

Abstract
The human histone deacetylase 8 (HDAC8) is a key hydrolase in gene regulation and has been identified as a drug target for the treatment of several cancers. Previously the HDAC8 enzyme has been extensively studied using biochemical techniques, X-ray crystallography, and computational methods. Those investigations have yielded detailed information about the active site and have demonstrated that the substrate entrance surface is highly dynamic. Yet it has remained unclear how the dynamics of the entrance surface tune and influence the catalytic activity of HDAC8. Using long time scale all atom molecular dynamics simulations we have found a mechanism whereby the interactions and dynamics of two loops tune the configuration of functionally important residues of HDAC8 and could therefore influence the activity of the enzyme. We subsequently investigated this hypothesis using a well-established fluorescence activity assay and a noninvasive real-time progression assay, where deacetylation of a p53 based peptide was observed by nuclear magnetic resonance spectroscopy. Our work delivers detailed insight into the dynamic loop network of HDAC8 and provides an explanation for a number of experimental observations.
Introduction
Post-translational histone modifications are essential cellular processes that regulate the accessibility of DNA to the cell’s transcriptional apparatus. One such modification is acetylation of lysine side-chains that renders these neutral in charge and thereby alters the electrostatic interactions between, for example, histone tails and DNA or bromodomains. As important as the enzymes that generate the post-translational modifications are the enzymes that reverse them. One class of enzymes that mediate the reversal of histone modifications is comprised of the histone deacetylases (HDACs),1 which catalyze deacetylations of acetylated lysine side-chains in histones1 and other cellular proteins.2 Deacetylation of histone tails leads to a condensed and inaccessible chromatin structure at the site of modification.3 Besides histones, (de)acetylations are now known to regulate a large body of enzymes in the cell,4,5 thus making HDACs key entities in the regulation of eukaryotic cells.6,7 In particular, HDACs are often up-regulated in cancers,8 and the inhibition of HDACs is now believed to be a promising way to improve the treatment of several cancers.9 Verinostat (suberoylanilide hydroxamic acid; SAHA) is one such HDAC inhibitor approved by the FDA for the treatment of T-cell lymphoma.
The family of human HDACs encompasses at least 11 classical zinc-dependent isoforms with various cellular functions9 and can be categorized into four major classes based on phylogenetic analysis and sequence similarity. For example, the classical class I HDACs include isoforms HDAC1, HDAC2, HDAC3, and HDAC8, which is the focus of this study. Crystal structures of HDACs and HDAC8 in particular have yielded detailed insight into the structure and conformational diversity of this protein10−13 when bound to inhibitors and substrates, whereas only one structure of HDAC8 without a binding partner is available.11 Crystal structures of HDAC8 complexed with inhibitors show different conformations of the HDAC8 surface, and computational studies have shown that HDAC8 can interconvert between different entrance topologies on the nanosecond time scale.14−18 Thus, the surface around the entrance tunnel to the active site is malleable, thereby allowing for interaction with different binding partners (Figure 1a).
Figure 1.
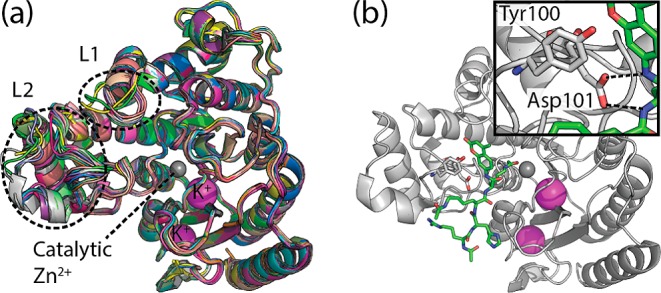
(a) Overlay of available crystal structures of HDAC8 represented as ribbons. Structural variation of the L1 and L2 loops is highlighted with a dashed line. (b) Ribbon representation of HDAC8 Tyr306Phe (PDB code 2V5W) in gray bound to a cleavable substrate in green licorice. Insert shows a zoom into the interaction of the binding rail residue Asp101 and the substrate backbone.
Despite the malleability of the surface that surrounds the entrance to the active site tunnel, crystal structures of HDAC8 exhibit a structurally highly conserved binding configuration formed by residues Tyr100 and Asp101,12 which we will refer to as the binding rail (Figure 1b). For example, Vannini et al. showed that Asp101 forms hydrogen bonds with the backbone of a substrate thereby positioning and stabilizing the substrate for the deacetylation reaction.10 When a substrate-like ligand is bound, Tyr100 and Asp101 stabilize the binding via stacking of aromatic groups or hydrogen bonds, respectively. The binding rail residues show this conformation in every available crystal structure where electron density is not missing for these residues. Moreover, mutating Asp101 to Ala renders HDAC8 inactive, which attests to the importance of this residue and the binding rail in general for the catalytic activity.10
The binding rail is located in the L2 loop (Figure 1). The L2 loop, which consists of residues 83–108, has two parts: residues 83–92 that are distal to the entrance tunnel and residues 93–108 that are proximal and include the binding rail, Tyr100 and Asp101. The distal part of the L2 loop is found in different conformations depending on the inhibitor bound, although electron density is often lacking for this part of the loop. In some crystal structures almost the entire L2 loop is missing electron density (PDB codes 3SFH, 1VKG) probably because no favorable interactions between the binding rail and the inhibitors can be formed. The ligands in these structures are not substrate-like and cannot form hydrogen bonds with Asp101 nor can they π-stack with Tyr100. The HDAC8 structure reported without a ligand bound shows no electron density for residues 85–103 (PDB code 3F07 chain C). Thus, previous studies suggest that the L2 loop is highly dynamic, the weaker the interaction between the ligand and the binding rail the more dynamic the loop. Moreover, in some crystal structures the L2 loop is involved in symmetry related crystal contacts, which affects the B-factors in this region, further confounding a concise interpretation of the loop flexibility from crystal structures alone.
