

Although the fundamental events underlying the pathogenesis of Langerhans cell histiocytosis (LCH) remain elusive, recent advances have significantly improved our understanding of this disease. In particular, the identification of activating mutations of BRAF in LCH patients, along with the recognition of the critical importance of the lesional microenvironment in tumor progression, have overturned the classical—and perhaps ambiguous—view of the disease, substantiating new opportunities for targeted therapies.
Keywords: Langerhans cell histiocytosis, BRAF kinases, Vemurafenib, Interleukin-17, Cancer microenvironment, Bone marrow
Learning Objectives
Explain the pathogenesis of Langerhans cell histiocytosis, with particular regard to recent advances in this field.
Better identify underdiagnosed disorders such as Langerhans cell histiocytosis.
Cite currently available therapeutic opportunities for patients with Langerhans cell histiocytosis.
Abstract
Langerhans cell histiocytosis (LCH) is a rare proliferative disorder characterized by an accumulation of cells sharing the major phenotypic features of cutaneous Langerhans cells. Given its variable clinical evolution, ranging from self-limiting lesions to multisystemic forms with a poor prognosis, in the last decades it has been debated whether LCH might not have a neoplastic rather than an inflammatory nature. However, although the fundamental events underlying the pathogenesis of LCH are still elusive, recent advances have strikingly improved our understanding of the disease. In particular, the identification of multiple interplays between LCH cells and their tumor microenvironment, along with the recognition of the lesional cytokine storm as a key determinant of LCH progression, has substantiated new opportunities for devising targeted therapeutic approaches. Strikingly, the detection of the rapidly accelerated fibrosarcoma isoform BV600E gain-of-function mutation as a genetic alteration recurring in more than 50% of patients has fueled the paradoxical picture of LCH as a tumor of the antigen-presenting cells that can evade rejection by the immune system. Thus, new evidence regarding the ontogeny of LCH cells, as well as a better understanding of the putative immune system frustrating strategy in LCH, may help to define the precise pathogenesis.
Implications for Practice:
Although many of the major pathogenic events underlying LCH remain elusive, recent advances have significantly improved our understanding of this disease. In particular, the identification of activating mutations of BRAF in LCH patients, along with the recognition of the critical importance of the lesional microenvironment in tumor progression, has overturned the classical—and perhaps ambiguous—view of the disease, substantiating new opportunities for targeted therapies. Despite the fact that no consensus currently exists for the optimal therapy of multisystem LCH, the detection of BRAF mutations may support the use of vemurafenib in patients with life-threatening disease.
Introduction
Langerhans cell histiocytosis (LCH) is a rare proliferative disorder of Langerhans cells (LCs), namely the cells primarily involved in the antigen-presenting function in the skin. Although familial cases with autosomal transmission have been reported [1, 2], LCH is a sporadic disease that occurs predominantly, but not exclusively, in children. It has an incidence of up to 9 cases per million children in Western Europe, dropping to 1–2 cases per million in adults [3]. The clinical presentation of LCH is variable and encompasses a spectrum of disorders ranging from eosinophilic granuloma, characterized by one or more bone lytic lesions as a result of eosinophil infiltrates, to the Hand-Schüller-Christian disease, identified by the typical triad of bone lesions, exophthalmos, and polyuria, and to the Letterer-Siwe disease, a fulminant clinical syndrome, including hepatosplenomegaly, lymphadenopathy, bone lesions, skin rash, and pancytopenia [4]. The severity of LCH is usually age-related, because extensive multisystem variants with a poor prognosis have been primarily described in early childhood, whereas multifocal single-system disease is often diagnosed in children aged up to 5 years and unifocal bone-restricted disease in older children [5]. Pulmonary LCH typically occurs in adults during the third and fourth decades of life and is apparently associated with cigarette smoking [6]. However, the observation of a polyclonal expansion of histiocytes within the lung lesions raised the hypothesis that adult pulmonary LCH could be a reactive, inflammatory, rather than a proliferative disease [7], and so a distinct disease from childhood LCH.
In view of both the variable nature and the rarity of this disease, treatment of LCH remains a challenge. Stratification of patients based on their prognosis appears to be a useful tool in planning therapies and improving clinical outcomes, increasing survival in multisystemic disease while avoiding overtreatment as well as short- and long-term complications in patients with low-risk LCH occurring in a single site [8]. Although the response rate to treatments rises to approximately 90% in patients with diffuse LCH after chemotherapy [9, 10], the mortality rate is as high as 20% of patients in the high-risk population. Furthermore, even when chemotherapy is effective, complications of the disease and its treatment, such as neurodegeneration, endocrine abnormalities, orthopedic disabilities, and second primary tumors, have been observed in approximately 50% of survivors [11, 12].
What major pathogenic events drive the development of LCH is still unclear. Recently, genetic and molecular advances have led to a better understanding of the disease, and the identification of a putative myeloid progenitor in the ontogeny of Langerhans cells (LCs) [13], together with the detection of a rapidly accelerated fibrosarcoma isoform B (BRAF)V600E gain-of-function mutation in the majority of patients with severe disease [14], has led to the general conclusion that LCH may be a myeloid neoplasm. Nevertheless, the pathogenic role of the intralesional “cytokine storm” [15] in the progression of LCH provides new opportunities for targeted therapy, contributing to change the clinical management of patients.
This review focuses on the molecular advances in the pathogenesis of LCH and explores the new therapeutic approaches available in this puzzling disease.
The Cell of Origin
In 1953, Lichtenstein [16] grouped the spectrum of clinical manifestations of LCH under the denomination histiocytosis X, thus identifying the histiocyte as the proliferating clonal cell that originates the disease. However, the ontogeny of histiocytes is controversial. They are large cells with a granulated cytoplasm deriving from bone marrow progenitors that differentiate into mononucleated phagocytes in response to both granulocyte-macrophage colony-stimulating factor (GM-CSF) and tumor necrosis factor (TNF)-α [17]. The histiocyte lineage includes two major cellular subsets, namely antigen-processing cells, that is, monocytes and macrophages, and antigen-presenting cells, that is, dendritic cells and LCs. As depicted in Figure 1, the current model of hematopoiesis postulates that monocytes, macrophage subsets, and most dendritic cells originate from CD34+CD38+ interleukin (IL)-3Rα+CD45RA− progenitors with a restricted potential for myeloid differentiation [18]. Monocytes enter tissues and generate both macrophages, which maintain tissue homeostasis, and dendritic cells, which drive immune functions through the activation of the T-cell response. Nevertheless, during inflammatory processes, the monocytes differentiate to activated macrophages as well as to inflammatory dendritic cells specialized in both processing and presenting the antigens to T cells [19]. Interestingly, in their steady state and, possibly, during mild inflammation, LCs undergo renewal independently from the bone marrow [20–22], whereas other evidence suggests that LCs develop from an embryonic precursor that expands within the epidermis before birth, and then differentiates to establish the self-renewing LC network [23]. The increased expression of early myeloid markers has also given rise to the “Misguided Myeloid Dendritic Cell Precursor” hypothesis, whereby LCH lesions might originate from early myeloid progenitors that have accumulated in bone marrow rather than from epidermal LCs [13].
