

Abstract
Alport syndrome (AS) is a heterogeneous basement membrane disease characterised by haematuria with progressive hereditary nephritis, high-frequency sensorineural hearing loss (SNHL) and pathognomonic ocular lesions. It is one of the spectra of diseases representing hereditary nephritis, which inevitably leads to end-stage renal disease (ESRD). Microscopic or frank haematuria persistent from childhood constitutes the clinical clue for its early recognition. It occurs as a result of genetically inherited or de novo mutations in type IV collagen genes. The most common mode of inheritance is X-linked and men are more severely affected. We report a case of a young woman, in her fourth decade of life presenting with overt nephropathy, having persistent haematuria associated with SNHL and lenticonus with dot and fleck retinopathy on detailed clinical examination, diagnosed as a previously undetected case of Alport syndrome.
Background
Alport syndrome (AS) is a rare disease of ultrastructural collagen abnormality responsible for 0.2% of all causes of end-stage renal disease (ESRD).1 2
Female participants are rarely affected except in cases of lyonisation of sex chromosomes in an X-linked inheritance (XLAS), which is the most common type (85%). In the rarer autosomal recessive AS (ARAS) or autosomal dominant AS (ADAS), both sexes are equally affected.
Women are less frequently affected with hearing loss.
Anterior lenticonus is a hallmark for ocular manifestation of AS. However, bilateral anterior as well as posterior lenticonus is a unique observation in classical AS, previously documented by very few reports in medical literature.
Case presentation
A 35-year-old woman presented to the medical emergency department with low-grade fever for 3 weeks, vomiting for 1 week and anuria for 3 days. She also reported dysuria and breathlessness for 1 week. There was no history of decreased urine output, dialysis, effort intolerance, chest pain or palpitation, dyspnoea and weight loss. Menstrual history was within normal limit but she reported gradually progressive loss of appetite. Family history included smoky urine in her younger brother in his childhood, who died in an accident.
On general survey, the patient was conscious and alert. She was dyspnoeic and febrile. Severe pallor was present with mild pedal oedema. Blood pressure was 180/100 mm Hg and pulse rate of 116/min regular. No evidence of jaundice, clubbing cyanosis or lymphadenopathy was found. Physical examination revealed bibasilar end-inspiratory crepitations in lungs and suprapubic tenderness. There was no hepatosplenomegaly or ascites. Cardiac examination was normal. She was found to have severe bilateral hearing loss, which was gradually progressive for 5 years. The fundi were bilaterally pale.
The patient was referred to the department of ophthalmology for a comprehensive eye examination. Her visual acuity was documented as 6/18 in both eyes with no obvious lenticular opacity. Slit-lamp examination showed bilateral anterior lentiglobus (figure 1) with posterior lenticonus (figure 2). Distant direct ophthalmoscopy revealed oil droplet sign (a suggestive confirmation of the presence of lenticonus); and peripheral retina revealed multiple yellowish white lesion-like flecks in the mid-periphery, and few blot haemorrhages indicative of hypertensive changes (figures 3 and 4).
Figure 1.
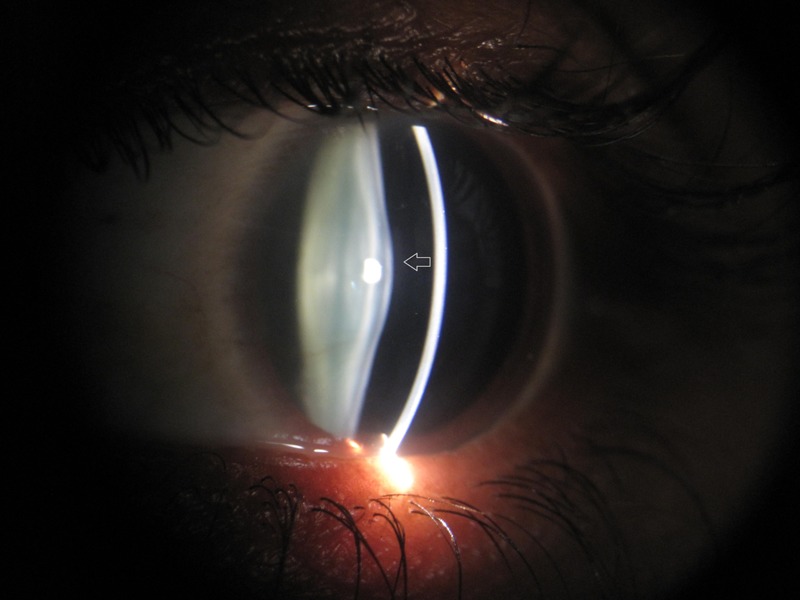
Slit-lamp examination of the left eye showing anterior lentiglobus (grey arrow).
