z, Gioeli D, Earp HS, Whang YE. Dasatinib inhibits site-specific tyrosine phosphorylation of androgen receptor by Ack1 and Src kinases. Oncogene. 2010;29:3208–16. doi: 10.1038/onc.2010.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gioeli D, Ficarro SB, Kwiek JJ, Aaronson D, Hancock M, Catling AD, et al. Androgen receptor phosphorylation. Regulation and identification of the phosphorylation sites. The Journal of biological chemistry. 2002;277:29304–14. doi: 10.1074/jbc.M204131200. [DOI] [PubMed] [Google Scholar]
- 25.Ponguta LA, Gregory CW, French FS, Wilson EM. Site-specific androgen receptor serine phosphorylation linked to epidermal growth factor-dependent growth of castration-recurrent prostate cancer. The Journal of biological chemistry. 2008;283:20989–1001. doi: 10.1074/jbc.M802392200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nilsson S, Strang P, Aksnes AK, Franzen L, Olivier P, Pecking A, et al. A randomized, dose-response, multicenter phase II study of radium-223 chloride for the palliation of painful bone metastases in patients with castration-resistant prostate cancer. European journal of cancer. 2012;48:678–86. doi: 10.1016/j.ejca.2011.12.023. [DOI] [PubMed] [Google Scholar]
- 27.Shelley MD, Mason MD. Radium-223 for men with hormone-refractory prostate cancer and bone metastases. Lancet Oncol. 2007;8:564–5. doi: 10.1016/S1470-2045(07)70180-8. [DOI] [PubMed] [Google Scholar]
- 28.Tu SM, Millikan RE, Mengistu B, Delpassand ES, Amato RJ, Pagliaro LC, et al. Bone-targeted therapy for advanced androgen-independent carcinoma of the prostate: a randomised phase II trial. Lancet. 2001;357:336–41. doi: 10.1016/S0140-6736(00)03639-4. [DOI] [PubMed] [Google Scholar]
- 29.Russell DW, Wilson JD. Steroid 5 alpha-reductase: two genes/two enzymes. Annu Rev Biochem. 1994;63:25–61. doi: 10.1146/annurev.bi.63.070194.000325. [DOI] [PubMed] [Google Scholar]
- 30.Sharifi N. The 5alpha-androstanedione pathway to dihydrotestosterone in castration-resistant prostate cancer. Journal of investigative medicine. 2012;60:504–7. doi: 10.231/JIM.0b013e31823874a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thompson IM, Goodman PJ, Tangen CM, Lucia MS, Miller GJ, Ford LG, et al. The influence of finasteride on the development of prostate cancer. The New England journal of medicine. 2003;349:215–24. doi: 10.1056/NEJMoa030660. [DOI] [PubMed] [Google Scholar]
- 32.Andriole GL, Bostwick DG, Brawley OW, Gomella LG, Marberger M, Montorsi F, et al. Effect of dutasteride on the risk of prostate cancer. The New England journal of medicine. 2010;362:1192–202. doi: 10.1056/NEJMoa0908127. [DOI] [PubMed] [Google Scholar]
- 33.Fleshner NE, Lucia MS, Egerdie B, Aaron L, Eure G, Nandy I, et al. Dutasteride in localised prostate cancer management: the REDEEM randomised, double-blind, placebo-controlled trial. Lancet. 2012;379:1103–11. doi: 10.1016/S0140-6736(11)61619-X. [DOI] [PubMed] [Google Scholar]
- 34.Roehrborn CG, Andriole GL, Wilson TH, Castro R, Rittmaster RS. Effect of dutasteride on prostate biopsy rates and the diagnosis of prostate cancer in men with lower urinary tract symptoms and enlarged prostates in the Combination of Avodart and Tamsulosin trial. European urology. 2011;59:244–9. doi: 10.1016/j.eururo.2010.10.040. [DOI] [PubMed] [Google Scholar]
- 35.Lin TM, Chang C. Cloning and characterization of TDD5, an androgen target gene that is differentially repressed by testosterone and dihydrotestosterone. Proc Natl Acad Sci U S A. 1997;94:4988–93. doi: 10.1073/pnas.94.10.4988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dadras SS, Cai X, Abasolo I, Wang Z. Inhibition of 5alpha-reductase in rat prostate reveals differential regulation of androgen-response gene expression by testosterone and dihydrotestosterone. Gene Expr. 2001;9:183–94. doi: 10.3727/000000001783992551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li J, Ding Z, Wang Z, Lu JF, Maity SN, Navone NM, et al. Androgen regulation of 5alpha-reductase isoenzymes in prostate cancer: implications for prostate cancer prevention. PLoS One. 2011;6:e28840. doi: 10.1371/journal.pone.0028840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mitsiades N, Sung CC, Schultz N, Danila DC, He B, Eedunuri VK, et al. Distinct patterns of dysregulated expression of enzymes involved in androgen synthesis and metabolism in metastatic prostate cancer tumors. Cancer research. 