Figure 1.

Development of monocytes, macrophages, and dendritic cells. In the bone marrow, hematopoietic stem cells produce myeloid and lymphoid precursors. Myeloid precursors give rise to monocytes/macrophages and dendritic cell (DC) progenitors that in turn generate monocytes and common DC precursors. DC precursors give rise to preclassical dendritic cells that circulate in the blood and enter lymphoid tissues as classical dendritic cells and nonlymphoid tissues as mucosal DCs in the lamina propria. Epidermal Langerhans cells replicate in situ and are independent from the bone marrow because they derive from an embryonic precursor invading the epidermis before birth and subsequently proliferate in situ to create a pool of Langerhans cells. Monocytes derived from monocytes/macrophages and dendritic cell progenitors give rise to macrophages, both in a steady state and during infection, and inflammatory monocyte-derived DCs. A contribution of lymphoid precursors to DC development cannot be excluded in view of their intrinsic myeloid potential (dashed line).
Abbreviations: cDC, classical dendritic cell; CDP, common dendritic cell precursor; DCP, dendritic cell precursor; HSC, hematopoietic stem cell; LCs, Langerhans cells; LP, lymphoid precursor; lpDCs, dendritic cells in the lamina propria; MØ, macrophage; MDP, monocytes/macrophages and dendritic cell progenitors; moDCs, monocyte-derived dendritic cells; MP, myeloid precursor; Pre-c DC, preclassical dendritic cell.
However, although commonly considered to be of myeloid origin, LCs have also been described to derive from CD4+ lymphoid precursors. Chen et al. [24] have, indeed, recently demonstrated rearrangements of the T-cell receptor gene in 30% of LCH lesions, thus suggesting a relationship between LCs and lymphoid cells or, alternatively, their potential derivation from lymphoid/myeloid precursors. This hypothesis is in apparent contrast with the classical model of a very early divergence of lymphoid and myeloid lineages from multipotent stem cells, whereas it corroborates recent evidence suggesting that CD34+CD38−Thy-1neg-lowCD45RA+ multilymphoid progenitors maintain a myeloid potential, while generating all lymphoid cell types as well as monocytes, macrophages, and dendritic cells [25]. Therefore, a role of lymphoid precursors in the ontogenesis of LCH cannot be excluded and warrants further investigations, because the precise definition of the derivation of LCs may drive the therapeutic approach to the disease. However, provided that B cells apparently undergo a lineage plasticity, transdifferentiation or dedifferentiation phenomena into uncommitted precursors redifferentiating toward a mature cell of different lineage may putatively account for the clonal relationship between LCs and lymphoid cells [26].
Regardless of their true origin, LCH cells are characterized by the expression of CD207, also known as Langerin, and CD1a. CD207 is a transmembrane receptor that binds both endogenous ligands, including components of the extracellular matrix, and pathogens expressing surface mannose-containing glycoconjugates, such as Mycobacterium leprae and human immunodeficiency virus (HIV) [27, 28]. After binding, microbial agents are internalized in Birbeck granules, namely the wide tubular or racquet-shaped endosomal structures involved in the antigen processing and presentation that are typical of LCH cells [29]. Although they were previously regarded as essential for the diagnosis of LCH, an ultrastructural demonstration of the presence of Birbeck granules is now considered obsolete [7]. In contrast with the original description, both CD207 and CD1a have recently been identified in a subset of cells resident within dermal and lymphoid tissue, as well as in mononuclear phagocyte precursors, thereby excluding their use as unique markers of LCs [30–33]. Thus, investigation of alternative LC-specific antigens has intensified, and the coexpression of CD68 and CD14, as markers of immature dendritic cells, with a concurrent defect of CD86, CD83, and dendritic cell-Lamp, as antigens of mature dendritic cells, has been described on CD1a+ LCH cells from both bone and lymph node lesions. By contrast, in patients with self-healing and/or isolated cutaneous disease, LCH cells showed a mature phenotype, being frequently CD14− and CD86+. Taken together, these findings suggest that maturation of LCH cells is apparently incomplete as compared with normal LCs, although few differences have been reported in relation to the site of the disease [34]. Recently, the JL1 epitope, which encompasses a unique nonglycosylated portion of the extracellular domain of CD43, has been described as a specific marker of neoplastic LCs. Thus, because posttranslational O-glycosylation of CD43 is tightly regulated during the maturation of hematopoietic cells, it has been suggested that JL1 may serve as both immunostaining marker of LC immaturity and candidate target for antibody-based immunotherapy [35].
The immature phenotype of LCH cells in bone lesions is presumably the result of a differentiation blockade induced by inhibitory signals from the microenvironment. In particular, IL-10, a cytokine produced by M2 macrophages within bone and lymph node LCH lesions but not in skin lesions, has been demonstrated to downregulate the expression of CD86 and major histocompatibility complex (MHC) class II antigens in LCs. Therefore, a potential role for IL-10 in restraining LCH cell maturation has been postulated. Based on these findings, the paradox of an antigen-presenting cell tumor that can evade its own rejection by the immune system seems plausible. As depicted in Figure 2, indeed, cocultures have demonstrated that CD40L-transfected fibroblasts upregulate the expression of both CD86 and MHC class II molecules in LCH cells, leading to a more mature phenotype in LCs featuring a proper function that promotes both antigen presentation and activation of the immune system. Thus, new attempts in vivo to improve the maturation of LCH cells and hence drive an efficient immune response seem to be called for [34].
Figure 2.

IL-10 prevents maturation of Langerhans cell histiocytosis (LCH) cells. LCH cells express CD40 at higher levels than normal Langerhans cells. When cocultured with CD40L-transfected fibroblasts, they become mature cells and express high levels of membrane MHC class II molecules that link antigens presented by T cells through both T-cell receptor and CD86, the costimulatory molecule binding CD28 for full activation. IL-10 produced by intralesional macrophages downregulates the expression of both molecules on the surface of LCH cells.
Abbreviations: IL10, interleukin 10; iLCH, immature Langerhans cell histiocytosis; MØ, macrophage; MHCII, major histocompatibility complex II; mLCH, mature Langerhans cell histiocytosis; T-reg, regulatory T cells; TCR, T-cell receptor; TH, T helper.
LCH: A Malignancy or a Reactive Disorder?
Although according to the World Health Organization classification LCH is a neoplasm deriving from either histiocytes or dendritic cells, there is a longstanding debate as to whether the disease has a malignant or an inflammatory nature. This is ascribable to the heterogeneous clinical manifestations of the disease, which range from spontaneously disappearing lesions to a life-threatening multisystem disorder featuring rapid progression and death. Certainly, the inflammatory or neoplastic pathogenesis of LCH is not just an academic debate because solving this controversy may dramatically change the clinical approach to the disease.