The L1 loop, which spans residues 31–35 (Figure 1a), is also located in the vicinity of the entrance to the catalytic site. The L1 loop is directly above an internal cavity, which is believed to play a role in the catalytic mechanism18 by functioning as an exit tunnel for the acetate product after catalysis. As for the L2 loop described above, the L1 loop is found in very different conformations in different crystal structures (Figure 1a). Specifically, the Cα position of Lys33 in this loop deviates by more than 0.5 nm when bound to different inhibitors (PDB codes 1T69 and 1T64), demonstrating its flexibility and ability to undergo large-scale motions. Both, the L1 and L2 loops are regions of the highest sequence variation among the class I HDACs.
Correlation between HDAC8 activity and loop flexibility has been suggested previously by Somoza et al.12 and Dowling et al.,11 among others. In particular, it was shown that a zinc ion can bind to the distal part of the L2 loop and thereby stabilize its conformation.11 This stabilization down-regulates the activity, which demonstrates that flexibility is important for function.11 Moreover, a recent crystal structure and study of HDAC319 shows strong evidence that flexibility of both the L1 and L2 loops is essential for catalytic function of HDAC3. HDAC3 and HDAC8 are both class I histone deacetylases, and they have high sequence and structural similarity (41% sequence identity and structure similarity Z-score of 55.8 for crystal structures 1T64 and 4A69), which indicate that flexibility of the L1 and L2 loops is also important for HDAC8 and possibly for class I HDACs in general.
A mechanism describing how the loop dynamics and different conformations tune the activity of HDACs and the involvement of dynamics in the catalytic cycle is, to the best of our knowledge, not present in the literature. To explore these questions and the possible function of the malleable surface we first carried out two 1.1 μs fully atomistic molecular dynamics (MD) simulations of HDAC8 (with and without inhibitor) in explicit solvent followed by a 13.2 μs simulation of HDAC8 on the Anton supercomputer.20,21 As these simulations showed interesting conformational transitions we designed mutants to interfere with such transitions and measured the resulting change in enzymatic activity. We employed a fluorogenic assay as well as a novel real-time progression NMR assay we have developed, where we use part of the acetylated N-terminal tail of p53 as a substrate. Overall, our data indicate that interconversions between different L1 and L2 loop conformations may be key for the enzymatic activity.
Results and Discussion
MD Simulation of the Free HDAC8 Shows Population of Distinct States
As expected from the previously solved structures and experimental results, the simulation of free HDAC8 shows that the surface in the vicinity of the entrance to the catalytic site is very flexible and in particular the L1 and L2 loops of HDAC8 interconvert between different states (Figure 2), some of which are closely related to different crystal structures (Figure S1). For example, in one of the L1 loop conformations present during the simulation an extra cavity and a large groove become accessible, as has been observed in crystal structures with different ligands10,12 and in other computational studies.14,15 For the L2 loop, the binding rail has two distinct conformations, which we will refer to as ‘in’ and ‘out’ (colored light- and dark-green, respectively, in Figure 2a). In the ‘in’ conformation Tyr100 and Asp101 build a rail toward the catalytic site as observed in crystal structures10,13 (Figures 1b and S1d and Video S1), which has been attributed to substrate binding and positioning (Figure 1b).10 On the contrary, in the ‘out’ conformation, the binding rail residues 100 and 101 are oriented away from the entrance to the catalytic site. The interconversion between ‘in’ and ‘out’ conformations takes place at Tyr100, as shown in the analysis of the backbone Φ angle (Figure 2b). The Φ angle is larger than −60° in the ‘in’ conformation, as illustrated in Figure S1d, where, for example, the binding rail is mainly in the ‘in’ conformation between 800 and 1000 ns.
Figure 2.
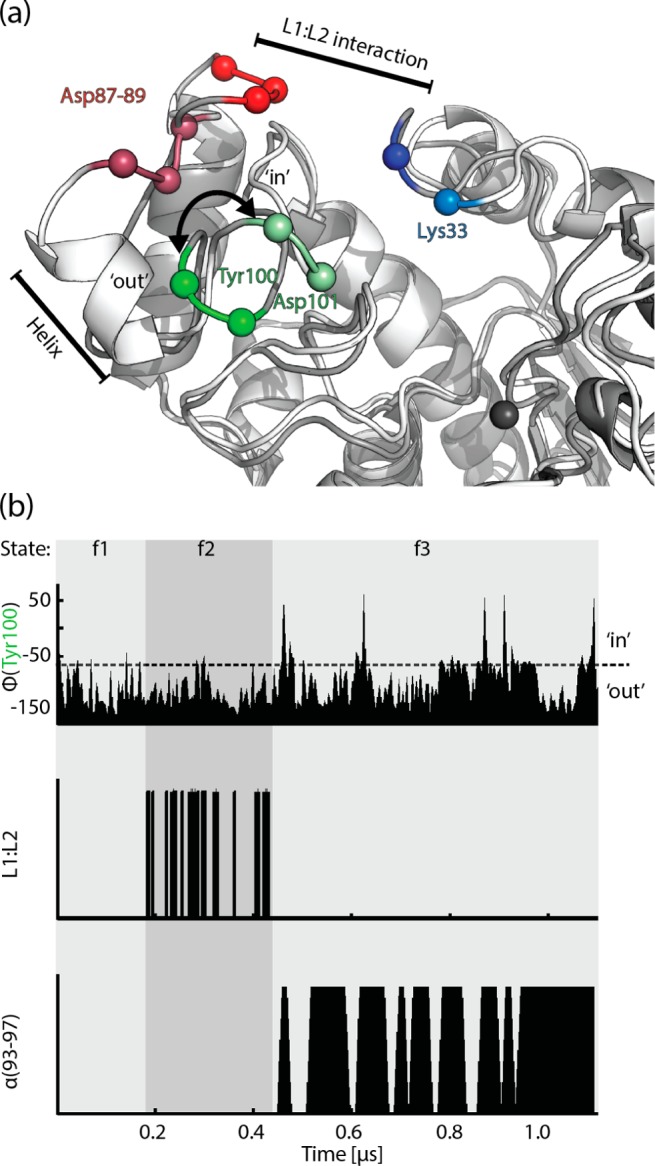
(a) Ribbon representation of snapshots during the simulation showing the ‘in’ (pale colors) and ‘out’ (dark colors) conformation of the binding rail (green residues). Microkinetic processes including their localization are annotated. (b) Microkinetic processes and states over the simulation time: (i) binding rail flips, measured via Φ of Tyr100 (ii) L1:L2 salt-bridge formation between Lys33 and Asp87–89 measured using a Lys Nζ-Asp Oδ distance cutoff of 0.35 nm (iii) presence of an α-helix at residues 93–97 as calculated by STRIDE.
