

Abstract
The Lyme disease agent Borrelia burgdorferi, which is transmitted via a tick vector, is dependent on its tick and mammalian hosts for a number of essential nutrients. Like other bacterial diderms, it must transport these biochemicals from the extracellular milieu across two membranes, ultimately to the B. burgdorferi cytoplasm. In the current study, we established that a gene cluster comprising genes bb0215 through bb0218 is cotranscribed and is therefore an operon. Sequence analysis of these proteins suggested that they are the components of an ABC-type transporter responsible for translocating phosphate anions from the B. burgdorferi periplasm to the cytoplasm. Biophysical experiments established that the putative ligand-binding protein of this system, BbPstS (BB0215), binds to phosphate in solution. We determined the high-resolution (1.3 Å) crystal structure of the protein in the absence of phosphate, revealing that the protein's fold is similar to other phosphate-binding proteins, and residues that are implicated in phosphate binding in other such proteins are conserved in BbPstS. Taken together, the gene products of bb0215-0218 function as a phosphate transporter for B. burgdorferi.
Keywords: ligand-binding protein, phosphate-binding protein, ABC transporter, lipoprotein, Borrelia burgdorferi, X-ray crystallography
Introduction
Borrelia burgdorferi, the etiological agent of Lyme disease, has a complex life cycle that involves an arthropod vector and mammalian hosts.1–2 As such, B. burgdorferi must undergo dramatic adaptive changes as it transitions between ticks and mammals.3,4 Despite this need for broad environmental adaptation, a remarkable feature of B. burgdorferi is its small genome and its consequent limited coding capacity for the biosynthesis of amino acids, fatty acids, nucleotides, and many other components.6 This extraordinarily limited biosynthetic capability mandates that B. burgdorferi acquire many of its essential nutrients from its diverse niches. To this end, it is predicted that B. burgdorferi encodes as many as 50 transporters, about 26% of which belong to the ATP-binding cassette (ABC) family of transport systems.6–7
Inorganic phosphate (Pi) is essential for all life forms and participates in a wide variety of cellular functions such as energy production, intracellular signaling, and the biosynthesis of nucleic acids and phospholipids. In Escherichia coli, the acquisition of Pi occurs via two major independent systems: the low-affinity transport system Pit and the high-affinity phosphate transporter Pst.8 The Pit system consists of only one transmembrane component (Pit); driven by the proton-motive force, Pi transport occurs when phosphate is present in excess. In contrast, under Pi starvation, E. coli primarily relies on the Pst system for Pi acquisition. As a typical ABC transporter,9–10 the Pst system is comprised of a periplasmic Pi-binding protein, one (or multiple) transmembrane permease(s), and an ATPase. In the E. coli Pst operon, pstS encodes the periplasmic substrate-binding protein (that binds Pi with high affinity), pstB encodes the ATPase (providing energy for the transport of Pi across the cytoplasmic membrane), and PstA and PstC are permeases (that comprise the transmembrane channel for Pi entry).8
BLAST analyses of the B. burgdorferi genome have revealed that B. burgdorferi may encode a putative ABC transport system with homology to the E. coli Pst transport system.6 This system, named BbPst, is composed of four genes including bb0215, -0216, -0217, and-0218. Specifically, bb0215 has been predicted to encode the periplasmic Pi-binding protein, bb0216 and bb0217 likely encode two permeases, and bb0218 putatively encodes an ATPase. Despite these predictions, whether the BbPst system is actually involved in Pi uptake has remained unsubstantiated. Furthermore, whereas bioinformatics can be useful for predicting ABC transport systems,7,11 sequence homologies alone often are not reliable for determining the actual ligand(s) of a putative transport system.13–16 For example, in the pathogenic spirochetes Treponema pallidum, Treponema denticola, and Borrelia burgdorferi, an RfuABCD system was predicted to be a sugar transport system.6,17 However, further structural and biochemical investigations by us led to the surprising finding that the RfuABCD operon of pathogenic spirochetes actually constitutes a new type of prokaryotic riboflavin transport system.13 To directly investigate the function(s) of the putative BbPst system in B. burgdorferi, we solved the three-dimensional structure of a recombinant version of the putative periplasmic substrate-binding protein (BB0215), and further substantiated those data with additional biochemical and biophysical studies. Our combined structural, biophysical, and biochemical data constitute compelling evidence that the BbPst system functions as a Pi transporter in B. burgdorferi. The mechanism(s) by which B. burgdorferi acquires and utilizes Pi can potentially explain the organism's wide host adaptation and how it survives during potentially stressful environmental conditions (e.g., during phosphate limitation).
Results
Identification of the Pst operon in B. burgdorferi
In a previous study, we identified a novel manganese transporter, BmtA (BB0219), in B. burgdorferi.19 This protein is essential for B. burgdorferi to survive in ticks and to infect mammalian hosts. Bioinformatics analyses revealed that, in the vicinity of bmtA, the B. burgdorferi chromosome encodes four additional genes (bb0215, -0216, -0217, and bb0218) that putatively constitute a ATP-binding cassette (ABC) phosphate transport system, Pst [Fig. 1(A)].6 In the genome, bb0215 is separated from bb0216 by 92 bp, bb0216 is separated from bb0217 by 107 bp, and an intergenic region of 3 bp is predicted between bb0217 and bb0218. Moreover, these four genes are oriented in the same direction, suggesting they may form a gene operon in B. burgdorferi. To determine whether these genes are cotranscribed, RT-PCR was performed as previously described.20 As shown in Figure 1(B), amplicons were amplified from borrelial cDNA by using the primer pairs (Table1) spanning the junctions of bb0215 and bb0216 (lane 1), bb0216 and bb0217 (lane 2), bb0217 and bb0218 (lane 4), respectively, but not in the amplification using RNA as the template (lane 5). These data indicate that bb0215, bb0216, bb0217 and bb0218 are cotranscribed.
Figure 1.
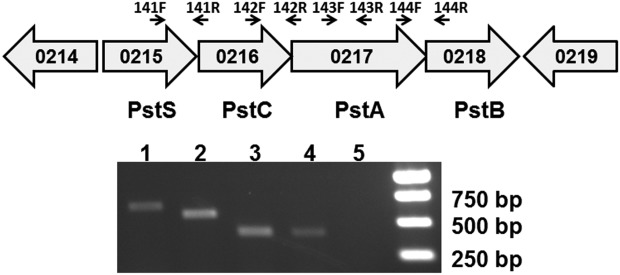
The bb0215 gene is co-transcribed with genes bb0216-bb0218. (A) Schematic representation of the putative bb0215-bb0218 operon in B. burgdorferi. Genes are shown as thick arrows circumscribing the respective gene numbers. The small arrows identify the primers used (see (B) and Table1). (B) Results from RT-PCR. Lane 1, primer pair 141F and 141R; lane 2, primer pair 142F and 142R; lane 3, primer pair 143F and 143R; lane 4, primer pair 144F and 144R; lane 5, primer pair 143F and 143R in ordinary PCR using RNA as template (no RT control); lane 6, molecular weight markers.
Table I.