The clonal derivation of nonpulmonary forms of LCH has been assessed in seminal studies [36, 37] using X chromosome-linked DNA probes to detect the pattern of X chromosome inactivation in female lesional specimens, according to the lyonization theory. Although clonality is a hallmark of malignancy, the presence of recurrent genetic aberrations may also support the definition of LCH as a neoplasm. Unfortunately, data on cytogenetic abnormalities in LCH are controversial, because the nonrecurrent genomic aberrations described by Betts et al. [38] have not been confirmed by subsequent studies exploring larger patients cohorts [39]. Similarly, widespread alterations of gene copy numbers and recurrent loss of heterozygosity were detected by comparative genomic hybridization [40], but subsequent analysis failed to confirm these abnormalities, suggesting that LCH stems from a restricted oligoclonal stimulation rather than an uncontrolled malignant proliferation [39]. However, a polymerase chain reaction-based analysis [41] has recently identified a higher degree of fractional allelic loss in high-risk multisystemic disease, as compared with the variants occurring as indolent forms of LCH. Molecular assessment of LCH is also useful to predict the clinical course, because several mutational events, albeit described only in a few cases, seem to recur more commonly in high-risk disease than in single-system LCH [41].
Based on the notion that LCs are immune cells that react to inflammatory stimuli by activation or proliferation, a purely reactive nature of LCH has been postulated and both clonal and polyclonal populations of LCH cells have been identified in pulmonary LCH [42], seeming to exclude its malignant origin. In contrast, epidemiological studies found an association between cigarette smoking and pulmonary LCH [6, 43], suggesting a pathogenic role for tobacco. Tobacco glycoproteins, indeed, are potent immunostimulants that inhibit IL-2 secretion by immune cells [44, 45]; IL-2 levels are inversely related to histiocyte recruitment and differentiation. Thus, the reactive accumulation and proliferation of LCH histiocytes appear to be dependent on smoke-induced alterations of the bronchiole microenvironment [6]. In this context, whether smoking cessation may affect the course of the disease will perhaps be assessed in future studies [43]. However, although pulmonary LCH is commonly considered an inflammatory disorder, no infective or environmental causes have yet been identified for multisystem LCH. In particular, no viral genomes have been detected in LCH lesions [46], nor have increased antibody titres against herpes viral antigens been demonstrated in LCH patients, as compared with age-matched controls [47].
Based on the notion that LCs are immune cells that react to inflammatory stimuli by activation or proliferation, a purely reactive nature of LCH has been postulated and both clonal and polyclonal populations of LCH cells have been identified in pulmonary LCH, seeming to exclude its malignant origin. In contrast, epidemiological studies found an association between cigarette smoking and pulmonary LCH, suggesting a pathogenic role for tobacco.
The Molecular Pathogenesis of LCH
Regardless of their malignant or inflammatory origin, both intrinsic aberrations of the LCH cells and perturbations of the lesional microenvironment contribute to the pathogenesis and progression of LCH [7]. However, peripheral blood monocytes from LCH patients also display functional abnormalities when undergoing differentiation to dendritic cells [48]. Thus, LCH can be considered as a systemic disease that originates in the bone marrow and affects the monocyte lineage [4, 8], so that underlying myeloid abnormalities may drive the formation of LCH lesions upon specific local triggering. Hence, further basic and translational studies are mandatory to define the interactions between peripheral LCs, lesional bystander cells, and their bone marrow precursors. A better picture of the LCH pathogenesis, in fact, will contribute in the clinical field to the design of new targeted therapies.
LCH Cell-Intrinsic Aberrations
Although the unbalanced translocation t(7;12)(q11.2;p13) was originally reported in LCH cells [38], no specific chromosomal abnormalities nor cellular ploidy alterations have been described [39]. Nevertheless, LCH cells display increased levels of cell cycle-related proteins, antiapoptotic factors, and cell growth-promoting cues. In particular, the p53-p21 and p16-Rb axes were described as hyperactive in LCH cells [49], suggesting a predominant cell exposure to stress events. In contrast, the proliferative marker ki-67 and the antiapoptotic protein Bcl-2 are upregulated in multisystem disease as compared with single system LCH, whereas a reduced expression of caspase-3 is typical of aggressive variants of the disease [50]. Intriguingly, because LCH infiltrates from patients with single-system disease showed significant levels of Fas-L, an autocrine apoptotic shortcut mediated by the Fas/Fas-L pathway has also been postulated to contribute to the spontaneous regression of lesions in LCH [51].
As in various cancers, telomere length shortening has been described during all stages of LCH [52]. However, immunohistochemistry studies of human telomere reverse transcriptase (hTERT) found active telomerase in LCH cells from patients with multisystem disease or skin involvement, whereas the bone lesions were hTERT-negative, suggesting a potential difference between the telomerase-positive phenotype, being a more aggressive disease than the telomerase-negative variants [53]. Based on these findings, LCH cells are suspected to escape apoptosis during the phase of critical telomere shortening by both triggering telomerase activity and inducing the p53 pathway [7]. This, in turn, results in an imbalance among chromosomal stability, death signal disabling, and cell survival mechanisms that, together, drive the clonal proliferation of LCH cells.
It has been reported that LCH cells differ from resting epidermis LCs because they lack peculiar dendritic protrusions as well as the expression of a number of chemokine receptors [54]. In particular, the adhesion molecules CD54 and CD58, also named intercellular adhesion molecule-1 and leukocyte function antigen-3, as well as integrin α4β1, are normally expressed by activated LCs and promote their migration from skin to draining lymph nodes. As a result of the upregulation of both molecules in LCH cells, an abnormal chemokine-mediated trafficking to aberrant anatomic sites has been presumed to play a major role in the pathological accumulation of LCs in LCH-affected tissues [55]. Interestingly, the activated phenotype of LCH cells has been associated with the multisystem variant of the disease and with a poor outcome, because it is characterized by the downregulation of E-cadherin, namely the molecule that mediates the homophilic adhesion of normal LCs to keratinocytes [56].
In physiological conditions, resting LCs express the chemokine receptor CCR6, which enables anchorage to CCL20, the surface ligand produced by epidermal keratinocytes. However, activated LCs downregulate CCR6 while upregulating CCR7, whose ligands, CCL19 and CCL21, are expressed in lymphoid tissue. Thus, the sequential expression of chemokine receptors drives the migratory capacities of activated LCs and enables their interaction with T cells within the regional lymph nodes [57–59], whereas an evident deregulation has been found in LCH, because pathological LCs constitutively express CCR6. However, although Fleming et al. [60] showed that the concurrent presence of CCR6 and CCR7 contributed to the LCH cell accumulation within the skin, bone, and lymphoid tissue, Annels et al. [61] found a striking upregulation of the CCR6/CCL20 axis and the absence of CCR7 on lesional LCs. Consistently, because both immature and mature dendritic cell markers are expressed by LCH cells, it has been postulated that a maturation defect in combination with a microenvironment-driven clonal heterogeneity may underlie the pathogenesis of LCH [4].