Figure 2.
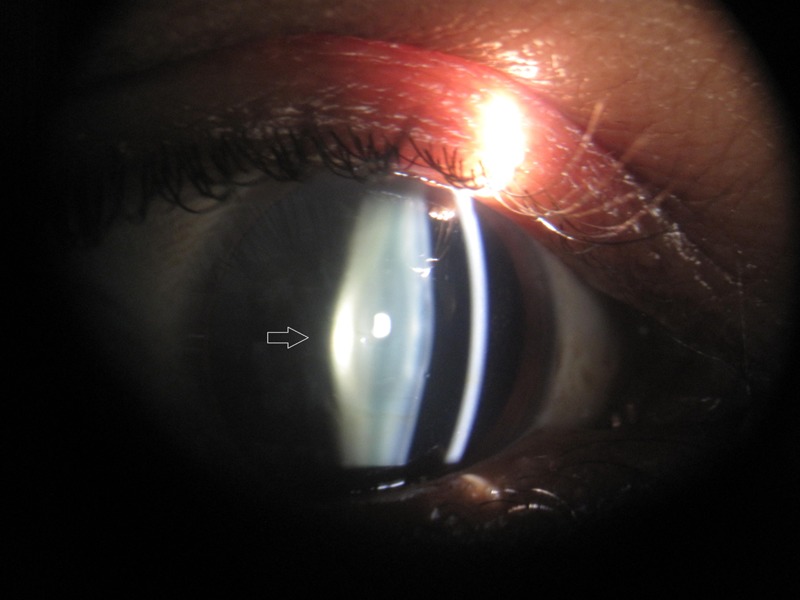
Slit-lamp examination of the right eye showing posterior lenticonus (grey arrow).
Figure 3.
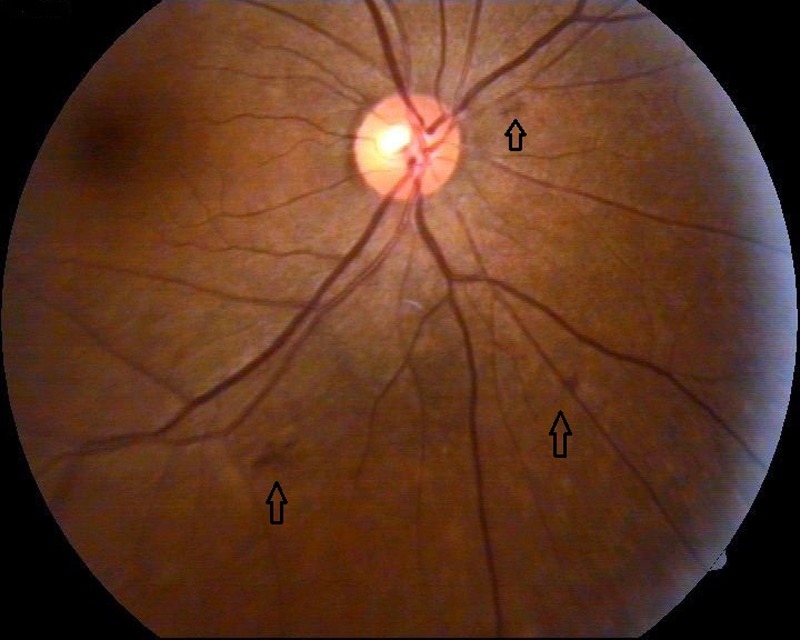
Fundus examination of the right eye demonstrating peri-macular flecks along with few blot haemorrhages (black arrow).
Figure 4.