Primer Sequences Used in this Study
| Primer | Sequence (5'-3') |
|---|---|
| ZM141F | GACAAGCTCAACTGGACAAG |
| ZM141R | CGGCTTTGAGCGATGAAT |
| ZM142F | TCGGGTGTTCACAGAGAAG |
| ZM142R | AGCCTACAGAGCCTATTG |
| ZM143F | CAGCACAATAGGCTCTGTAG |
| ZM143R | ACCATATGCGCCAGAATCTC |
| ZM144F | AGCAGCTATTCCCGGAATAC |
| ZM144R | AGCCAGATGGGCCTATTAAG |
In this gene cluster, bb0215 was hypothesized to encode a 31.1-kDa periplasmic phosphate-binding protein PstS. The next two genes, bb0216 and bb0217, were predicted to encode two permeases, PstC and PstA, of 33.1 and 53.3 kDa, respectively. Based on SOSUI analysis,21 PstC has an average hydrophobicity of 0.84, and apparently contains seven transmembrane α-helices. PstA was also predicted to be strongly hydrophobic (with an average hydrophobicity of 0.59) and likely contains four transmembrane helices. In addition, bb0218 encodes a 29.4-kDa putative ATPase (PstB). The characteristic Walker A motif [GxxxxGK(T/S)] and Walker B motif (hhhhDE),22–23 potentially involved in ATP binding and hydrolysis, respectively, were located in this protein. B. burgdorferi PstB also contains the signature sequence of the ABC superfamily of proteins (LSGGQQQRLC).24–25 This sequence, also known as the “Walker C motif,” is predicted to be involved in ATP hydrolysis. These four structural components, PstS, PstC, PstA, and PstB thus constitute a predicted bacterial phosphate transport system. Amino acid sequence comparisons demonstrated that this entire gene cluster in B. burgdorferi strain B31 was almost identical (93%) to those of the two other borrelial strains, B. garinii PBi and B. afzelii PKo (FLI).26–27
To assess the role of the BbPst system in B. burgdorferi, we attempted to inactivate this system in laboratory strains (not shown). However, all efforts failed, suggesting that this system is essential for the growth of B. burgdorferi in vitro. This hypothesis is also supported by two other observations. First, B. burgdorferi lacks an obvious homolog of the Pit system.5–27 As such, the Pst system is the only gene cluster predicted to transport phosphate in this bacterium. Second, a previous transposon mutagenesis study28 failed to obtain a mutant deficient in the BbPst system. Thus, both our experiments and other available data imply that BbPst is important, if not vital, for the growth of this organism.
Purification and solution characterization of BbPstS (BB0215) lipoprotein
As a step toward confirming the putative function of the Pst operon in Borrelia, we cloned the gene for BbPstS (the likely substrate-binding component). The bb0215 gene from the B. burgdorferi chromosome was modified such that the amino-terminal 19 amino acids, which comprise the putative lipidation signal for this protein, were replaced with an affinity tag, a protease site, and a five amino acid vector-derived sequence (GAMGS). We hyperexpressed this modified gene product (rBbPstS) in the heterologous organism E. coli. After lysing the cells, two chromatographic steps, and proteolysis (see Materials and Methods), the protein was purified to homogeneity as assessed by SDS-PAGE (not shown).
We sought to confirm the purity and to establish the oligomeric state of rBbPstS using sedimentation velocity (SV) analytical ultracentrifugation. We centrifuged three concentrations of rBbPstS (3, 27, and 45 μM) and analyzed the measured absorption profiles as a function of radius and time using the c(s) distribution (Fig. 2).29 This data treatment detects trace contaminants with a very high sensitivity.30 We found that the percentage of the absorbance signal in the main c(s) peak at ca. 2.5 S ranged from 99.2 to 99.9. Thus, our preparation of rBbPstS was essentially free of detectable contaminants. We cannot rule out the existence of contaminants having the same sedimentation coefficient as rBbPstS, but this possibility is unlikely.
Figure 2.
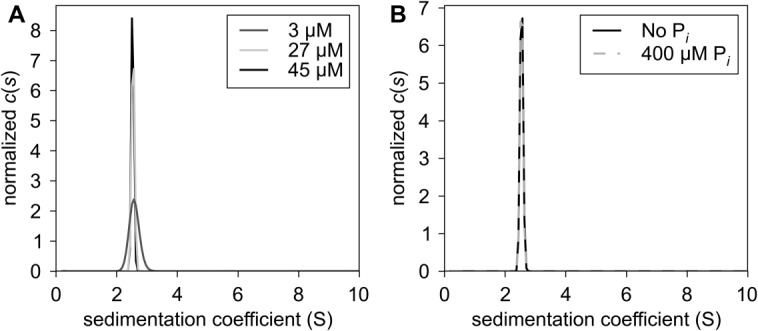
Hydrodynamic behavior of rBbPstS. (A) Sedimentation in the absence of phosphate. The c(s) distributions, showing normalized signal populations of species as a function of sedimentation coefficient, are shown for three concentrations of rBbPstS, which are denoted in the legend. (B) Comparison of sedimentation in the absence and presence of phosphate. The concentrations rBbPstS in both experiments was 27 μM. The components present in the respective curves are given in the legend. All distributions have been normalized by their respective total signals.
The molar mass of rBbPstS was also examined using SV. The refined frictional ratio (fr) of rBbPstS ranged from 1.27 to 1.33, which is in the expected range for a globular, hydrated protein. Given these ratios, the diffusional scaling law used by the analysis program,29 and the Svedberg equation, we calculated that the average molar mass of the protein was 30,740 g/mol, close to the calculated value of a monomer (29,309 g/mol) based on the known amino acid sequence of rBbPstS. Further, the observed sedimentation coefficient was essentially unchanged through more than a ten-fold change in protein concentration, indicating no propensity to form oligomers. These data therefore demonstrate rBbPstS is a monomer under these solution conditions.
Because phosphate anion was suspected to be the ligand of this protein, we conducted separate SV experiments in the presence of phosphate. As shown in Figure 2(B), the presence of 400 μM of sodium phosphate had no discernible effect on the sedimentation of rBbPstS. Although this result does not rule out a conformational change upon phosphate binding, it does eliminate the possibility that the presence of phosphate causes changes in the oligomerization state of rBbPstS under these experimental conditions.
Substrate-binding properties of rBbPstS
The sequence analyses performed above suggested that BbPstS is a phosphate-binding protein. However, sequence homologies can be misleading regarding ligand identity for ligand-binding proteins (LBPs) (e.g. see Refs.13 and15). In order to establish the phosphate-binding activity of BbPstS, we performed microscale thermophoresis experiments. rBbPstS was labeled with a fluorescent dye, and its diffusion in a thermal gradient was monitored as a function of phosphate concentration. These experiments were carried out at pH 8.5 so that most of the phosphate anion would be in the dibasic state. The resulting data demonstrated that rBbPstS bound to phosphate with a Kd of 1.2 ± 0.1 μM (Fig. 3). This dissociation constant is comparable to those obtained using the homologous phosphate-binding proteins from E. coli and Mycobacterium tuberculosis.31,32 As a control, we performed similar experiments on rBbPstS using sulfate anion as the ligand. Using the same ligand concentrations as in the phosphate experiment, we were unable to obtain a definitive binding constant for SO42−. We estimate that the dissociation constant is at least 20-fold higher for sulfate compared with phosphate.
Figure 3.
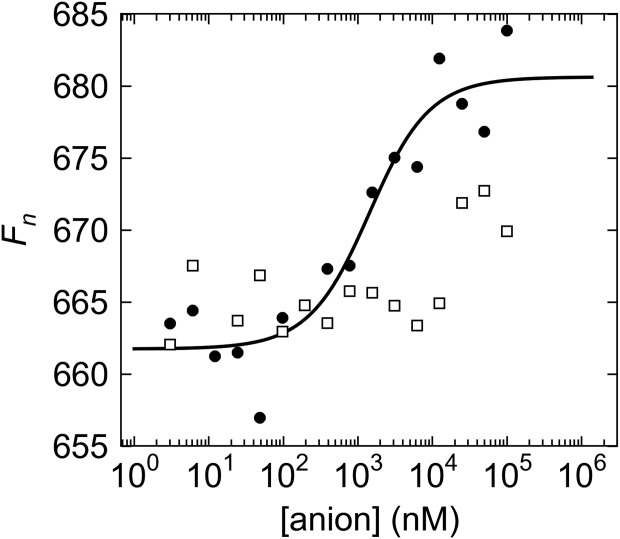
Microscale thermophoretic evidence of phosphate binding in rBbPstS. The markers represent the data points; black circles for the phosphate data, and white squares for the sulfate data. The black line is the fit to the phosphate data. The sulfate data can be fitted, but the value for the maximum Fn in this case is very uncertain; the fit is therefore not shown here.