Recently, Badalian-Very et al. [14] have provided interesting insights into the molecular pathogenesis of LCH by identifying BRAFV600E gain-of-function mutations as recurrent genetic alterations in 57% of 61 patients. Although recurring in younger patients, the BRAFV600E mutation was not associated with a specific site, nor with the disease stage. As depicted in Figure 3, BRAF, a serine threonine kinase, is an essential component of the RAS-RAF-mitogen-activated protein kinase kinase (MEK)-extracellular signal-regulated kinase (ERK) signaling cascade. This pathway is triggered by extracellular mitogens, including growth factors, cytokines, and hormones that bind the tyrosine kinase receptors, thereby enhancing proliferation, cell survival, differentiation, and motility [62]. Several BRAF-activating somatic mutations have been described in almost all cases of hairy cell leukemia, in more than 50% of patients with malignant melanoma, as well as in 60% of the thyroid papillary carcinoma and 10% of colo-rectal cancer patients. The single nucleotide substitution resulting in a Val-to-Glu replacement in the kinase domain of BRAF at position 600 is by far the most common mutation affecting the protein, and drives its constitutive activation regardless of extracellular stimuli [63]. An immunohistochemical study applying a BRAFV600E mutation-specific antibody has recently characterized the phenotype of cells that harbor the mutation and that are responsible for proliferation in LCH. The majority of these cells coexpressed S-100, CD1a, CD14, CD36, CD80, CD86, and, at least in part, CD207, thus exhibiting a heterogeneous maturation ranging from myeloid immature cells to dedifferentiated Langerhans cells [64]. However, regardless of the BRAF mutation status, the activation of the RAS-RAF-MEK-ERK pathway was shown to uniformly occur in LCH cells [14], thereby suggesting that concurrent molecular events, such as the overexpression of BRAF itself, an autocrine or paracrine upregulation of its ligand, or a pathway transactivation by alternative signaling cascades, may play a key role in the pathogenesis of LCH. Mutations of BRAF have been also described in congenital LCH [65] as well as in pulmonary LCH [66], in contrast with the classical opinion depicting the disease as a reactive process. Interestingly, identical BRAFV600E mutational status has been detected by the next generation sequencing technology in all concurrent nodules of each patient, thus suggesting that pulmonary LCH is a clonal process in which BRAF alterations may occur as early mutational event following the tobacco inhalation. However, the molecular events related to cigarette smoking that promote the formation of a multifocal nodular although clonal pulmonary disease, are still unclear and potentially involve a germline constitutive BRAFV600E transversion or a selective mutagenic effect of tobacco on marrow myeloid progenitors.
Figure 3.

The BRAFV600E mutated protein and its pathogenic role in LCH. (A): In normal LCs, the binding of ligands such as hormones, cytokines, and growth factors to the cell-surface RTK leads to dimerization and autophosphorylation. Activation of RTK induces formation of the active RAS-GTP complex, which binds and activates BRAF that, in turn, triggers the phosphorylation of both MEK and ERK. By altering the levels and activities of nuclear trascription factors, ERK drives the transcription of genes involved in cell survival, proliferation, motility, and differentiation. (B): In LCH, the BRAFV600E mutated gene constitutively activates the MEK-ERK pathway that reinforces cell survival, proliferation, motility, and differentiation. (C): In normal melanocytes, the expression of a BRAFV600E mutated protein can lead to senescence after an initial phase of naevus growth, as confirmed by the intense activity of the stress marker SA-β-Gal and the increased levels of the tumor suppressor p16INK4a.
Abbreviations: BRAF, rapidly accelerated fibrosarcoma isoform B; ERK, extracellular signal-regulated kinase; LC, Langerhans cell; LCH, Langerhans cell histiocytosis; MEK, mitogen-activated protein kinase kinase; RAS, Rat Sarcoma; RTK, receptor tyrosine kinase.
To verify whether the spectrum of histiocytoses shares similar oncogenic pathways, the frequency of BRAF mutations has also been investigated by pyrosequencing in 127 patients with LCH, Erdheim-Chester disease (ECD), Rosai-Dorfman disease, juvenile xanthogranuloma, or other rare histiocyte-related malignancies. Intriguingly, the BRAFV600E mutation was detected in 38% and 54% of patients with LCH and ECD, respectively, whereas none of the other histiocytoses was shown to harbor the mutation. Thus, LCH and ECD have been postulated to derive from a common cellular progenitor [67]. Based on these studies, targeted therapy with the inhibitor of mutated BRAF, vemurafenib, has been used in life-threatening LCH and ECD and yielded strikingly efficacious results [68]. In particular, all three patients treated with vemurafenib showed remarkable clinical and biological improvement within a few days of initiation of treatment, undergoing rapid and substantial regression of lesions on imaging, both in terms of extension and 18F-flurodeoxyglucose uptake. Treatment was well tolerated, with minor cutaneous adverse effects, including erythema and itching as main toxicities. However, the extent and durability of the therapeutic response to this tyrosine kinase inhibitor should be evaluated in larger patient populations and for longer times. Furthermore, because resistance to therapy with BRAF inhibitors is often associated with a rapid recovery of the mitogen-activated protein kinase pathway [69], the combination of selective BRAF inhibitors such as vemurafenib or dabrafenib and the MEK inhibitor, trametinib, should be evaluated in LCH in future multicentric controlled clinical trials. In this context, the recent recognition of an oncogenic NRAS mutation in ECD [70] may support the relevance of the RAS-RAF-MEK-ERK pathway in the pathogenesis of histiocytoses, as well as suggest its molecular characterization for pharmacogenomic and targeted therapies.
Perturbations of the LCH Microenvironment
LCH lesions contain multiple cell types such as LCH cells (36%–58%), T cells (13%–18%), macrophages (2%–30%), eosinophils (1%–10%), rare B cells (1%–3%), and, surprisingly, multinucleated giant cells (MGCs) [71]. Furthermore, a relative abundance of locally produced cytokines, including IL-1, IL-3, IL-4, IL-8, IL-11, GM-CSF, interferon (IFN)-γ, TNF-α, and transforming growth factor (TGF)-β, has been described within LCH infiltrates [72–74], suggesting they may be inflammatory niches that facilitate both the recruitment and the retention of inflammatory cells lacking a role in immune surveillance [7]. Therefore, a complex cross-talk between cells housed within the LCH lesions occurs and may drive the disease progression, as depicted in Figure 4.
Figure 4.

The cytokine storm: possible interplays with the LCH microenvironment. The LCH microenvironment plays a critical role in the disease progression, both by inducing a local state of immune system frustration and by enhancing the proliferation of pathological LCH cells. Arrows indicate stimulating effects, whereas lines designate inhibitory effects.