2012;72:6142–52. doi: 10.1158/0008-5472.CAN-12-1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mostaghel EA, Geng L, Holcomb I, Coleman IM, Lucas J, True LD, et al. Variability in the androgen response of prostate epithelium to 5alpha-reductase inhibition: implications for prostate cancer chemoprevention. Cancer research. 2010;70:1286–95. doi: 10.1158/0008-5472.CAN-09-2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim J, Amos CI, Logothetis C. 5alpha-Reductase inhibitors for prostate-cancer prevention. The New England journal of medicine. 2011;365:2340. doi: 10.1056/NEJMc1112146. [DOI] [PubMed] [Google Scholar]
- 41.Shen MM, Abate-Shen C. Molecular genetics of prostate cancer: new prospects for old challenges. Genes & development. 2010;24:1967–2000. doi: 10.1101/gad.1965810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gallick GE, Corn PG, Zurita AJ, Lin SH. Small-molecule protein tyrosine kinase inhibitors for the treatment of metastatic prostate cancer. Future Med Chem. 2012;4:107–19. doi: 10.4155/fmc.11.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deocampo ND, Huang H, Tindall DJ. The role of PTEN in the progression and survival of prostate cancer. Minerva Endocrinol. 2003;28:145–53. [PubMed] [Google Scholar]
- 44.Zhu ML, Kyprianou N. Role of androgens and the androgen receptor in epithelial-mesenchymal transition and invasion of prostate cancer cells. FASEB journal. 2010;24:769–77. doi: 10.1096/fj.09-136994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jennbacken K, Tesan T, Wang W, Gustavsson H, Damber JE, Welen K. N-cadherin increases after androgen deprivation and is associated with metastasis in prostate cancer. Endocr Relat Cancer. 2010;17:469–79. doi: 10.1677/ERC-10-0015. [DOI] [PubMed] [Google Scholar]
- 46.Lee YC, Cheng CJ, Huang M, Bilen MA, Ye X, Navone NM, et al. Androgen depletion up-regulates cadherin-11 expression in prostate cancer. J Pathol. 2010;221:68–76. doi: 10.1002/path.2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Taplin ME, Balk SP. Androgen receptor: a key molecule in the progression of prostate cancer to hormone independence. Journal of cellular biochemistry. 2004;91:483–90. doi: 10.1002/jcb.10653. [DOI] [PubMed] [Google Scholar]
- 48.Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. The New England journal of medicine. 2012;367:1187–97. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 49.Zhao XY, Malloy PJ, Krishnan AV, Swami S, Navone NM, Peehl DM, et al. Glucocorticoids can promote androgen-independent growth of prostate cancer cells through a mutated androgen receptor. Nat Med. 2000;6:703–6. doi: 10.1038/76287. [DOI] [PubMed] [Google Scholar]
- 50.Andersen RJ, Mawji NR, Wang J, Wang G, Haile S, Myung JK, et al. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell. 2010;17:535–46. doi: 10.1016/j.ccr.2010.04.027. [DOI] [PubMed] [Google Scholar]