The crystal structure of rBbPstS
To buttress our hypothesis that BbPstS is the phosphate-binding protein of a phosphate-importing ABC transporter, we crystallized rBbPstS and determined its crystal structure at a resolution of 1.3 Å (Fig. 4; Table2). The electron-density maps resolve amino acid residues 7–227 and 230–261 (mature-protein numbering). The protein comprises two domains that have similar folds (r.m.s.d. = 3.6 Å over 64 comparable Cα atoms). Domain I contains residues 7–81 and 222–260, and domain II houses residues 85–218. Both domains are respectively composed of a central, five stranded β-sheet in which four of the five β-strands are parallel. Using numbering that is local to the respective domains, the overall topology of both β-sheets is β2-β1-β3-βn-β4, where “n” represents a strand that occurs just after a crossover from the other domain. At least two α-helices are packed against each side of both sheets. Additionally, there are two short spans of amino acids (residues 82–84 and 219–221) that connect the two domains.
Figure 4.
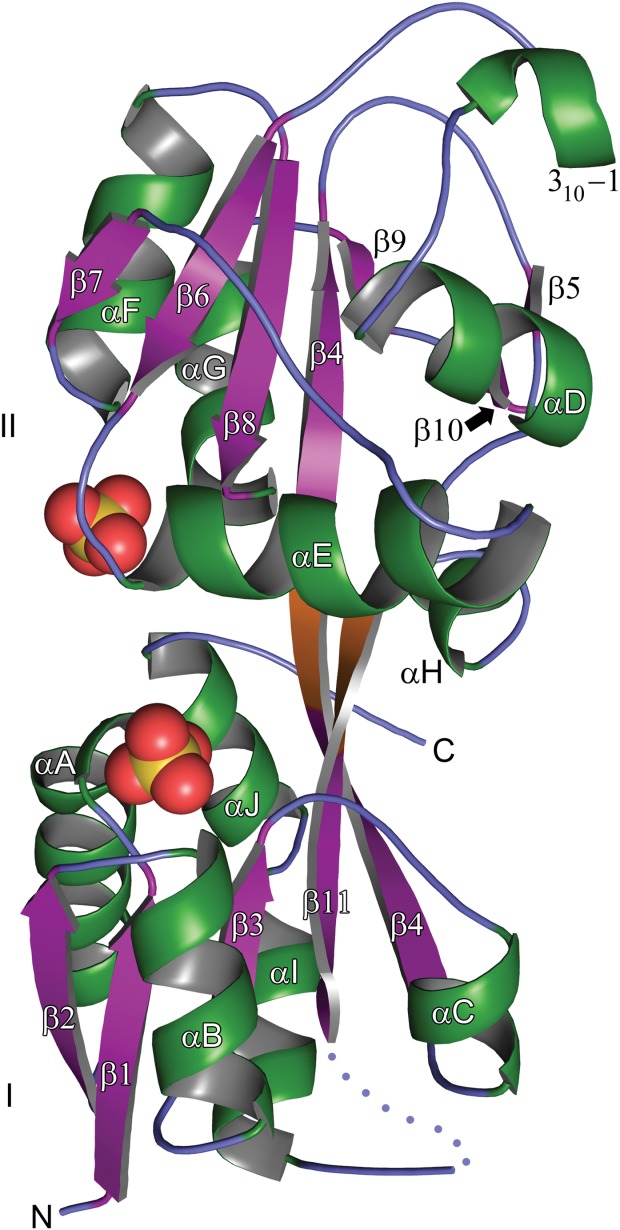
The crystal structure of rBbPstS. A ribbons-style representation of the final, refined model of the structure is shown. The β-strands are purple, the α-helices and 310 helices are green, and the regions without any regular secondary structure are slate blue. The connector region is shown in orange. The designations of all of the regular secondary structural elements are shown, as are the identities of the two domains (I and II). The amino-and carboxyl-termini are marked “N” and “C,” respectively. The two sulfate anions that bind near to the putative phosphate-binding site are shown with yellow spheres denoting the sulfur atoms and red spheres the oxygen atoms. The two distal sulfate anions mentioned in the text are omitted from this figure for clarity.
Table II.
Data Collection and Refinement Statistics for BbPstS
| PDB accession no. | 4N13 |
| Space group | P21 |
| Unit cell dimensions (Å) | |
| a | 32.3 |
| b | 84.6 |
| c | 42.9 |
| α, β, γ | 90, 105.6, 90 |
| Resolution (Å) | 42.3–1.30 (1.32–1.30) |
| Completeness (%) | 98.9 (98.5) |
| Multiplicity | 5.0 (4.9) |
| Unique reflections | 53,905 (2643) |
| Rsyma | 0.053 (0.465) |
| I/σI | 29.1 (4.2) |
| Wilson B (Å2) | 7.7 |
| Refinement | |
| Resolution (Å) | 41.4-1.3 |
| No. nonsolvent, non-H atoms | 2,223 |
| No. solvent atoms | 135 |
| No. sulfate atoms | 20 |
| Cutoff Fo/σFo | 0 |
| Maximum-likelihood coordinate error (Å) | 0.10 |
| Avg. B-factors | |
| Nonsolvent (Å2) | 12.4 |
| Solvent (Å2) | 20.2 |
| R-values | |
| Rwork | 0.138 |
| Rfree | 0.161 |
| Ramachandran statisticsb | |
| Outliers (%) | 0.0 |
| Most favored region (%) | 97.9 |
| r.m.s. deviations | |
| Bonds (Å) | 0.010 |
| Angles (°) | 1.3 |
Values in parentheses are for the highest resolution shell.
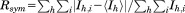 where the outer sum (h) is over the unique reflections and the inner sum (i) is over the set of independent observations of each unique reflection.
where the outer sum (h) is over the unique reflections and the inner sum (i) is over the set of independent observations of each unique reflection.
From MolProbity.34
Near to the connector regions is a cleft that separates the two domains and houses two sulfate anions (Figs. 5 and 6). These tetrahedral anions were modeled as sulfates because of the “open” disposition of the protein (see below) and the high concentration (2M) of ammonium sulfate present in the crystallization medium. Sulfate I, so called because it associates only with amino acids from Domain I, has apparent hydrogen bonds to the side chains of S15, S45, and S63 [Fig. 5(A)]. Additionally, the main-chain amides of residues T16 and S45 have apparent hydrogen bonds with this anion, as does a water molecule (“Wat 39”). Finally, the guanidinium group of R64 approaches the sulfate, but this side chain has two clear conformations: one that is close to Sulfate I, whereas the other is oriented away. Sulfate II, which has interactions only with Domain II, has apparent hydrogen bonds to the side chains of S135 and R129, and the main-chain amide atoms of G134 and S135 appear to contribute other hydrogen bonds [Fig. 5(B)].
Figure 5.
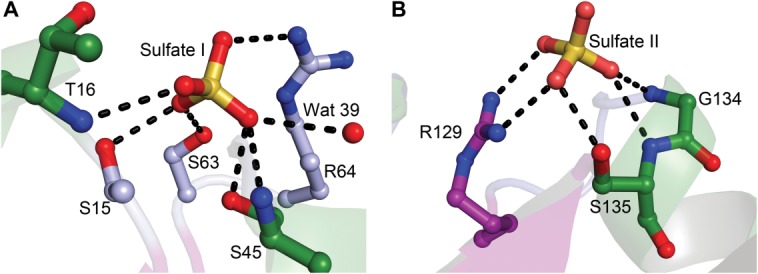
Sulfate anions bound to rBbPstS. (A) Sulfate I. (B) Sulfate II. Secondary structural elements are shown faded for clarity. All contacts shown (black dashed lines) are less than 3.2 Å in length. Carbon atoms are colored according to the secondary structural element they emanate from (Fig. 4). Oxygen atoms are colored red, nitrogen atoms are blue, and sulfur atoms are yellow.
Figure 6.