Abbreviations: CAT K, cathepsin K; Eo, eosinophils; GM-CSF, granulocyte-macrophage colony-stimulating factor; IFN, interferon; IL, interleukin; LCH, Langerhans cell histiocytosis; MØ, macrophage; MØ M2, macrophages M2 phenotype; M-CSF, macrophage colony-stimulating factor; MGC, multinucleated giant cell; MMP, metalloproteinases; T reg, T regulatory cell; TGF, transforming growth factor; Th17, T helper 17; TNF, tumor necrosis factor; TRAP, tartrate resistant acid phosphatase; VNR, vitronectin receptor.
Notably, MGCs displaying an osteoclast-like phenotype have been reported not only within LCH bone infiltrates but also in nonostotic lesional sites such as both skin and lymph nodes. However, even in ostotic LCH lesions, MGCs appear disseminated within the cellular infiltrate rather than adherent to the bone matrix, as normally occurs. To investigate the origin of MGCs in LCH, da Costa et al. [48] performed a multicolor immunohistochemical analysis of either ostotic or nonostotic lesional sites and showed that LCH polykarions express typical osteoclast markers, including tartrate-resistant acid phosphatase, cathepsin K, vitronectin receptor, as well as the matrix metalloproteinase-9. However, despite CD68 expression, as a typical marker of the monocyte-macrophage lineage by osteoclast-like MGCs in both bone and nonbone lesions, only the polykarions from both skin and lymph nodes coexpressed CD1a, supporting the hypothesis that fusion events between LCH cells themselves occur and, probably, prime the formation of MGCs. Furthermore, because a number of osteoclastogenic factors, including IL-1, IL-6, IL-17, TNF-α, macrophage colony-stimulating factor (M-CSF), and osteopontin, are highly expressed in LCH lesions [48, 72, 75, 76], it has been hypothesized that, as for other malignancies [77, 78], an excess of osteoclast-differentiating cytokines may induce the formation of MGCs from unrelated precursors [7]. In this context, we have previously reported that in multiple myeloma, myeloid progenitors, dendritic cells, and malignant plasma cells can undergo homotypic or heterotypic fusion events, showing an intrinsic fusogenicity and osteoclastogenic potential as a result of the expression of a number of fusogenic proteins, as well as the high sensitivity to fusogenic factors produced within an inflamed bone marrow [78, 79]. Thus, it is conceivable that similar mechanisms drive the formation of multinucleated giant cells, as well as the concomitant osteoclast-like transdifferentiation of LCH cells. In this view, the efficacy of the immunomodulatory drug Lenalidomide in LCH (see below) may be the result of the documented [80] ability in restraining the expression of osteoclast-like enzymes by polykarions, at least in myeloma.
Because LCH is characterized by a number of symptoms that are also observed in several IL-17A-related diseases, such as tuberculosis, rheumatoid arthritis, or Crohn’s disease, the role of IL-17A has been thoroughly investigated. IL-17A is a well-known T-cell-derived cytokine involved in bone resorption through receptor activator for nuclear factor κB ligand (RANKL) induction. A seminal study described high serum levels of this cytokine in patients with active LCH and identified an autocrine IL-17A-dependent pathway enrolled in the fusion of dendritic cells. In this context, whereas a synergistic effect of IFN-γ was described, the IL-17A-dependent fusion proficiency was shown to parallel the activity of LCH, being higher in more aggressive disease [81]. Based on these findings, a potential IL-17A-targeted therapy was proposed for the future treatment of LCH, whereas IL-17A appeared to be a possible biomarker for monitoring the disease activity. However, subsequent studies failed to identify any IL-17A gene expression in LCH cells, nor the IL-17A protein in LCH lesion lysates [82, 83], thus raising several concerns regarding the specificity of the enzyme-linked immunosorbent assay antibody used in those studies [84]. Recently, the absence of IL-17A in LCH cells has been further confirmed, even if higher levels of the cytokine were found in the serum of LCH patients as compared with healthy controls. In particular, a clear-cut correlation between the expression of the IL-17A receptor in LCH tissue and the disease stage has been described, thus settling the controversy about the role of IL-17A as a biomarker of LCH [84]. However, given that IL-17A targeting drugs are still only in preclinical development for applications in LCH [85], the precise function of this cytokine in the disease needs to be elucidated to define the molecular pathways involved in the osteoclast-like transdifferentiation of dendritic cells. This phenomenon has been well established in other malignancies; we ourselves reported it in multiple myeloma, in which it is dependent on the activation of the receptor activator for nuclear factor κB (RANK)/RANKL, CD47/thrombospondin-1, and, overall, IL-17A/IL-17A receptor axes [86–88]. However, definite evidence in LCH is still lacking.
As recently reported [89], IL-17A is not uniquely involved in the formation of so-called giant myeloid inflammatory polykarions, because it regulates long-term survival pathways of myeloid dendritic cells by activating the NF-κB signaling. This, in turn, promotes the expression of the BCL2A1 gene, a prosurvival Bcl-2 family member, thus providing a molecular basis for dendritic cell accumulation, maintenance, and chemoresistance within the LCH infiltrates. Of note, the antimicrotubuli agent Vinblastine, namely the first in-class chemotherapeutic drug against LCH, has been shown to overcome the chemoresistance of IL-17A-treated dendritic cells by downregulating the expression of MCL1, a further prosurvival Bcl-2 familiy member that is constitutively expressed by dendritic cells. Because similar results can be achieved upon treatment with IL-17A-neutralizing antibodies that selectively reduces the expression of BCL2A1, it is conceivable that manipulation of the Bcl-2 family will provide a novel therapeutic avenue for LCH [90].
Consistent with the hypothesis that a state of immune frustration takes place in LCH because of the dysregulated intralesional cytokine production [91], accumulating evidence indicates that macrophages are probably polarized to the anti-inflammatory M2 phenotype within the LCH infiltrates [92]. In particular, a significant number of tumor-associated macrophages has been shown to produce IL-10 and suppress the proliferation of LCH cells via both signal transducer and activator of transcription (STAT)3 activation and cyclin-dependent kinase inhibition. In contrast, because IL-10 is known to induce an aberrant maturation of dendritic cells, it has been hypothesized that this cytokine may prime the typical tolerogenic properties of LCH cells [93], and it may thus be a possible therapeutic target.
Consistent with the hypothesis that a state of immune frustration takes place in LCH because of the dysregulated intralesional cytokine production, accumulating evidence indicates that macrophages are probably polarized to the anti-inflammatory M2 phenotype within the LCH infiltrates.