- 51.Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009;69:16–22. doi: 10.1158/0008-5472.CAN-08-2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hu R, Lu C, Mostaghel EA, Yegnasubramanian S, Gurel M, Tannahill C, et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer research. 2012;72:3457–62. doi: 10.1158/0008-5472.CAN-11-3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, Mostaghel EA, et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. The Journal of clinical investigation. 2010;120:2715–30. doi: 10.1172/JCI41824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cai C, He HH, Chen S, Coleman I, Wang H, Fang Z, et al. Androgen receptor gene expression in prostate cancer is directly suppressed by the androgen receptor through recruitment of lysine-specific demethylase 1. Cancer Cell. 2011;20:457–71. doi: 10.1016/j.ccr.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tanaka H, Kono E, Tran CP, Miyazaki H, Yamashiro J, Shimomura T, et al. Monoclonal antibody targeting of N-cadherin inhibits prostate cancer growth, metastasis and castration resistance. Nat Med. 2010;16:1414–20. doi: 10.1038/nm.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Smith DC, Smith MR, Sweeney C, Elfiky AA, Logothetis C, Corn PG, et al. Cabozantinib in Patients With Advanced Prostate Cancer: Results of a Phase II Randomized Discontinuation Trial. Journal of clinical oncology. 2013;31:412–9. doi: 10.1200/JCO.2012.45.0494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cai H, Babic I, Wei X, Huang J, Witte ON. Invasive prostate carcinoma driven by c-Src and androgen receptor synergy. Cancer research. 2011;71:862–72. doi: 10.1158/0008-5472.CAN-10-1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gleave M, Hsieh JT, Gao CA, von Eschenbach AC, Chung LW. Acceleration of human prostate cancer growth in vivo by factors produced by prostate and bone fibroblasts. Cancer research. 1991;51:3753–61. [PubMed] [Google Scholar]
- 59.Wu TT, Sikes RA, Cui Q, Thalmann GN, Kao C, Murphy CF, et al. Establishing human prostate cancer cell xenografts in bone: induction of osteoblastic reaction by prostate-specific antigen-producing tumors in athymic and SCID/bg mice using LNCaP and lineage-derived metastatic sublines. International journal of cancer. 1998;77:887–94. doi: 10.1002/(sici)1097-0215(19980911)77:6<887::aid-ijc15>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 60.Thalmann GN, Sikes RA, Wu TT, Degeorges A, Chang SM, Ozen M, et al. LNCaP progression model of human prostate cancer: androgen-independence and osseous metastasis. The Prostate. 2000 Jul 1;44(2):91–103. doi: 10.1002/1097-0045(20000701)44:2<91::aid-pros1>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 61.Lee YC, Cheng CJ, Bilen MA, Lu JF, Satcher RL, Yu-Lee LY, et al. BMP4 promotes prostate tumor growth in bone through osteogenesis. Cancer research. 2011;71:5194–203. doi: 10.1158/0008-5472.CAN-10-4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dai J, Keller J, Zhang J, Lu Y, Yao Z, Keller ET. Bone morphogenetic protein-6 promotes osteoblastic prostate cancer bone metastases through a dual mechanism. Cancer Res. 2005;65:8274–85. doi: 10.1158/0008-5472.CAN-05-1891. [DOI] [PubMed] [Google Scholar]
- 63.Dai J, Kitagawa Y, Zhang J, Yao Z, Mizokami A, Cheng S, et al. Vascular endothelial growth factor contributes to the prostate cancer-induced osteoblast differentiation mediated by bone morphogenetic protein. Cancer research. 2004;64:994–9. doi: 10.1158/0008-5472.can-03-1382. [DOI] [PubMed] [Google Scholar]
- 64.Kitagawa Y, Dai J, Zhang J, Keller JM, Nor J, Yao Z, et al. Vascular endothelial growth factor contributes to prostate cancer-mediated osteoblastic activity. Cancer research. 2005;65:10921–9. doi: 10.1158/0008-5472.CAN-05-1809. [DOI] [PubMed] [Google Scholar]
- 65.Li ZG, Mathew P, Yang J, Starbuck MW, Zurita AJ, Liu J, et al. Androgen receptor-negative human prostate cancer cells induce osteogenesis in mice through FGF9-mediated mechanisms. J Clin Invest. 2008;118:2697–710. doi: 10.1172/JCI33093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen N, Ye XC, Chu K, Navone NM, Sage EH, Yu-Lee LY, et al. A secreted isoform of ErbB3 promotes osteonectin expression in bone and enhances the invasiveness of prostate cancer cells. Cancer research. 2007;67:6544–8. doi: 10.1158/0008-5472.CAN-07-1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jacob K, Webber M, Benayahu D, Kleinman HK. Osteonectin promotes prostate cancer cell migration and invasion: a possible mechanism for metastasis to bone. Cancer research. 1999;59:4453–7. [PubMed] [Google Scholar]