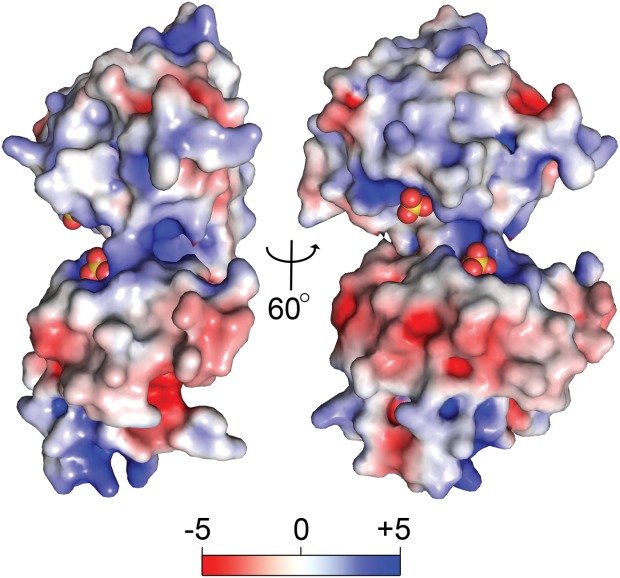
Two views of the surface of rBbPstS. The solvent-accessible surface of rBbPstS is shown, colored according to its surface electrostatic potential. A color legend is shown in kT units.
The electropositive nature [Fig. 6] of most of the cleft region accounts for the propensity of anions to bind there. Away from the binding cleft, the surface electrostatic potential of rBbPstS is mixed, with a notable negative area on the side opposite to the cleft and a positive area near to the amino-terminus (not shown).
Comparisons with other protein structures
Comparisons of the structure of rBbPstS with those of other proteins indicate that the protein is structurally similar to the LBPs of ABC transporters. Both DALI35 and SSM36 were used to query the Protein Data Bank for structural matches to rBbPstS. The highest-scoring results from both searches are LBPs; most of the top hits are known or putative phosphate-binding proteins (e.g. 1TWY, 4EXL, 4JWO, 4GD5). Analysis of the topologies of the two domains of rBbPstS and querying the SCOP37 database indicates that this protein belongs to “Class II” of the LBPs, using the nomenclature of Fukami-Kobayashi et al.38 Using a more recent classification scheme,39 rBbPstS belongs to “Cluster D-III,” a class of LBPs that comprises proteins known to bind tetrahedral anions (e.g. phosphate, sulfate, and tungstate).
The highest-scoring structural match to rBbPstS is a protein from Vibrio cholerae (VCA0807; PDB accession code 1TWY; no attendant publication), which shares 31% amino acid sequence identity with rBbPstS. A comparison of the two structures reveals a close correspondence: for 236 comparable Cα positions, the r.m.s. deviations are 2.35 Å (Fig. 7). Indeed, the chief difference between the structures is that the V. cholera protein (termed “VcPstS” herein) has a small insertion between αE and β7 (rBbPstS nomenclature) that forms a small β-hairpin (Fig. 7).
Figure 7.
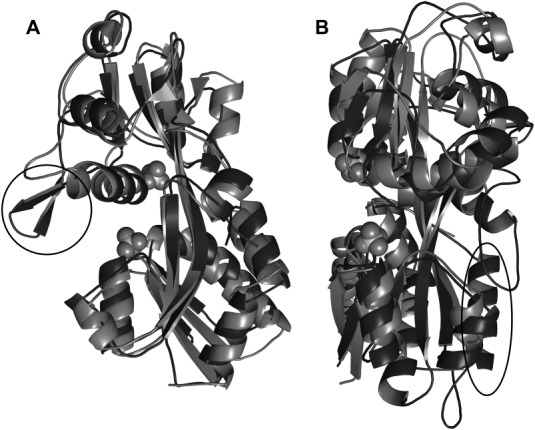
Comparisons of other structures to rBbPstS. (A) VcPstS superposed on rBbPstS. The rBbPstS molecule is light gray, and VcPstS (PDB accession code 1TWY) is shown in darker gray. The β-hairpin that is present in VcPstS but not in rBbPstS is circled. This view is rotated from that of Figure 4 to emphasize the position of the hairpin. (B) EcPBP superposed on rBbPstS. The coloration of rBbPstS is as in part A, while EcPBP (open form, PDB accession code 1OIB) is shown in the darker shade. The orientation is the same as that shown in Fig. 4. The C-terminal extension present in EcPBP but not in rBbPstS is circled.
Many LBPs are known to employ a “Venus flytrap” mechanism for substrate binding.40–41 In the absence of ligand, the LBP adopts an “open” conformation suited to accepting the ligand. Once the ligand has bound, the protein “closes,” i.e. the two domains undergo rigid-body motions that bring them closer together, with the connector regions acting as a hinge. Typically, both domains participate in binding the ligand by providing specific contacts to the ligand. The first few structures of phosphate-binding Cluster D-III proteins were in the closed conformation with a copurified phosphate anion bound.42,43 It is therefore remarkable that both rBbPstS and VcPstS are in the open, unliganded conformation. Indeed, the structures of both the open (unliganded45) and closed (liganded42) forms of E. coli phosphate-binding protein (EcPBP, an LBP for phosphate) are known, and rBbPstS has a closer structural correspondence to the open form (r.m.s.d. of 2.98 Å over 219 comparable Cα atoms) than to the closed (r.m.s.d of 4.05 Å over 187 comparable Cα atoms) (Fig. 7). In the current work, no steps were taken specifically to remove bound ligand from rBbPstS. Thus, this protein either did not bind the phosphate available to it in the E. coli cytoplasm or the bound phosphate dissociated as a consequence of the purification or crystallization procedures.
Another notable feature of this open conformation is the fact that it occurs in the presence of 2M sulfate anion. If this anion were an effective competitor for the rBbPstS binding site, it would be expected to bind the protein at this high concentration and induce the “closed” conformation. Instead, the open conformation prevails, and the residues expected (by homology to EcPBP) to participate in the binding of phosphate are divided into two half-sites: one in Domain I and one in Domain II. Indeed, the two sulfate anions described above (Sulfate I and Sulfate II) respectively bind at these two half sites (Fig. 5). Presumably, in a ligand-free rBbPstS, a single phosphate anion binding at one of these half sites would cause the protein to close, bringing the two half-sites together and resulting in high-affinity phosphate binding. The fact that this has not occurred in the presence of sulfate may be due to the exquisite selectivity of phosphate-binding proteins.33,42
The structure of the putative phosphate-binding LBP from Clostridium perfringens (CpPstS) exemplifies the closed, phosphate-bound form of this newer class of proteins (accession number 4GD5; no attendant publication). CpPstS engages every oxygen atom in this structure's bound phosphate anion with multiple hydrogen bonds (Table3). There is no obligate hydrogen-bond acceptor in the binding site; in place of EcPBP's D56, CpPstS has S89 (Table3). Notably, compared with EcPBP, there are extra hydrogen bonds to O4 of the phosphate, made by S41 and S43 of CpPstS. Because in rBbPstS nearly all of these residues are conserved (the only substitution is conservative, i.e. threonine to serine) and the overall 26% identity between the two proteins, we expect that the phosphate-binding mode of rBbPstS mimics that of CpPstS. While it is likely that at least one of the phosphate oxygen atoms is protonated in the CpPstS structure (crystallization occurred at pH 7.0), the positions of any protons are not evident because of resolution of the data (1.7 Å) and the lack of an obligate hydrogen bond acceptor. This structure shows that, despite the absence of the acidic hydrogen bond acceptor, this class of proteins may still bind a phosphate anion. Our solution evidence for phosphate binding to rBbPstS also demonstrates this point (Fig. 3).
Table III.