Since, as outlined above, the binding rail is very important for function, we examined the simulations with the aim of revealing possible mechanisms that interfere with the binding rail conformation. We observed three features of the L1 and L2 dynamics that can be represented by three microkinetic processes, that is, (1) the binding rail flips between ‘in’ and ‘out’ conformations, (2) L1:L2 interaction, dominated by salt-bridge formation between Lys33 of L1 and the triple Asp repeat of L2 (residues 87–89), and (3) formation of a helix in the L2 loop. These processes seem to be correlated (Figure 2b), and hence we divide the trajectory into three consecutive states: f1, f2 and f3, which each show a characteristic pattern regarding the microkinetic processes.
The binding rail initially starts in the ‘in’ conformation and flips to the ‘out’ conformation after a couple of nanoseconds. After the initial flipping out, the binding rail is only found in its ‘out’ conformation for the next 420 ns. This behavior of the binding rail appears to be cooperative with conformational changes along the L2 loop from Tyr100 toward the triple Asp repeat and the formation of salt bridges between the triple Asp repeat of the L2 loop and the Lys33 of the L1 loop, characteristic for the L1:L2 interaction. Thus, the binding rail is trapped in the ‘out’ state between 160 and 420 ns where the L1 and L2 loop interact (Video S1). These electrostatic interactions are not present in the subsequent f3 state, which allows the L2 loop to adopt conformations where the binding rail becomes loose and can flip between the ‘in’ and ‘out’ conformations. Thus, after 420 ns of simulation time, when reaching the f3 state, we see several ‘in’ to ‘out’ and ‘out’ to ‘in’ flipping events (Figure S1d).
It seems that the binding rail behavior can be steered by the presence or absence of the L1:L2 interactions. We observe a number of transitions between the microkinetic states, which suggests that the energy landscape between the states is shallow. Hence, a small perturbation to the energy landscape, such as point mutations or binding of small regulators and inhibitors could stabilize one of the states and thereby shift the populations, as has been shown for low-lying thermally excited states in other proteins.22−24 To further investigate a possible correlation between the binding rail and the L1:L2 interaction, we perturbed the energy landscape of HDAC8 by simulating the response to a small and flexible HDAC inhibitor, SAHA, followed by the experimental investigation of the L1:L2 interactions using point mutations assessed by in vitro biochemical assays. Overall, we seek to characterize the response of the interactions, dynamics, and structure to perturbations and relate this to the catalytic function of HDAC8.
MD Simulation of the HDAC8:SAHA Complex
The structure of the HDAC8:SAHA complex is shown in Figure 3a. Flexible behavior of the ligand and the loops at the entrance surface in the HDAC8:SAHA complex is expected, since crystallographic B-factors are large in these regions, while high R values are also observed for this ligand (Video S2).
Figure 3.
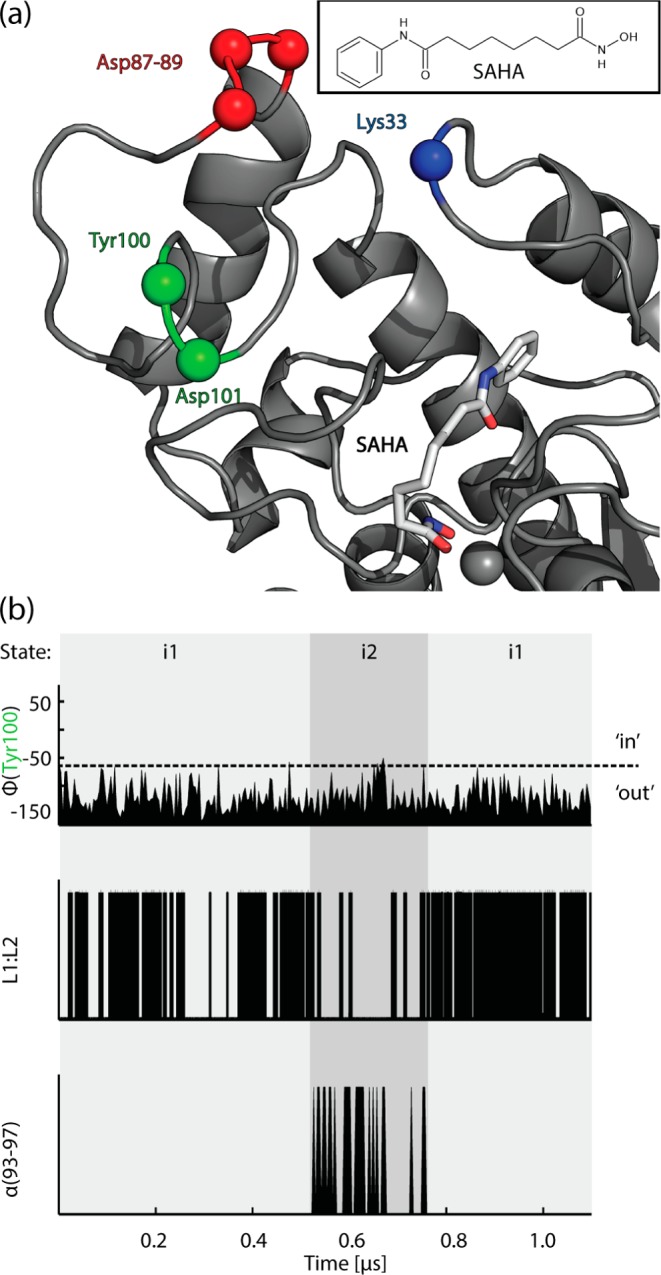
States of the L1 and L2 loop conformations when interacting with SAHA (chemical structure shown in the insert). (a): HDAC8:SAHA complex snapshot of the simulation with SAHA (licorice) and HDAC8 (gray cartoon) where the binding rail is in its ‘out’ conformation. Positions of Lys33, Asp87–89, Tyr100, and Asp101 are illustrated with colored spheres. (b): Microkinetic processes over the simulation time as defined in Figure 2: (i) Φ angle of Tyr100, indicating binding-rail conformation; (ii) L1:L2 salt bridge presence between Lys33 and Asp87–89; (iii) presence of an α-helix at residues 93–97.