- 68.Chen G, Sircar K, Aprikian A, Potti A, Goltzman D, Rabbani SA. Expression of RANKL/RANK/OPG in primary and metastatic human prostate cancer as markers of disease stage and functional regulation. Cancer. 2006;107:289–98. doi: 10.1002/cncr.21978. [DOI] [PubMed] [Google Scholar]
- 69.Jung K, Lein M, Stephan C, Von Hosslin K, Semjonow A, Sinha P, et al. Comparison of 10 serum bone turnover markers in prostate carcinoma patients with bone metastatic spread: diagnostic and prognostic implications. International journal of cancer. 2004;111:783–91. doi: 10.1002/ijc.20314. [DOI] [PubMed] [Google Scholar]
- 70.Saad F, Lipton A. Bone-marker levels in patients with prostate cancer: potential correlations with outcomes. Curr Opin Support Palliat Care. 2010;4:127–34. doi: 10.1097/SPC.0b013e32833ac6d6. [DOI] [PubMed] [Google Scholar]
- 71.Oreffo RO, Mundy GR, Seyedin SM, Bonewald LF. Activation of the bone-derived latent TGF beta complex by isolated osteoclasts. Biochem Biophys Res Commun. 1989;158:817–23. doi: 10.1016/0006-291x(89)92795-2. [DOI] [PubMed] [Google Scholar]
- 72.Abrahamsson PA. Neuroendocrine differentiation in prostatic carcinoma. Prostate. 1999;39:135–48. doi: 10.1002/(sici)1097-0045(19990501)39:2<135::aid-pros9>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 73.Clegg N, Ferguson C, True LD, Arnold H, Moorman A, Quinn JE, et al. Molecular characterization of prostatic small-cell neuroendocrine carcinoma. Prostate. 2003;55:55–64. doi: 10.1002/pros.10217. [DOI] [PubMed] [Google Scholar]
- 74.Tetu B, Ro JY, Ayala AG, Johnson DE, Logothetis CJ, Ordonez NG. Small cell carcinoma of the prostate. Part I. A clinicopathologic study of 20 cases. Cancer. 1987;59:1803–9. doi: 10.1002/1097-0142(19870515)59:10<1803::aid-cncr2820591019>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 75.Humphrey PA. Histological variants of prostatic carcinoma and their significance. Histopathology. 2012;60:59–74. doi: 10.1111/j.1365-2559.2011.04039.x. [DOI] [PubMed] [Google Scholar]
- 76.Aparicio A, Tzelepi V, Araujo JC, Guo CC, Liang S, Troncoso P, et al. Neuroendocrine prostate cancer xenografts with large-cell and small-cell features derived from a single patient’s tumor: morphological, immunohistochemical, and gene expression profiles. The Prostate. 2011;71:846–56. doi: 10.1002/pros.21301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Guo CC, Wang Y, Xiao L, Troncoso P, Czerniak BA. The relationship of TMPRSS2-ERG gene fusion between primary and metastatic prostate cancers. Human pathology. 2012;43:644–9. doi: 10.1016/j.humpath.2011.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mosquera JM, Mehra R, Regan MM, Perner S, Genega EM, Bueti G, et al. Prevalence of TMPRSS2-ERG fusion prostate cancer among men undergoing prostate biopsy in the United States. Clinical cancer research. 2009;15:4706–11. doi: 10.1158/1078-0432.CCR-08-2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Aparicio A, Logothetis CJ, Maity SN. Understanding the lethal variant of prostate cancer: power of examining extremes. Cancer discovery. 2011;1:466–8. doi: 10.1158/2159-8290.CD-11-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Beltran H, Rickman DS, Park K, Chae SS, Sboner A, MacDonald TY, et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer discovery. 2011;1:487–95. doi: 10.1158/2159-8290.CD-11-0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Qin J, Liu X, Laffin B, Chen X, Choy G, Jeter CR, et al. The PSA(-/lo) prostate cancer cell population harbors self-renewing long-term tumor-propagating cells that resist castration. Cell Stem Cell. 2012;10:556–69. doi: 10.1016/j.stem.