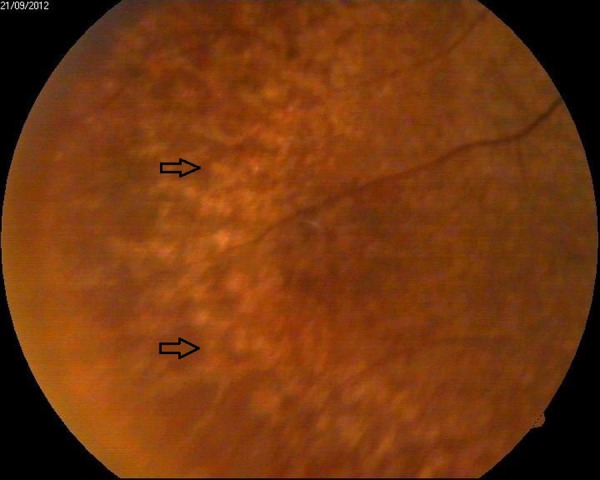
Indirect ophthalmoscopy depicting typical yellowish mid-periphery retinal flecks (black arrow).
Investigations
Haemogram showed haemoglobin (Hb) 5.7 g/dL, erythrocyte sedimentation rate 15 mm in first hour, white cell count 17 200/µL with 82% polymorphs, adequate platelets and mean corpuscular volume 83.3 fL. Peripheral smear showed normocytic normochromic anaemia with mild anisocytosis. Fasting sugar 78 mg/dL, blood urea 325 mg/dL, serum creatinine 11.2 mg/dL and uric acid 8.3 mg/dL. Liver function tests were within normal limit as were serum electrolytes, except serum calcium (conc.) 5.8 mg/dL (adjusted with serum albumin). Lipid profile and iron profile were also normal. HIV and viral markers for HbsAg and hepatitis C virus were non-reactive. ECG showed sinus tachycardia with features of left ventricular hypertrophy and chest X-ray posteroanterior view revealed cardiomegaly. Urinalysis showed full field of pus cells with 35–40 RBCs/hpf and 3(+) proteinuria. Urine samples for cultures were sent which reported pure growth of Escherichia coli. Spot urine for protein:creatinine ratio was 2.07 g/g Cr.
She underwent pure tone audiometry which revealed features suggestive of severe bilateral sensorineural hearing loss (SHNL). Ultrasound of the abdomen showed bilateral contracted kidneys: right measured 6.7×2.3 cm and left 7.8×3 cm, with increased cortical echogenecity and loss of corticomedullary differentiation, suggestive of medical renal disease. Two-dimensional Echo reported dilated left ventricular cavity with mild mitral regurgitation and ejection fraction of 55%.
Renal and skin biopsies were conducted and specimens were sent for light and electron microscopy (EM). Renal tissue on H&E stain (figure 5) was reported as focal segmental glomerulonephritis (FSGS). Ultrathin sections of EM study of renal tissue revealed disruption of glomerular basement membrane (GBM) with diffuse thickening of glomerular capillary wall (figure 6). Dermal tissue depicted discontinuity of lamina densa with basket weaving pattern under EM (figures 7 and 8).
Figure 5.
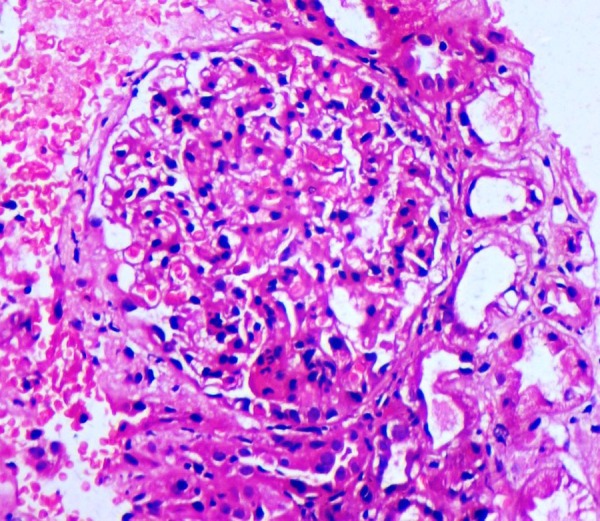
Light microscopy with H&E staining, suggestive of focal segmental glomerulonephritis.
Figure 6.