Phosphate Contacts in Other Protein Structures
| Organism |
||||
|---|---|---|---|---|
| Phosphate atom | E. coli | C. perfringens | ||
| Residue | Atom | Residue | Atom | |
| O1 | T10 | OG1 | T42 | OG1 |
| T10 | N | T42 | N | |
| R135 | NH2 | R155 | NH2 | |
| O2 | S139 | OG | S159 | OG |
| R135 | NH1 | R155 | NH1 | |
| T141 | OG1 | T161 | OG1 | |
| T141 | N | T161 | N | |
| O3 | S38 | OG | S71 | OG |
| S38 | N | S71 | N | |
| G140 | N | G160 | N | |
| O4 | D56a | OD2a | S89 | OG1 |
| F11 | N | S43 | N | |
| S41 | OG | |||
| S43 | OG | |||
These two atoms are hypothesized to be involved in a low-barrier hydrogen bond.
Another distinguishing aspect of rBbPstS is its size. At 261 amino acids (in the mature form), the protein is 20% less massive than phosphate-binding proteins from E. coli and M. tuberculosis. A sequence alignment (Fig. 8) demonstrates that, compared with these larger proteins, rBbPstS has deletions, most of which occur in loop regions. Further, rBbPstS lacks a C-terminal α-helix that is found in the larger proteins. The functional consequences of the minimization of this phosphate-binding motif are unknown.
Figure 8.

Structure-guided sequence alignment of phosphate-binding LBPs. The species codes are: Bb, Borrelia burgdorferi; Vc, Vibrio cholera; Cp, Clostridium perfringens; Lb, Lactobacillus brevis; Ec, Escherichia coli; Mt, Mycobacterium tuberculosis. The secondary structure of rBbPstS is shown above the alignment, with cylinders representing α-helices and arrows depicting β-sheets. The residues highlighted with a gray background are those in contact with the bound phosphate in the E. coli PBP structure (PDB accession code 1IXH). Boxes denote areas of the E. coli and M. tuberculosis proteins that are not present in the B. burgdorferi protein.
Discussion
In this report, our data indicate that Borrelia burgdorferi lipoprotein BbPstS is the LBP of an ABC transporter that is specific for phosphate. This conclusion is supported by the fact that BbPstS exists in an operon (Fig. 1) that includes other proteins bearing sequence markers for other components of an ABC transporter. Further, a recombinant version of the protein binds to phosphate in solution (Fig. 3). Finally, sequence and structural characterization of rBbPstS show that it is structurally similar to known phosphate-binding proteins (Fig. 7) and that residues homologous to those that bind phosphate in similar proteins are mostly conserved (Fig. 8). Thus, the evidence favors the proposed phosphate-binding function of BbPstS.
However, this proposed function raises a salient question: how do phosphate-binding proteins distinguish phosphate from sulfate? Our results demonstrate that, at very high (i.e. nonphysiological) concentrations, sulfate can bind at the exposed “half sites” that are present in the open form of the protein (Figs. 6). Thus, an apt refinement of the above question is: why does sulfate binding fail to lock this phosphate-specific binding protein into the closed, high-affinity conformation? Previously, the presence of one or more obligate hydrogen bond acceptors in the binding site was thought to impart this selectivity,42–43 because, at physiological pH, the phosphate anion should have at least one proton, while sulfate has none. However, BbPstS does not have any such acceptors, and the phosphate-bound structure CpPstS demonstrates that phosphate binding is not impeded in the absence of obligate hydrogen-bond acceptors.
Several potential answers to the open conformation of rBbPstS exist. One possibility is that a mixed population of open and closed forms exists in the crystallization solution, and the open form was selected because of favorable crystal contacts. A related possibility is that the sulfate concentration is actually too high; the simultaneous occupancy of both half-sites has inhibited closure of a subpopulation of the protein (i.e. the population that crystallized). Additionally, ionic strength has been reported to inhibit binding to EcPBP.32 The very low pH of the crystallization buffer (3.5) could influence the protein's ability to assume the closed state. Finally, differences in size or charge density of phosphate vs. sulfate could explain the protein's apparent rejection of the sulfate anion.
Whereas we cannot rule out any of the above explanations, none seems to fully explain the lack of closure. If sulfate binds and induces closure, the 2M concentration would very likely ensure that the population of open protein is very small. Although both half-sites have a sulfate anion in them, the sites are not fully occupied (i.e. the best-fit occupancies were below 1.0). Although the ionic strength of the crystallization medium is quite high, presumably this effect is due to the competition of anions for the binding site, but sulfate anion is present at a much higher concentration (2M vs. 100 mM) than the second-most prevalent anion, citrate. We observed no ionizable side chain on the surface of rBbPstS whose protonation would impede closure. Also, the respective sizes and oxygen charge densities of the phosphate and sulfate anions are very similar.46
From these data and considerations, we conclude that further study is necessary to address the phosphate specificity of rBbPstS and similar proteins. Structural data of rBbPstS with bound phosphate anion would be particularly illuminating; unfortunately, although several phosphate-containing crystallization conditions were screened, none have yielded diffraction-quality crystals (see Materials and Methods). If such data can be obtained, it seems likely that the source of the specificity will lie in geometric and chemical properties of the anions, as was recently surmised for the phosphate specificity of the phosphate-binding protein from Pseudomonas fluorescens.47–48
In conclusion, the data in this report demonstrate that B. burgdorferi likely satisfies its metabolic needs for Pi via the action of an ABC transporter that is specific for this anion. The structural and biophysical data, coupled with data from recent crystal structures, augment the current knowledge of how phosphate-binding proteins achieve their specificity. This feature of this class of proteins is of renewed interest due to the discovery of bacteria that can grow in milieus containing high concentrations of similar, potentially toxic tetrahedral anions.49–50
Materials and Methods
Bacterial strains and culture conditions
Infectious B. burgdorferi strain B3151 was used as the wild-type strain throughout this study. B. burgdorferi was cultured at 37°C in BSK-II medium52 supplemented with 6% rabbit serum (Pel-Freez, Rogers, AR). Spirochetes were enumerated by dark-field microscopy.
Reverse transcriptase-PCR (RT-PCR)
Total RNA was extracted from B. burgdorferi strains B31 as previously described.20 Briefly, spirochetes were grown in BSK-II at 37°C under 5% CO2, and harvested when bacterial growth reached a density of 5 × 107 cells per mL. Total RNA was isolated using Trizol (Invitrogen, Grand Island, NY) according to the instructions. Genomic DNA was removed from the RNA samples using RNase-free DNase I (GenHunter Corporation, Nashville, TN). Following digestion, RNA was further purified using a RNeasy Mini Kit (Qiagen, Valencia, CA). For RT-PCR, cDNA was generated from 1 µg of RNA using the SuperScript III Platinum Two-step qRT-PCR kit according to the manufacturer's protocol (Invitrogen). The sequences of the primers used in this report are shown in Table1.
Protein expression and purification
To produce a nonlipidated, recombinant derivative of BbPstS in E. coli, the DNA fragment encoding amino acid residues 2–261 (cloned without the post-translationally modified N-terminal Cys; the numbering reflects the assignment of this Cys as residue 1 of the processed protein) of BbPstS was PCR amplified from B. burgdorferi genomic DNA using primer pairs encoding the predicted 5'-and 3'-termini. The “forward” primer had the sequence 5'-gaccGGATCCAAAAATCAAGACAATGAAAAAATTGTATC-3', and the “backward” primer sequence was 5'-gaccAAGCTTTTATGTTTTTATCCCTAAAAAGCCTTGTT-3'. In the preceding sequences, the region of each primer complementary to the BbPstS sequence is underlined. The forward primer contained both a gacc overhang (lowercase) and a BamHI site (italicized); the reverse primer contained a gacc overhang (lowercase) and an HindIII site (italicized). The PCR product was cloned into BamHI/HindIII digested pProEx HTb-based bacterial expression vector containing an N-terminal His-tag (Invitrogen). The plasmid construct was confirmed by DNA sequencing. A verified plasmid in E. coli XL1-Blue cells were grown at 37°C in LB medium containing 100 µg/mL of ampicillin until the cell density reached an A600 of 0.5. The culture was then induced for 3 h with 0.6 mM IPTG. Cells derived from 1 L of culture were harvested by centrifugation and lysed at room temperature with gentle rocking for 20 min using 50 mL of B-PER II (Thermo Scientific, Lafayette, CO). The resulting suspension was centrifuged at 25,000g for 15 min to remove cell debris. The recombinant BbPstS (rBbPstS) was isolated from the supernatant by affinity chromatography using Ni-NTA Agarose (Qiagen). The His-tag was removed by the proTEV Plus protease (Promega), and the digestion mixture was purified by Ni2+-affinity column to remove the cleaved His-tag and His-tagged-proTEV protease. The protein without the His-tag was further purified by size-exclusion chromatography using a HiLoad 16/60 Superdex 200 prep grade column (GE Healthcare, Pittsburgh, PA) equilibrated with buffer A (20 mM Hepes, 0.1M NaCl, pH 7.5, 2 mM n-octyl β-d-glucoside). Peak fractions were analyzed by SDS-PAGE. Fractions containing purified rBbPstS (>98% homogeneity) were pooled and concentrated using an Amicon concentrator (Millipore, Billerica, MA). The purified protein was stored at 4°C in buffer A for 2 weeks. Protein concentrations were determined spectrophotometrically using an extinction coefficient of 20,400M−1 cm−1 at 280 nm (calculated using the ProtParam utility of ExPASy available at http://www.expasy.org).