Whereas eosinophils have been considered innocent bystander cells in LCH, and their recruitment ascribed to the high intralesional levels of CCL5 [61], T cells seem to play a major role in the pathogenesis of the disease. T cells are attracted within LCH granulomas by LCH cell-produced cytokines such as CCL20 and CXCL11 [61] and express a wide repertoire of T-cell receptor γ rearrangements, suggesting they may have a polyclonal nature [94]. Although the occurrence of antigenic stimulation needs further confirmation, LCH-infiltrating T cells have been shown to express activation markers, including CD40L [95], CD45RO [61], and RANKL [48]. Of note, both T cells and LCH cells interact in a cytokine amplification cascade, resulting in the stimulation of autocrine and paracrine stimulatory loops that induce characteristic features of the disease, such as fibrosis, necrosis, and osteolysis [15]. In this context, it has been reported [94] that approximately 20% of intralesional T cells are phenotypically CD4+CD25highFoxP3high regulatory T cells (Treg) and are in close contact with LCH cells. Along with intralesional TGF-β [92], RANK-expressing LCH cells may also have a role in the local expansion of Treg cells [93]. However, expansion of the FoxP3+ Treg compartment was also found in the blood of patients with active LCH as compared with those in clinical remission and with healthy subjects. In keeping with the inflammation-modulating properties of Treg cells, the delayed-type hypersensitivity response in LCH was shown to be impaired, suggesting that underlying perturbations of the immune system are involved, at least in part, in the pathophysiology of the disease [95]. Although an increased percentage of B7, DR4, or Cw7 HLA haplotypes was found in LCH patients [96, 97], further studies are needed to clarify the intimate relationship between immune system dysfunctions and LCH.
Abnormalities of the Marrow Myeloid Compartment in LCH
Based on the hypothesis that LCH cells are clonally derived from the marrow stem cell compartment, the alternative issue has been raised as to whether multisystem LCH is to be considered a myeloid dendritic stem cell disorder [13, 14]. This could bring the classification of LCH closer to that of hematologic malignancies such as leukemias or multiple myeloma in which the malignant clone, despite its origin in bone marrow, may colonize visceral sites. Bone marrow changes have been assessed in LCH and include the presence of CD1a+ LCH cells, whose levels have been strikingly correlated to the disease severity [98, 99]. Moreover, although chemoimmunotherapy allows a temporary control of the disease, bone marrow transplantation is considered to be the only procedure with a potential therapeutic effect, particularly in childhood histiocytoses [100, 101].
Given its ability to induce the proliferation, activation, and differentiation of LCs from marrow progenitors, GM-CSF has been viewed as a possible key player in the pathogenesis of LCH. In this regard, studies by Emile et al. [102] documented a clear correlation between serum levels of the growth factor and the disease extent and activity. Moreover, the expression of both GM-CSF and its receptor has been demonstrated in LCH cells [103, 104]. However, other early hemopoietic cytokines, including M-CSF, stem cell factor, and fms-like tyrosine kinase ligand, are upregulated in the serum of LCH patients and possibly drive the expansion of the circulating blood pool of lineage-negative (lin−)HLA-DR+CD11c+ precursors of dendritic cells [105]. Of interest, the increased levels of CD34+ cells in the blood of LCH patients are also considered to be the result of the presence of circulating LCH cell progenitors, supporting the hypothesis that LCH is a myeloid neoplasm [106].
Also, LCH cells lack the functional β-catenin signaling cascade that modifies their development as compared with normal LCs [107]. In fact, the β-catenin-Wnt pathway seems to be involved in all stages of dendritic cell development, from the earliest dichotomy in marrow progenitor cells to the subsequent lineage determination as monocytes in the intermediate stage and finally as differentiated LCs. Therefore, a defective β-catenin signaling may not only produce a maturation arrest of LCH cells but also separate them from other myeloid elements. Consistently, molecular analyses have recently revealed that LCH cells are the only dendritic cells that express both the Notch ligand Jagged 2 and its receptor [108], substantiating the concept that the marrow development of LCH cells follows unique, still poorly understood routes.
The Therapeutic Management of LCH: Future Directions
No universally accepted guidelines for the treatment of LCH are currently available, as a result of the heterogeneity of the disease in terms of location, severity, and molecular biology. In fact, whereas careful observation and local therapy, including curettage or intralesional injection of steroids or surgical excision, are indicated for single-system LCH with unifocal bone or skin involvement, systemic therapy should be considered for either multisystem disease or single-system forms with multifocal or peculiar site lesions. In this context, regimens including vinblastine plus prednisolone are considered the treatment of choice in the first-line setting, as recently assessed by the LCH-III trial [109]. Thus, given their minor toxicity, several chemotherapeutics, including cladribine, clofarabine, cytarabine, or etoposide, are emerging as reliable alternatives to the chemodoublet [110]. In particular, the second-generation purine analog clofarabine and its prototypic compound cladribine are under intensive investigation in LCH, and exploratory studies have reported promising results in both upfront and refractory setting [111, 112]. Nevertheless, intensive combination chemotherapy exploiting the MACOP-B regimen or hematopoietic stem cell transplantation has been used in aggressive LCH, yielding encouraging results [110].
Based on the recognition that both cell-intrinsic aberrations and microenvironmental perturbations are crucial events in the pathogenesis of LCH, new targeted drugs are currently under intensive investigation for this disease. In particular, the antiangiogenic agents thalidomide and lenalidomide have a proven therapeutic efficacy in low-risk mucocutaneous and/or bone forms of LCH [113, 114]. Both drugs exhibit pleiotropic properties, exerting antiangiogenic, anti-inflammatory, and immunomodulatory effects, and alter the chemokine network of LCH lesions by downregulating the expression of TNF-α. In this context, McClain and Kozinetz [114] have recently carried out a phase II study of thalidomide for the salvage treatment of LCH, recruiting 10 low-risk and 6 high-risk patients. No high-risk LCH patients responded, whereas within the low-risk group 7 complete or partial responses were recorded. Based on this latter promising outcome, other TNF-α inhibitors, including etanercept or infliximab, have been used in LCH patients, showing contrasting results [115–117]. As summarized in Table 1, ongoing clinical trials are currently investigating second-generation purine nucleoside analogs, monoclonal antibodies (moAbs), as well as tyrosine-kinase inhibitors for the treatment of LCH patients. Alemtuzumab is a humanized moAb directed against CD52, a protein that is differentially expressed by normal LCs and LCH cells [118]. Despite anecdotal experiences of promising results in terms of efficacy and tolerability [101], no preliminary data are available from clinical trials. By contrast, the recent discovery of the oncogenic BRAFV600E mutation in LCH patients has raised the possibility that BRAF inhibitors such as vemurafenib or dabrafenib could be applied in the treatment of patients bearing the mutation. Because remarkable effects of imatinib have been described in some patients with histiocytic disorders [119], probably because of inhibition of the differentiation of CD34+ progenitors into dendritic cells, the tyrosine-kinase inhibitor afuresertib is currently under investigation in a phase II clinical trial whose results are eagerly awaited. In this context, immunohistochemical and molecular cytogenetic evaluation of potential targets for tyrosine kinase inhibitors as platelet-derived growth factor receptor (PDGFR), c-KIT, and c-Abl needs to be carried out to identify the optimal subset of responsive patients [120].
Table 1.