Again we decomposed the trajectory into the three microkinetic processes (Figure 3b), clearly revealing two distinct states, which we name i1 and i2. In the i1 state, after the initial interaction of the binding rail with SAHA is lost, the aromatic ring of SAHA adopts a number of binding modes different from the crystal structure, although the hydroxamate group always remains bound to the catalytic zinc (Video S2). Overall, in the i1 state SAHA can adopt a conformation where its aromatic cap group is buried in a pocket that becomes accessible in the vicinity of the entrance tunnel and the L1 loop. In this conformation, where SAHA is buried in the pocket at the L1 loop, Lys33 exhibits frequent electrostatic contacts with the triple Asp repeat of the distal part of the L2 loop, while the binding rail is in the ‘out’ conformation, preventing any contacts with the inhibitor. Thus, the frequent interactions of L1 and L2 loops prevent helix formation and keep the binding rail trapped in its ‘out’ conformation. This is the same mechanism that we observed for the free form in the f2 state, where the distal part of the L2 loop interacts with the L1 loop. In the i2 state, which is reached after ∼500 ns, the L1:L2 interactions are less frequent, allowing the formation of a helix in the L2 loop (residues 93–97). During the i2 to i1 transition at 780 ns the L1:L2 interactions again increase, coinciding with an unraveling of the helix.
The L1:L2 interactions in the HDAC8:SAHA complex are much more pronounced than in free HDAC8, trapping the binding rail in its ‘out’ conformation throughout the simulation. Nevertheless, it is clear that the states sampled by the perturbed form (HDAC8:SAHA) and the free form of HDAC8 show many similarities, in particular, the f2 and the i1 state have many common features, such as the conformation of the L1 loop and the formation of salt bridges between Lys33 and the triple Asp repeat in the L2 loop. Hence, we conclude that the mechanism of L1:L2 interaction steering the binding rail behavior we observe is robust and can be triggered by small perturbations such as the binding of a small molecule.
In the 1.1 μs duration simulations, we have only observed a few transitions between the states. To enhance the sampling of the transitions we used metadynamics simulations. However, these proved unsuccessful as using collective variables such as the L1–L2 loop distance and backbone angle of Tyr100 did not enhance sampling of the transitions. We infer that there are unknown processes transverse to the chosen collective variables that limit the sampling rate and lead to strong hysteresis.
As an alternative route to improve the sampling of the state transitions, we used the Anton supercomputer to carry out a much longer, unbiased, MD simulation. While simulating free HDAC8 for 13.2 μs did not yield proportionally more sampling of the state transitions, the longer simulation confirms our previous observation that L1:L2 interactions steer the binding rail backbone conformation (Figure S2), and we can see several states where there are L1:L2 interactions present or absent.
Since these microkinetic processes steer a functionally important part of the protein, we hypothesize that the observed processes play a role in the catalytic cycle of HDAC8. It is unfortunately not feasible to simulate the whole catalytic mechanism including binding, cleaving of substrate and product egression. Even obtaining statistically robust measurements of the loop state transition is barely achievable, as we have shown using Anton. Given these limitations, only experimental evidence can substantiate this hypothesis. Hence, we expressed HDAC8 mutants in which the L1:L2 interactions are altered and measured their activity.
Enzymatic Activity of HDAC8 Mutants
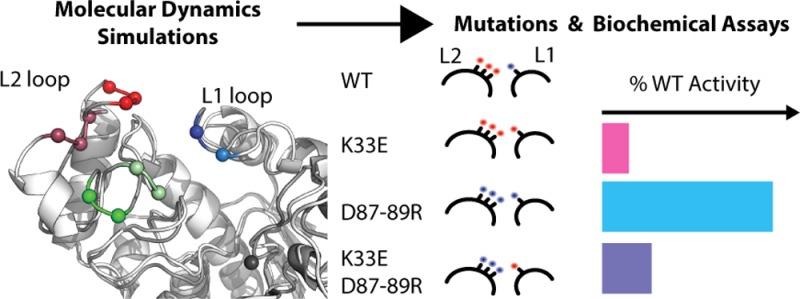
Salt-bridge formation between the L1 and L2 loop is characteristic of the L1:L2 interaction, which led us to design three mutants that change the charges of the loops in a systematic manner and thereby modulate the L1:L2 interactions. The three mutants are: Lys33Glu; Asp87Arg/Asp88Arg/Asp89Arg, which we abbreviate as Asp87–89Arg; and a charge-swap mutant Lys33Glu/Asp87–89Arg. We first assessed the enzymatic activity of the wild-type HDAC8 and these mutants with a fluorogenic assay using Boc-Lys(Ac)-7-amino-4-methylcoumarin (MAL) as a substrate.25 The wild-type HDAC8 has a kcat/Km of 38 ± 4 M–1 s–1 for the MAL substrate while, as shown in Table 1, mutating Lys33 to Glu leaves the enzyme with only 8% activity and mutation of the three Asp residues 87–89 to Arg results in 54% residual activity. Now, if the L1:L2 interaction were irrelevant for enzymatic activity, then one would expect that the charge-swap mutant Lys33Glu/Asp87–89Arg should have a relative activity given by the product of the two individual relative activities of the mutants, Lys33Glu and Asp87–89Arg. Interestingly, the charge-swap mutant shows 15% of wild-type activity, which is significantly larger than what would be expected in a model where the enzyme activity were independent of the Lys33:Asp87–89 interaction.