Assay for phosphate binding
Microscale thermophoresis53 was used to monitor the binding of phosphate anions to rBbPstS. First, the protein in buffer A was labeled with the “Blue-NHS” fluorescent dye according to the manufacturer's instructions (NanoTemper, Munich, Germany). The free dye was removed by passing the labeling mixture over a small desalting column (provided by the NanoTemper) that had been equilibrated with MST Buffer (20 mM Tris pH 8.5, 20 mM NaCl, 0.05% (v/v) Tween 20). MST Buffer was also used to make two stock ligand solutions: 200 mM Na2HPO4 and 200 mM Na2SO4. A 1000-fold dilution of these solutions (i.e. 200 μM) was used as the first in a 1:1 dilution series using MST buffer. Thus, the concentrations were 200 μM, 100 μM, 50 μM, etc. Sixteen such solutions of 10 μL each were prepared. Each of these solutions was further diluted by the addition of 10 μL of the labeled protein at a concentration of 800 nM. The resulting concentration of protein was therefore 400 nM, and the 16 concentrations of ligand that were examined were (rounded to the nearest nM) 100, 50, 25, 12.5, 6.25, 3.125, 1.562, 0.781, 0.391, 0.195, 0.098, 0.049, 0.024, 0.012, 0.006, and 0.003 μM. These mixtures were allowed to incubate at room temperature for at least 30 min before being subjected to thermophoresis. Each mixture was loaded into a NanoTemper “Standard” capillary, then was subjected to thermophoresis with an LED power (i.e. excitation intensity) of 70% and an MST power (i.e. strength of the temperature gradient; about 10°C in this case) of 100%. The protocol for each capillary was to illuminate the excitation LED and collect data for 5 s, then illuminate the IR laser and collect 30 s of thermophoresis data, then terminate the IR illumination and collect 5 s of data. The data are thus time-dependent fluorescence signal. The thermophoretic signal was taken as:
where Fn represents “normalized” fluorescence, F0 is the initial fluorescence intensity, Ff is the final fluorescence intensity, and the brackets represent mean values taken over approximately 1 s of data. The Fn data therefore include intensity changes due to response of the fluorophore at the higher temperature and those due to labeled rBbPstS molecules diffusing out of the illuminated area (i.e. “positive thermophoresis”). The Kd of the association was obtained using the NanoAnalyze software, which uses the equation system:
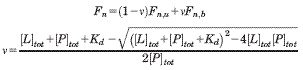 |
where ν is the fraction of ligand bound to the protein, Fn,b is the normalized fluorescence of the bound state, Fn,u is the normalized fluorescence of the unbound state, L is the ligand, and P is the protein. Because the concentrations of the ligand and protein are known, three parameters are refined in this analysis: Fn,b, Fn,u, and Kd. The reported Kd was the weighted average of three separate experiments, where the inverse squares of the respective fitting errors were used as the weighting terms.
Crystallization and X-ray diffraction data collection
The crystallization condition was obtained by screening the purified protein against the commercial crystallization screens Wizard I and Wizard II (Emerald BioSystems, Bedford, MA) and Index (Hampton Research, Aliso Viejo, CA) screens. Several of these conditions contained phosphate salts as additives or precipitants. However, no diffraction-quality crystals resulted from the phosphate-containing trials. Instead, the plate-like crystals of rBbPstS were obtained in a sulfate-containing medium: using the hanging-drop vapor-diffusion method, they appeared at 20°C 7 to 10 days after mixing 3 µL protein (∼18 mg/mL in buffer A) and 3 µL of the precipitant solution (2M (NH4)2SO4, 0.1M citric acid, pH 3.5). The crystals were cryoprotected in 30% (v/v) ethylene glycol, 2M (NH4)2SO4, 0.1M citric acid, pH 3.5, mounted into a nylon loop, flash-cooled in liquid nitrogen, and used for data collection.
When irradiated at beamline 19-ID of the Structural Biology Center at the Advanced Photon Source in Argonne National Laboratories, the crystals diffracted X-rays to a dmin spacing of 1.3 Å. Examination of the diffraction data demonstrated that the crystals exhibited the symmetry of space group P21, with unit cell dimensions a = 32.3 Å, b = 84.6 Å, c = 42.9 Å, and β = 105.6°. Further information on the quality of the diffraction data is found in Table2. These data were collected using a Quantum 315r CCD detector (ADSC) and were processed using HKL2000.54 The diffraction data were put on a positive, absolute scale using the TRUNCATE procedure55 of CCP4.56
Structure determination and refinement
The structure of rBbPstS was determined using molecular replacement. Using a hidden-Markov search method, we queried the Protein Data Bank for sequences matching that of rBbPstS. The best match was the sequence of the V. cholera protein VcPstS (accession code 1TWY). We used this model as the template for the search model. With a ClustalW57 alignment of the two protein sequences as a guide, we used SEAMAN (http://xray.bmc.uu.se/usf) to modify the model using the following rules:
Homologous residues were retained.
-
At nonhomologous sites:
2.1 Positions where both side-chains were as long as or longer than serine were changed to serine.
2.2 Positions where the rBbPstS residue was alanine were changed to alanine.
2.3 Positions where the rBbPstS residue was glycine were changed to glycine.
The β-hairpin loop region of VcPstS was removed.
Using this modified model, a molecular-replacement solution was readily obtained using CNS version 1.2.58 The model was subjected to rigid-body and simulated-annealing refinement in PHENIX.59 Coot60 was used to build the missing parts of the rBbPstS model, and subsequent rounds of positional and individual anisotropic B-factor refinement using PHENIX allowed most of the model to be built. Riding hydrogen atoms were employed in refinement. The current model contains amino acid residues 7–227 and 230–260, 135 water molecules, and four sulfate anions. Some regions of the difference electron-density maps contain unidentifiable features; these remain unmodeled. The final R-and Rfree-values for the model are 0.138 and 0.161, respectively. Other statistics regarding the model are found in Table2.
Analytical Ultracentrifugation
Analytical ultracentrifugation was carried out as in ref. 13. Briefly, solutions of the protein (400 µL) at the referenced concentrations in Buffer A were placed in dual-sector Epon centerpieces that were sandwiched between sapphire windows. After 2.5 h of equilibration at 20°C, the samples were centrifuged at 50,000 rpm in an An50-Ti rotor. Data were acquired using absorbance optics tuned to 280 nm. All data were analyzed using SEDFIT (http://www.analyticalultracentrifugation.com), and the panels of Fig. 2 were generated using GUSSI (http://biophysics.swmed.edu/MBR/software.html).
Acknowledgments
The authors express their thanks to Martin Goldberg and Dr. Wei Liu for technical assistance. Some results shown in this report are derived from work performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. Argonne is operated by UChicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357.