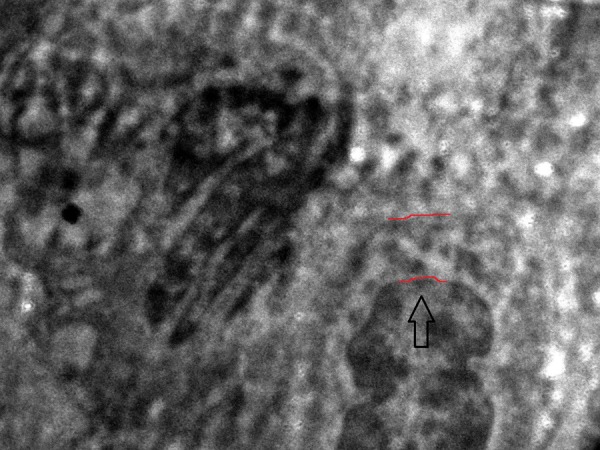
Ultrathin section of electron microscopic view of glomerular basement membrane (black arrow) showing focal disruption with diffuse thickening of glomerular capillary wall (as outlined by red borders).
Figure 7.
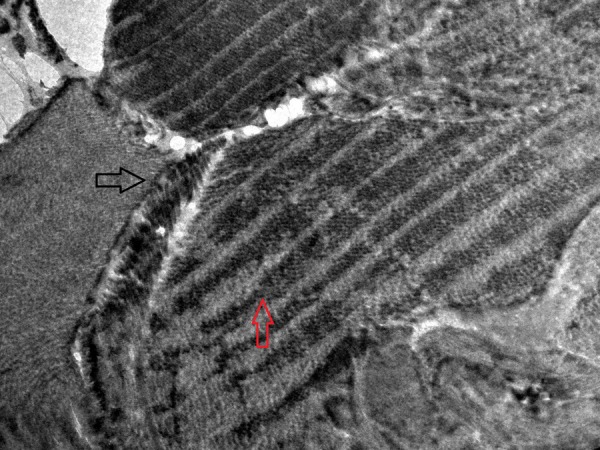
Electron microscopic study of patient's skin tissue depicting discontinuity of lamina densa (black arrow) with basket weave appearance (red arrow).
Figure 8.
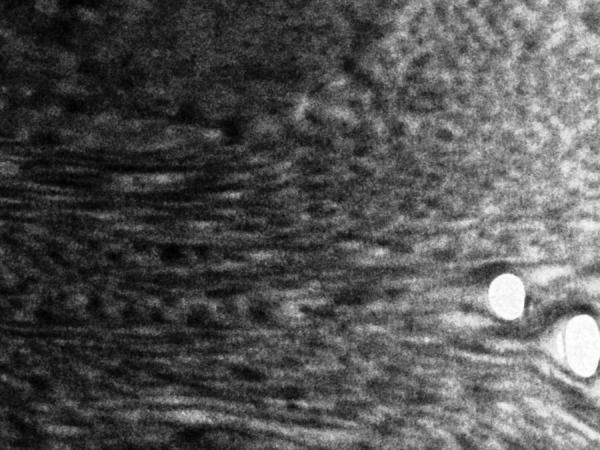
Normal dermal collagen as viewed under electron microscope.
Differential diagnosis
Thus a diagnosis of XLAS, associated with urinary tract infection and acute kidney injury on ESRD was established. The above diagnosis of AS was substantiated on the basis of early renal involvement, typical ocular signs with bilateral SNHL, a family history of a severely affected brother and histological/EM findings.
The differential diagnoses considered were:
Thin basement membrane disease (TBMD)
Mesangial IgA nephropathy
Drug-induced renal and ototoxicity (eg, aminoglycosides)
Branchio-otorenal syndrome.
TBMD presents as an asymptomatic microscopic haematuria since childhood, as a differential to AS, without any other systemic manifestations. It has a benign course and progression to ESRD is unlikely. Pathologically, it is confirmed by EM, demonstrating thinning of GBM to 150–200 nm. Although AS and TBMD are believed to be COL4A nephropathy, a family history of renal failure or hearing loss, or sex predilection, is not found in the latter. However, it has been ascertained that TBMD can be a manifestation of a carrier state of ARAS in certain kindred.3 Similarly, IgA nephropathy may present with acute renal failure and haematuria, often following a sore throat, with no family history and characteristic renal biopsy picture of mesangial proliferation and IgA deposits on immunofluorescence. Aberrant branchial cleft and auricular abnormality are evident in branchio-oto-renal syndrome.