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Deeraksa A, Pan J, Sha Y, Liu XD, Eissa NT, Lin SH, et al. Plk1 is upregulated in androgen-insensitive prostate cancer cells and its inhibition leads to necroptosis. Oncogene. 2012 doi: 10.1038/onc.2012.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mosquera JM, Beltran H, Park K, Macdonald TY, Robinson BD, Tagawa ST, et al. Concurrent AURKA and MYCN Gene Amplifications Are Harbingers of Lethal Treatment-Related Neuroendocrine Prostate Cancer. Neoplasia. 2013;15:1–10. doi: 10.1593/neo.121550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Otto T, Horn S, Brockmann M, Eilers U, Schuttrumpf L, Popov N, et al. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell. 2009;15:67–78. doi: 10.1016/j.ccr.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 85.Lens SM, Voest EE, Medema RH. Shared and separate functions of polo-like kinases and aurora kinases in cancer. Nature reviews Cancer. 2010;10:825–41. doi: 10.1038/nrc2964. [DOI] [PubMed] [Google Scholar]
- 86.Weichert W, Schmidt M, Gekeler V, Denkert C, Stephan C, Jung K, et al. Polo-like kinase 1 is overexpressed in prostate cancer and linked to higher tumor grades. The Prostate. 2004;60:240–5. doi: 10.1002/pros.20050. [DOI] [PubMed] [Google Scholar]
- 87.Liu XS, Song B, Elzey BD, Ratliff TL, Konieczny SF, Cheng L, et al. Polo-like kinase 1 facilitates loss of Pten tumor suppressor-induced prostate cancer formation. The Journal of biological chemistry. 2011;286:35795–800. doi: 10.1074/jbc.C111.269050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang W, Epstein JI. Small cell carcinoma of the prostate. A morphologic and immunohistochemical study of 95 cases. Am J Surg Pathol. 2008;32:65–71. doi: 10.1097/PAS.0b013e318058a96b. [DOI] [PubMed] [Google Scholar]
- 89.Meulenbeld HJ, Bleuse JP, Vinci EM, Raymond E, Vitali G, Santoro A, et al. Randomized phase II study of danusertib in patients with metastatic castration-resistant prostate cancer after docetaxel failure. BJU international. 2012 doi: 10.1111/j.1464-410X.2012.11404.x. [DOI] [PubMed] [Google Scholar]
- 90.Reagan-Shaw S, Ahmad N. Polo-like kinase (Plk) 1 as a target for prostate cancer management. IUBMB Life. 2005;57:677–82. doi: 10.1080/15216540500305910. [DOI] [PubMed] [Google Scholar]
- 91.Luo J, Liu X. Polo-like kinase 1, on the rise from cell cycle regulation to prostate cancer development. Protein Cell. 2012;3:182–97. doi: 10.1007/s13238-012-2020-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Eckerdt F, Yuan J, Strebhardt K. Polo-like kinases and oncogenesis. Oncogene. 2005;24:267–76. doi: 10.1038/sj.onc.1208273. [DOI] [PubMed] [Google Scholar]
- 93.Schoffski P, Blay JY, De Greve J, Brain E, Machiels JP, Soria JC, et al. Multicentric parallel phase II trial of the polo-like kinase 1 inhibitor BI 2536 in patients with advanced head and neck cancer, breast cancer, ovarian cancer, soft tissue sarcoma and melanoma. The first protocol of the European Organization for Research and Treatment of Cancer (EORTC) Network Of Core Institutes (NOCI) European journal of cancer. 2010;46:2206–15. doi: 10.1016/j.ejca.2010.03.039. [DOI] [PubMed] [Google Scholar]
- 94.Schoffski P, Awada A, Dumez H, Gil T, Bartholomeus S, Wolter P, et al. A phase I, dose-escalation study of the novel Polo-like kinase inhibitor volasertib (BI 6727) in patients with advanced solid tumours. European journal of cancer. 2012;48:179–86. doi: 10.1016/j.ejca.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 95.Beltran H, Rubin MA. New strategies in prostate cancer: translating genomics into the clinic. Clinical cancer research. 2013;19:517–23. doi: 10.1158/1078-0432.CCR-12-1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sowalsky AG, Ye H, Bubley GJ, Balk SP. Clonal progression of prostate cancers from Gleason grade 3 to grade 4. Cancer research. 2013;73:1050–5. doi: 10.1158/0008-5472.CAN-12-2799. [DOI] [PMC free article] [PubMed] [Google Scholar]