Table 1. Relative Enzymatic Activity of HDAC8 Mutants.
| variant | MAL assay [relative] | NMR p53 assay [relative] |
|---|---|---|
| WT | 100% | 100% |
| Lys33Glu | 8% ± 2% | 1.8% ± 0.2% |
| Asp87–89Arg | 54% ± 7% | 10.1% ± 0.5% |
| Lys33Glu/Asp87–89Arg | 15% ± 3% | 11.5% ± 0.5% |
Although the three mutations alter the L1:L2 interaction, they could also affect substrate binding and cause other changes due to the change of charge and steric effects. Specifically, Lys33 is not far from the binding rail itself, and mutation of this residue could directly interfere with substrate binding. Moreover, the MAL substrate is by no means a perfect mimic of a natural substrate since the methylcoumarin fluorophore is attached directly to the C-terminus of the acetylated lysine, which like the substrate in the crystal structure 2V5W probably points directly toward Lys33. Also, the amino acids introduced by mutation (up to four) are of different sizes to those in the wild-type enzyme; it is therefore very likely that we observe a combination of an alteration of the L1:L2 interaction together with other secondary effects.
To address these issues and to confirm that the L1:L2 interaction generally tunes the enzymatic activity of HDAC8, we established a real-time NMR assay with a more natural-like substrate. In this assay the activity of the HDAC8 mutants was measured against an acetylated lysine embedded in a 21 amino acid residue peptide based on a part of the N-terminal tail of the tumor suppressor p53 GSHLKSKKGQSTSRHK-K(Ac)-LMFK, where the acetylated Lys (K(Ac)) corresponds to the 382 position in p53. Monitoring the amount of substrate and product over time followed by fitting to the Michaelis–Menten differential equations allows us to determine kcat/Km values for HDAC8 with this natural-like substrate (see Supporting Information).
For wild-type HDAC8, kcat/Km = 289 ± 6 M–1 s–1 in the real-time p53 based assay, which is approximately one-third of that reported for the Fluor-de-Lys assay (BIOMOL) previously.11 As shown in Table 1, the activity of the Lys33Glu mutant is minimal, with only ∼2% of the wild-type activity. The Asp87–89Arg mutant and the charge-swap mutant Lys33Glu/Asp87–89Arg show very similar kcat/Km values, at ∼10% of the wild-type activity. Hence, for this more natural-like substrate the rescue effect resulting from reintroducing attracting charges on the L1 and L2 loops is larger than in the fluorogenic assay and yields an activity which exceeds that of the Asp87–89Arg mutation alone. Again, the importance of the Lys33:Asp87–89 interaction for activity is further emphasized as the product of the relative activity of the two individual mutants, 0.018 × 0.101 ≈ 0.002, is significantly lower than the relative activity of the charge-swap Lys33Glu/Asp87–89Arg mutant, 0.115.
The influence on activity and the rescue effect that we observe above for the L1:L2 interaction is in the same range as that of other mechanisms that have been shown to down-regulate HDAC8, such as Zn2+ binding to the L2 loop and phosphorylation of Ser39.11,13,26,27 The latter has been linked to cellular regulation of HDAC8, where the activity of the phosphorylated Ser39p and the mimicking mutant Ser39Glu is 2–6-fold less than that of the wild-type.27
The above results show that there are substrate-specific contributions to the effects of the mutations and that the charge-altering mutations in the L1 and L2 loops are not independent. Hence, the L1:L2 interaction is an important component within the catalytic cycle of HDAC8. The explicit influence on the kon, koff, and kcat rates is hidden in the (kcat/Km) ratio, but we consistently see a rescue effect by swapping the charges on both loops, supporting our hypothesis that this mechanism has a direct influence on the catalytic cycle. Note that the residues mutated are not conserved, while a direct interaction between the L1 and L2 loop has not been observed before.
A detailed analysis of the MD and interactions present in the three mutants would shed further light on the enzymatic mechanism. Unfortunately, the sampling issues in such MD simulations would be worse for the mutations as compared to the wild-type, since certain states are predicted to be depopulated. Achieving converged sampling thus seems out of reach for such mutants. We therefore developed an alternative approach to further bridge the experimental and theoretical results, wherein the mutation is performed in silico. Specifically, as shown in Supporting Information, mutating Lys33 to Glu leads to a depopulation of the f2 state due to its higher estimated free energy (Figure S3). Hence, our experimental data supports the MD simulations, thereby substantiating our hypothesis.
Enzymatic Function of the Dynamic Loop Interactions
The catalytic cycle of HDAC8 involves at least three steps, including binding of substrate, cleaving of the acetylated lysine, and release of the products. Characterization of the interconversions between these states during catalysis is central to both understanding the underlying mechanism of HDAC8 and facilitating development of novel inhibitors of HDACs.
The structure of an inactive mutant of HDAC8 bound to a cleavable substrate (PDB code 2V5W) has been previously determined, and we will assume that this structure largely represents a substrate binding state. A particular feature of this structure is that the binding rail adopts the ‘in’ conformation, forming hydrogen bonds with the substrate, there being no obvious contacts between the L1 loop and the triple Asp repeat of the L2 loop. Moreover, the L2 loop is structured in the binding state with electron density for both backbone and side-chains (Figure 1b).
Our simulations suggest that the L1 loop conformations and dynamic interactions are able to steer the structure of the L2 loop and thereby influence the conformational sampling of the binding rail. Only in the free form is there an effective sampling of the binding conformation of the L2 loop (Figure 2b), where specifically in the f3 state the binding rail frequently samples the ‘in’ conformation. When the distal part of L2 interacts with L1, it traps the binding rail in the ‘out’ state. Conversely, when there is no interaction with the L1 loop, the binding rail can dynamically sample the ‘in’ conformation, i.e., a binding state.