Novel treatments in patients with Langerhans cell histiocytosis resistant to conventional therapy; ongoing clinical trials
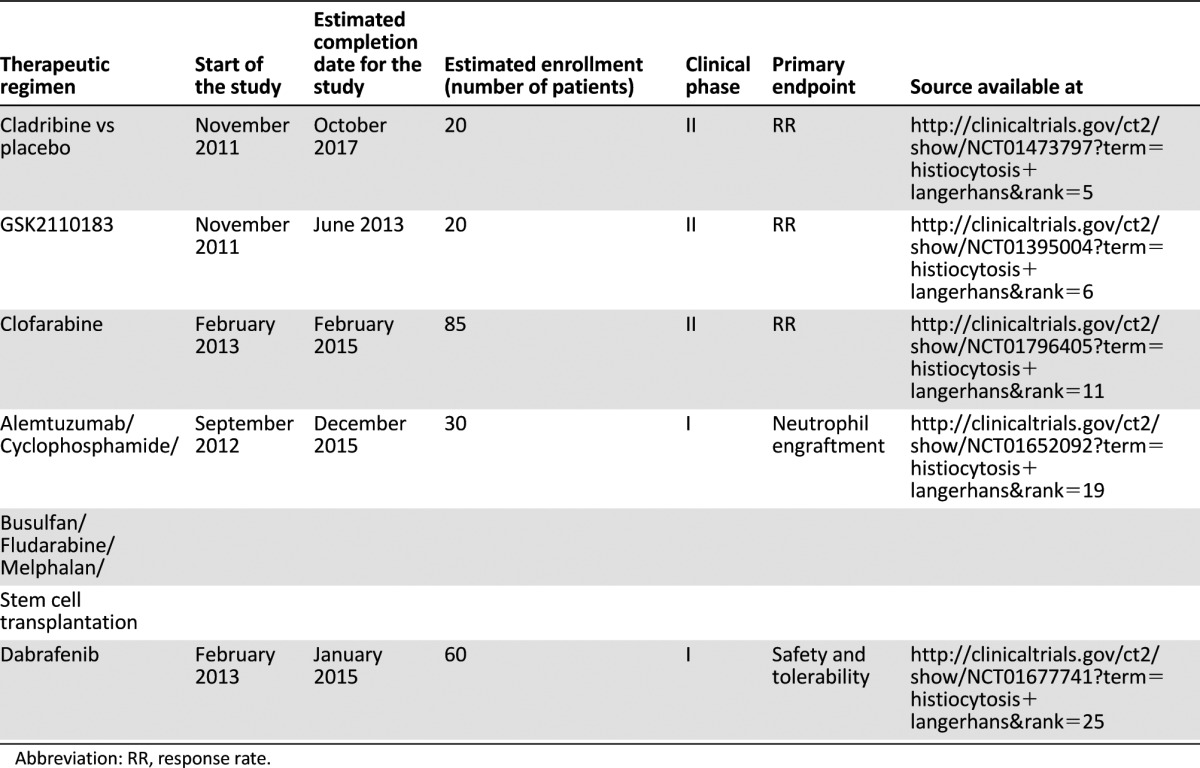
Based on the recognition that both cell-intrinsic aberrations and microenvironmental perturbations are crucial events in the pathogenesis of LCH, new targeted drugs are currently under intensive investigation for this disease. In particular, the antiangiogenic agents thalidomide and lenalidomide have a proven therapeutic efficacy in low-risk mucocutaneous and/or bone forms of LCH.
Intriguingly, based on the recent identification of the role of several Bcl-2 family members in the biology of long-term surviving inflammatory dendritic cells, it has been proposed that both prevention of the IL-17A signal transduction and inhibition of the intracytoplasmic activity of BCL2A1 by small molecule inhibitors loaded into biodegradable nanoparticles may contribute to extend the therapeutic horizon in LCH. However, these innovative strategies, particularly if associated with a therapeutic vaccination with autologous tolerogenic dendritic cells, should be fine-tuned in preclinical studies before assessing their relevance to the clinical practice [89]. In contrast, a fully human anti-CD1a moAb, namely CR2113, was recently produced and provided powerful antibody-dependent cell cytotoxicity activity in vitro, whereas only a modest antitumor effect was shown in CD1a-expressing xenograft models [121]. However, the highly specific targeting capacity found in vivo can provide the basis for future development of CR2113 for diagnostic imaging and/or treatment modality, thus fostering studies aimed at improving its antitumor activity by radiolabeling and/or by conjugation with immunotoxins or BRAF inhibitors. Finally, based on the high expression of metalloproteinase-12 in multisystem LCH and its putative role in the disease progression [122], concurrent studies investigating metalloproteinase inhibitors such as GM6001 are presently in progress [123].
Conclusion
Based on the currently available findings, LCH may include a wide spectrum of neoplastic disorders characterized by a variable degree of clinical severity and, probably, by a number of unidentified genetic abnormalities. In this context, Badalian-Very et al. [4] have recently proposed a model encompassing hyperactivated BRAF as the initiating factor of self-limiting forms of LCH, whereas additional genetic changes would be essential for the development of more aggressive LCH. However, because cancers harboring the BRAFV600E mutation also display additional genomic aberrations [124], a causative rather than merely concurrent role of mutated BRAF should be carefully assessed in LCH and the Badalian-Very hypothesis needs further confirmation in animal models mimicking the disease evolution.
As depicted in Figure 3, it has been hypothesized that expression of the BRAFV6OOE protein may also protect from a malignant transformation of LCH [4]. Indeed, like naevi carrying the mutated form of BRAF [125, 126], the presence of this activating oncogene mutation may trigger cellular senescence as an escape response to cancerogenesis. This, in turn, may explain the ostensibly spontaneous regression of early LCH lesions, particularly in the pulmonary disease form. However, what kind of molecular events orchestrate the fate of LCH cells downstream of the BRAF mutation remains to be identified, and differences in the microenvironment “cytokine storm” should be evaluated as possible determinants of the alternative routes of malignant transformation or senescence of neoplastic BRAFV600E LCH lesions.
Despite overwhelming efforts, the LCH cell of origin is still a matter of debate. Its identification will surely be a milestone in the understanding of the disease. In fact, the coexpression of immunophenotypic markers of both mature LCs and their myeloid precursors has hampered recognition of the maturation stage when the neoplastic transformation occurs. Nonetheless, as recently reported [4], a reactivation of early myeloid genes as a consequence of a transformation-induced reprogramming of the LC transcriptional profile could also explain the apparent paradox of the heterogeneous phenotype. Thus, a dedifferentiation process similar to the epithelial-mesenchymal transition [127] fostered by the inflammatory milieu generated by tumor-associated cells has been hypothesized. However, at present it is still undefined whether underlying myeloid abnormalities drive the onset of LCH. The attribution of a pathogenic role to BRAFV600E or other recurrent genetic abnormalities, and their identification in a subset of LC precursors, will help to define the role of the bone marrow in the pathophysiology of the disease. Meanwhile, the hypothesis depicting LCH as an “inflammatory myeloid neoplasm” in which BRAF activation influences lineage commitment, migration, and proliferation of myeloid dendritic cell precursors recruiting bystander cells within the lesions cannot be excluded [128]. This would combine the two distinct entities of immune disorder and neoplasia to be reconciled under the LCH multifactorial clinical disease.