Treatment
She was given urgent haemodialysis on two consecutive days and started with injections piperacillin and tazobactam according to culture sensitivity reports, and other supportive medications for acute-on-chronic kidney disease. Even after successful treatment of the underlying urinary tract infection and improvement from the status of uraemia, the patient persistently had 30–40 RBCs/hpf and a nephrotic range proteinuria.
Outcome and follow-up
After dialysis and supportive care treatment, from inpatient department, her creatinine level declined, and was maintained at 6.3 mg/dL at the time of discharge.
She was advised regular maintenance haemodialysis as a bridging therapy for her ESRD status, while waiting and preparing for an HLA-matched renal transplant.
Discussion
AS was first identified in a British family by Dr Cecil A Alport in 1927.4 However, it was William Howship Dickinson who made significant contributions for diagnostic characterisation of the disease5 and proposed that at least three of the following four criteria to establish the diagnosis of AS including family history of haematuria, evidence of GBM thickening and splitting in kidneys, progressive, high-tone SNHL and anterior lenticonus with retinal flecks; a disease complex attributed to basement membrane structural defect.6
In 1996, Gregory et al7 revised the criteria for its diagnosis, for which at least 4 of the following 10 criteria should be fulfilled:
A family history of nephritis in a first-degree relative male linked to the index case.
A history of persistent haematuria.
Bilateral SNHL involving higher frequencies.
Widespread GBM ultrastructural abnormalities.
Ocular findings such as anterior lenticonus and retinal flecks.
Mutation in COL4A gene.
Immunohistochemical evidence of partial or complete loss of Alport epitope.
Gradual progression to ESRD in at least relatives of index case.
Macrothrombocytopenia.
Diffuse leiomyomatosis.
Our patient satisfied the former five criteria for diagnosing AS. Molecular and genetic analysis for Alport epitope or type IV collagen genes could not be carried out due to lack of facility.
The prevalence of Alport syndrome has been estimated at 1:10 000 live-births for XLAS and 1:50 000 live-births for ARAS.8 The median age of onset of ESRD due to AS in young untreated patients has been reported to be 22 years.9 10 The prevalence of ESRD in women, were although, considered a rarity, but as many as 12% of them with AS may progress to ESRD by the age of 40.11
Approximately 60–70% of heterozygous women exhibit discontinuous staining of the collagen α5 (IV) chain in the skin biopsy specimen when incubated with monoclonal antibody against collagen. EM of dermal tissue also showed discontinuity of lamina densa as described by Kuroki et al.12 We present a similar case of a female patient of AS with probably XLAS, showing discontinuous disposition of collagen pattern of skin EM, supporting possible lyonisation in her sex chromosomes. This unique observation is emphasised in the present study.
Anterior lenticonus, corneal posterior polymorphous dystrophy and retinal flecks in the macula and mid-peripheral fundus are the commonest and specific ocular findings of AS.13 In the present case, yellowish-white flecks were noted in bilateral mid-periphery of the fundus and also extending to peripheral retina, which have been less frequently reported. The flecks in the macula and mid-periphery probably have separate pathogenesis.14 Visualisation of ‘oil droplet sign’, a real-time optical sign clinically confirmed anterior lenticonus. Presence of anterior and posterior lenticonus simultaneously, as evident in our case by slit-lamp examination, is also a rare manifestation of AS, as suggested by Vedantham et al.15
Disease management entails prescribing ACE inhibitor drugs to decrease proteinuria, renal transplantation and hearing aids. As our patient already had ESRD with nephrotic range proteinuria, ACE inhibitors were avoided. Renal transplantation was advocated in our patient to achieve a better quality of life as she was only in her 30s, anticipating an excellent allograft survival as compared with general population.
On contrary, renal transplant is unsuccessful in those who develop rejection reaction secondary to post-transplant anti-GBM nephritis. It especially occurs in male patients of AS who present with ESRD and deafness early (within the second decade of life) and have detectable circulating anti-GBM antibodies16 due to absolute lack of the normal ‘Alport antigen’.17 A very low incidence of clinical anti-GBM disease18 is seen in female patients as they have one normal allele due to their inherent heterozygosity of the mutational chromosomal defect.