We therefore hypothesize that the observed microkinetic processes, in particular the L1:L2 interaction, are able to tune the binding rail availability, hence tuning enzymatic activity.
Conclusions
Based on our MD simulations we propose a model in which dynamic interactions of the L1 and L2 loops steer the behavior of the binding rail of HDAC8 and should thus be regarded as part of the catalytic process of the enzyme. Informed by these simulations we designed mutations that interfere with this interaction and were able to show, using two different assays, that this interaction has an effect on the activity of the enzyme.
Moreover, it has been shown that zinc binding to a second site at the distal L2 loop down regulates the enzyme. In the structure where this site is occupied by zinc it is clear that the Asp87–89 residues are not available to form salt-bridges with the L1 loop. The mechanism proposed here helps to explain this allosteric down regulation. Interestingly, the regions that we have identified to be important for catalytic activity of HDAC8 were also recently shown to be important in HDAC3, where two binding partners are needed for activation.19 In the recently published crystal structure of HDAC3, the activating binding partners clamp the interface adjacent to the L1 and L2 loop. This clamping yields a structure of HDAC3 similar to that of HDAC8, possibly allowing for a similar L1:L2 interaction and allosteric mechanism. A similar feature can also be observed in HDAC2, where a recent crystal structure revealed a more helical L2 loop, but a longer L1 loop, again allowing for interloop interactions.28
Our theoretical work on the atomistic scale has yielded a hypothesis we have been able to substantiate using biochemical experiments on the macroscopic scale. Therefore, the mechanism we propose furnishes a conceptual platform by means of which one can rationalize the influence of L1 and L2 dynamic interactions on enzymatic activity of HDAC8 and possibly other HDACs.
Methods
Unbiased MD Simulations
The first set of unbiased MD simulations were performed using GROMACS 4.5.3,29−32 the AMBER99SB-ILDN33 force field, and TIP3P water. Five missing residues in 1T69 were initially modeled using Modeller 9.09.34 The solvated systems consisted of ∼57 000 atoms. 0.9 nm cutoffs were used for electrostatic and van der Waals interactions and the particle mesh Ewald (PME) method was applied for long-range electrostatics.35 We used an extensive equilibration protocol, where each system was simulated for 5 ns in an NVT ensemble followed by 5 ns in an NpT ensemble during which we applied positional restraints of 1000 kJ mol–1 nm–2 on heavy atoms of the protein and increased the temperature. All production runs were performed in an NpT ensemble using a Nosé–Hover thermostat at 300 K, an isotropic Parrinello–Rahman pressure coupling and periodic boundary conditions. The neighbor list was updated every 5 steps using 2 fs integration time steps while keeping bonds involving hydrogens and heavy atoms constrained using the P-LINCS algorithm.36 The topology for SAHA was generated using AmberTools 1.5 with the RESP methodology after geometry optimization at the B3LYP 6-31G* level using ORCA 2.8.0.37 Protonation states of histidines were calculated using the H++ server38 and the protonation states of histidines 142 and 143 close to the catalytic site were taken from (ref (16)) as protonated on the δ nitrogen.
Analysis of backbone angles was done in VMD 1.9.39 Data was analyzed using GROMACS, VMD 1.9, and MATLAB (MathWorks). Sequence similarities and structure similarity Z-score of crystal structures were obtained from the Dali server.40 Secondary structure analysis was done using STRIDE.41 Protein structures were rendered in PyMOL 1.4.1 (Schrödinger LLC).
The Anton20,21 production simulation was run in the NpT ensemble with the same force field and water model as the GROMACS simulations. 1.1 nm cutoffs were used for short-range interactions, and a 64 × 64 × 64 PME mesh was used for long-range electrostatic interactions. A Berendsen thermostat and barostat42 were applied to maintain 300 K and 1 bar as the simulation temperature and pressure, respectively.
Protein Expression
HDAC8 was expressed as described elsewhere.43 In brief, Escherichia coli codon-optimized coding sequence of HDAC8 wild-type was obtained from GenScript (Piscataway, USA) in a pET-29b+ vector containing an N-terminal His-NusA-tag44 separated from the HDAC8 coding sequence by a linker containing a specific TEV cleavage site. Mutations were introduced by the Quikchange protocol using codon-optimized primers. Wild-type and mutant constructs were expressed using BL21(DE3) cells in 1 or 2 L of LB medium at 21 °C overnight. Cells were harvested, resuspended in lysis buffer [50 mM Tris pH 8.0, 3 mM MgCl2, 500 mM KCl, 5 mM β-mercaptoethanol, 5 mM imidazole, 5% glycerol, 0.25% IGEPAL, 1 tablet of complete protease inhibitor (Roche) per 50 mL, traces of DNase I (Roche), and lysozyme (Sigma)], before lysis through sonication and subsequent centrifugation of the lysate at 25 000 g for 45 min. Purification over a first Ni-NTA column (GE Healthcare) using an imidazole gradient (5–200 mM) was followed by dialysis into cleavage buffer (50 mM Tris pH 8.0, 150 mM KCl, 5 mM β-mercaptoethanol, 5% glycerol) and cleavage by His-tagged TEV-protease. Cleaved HDAC8 was separated from the His-NusA-tag, the TEV-protease and nonspecific contaminants by passage through a second Ni-NTA column. The flow-through was pooled, concentrated, and subjected to a gel filtration column (S75, GE Healthcare) in gel-filtration buffer (50 mM Tris pH 8.0, 150 mM KCl, 1 mM TCEP, 5% glycerol). Samples were concentrated and flash frozen in liquid N2.
Fluorogenic HDAC8 Activity Assay
Trichostatin A (TSA) was purchased from Enzo Life Sciences and porcine pancreatic trypsin (type IX-S) from Sigma. Boc-Lys(Ac)-7-amino-4-methylcoumarin (MAL) was synthesized according to the literature.45 The in vitro HDAC assay used is based on a homogeneous fluorogenic HDAC assay.46 Aliquots were prepared for HDAC8 wild-type and each mutant to yield a 0.4, 1, and 2 μM final concentration in the total reaction volume of 60 μL in assay buffer (50 mM Tris pH 8.0, 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 1 mg/mL BSA). MAL substrate solution of 50 mM in DMSO was diluted in assay buffer and added to the enzyme solution to yield a final concentration of 250 μM in the reaction volume.