Glossary
- BbPstS
PstS protein from B. burgdorferi
- CpPstS
PstS protein from C. perfringens
- EcPBP
phosphate-binding protein from E. coli
- LBP
ligand-binding protein
- Pi
inorganic phosphate
- SV
sedimentation velocity
- VcPstS
PstS protein from V. cholerae.
References
- Burgdorfer W, Barbour AG, Hayes SF, Benach JL, Grunwaldt E, Davis JP. Lyme disease-a tick-borne spirochetosis? Science. 1982;216:1317–1319. doi: 10.1126/science.7043737. [DOI] [PubMed] [Google Scholar]
- Steere AC, Grodzicki RL, Kornblatt AN, Craft JE, Barbour AG, Burgdorfer W, Schmid GP, Johnson E, Malawista SE. The spirochetal etiology of Lyme Disease. N Engl J Med. 1983;308:733–740. doi: 10.1056/NEJM198303313081301. [DOI] [PubMed] [Google Scholar]
- Radolf JD, Caimano MJ, Stevenson B, Hu LT. Of ticks, mice and men: understanding the dual-host lifestyle of Lyme disease spirochaetes. Nat Rev Microbiol. 2012;10:87–99. doi: 10.1038/nrmicro2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels DS. Gene regulation in Borrelia burgdorferi. Ann Rev Microbiol. 2011;65:479–499. doi: 10.1146/annurev.micro.112408.134040. [DOI] [PubMed] [Google Scholar]
- Samuels DS, Radolf JD, editors. Borrelia: molecular biology, host interaction and pathogenesis. Norfolk, UK: Caister Academic Press; 2010. [Google Scholar]
- CM Fraser, S Casjens, WM Huang, GG Sutton, R Clayton, R Lathigra, O White, KA Ketchum, R Dodson, EK Hickey, M Gwinn, B Dougherty, JF Tomb, RD Fleischmann, D Richardson, J Peterson, AR Kerlavage, J Quackenbush, S Salzberg, M Hanson, R vanVugt, N Palmer, MD Adams, J Gocayne, J Weidman, T Utterback, L Watthey, L McDonald, P Artiach, C Bowman, S Garland, C Fuji, MD Cotton, K Horst, K Roberts, B Hatch, HO Smith, JC Venter. Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi. Nature. 1997;390:580–586. doi: 10.1038/37551. [DOI] [PubMed] [Google Scholar]
- Saier MH, Paulsen IT. Whole genome analyses of transporters in spirochetes: Borrelia burgdorferi and Treponema pallidum. J Mol Microbiol Biotechnol. 2000;2:393–399. [PubMed] [Google Scholar]
- Rao NN, Torriani A. Molecular aspects of phosphate transport in Escherichia coli. Mol Microbiol. 1990;4:1083–1090. doi: 10.1111/j.1365-2958.1990.tb00682.x. [DOI] [PubMed] [Google Scholar]
- Higgins CF. ABC transporters: from microorganisms to man. Ann Rev Cell Biol. 1992;8:67–113. doi: 10.1146/annurev.cb.08.110192.000435. [DOI] [PubMed] [Google Scholar]
- Van der Heide T, Poolman B. ABC transporters: one, two or four extracytoplasmic substrate-binding sites? EMBO Rep. 2002;3:938–943. doi: 10.1093/embo-reports/kvf201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson AL, Dassa E, Orelle C, Chen J. Structure, function, and evolution of bacterial ATP-Binding cassette systems. Microbiol Mol Biol Rev. 2008;72:317–364. doi: 10.1128/MMBR.00031-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomii K, Kanehisa M. A comparative analysis of ABC Transporters in complete microbial genomes. Genome Res. 1998;8:1048–1059. doi: 10.1101/gr.8.10.1048. [DOI] [PubMed] [Google Scholar]
- Deka RK, Brautigam CA, Biddy BA, Liu WZ, Norgard MV. Evidence for an ABC-type riboflavin transporter system in pathogenic spirochetes. MBio. 2013;4:e00615–12. doi: 10.1128/mBio.00615-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deka RK, Brautigam CA, Goldberg M, Schuck P, Tomchick DR, Norgard MV. Structural, bioinformatic, and in vivo analyses of two Treponema pallidum lipoproteins reveal a unique TRAP transporter. J Mol Biol. 2012;416:678–696. doi: 10.1016/j.jmb.2012.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deka RK, Brautigam CA, Yang XF, Blevins JS, Machius M, Tomchick DR, Norgard MV. The PnrA (Tp0319; TmpC) lipoprotein represents a new family of bacterial purine nucleoside receptor encoded within an ATP-binding cassette (ABC)-like operon in Treponema pallidum. J Biol Chem. 2006;281:8072–8081. doi: 10.1074/jbc.M511405200. [DOI] [PubMed] [Google Scholar]
- Deka RK, Neil L, Hagman KE, Machius M, Tomchick DR, Brautigam CA, Norgard MV. Structural evidence that the 32-kilodalton lipoprotein (Tp32) of Treponema pallidum is an L-methionine-binding protein. J Biol Chem. 2004;279:55644–55650. doi: 10.1074/jbc.M409263200. [DOI] [PubMed] [Google Scholar]
- CM Fraser, SJ Norris, GM Weinstock, O White, GG Sutton, R Dodson, M Gwinn, EK Hickey, R Clayton, KA Ketchum, E Sodergren, JM Hardham, MP McLeod, S Salzberg, J Peterson, H Khalak, D Richardson, JK Howell, M Chidambaram, T Utterback, L McDonald, P Artiach, C Bowman, MD Cotton, C Fujii, S Garland, B Hatch, K Horst, K Roberts, M Sandusky, J Weidman, HO Smith, JC Venter. Complete genome sequence of Treponema pallidum, the syphilis spirochete. Science. 1998;281:375–388. doi: 10.1126/science.281.5375.375. [DOI] [PubMed] [Google Scholar]
- Seshadri R, Myers GSA, Tettelin H, Eisen JA, Heidelberg JF, Dodson RJ, Davidsen TM, DeBoy RT, Fouts DE, Haft DH, Selengut J, Ren Q, Brinkac LM, Madupu R, Kolonay J, Durkin SA, Daugherty SC, Shetty J, Shvartsbeyn A, Gebregeorgis E, Geer K, Tsegaye G, Malek J, Ayodeji B, Shatsman S, McLeod MP, Smajs D, Howell JK, Pal S, Amin A, Vashisth P, McNeill TZ, Xiang Q, Sodergren E, Baca E, Weinstock GM, Norris SJ, Fraser CM, Paulsen IT. Comparison of the genome of the oral pathogen Treponema denticola with other spirochete genomes. Proc Natl Acad Sci USA. 2004;101:5646–5651. doi: 10.1073/pnas.0307639101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang Z, He M, Oman T, Yang XF, Norgard MV. A manganese transporter, BB0219 (BmtA), is required for virulence by the Lyme disease spirochete, Borrelia burgdorferi. Proc Natl Acad Sci USA. 2009;106:3449–3454. doi: 10.1073/pnas.0812999106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang Z, Deka RK, Norgard MV. BosR (BB0647) controls the RpoN-RpoS regulatory pathway and virulence expression in Borrelia burgdorferi by a novel DNA-binding mechanism. PLoS Pathog. 2011;7:e1001272. doi: 10.1371/journal.ppat.1001272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa T, Boon-Chieng S, Mitaku S. SOSUI: classification and secondary structure prediction system for membrane proteins. Bioinformatics. 1998;14:378–379. doi: 10.1093/bioinformatics/14.4.378. [DOI] [PubMed] [Google Scholar]
- Hanson PI, Whiteheart SW. AAA+ proteins: have engine, will work. Nat Rev Mol Cell Biol. 2005;6:519–529. doi: 10.1038/nrm1684. [DOI] [PubMed] [Google Scholar]
- Walker JE, Saraste M, Runswick MJ, Gay NJ. Distantly related sequences in the a-and b-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a comman nucleotide binding fold. EMBO J. 1982;1:945–951. doi: 10.1002/j.1460-2075.1982.tb01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudet R, Wiley DC. Structure of the ABC ATPase domain of human TAP1, the transporter associated with antigen processing. EMBO J. 2001;20:4964–4972. doi: 10.1093/emboj/20.17.4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuwald AF, Aravind L, Spouge JL, Koonin EV. AAA +: a class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 1999;9:27–43. [PubMed] [Google Scholar]