Learning points.
Unrelated multiorgan system pathology on clinical examination merits a syndromic approach.
Young individuals presenting with progressive renal failure or end-stage renal disease may often have a genetic aetiology.
Although males are more severely and frequently affected, renal failure due to Alport syndrome can also occur in young women.
The role of an internist is prudent in the diagnosis of such interdisciplinary pathological entities.
Footnotes
Contributors: SG did the workup of the case. MS described the ocular changes. RS managed the case and SR wroteup the manuscript.
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Heidet L, Gubler MC. The renal lesions of Alport syndrome. J Am Soc Nephrol 2009;20:1210–15 [DOI] [PubMed] [Google Scholar]
- 2.Williamson DA. Alport syndrome of hereditary nephritis with deafness. Lancet 1961;2:1321–3 [DOI] [PubMed] [Google Scholar]
- 3.Buzza M, Wang YY, Dagher H, et al. COL 4A4 mutation in thin basement membrane disease previously described in Alport syndrome. Kidney Int 2001;60:480–3 [DOI] [PubMed] [Google Scholar]
- 4.Alport AC. Hereditary familial congenital haemorrhagic nephritis. BMJ (London) 1927;I:504–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lagona E, Tsartsali L, Kostaridou S, et al. Skin biopsy for the diagnosis of Alport syndrome. Hippokratia 2008;12:116–18 [PMC free article] [PubMed] [Google Scholar]
- 6.Kashtan CE. Familial haematuria. Pediatr Nephrol 2009;24:1951–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gregory MC, Terreros DA, Barker DF, et al. Alport syndrome-clinical phenotypes, incidence, and pathology. Contrib Nephrol 1996;117:1–28 [DOI] [PubMed] [Google Scholar]
- 8.Levy M, Feingold J. Estimating prevalence in single-gene kidney diseases progressing to renal failure. Kidney Int 2000;58:925–43 [DOI] [PubMed] [Google Scholar]
- 9.Flinter FA, Cameron JS, Chantler C, et al. Genetics of classic Alport syndrome. Lancet 1988;2:1005–7 [DOI] [PubMed] [Google Scholar]
- 10.Gross O, Licht C, Anders HJ, et al. Early angiotensin-converting enzyme inhibition in Alport syndrome delays renal failure and improves life expectancy. Kidney Int 2012;81:494–501 [DOI] [PubMed] [Google Scholar]
- 11.Kashtan CE. Alport syndrome and thin basement membrane nephropathy. 2001. [Updated 2013 Feb 28] [PubMed]
- 12.Kuroki A, Ito J, Yokochi A, et al. Diagnosing Alport syndrome using electron microscopy of the skin. Kidney Int 2008;73:364–5 [DOI] [PubMed] [Google Scholar]
- 13.Vanderschueren A, Stalmans I, Lombaerts R, et al. Ocular abnormalities in patients with Alport syndrome. Nephrol Dial Transplant 1996;11:2355–8 [DOI] [PubMed] [Google Scholar]
- 14.Jayaprasad B, Sathish KR, Chandrasekhar N, et al. Alport syndrome: a case report. IJO 1992;42:211–12 [PubMed] [Google Scholar]
- 15.Vedantham V, Rajagopa J, Ratnagiri PK. Bilateral simultaneous anterior and posterior lenticonus in Alport's syndrome. Indian J Ophtalmol 2005;53:212–13 [DOI] [PubMed] [Google Scholar]
- 16.Dehan P, Van Den Heuvel LPWJ, Smeets HJM, et al. Identification of post-transplant anti-α5(IV) collagen allo-antibodies in X-linked Alport syndrome. Nephrol Dial Transplant 1996;11:1983–8 [DOI] [PubMed] [Google Scholar]
- 17.Kleppel MM, Fan WW, Cheong HI, et al. Immunochemical studies of Alport syndrome. Kidney Int 1992;41:1629–37 [DOI] [PubMed] [Google Scholar]
- 18.Olaru F, Luo W, Wang XP, et al. Quaternary epitopes of a345(IV) collagen initiate Alport post-transplant anti-GBM nephritis. J Am Soc Nephrol 2013;24:889–95 [DOI] [PMC free article] [PubMed] [Google Scholar]