The HDAC8:MAL solution was incubated at 25 °C for 30 or 60 min, after which 50 μL of the reaction solution was pipetted on a 96-well white NBS microplate, where the wells had been preloaded with 50 μL developer solution (10 mg/mL trypsin and 4 μM TSA in assay buffer). The microplate was left for 30 min at ambient temperature before the fluorescence was measured on a BMG FLUOstar Optima plate reader with excitation at 380 nm and emission at 460 nm. Activities for different reaction times and concentrations were compared to the wild-type enzyme and averaged as summarized in Table 1. Errors were estimated as the root-mean-square deviation of the activity measured in the six assays for each mutant (two time points and three concentrations).
The kcat/Km for the wild-type enzyme was determined using substrate concentrations of 25, 50, 100, 200, 400, and 600 μM and an enzyme concentration of 400 nM. Reactions were incubated for 15, 30, and 45 min (Figure S4). The error given represents the 2σ interval for the χ2 fitting.
Real-Time NMR p53 Assay
The 21 amino acid peptide used as substrate in the assay was purchased from PEPCEUTICALS (Leicestershire, UK) with the sequence based on the N-terminal tail of p53: GSHLKSKKGQSTSRHK-K(Ac)-LMFK, where K(Ac) denotes acetylated lysine, which corresponds to the Lys382 position in p53. The substrate was dissolved in NMR assay buffer (25 mM Tris pH 8.0, 137 mM NaCl, 2.7 mM KCl, and 1 mM MgCl2) and 10% (v/v) D2O was added.
The substrate concentration was determined by NMR using a potassium acetate standard. Enzyme was added to start the reaction with a final enzyme concentration of 500 nM in a total volume of 550 μL for each NMR sample. 1D 1H NMR spectra were recorded (each 231 s) over a time course of typically 16–24 h. Substrate and product peaks were integrated for each spectrum to yield the progression of substrate to product conversion.
Michaelis–Menten differential equations were solved with known starting conditions for a grid of kcat/Km values yielding a first estimate of kcat/Km by fitting a first-order polynomial through the minimum of the χ2 surface. The fit was refined by calculating χ2 on a line perpendicular to first determined kcat/Km. kcat/Km was taken corresponding to χ2min and refined again. Errors reported represent the 1σ confidence interval assuming a 5% uncertainty in measuring the NMR signal, based on the signal-to-noise ratio of the spectra. See Supporting Information and Figure S5.
NMR spectra were recorded on a Bruker Avance 500 (Karlsruhe, Germany). Data were processed using the TopSpin software (Bruker). Analysis was done using MATLAB (Mathworks).
Acknowledgments
M.B.A.K.’s Ph.D. studentship is funded by the Wellcome Trust, N.D.W. is a FEBS long-term Fellow and D.F.H. is a BBSRC David Phillips Fellow. We are grateful to Dr. Shunzhou Wan for critical reading of the manuscript. Prof. Charles Marson and Christopher Matthews are acknowledged for generously providing the MAL substrate. Furthermore, we acknowledge the use of the Legion III cluster at UCL and thank the UCL Research Computing support team for all their help. Anton computer time was provided by the National Resource for Biomedical Supercomputing (NRBSC), the Pittsburgh Supercomputing Center (PSC), and the BTRC for Multiscale Modeling of Biological Systems (MMBioS) through grant P41GM103712-S1 from the National Institute of Health. The Anton machine at NBRSC/PSC was generously made available by D. E. Shaw Research. This research is supported by the BBSRC.
Supporting Information Available
Videos S1 and S2 and Figures S1–S5. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Taunton J.; Hassig C. A.; Schreiber S. L. Science 1996, 272, 408–11. [DOI] [PubMed] [Google Scholar]
- Lin H.-Y.; Chen C.-S.; Lin S.-P.; Weng J.-R.; Chen C.-S. Med. Res. Rev. 2006, 26, 397–413. [DOI] [PubMed] [Google Scholar]
- Luger K.; Mader A.; Richmond R.; Sargent D. Nature 1997, 7, 251. [DOI] [PubMed] [Google Scholar]
- Minucci S.; Pelicci P. G. Nat. Rev. Cancer 2006, 6, 38–51. [DOI] [PubMed] [Google Scholar]
- Lin Y.; Kiihl S.; Suhail Y.; Liu S.-Y.; Chou Y.; Kuang Z.; Lu J.; Khor C. N.; Lin C.-L.; Bader J. S.; Irizarry R.; Boeke J. D. Nature 2012, 482, 251–255. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Yang X.-J.; Seto E. Nat. Rev. Mol. Cell Biol. 2008, 9, 206–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Zang C.; Cui K.; Schones D. E.; Barski A.; Peng W.; Zhao K. Cell 2009, 138, 1019–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J.; Noh J. H.; Lee J. H.; Eun J. W.; Ahn Y. M.; Kim S. Y.; Lee S. H.; Park W. S.; Yoo N. J.; Lee J. Y.; Nam S. W. APMIS 2005, 113, 264–8. [DOI] [PubMed] [Google Scholar]
- Witt O.; Deubzer H. E.; Milde T.; Oehme I. Cancer Lett. 2009, 277, 8–21. [DOI] [PubMed] [Google Scholar]