- Casjens SR, Mongodin EF, Qiu W-G, Dunn JJ, Luft BJ, Fraser-Liggett CM, Schutzer SE. Whole-genome sequences of two Borrelia afzelii and two Borrelia garinii Lyme disease agent isolates. J Bacteriol. 2011;193:6995–6996. doi: 10.1128/JB.05951-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glöckner G, Lehmann R, Romualdi A, Pradella S, Schulte-Spechtel U, Schilhabel M, Wilske B, Sühnel J, Platzer M. Comparative analysis of the Borrelia garinii genome. Nucleic Acids Res. 2004;32:6038–6046. doi: 10.1093/nar/gkh953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin T, Gao L, Zhang C, Odeh E, Jacobs MB, Coutte L, Chaconas G, Philipp MT, Norris SJ. Analysis of an ordered, comprehensive STM mutant library in infectious Borrelia burgdorferi: insights into the genes required for mouse infectivity. PloS one. 2012;7:e47532. doi: 10.1371/journal.pone.0047532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck P. Size distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekar A, Sukumar M. Quantitation of aggregates in therapeutic proteins using sedimentation velocity analytical ultracentrifugation: practical considerations that affect precision and accuracy. Anal Biochem. 2007;367:225–237. doi: 10.1016/j.ab.2007.04.035. [DOI] [PubMed] [Google Scholar]
- Yao N, Ledvina PS, Choudhary A, Quiocho FA. Modulation of a salt link does not affect binding of phosphate to its specific active transport receptor. Biochemistry. 1996;35:2079–2085. doi: 10.1021/bi952686r. [DOI] [PubMed] [Google Scholar]
- Wang Z, Choudhary A, Ledvina PS, Quiocho FA. Fine tuning the specificity of the periplasmic phosphate transport receptor. J Biol Chem. 1994;269:25091–25094. doi: 10.2210/pdb1pbp/pdb. [DOI] [PubMed] [Google Scholar]
- Jacobson BL, Quiocho FA. Sulfate-binding protein dislikes protonated oxyacids: a molecular explanation. J Mol Biol. 1988;204:783–787. doi: 10.1016/0022-2836(88)90369-5. [DOI] [PubMed] [Google Scholar]
- Chen VB, Arendall III WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L, Rosenstrōm P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: A structural classification of proteins database for the investigation of seqences and structures. J Mol Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- Fukami-Kobayashi K, Tateno Y, Nishikawa K. Domain dislocation: a change of core structure in periplasmic binding proteins in their evolutionary history. J Mol Biol. 1999;286:279–290. doi: 10.1006/jmbi.1998.2454. [DOI] [PubMed] [Google Scholar]
- Berntsson RP-A, Smits SHJ, Schmitt L, Slotboom D-J, Poolman B. A structural classification of substrate-binding proteins. FEBS Lett. 2010;584:2606–2617. doi: 10.1016/j.febslet.2010.04.043. [DOI] [PubMed] [Google Scholar]
- Felder CB, Graul RC, Lee AY, Merkle H-P, Sadee W. The venus flytrap of periplasmic binding proteins: an ancient protein module present in multiple drug receptors. AAPS J. 1999;1:7–26. doi: 10.1208/ps010202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao B, Pear MR, McCammon JA, Quiocho FA. Hinge-bending in L-arabinose-binding protein. J Biol Chem. 1982;257:1131–1133. [PubMed] [Google Scholar]
- Luecke H, Quiocho F. High specificity of a phosphate transport protein determined by hydrogen bonds. Nature. 1990;347:402–406. doi: 10.1038/347402a0. [DOI] [PubMed] [Google Scholar]
- Vyas NK, Vyas MN, Quiocho FA. Crystal structure of M. tuberculosis ABC phosphate transport receptor. Structure. 2003;11:765–774. doi: 10.1016/s0969-2126(03)00109-6. [DOI] [PubMed] [Google Scholar]
- Kubena BD, Luecke H, Rosenberg H, Quiocho FA. Crystallization and X-ray diffraction studies of a phosphate-binding protein involved in active transport in Escherichia coli. J Biol Chem. 1986;261:7995–7996. [PubMed] [Google Scholar]
- Ledvina PS, Yao N, Choudhary A, Quiocho FA. Negative electrostatic surface potential of protein sites specific for anionic ligands. Proc Natl Acad Sci USA. 1996;93:6786–6791. doi: 10.1073/pnas.93.13.6786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kish MM, Viola RE. Oxyanion specificity of aspartate-beta-semialdehyde dehydrogenase. Inorg Chem. 1999;38:818–820. doi: 10.1021/ic981082j. [DOI] [PubMed] [Google Scholar]
- Liebschner D, Elias M, Moniot S, Fournier B, Scott K, Jelsch C, Guillot B, Lecomte C, Chabrière E. Elucidation of the phosphate binding mode of DING proteins revealed by subangstrom X-ray crystallography. J Am Chem Soc. 2009;131:7879–7886. doi: 10.1021/ja901900y. [DOI] [PubMed] [Google Scholar]
- Elias M, Wellner A, Goldin-Azulay K, Chabriere E, Vorholt JA, Erb TJ, Tawfik DS. The molecular basis of phosphate discrimination in arsenate-rich environments. Nature. 2012;491:134–137. doi: 10.1038/nature11517. [DOI] [PubMed] [Google Scholar]
- Erb TJ, Kiefer P, Hattendorf B, Günther D, Vorholt JA. GFAJ-1 is an arsenate-resistant, phosphate-dependent organism. Science. 2012;337:467–470. doi: 10.1126/science.1218455. [DOI] [PubMed] [Google Scholar]
- Butt AS, Rehman A. Isolation of arsenite-oxidizing bacteria from industrial effluents and their potential use in wastewater treatment. World J Microbiol Biotechn. 2011;27:2435–2441. [Google Scholar]
- Johnson RC, Schmid GP, Hyde FW, Steigerwalt AG, Brenner DJ. Borrelia burgdorferi sp. nov.: etiologic agent of Lyme disease. Int J Syst Bacteriol. 1984;34:496–497. [Google Scholar]
- Pollack RJ, Telford SR, Spielman A. Standardization of medium for culturing Lyme disease spirochetes. J Clin Microbiol. 1993;31:1251–1255. doi: 10.1128/jcm.31.5.1251-1255.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wienken CJ, Baaske P, Rothbauer U, Braun D, Duhr S. Protein-binding assays in biological liquids using microscale thermophoresis. Nat Commun. 2010;1:100. doi: 10.1038/ncomms1093. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- French S, Wilson K. On the treatment of negative intensity observations. Acta Crystrallogr A. 1978;34:517–525. [Google Scholar]
- Collaborative Computational Project Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- MA Larkin, G Blackshields, NP Brown, R Chenna, PA McGettigan, H McWilliam, F Valentin, IM Wallace, A Wilm, R Lopez, JD Thompson, TJ Gibson, DG Higgins. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Brunger AT. Version 1.2 of the crystallography and NMR system. Nat Protoc. 2007;2:2728–2733. doi: 10.1038/nprot.2007.406. [DOI] [PubMed] [Google Scholar]
- PD Adams, PV Afonine, G Bunkóczi, VB Chen, IW Davis, N Echols, JJ Headd, LW Hung, GJ Kapral, RW Grosse-Kunstleve, AJ McCoy, NW Moriarty, R Oeffner, RJ Read, DC Richardson, JS Richardson, TC Terwilliger, PH Zwart. PHENIX: a comprehensive Python-based system for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]