- Vannini A.; Volpari C.; Gallinari P.; Jones P.; Mattu M.; Carfí A.; De Francesco R.; Steinkühler C.; Di Marco S. EMBO Rep. 2007, 8, 879–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling D. P.; Gantt S. L.; Gattis S. G.; Fierke C. A.; Christianson D. W. Biochemistry 2008, 47, 13554–13563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somoza J. R.; Skene R. J.; Katz B. A.; Mol C.; Ho J. D.; Jennings A. J.; Luong C.; Arvai A.; Buggy J. J.; Chi E.; Tang J.; Sang B.-C.; Verner E.; Wynands R.; Leahy E. M.; Dougan D. R.; Snell G.; Navre M.; Knuth M. W.; Swanson R. V; McRee D. E.; Tari L. W. Structure 2004, 12, 1325–34. [DOI] [PubMed] [Google Scholar]
- Gantt S. L.; Joseph C. G.; Fierke C. A. J. Biol. Chem. 2010, 285, 6036–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D.-F.; Helquist P.; Wiech N. L.; Wiest O. J. Med. Chem. 2005, 48, 6936–47. [DOI] [PubMed] [Google Scholar]
- Estiu G.; West N.; Mazitschek R.; Greenberg E.; Bradner J. E.; Wiest O. Bioorg. Med. Chem. 2010, 18, 4103–4110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu R.; Wang S.; Zhou N.; Cao Z.; Zhang Y. J. Am. Chem. Soc. 2010, 132, 9471–9479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyaanamoorthy S.; Chen Y.-P. P. J. Chem. Inf. Model. 2012, 52, 589–603. [DOI] [PubMed] [Google Scholar]
- Haider S.; Joseph C.; Neidle S.; Fierke C. A. Bioorg. Med. Chem. Lett. 2011, 21, 2129–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson P. J.; Fairall L.; Santos G. M.; Schwabe J. W. R. Nature 2012, 481, 335–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw D.; Deneroff M.; Dror R. ACM 2007, 1–12. [Google Scholar]
- Shaw D. E.; Bowers K. J.; Chow E.; Eastwood M. P.; Ierardi D. J.; Klepeis J. L.; Kuskin J. S.; Larson R. H.; Lindorff-Larsen K.; Maragakis P.; Moraes M. A.; Dror R. O.; Piana S.; Shan Y.; Towles B.; Salmon J. K.; Grossman J. P.; Mackenzie K. M.; Bank J. A.; Young C.; Deneroff M. M.; Batson B. In Proceedings of the Conference on High Performance Computing Networking, Storage and Analysis - SC ’09; ACM Press: New York, 2009; p 1. [Google Scholar]
- Bouvignies G.; Vallurupalli P.; Hansen D. F.; Correia B. E.; Lange O.; Bah A.; Vernon R. M.; Dahlquist F. W.; Baker D.; Kay L. E. Nature 2011, 477, 111–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall B. M.; Roberts S. a; Heroux A.; Cordes M. H. J. J. Mol. Biol. 2008, 375, 802–11. [DOI] [PubMed] [Google Scholar]
- Werbeck N. D.; Itzhaki L. S. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 7863–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marson C. M.; Matthews C. J.; Yiannaki E.; Atkinson S. J.; Soden P. E.; Shukla L.; Lamadema N.; Thomas N. S. B. J. Med. Chem. 2013, 56, 6156–74. [DOI] [PubMed] [Google Scholar]
- Gantt S. L.; Gattis S. G.; Fierke C. A. Biochemistry 2006, 45, 6170–6178. [DOI] [PubMed] [Google Scholar]
- Lee H.; Rezai-Zadeh N.; Seto E. Mol. Cell. Biol. 2004, 24, 765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bressi J. C.; Jennings A. J.; Skene R.; Wu Y.; Melkus R.; De Jong R.; O’Connell S.; Grimshaw C. E.; Navre M.; Gangloff A. R. Bioorg. Med. Chem. Lett. 2010, 20, 3142–5. [DOI] [PubMed] [Google Scholar]
- Berendsen H. J. C.; van der Spoel D.; van Drunen R. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar]
- van der Spoel D.; Lindahl E.; Hess B.; Groenhof G.; Mark A. E.; Berendsen H. J. C. J. Comput. Chem. 2005, 26, 1701–18. [DOI] [PubMed] [Google Scholar]
- Lindahl E.; Hess B.; van der Spoel D. J. Mol. Model. 2001, 7, 306–317. [Google Scholar]
- Hess B.; Kutzner C.; van der Spoel D.; Lindahl E. J. Chem. Theory Comput. 2008, 4, 435–447. [DOI] [PubMed] [Google Scholar]
- Lindorff-Larsen K.; Piana S.; Palmo K.; Maragakis P.; Klepeis J. L.; Dror R. O.; Shaw D. E. Proteins 2010, 78, 1950–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A.; Blundell T. Protein Struct. Distance Anal. 1994, 64–86. [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. J. Chem. Phys. 1995, 103, 8577. [Google Scholar]
- Hess B. J. Chem. Theory Comput. 2008, 4, 116–122. [DOI] [PubMed] [Google Scholar]
- Neese F. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 2, 73–78. [Google Scholar]
- Gordon J. C.; Myers J. B.; Folta T.; Shoja V.; Heath L. S.; Onufriev A. Nucleic Acids Res. 2005, 33, W368–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. J. Mol. Graphics 1996, 14, 33–8. [DOI] [PubMed] [Google Scholar]
- Hasegawa H.; Holm L. Curr. Opin. Struct. Biol. 2009, 19, 341–8. [DOI] [PubMed] [Google Scholar]
- Frishman D.; Argos P. Proteins 1995, 23, 566–79. [DOI] [PubMed] [Google Scholar]
- Berendsen H. J. C.; Postma J. P. M.; van Gunsteren W. F.; DiNola A.; Haak J. R. J. Chem. Phys. 1984, 81, 3684. [Google Scholar]
- Werbeck N. D.; Kirkpatrick J.; Hansen D. F. Angew. Chem., Int. Ed. 2013, 52, 3145–3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Marco V.; Stier G.; Blandin S.; de Marco A. Biochem. Biophys. Res. Commun. 2004, 322, 766–71. [DOI] [PubMed] [Google Scholar]
- Hoffmann K.; Brosch G.; Loidl P.; Jung M. Pharmazie 2000, 55, 601–6. [PubMed] [Google Scholar]
- Wegener D.; Wirsching F.; Riester D.; Schwienhorst A. Chem. Biol. 2003, 10, 61–68. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.