Based on such a novel picture of the LCH pathogenesis, the therapeutic landscape of this likely neoplasm is actually changing. Targeted therapeutics including antibodies aimed at modulating the microenvironment production of cytokines, including TNF-α, IL-1, IL-17A, IL-17A receptor, RANKL, as well as novel agents targeting the CD1a+ LCs, are potential candidates for tests in clinical trials. Therefore, bench-to-bedside-and-back studies are warranted to improve clinical outcomes in LCH patients.
This article is available for continuing medical education credit at CME.TheOncologist.com.
Acknowledgments
We are grateful to Dr. Francesca Bisceglia for the editorial assistance in the manuscript preparation. This work was supported by a grant from the Italian Association for Cancer Research (IG11647) and from the Italian Ministry for the University and Research (PRIN 2010). F.M.R. and M.C. equally contributed to the preparation of this article.
Footnotes
For Further Reading: Jun Yin, Feng Zhang, Huizhen Zhang et al. Hand-Schüller-Christian Disease and Erdheim-Chester Disease: Coexistence and Discrepancy. The Oncologist 2013;18:19–24.
Implications for Practice: Central diabetes insipitus (CDI) is usually the first or one of the first symptoms of Hand-Schüller-Christian disease (HSC). It is difficult to determine whether CDI is part of HSC at its onset. We propose a new triad of symptoms including central diabetes insipitus, hyperprolactinemia, and pituitary stalk thickening on MRI. If a patient is present with the triad, HSC should be considered. Bone scans are very useful to reveal HSC in the absence of bone pain. Langerhans cell histiocytosis (LCH) and Erdheim-Chester disease (ECD) are featured with osteolytic lesions and osteosclerosis, respectively. If osteosclerosis is observed in a patient with LCH, coexistence of ECD should be considered. A new biopsy is helpful for the diagnosis.
Author Contributions
Conception/Design: Mauro Cives, Francesca M. Rizzo, Franco Silvestris
Collection and/or assembly of data: Francesca M. Rizzo, Valeria Simone
Data analysis and interpretation: Mauro Cives, Francesca M. Rizzo, Valeria Simone, Franco Silvestris
Manuscript writing: Mauro Cives, Francesca M. Rizzo, Franco Silvestris
Final approval of manuscript: Mauro Cives, Francesca M. Rizzo, Valeria Simone, Franco Silvestris
Disclosures
The authors indicated no financial relationships.
Section Editors: Fred Hirsch: Abbott (P – EGFR FISH); Genentech/Roche, Pfizer, Boehringer Ingelheim, BMS, Celgene, Novartis, Imclone/Lilly (C/A); Amgen, Genentech, Celgene, Ventana, Imclone/Lilly (RF); Jeffrey Ross: leadership position with Foundation Medicine; Foundation Medicine, Syfr (O); Genentech/Roche, Bristol-Myers Squibb, Boehringer-Ingelheim (H); research funding received from Foundation Medicine (RF); Foundation Medicine, Affiliated Pathology Services at Albany Medical Center (E)
Reviewer “A”: None
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1.de Chadarévian JP, Pawel BR. Hereditary Langerhans cell histiocytosis: Instances of apparent vertical transmission. Med Pediatr Oncol. 1998;31:559. doi: 10.1002/(sici)1096-911x(199812)31:6<559::aid-mpo27>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 2.Aricò M, Nichols K, Whitlock JA, et al. Familial clustering of Langerhans cell histiocytosis. Br J Haematol. 1999;107:883–888. doi: 10.1046/j.1365-2141.1999.01777.x. [DOI] [PubMed] [Google Scholar]
- 3.Guyot-Goubin A, Donadieu J, Barkaoui M, et al. Descriptive epidemiology of childhood Langerhans cell histiocytosis in France, 2000-2004. Pediatr Blood Cancer. 2008;51:71–75. doi: 10.1002/pbc.21498. [DOI] [PubMed] [Google Scholar]
- 4.Badalian-Very G, Vergilio JA, Fleming M, et al. Pathogenesis of Langerhans cell histiocytosis. Annu Rev Pathol. 2013;8:1–20. doi: 10.1146/annurev-pathol-020712-163959. [DOI] [PubMed] [Google Scholar]
- 5.Huang F, Arceci R. The histiocytoses of infancy. Semin Perinatol. 1999;23:319–331. doi: 10.1016/s0146-0005(99)80040-8. [DOI] [PubMed] [Google Scholar]
- 6.Suri HS, Yi ES, Nowakowski GS, et al. Pulmonary Langerhans cell histiocytosis. Orphanet J Rare Dis. 2012;7:16. doi: 10.1186/1750-1172-7-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Egeler RM, van Halteren AG, Hogendoorn PC, et al. Langerhans cell histiocytosis: Fascinating dynamics of the dendritic cell-macrophage lineage. Immunol Rev. 2010;234:213–232. doi: 10.1111/j.0105-2896.2009.00883.x. [DOI] [PubMed] [Google Scholar]
- 8.Badalian-Very G, Vergilio JA, Degar BA, et al. Recent advances in the understanding of Langerhans cell histiocytosis. Br J Haematol. 2012;156:163–172. doi: 10.1111/j.1365-2141.2011.08915.x. [DOI] [PubMed] [Google Scholar]
- 9.Gadner H, Grois N, Arico M, et al. Histiocyte Society A randomized trial of treatment for multisystem Langerhans’ cell histiocytosis. J Pediatr. 2001;138:728–734. doi: 10.1067/mpd.2001.111331. [DOI] [PubMed] [Google Scholar]
- 10.Minkov M, Grois N, Heitger A, et al. DAL-HX Study Group Treatment of multisystem Langerhans cell histiocytosis: Results of the DAL-HX 83 and DAL-HX 90 studies. Klin Padiatr. 2000;212:139–144. doi: 10.1055/s-2000-9667. [DOI] [PubMed] [Google Scholar]
- 11.Haupt R, Nanduri V, Calevo MG, et al. Permanent consequences in Langerhans cell histiocytosis patients: A pilot study from the Histiocyte Society-Late Effects Study Group. Pediatr Blood Cancer. 2004;42:438–444. doi: 10.1002/pbc.20021. [DOI] [PubMed] [Google Scholar]
- 12.Egeler RM, Neglia JP, Puccetti DM, et al. Association of Langerhans cell histiocytosis with malignant neoplasms. Cancer. 1993;71:865–873. doi: 10.1002/1097-0142(19930201)71:3<865::aid-cncr2820710334>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 13.Allen CE, Li L, Peters TL, et al. Cell-specific gene expression in Langerhans cell histiocytosis lesions reveals a distinct profile compared with epidermal Langerhans cells. J Immunol. 2010;184:4557–4567. doi: 10.4049/jimmunol.0902336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Badalian-Very G, Vergilio JA, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116:1919–1923. doi: 10.1182/blood-2010-04-279083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Egeler RM, Favara BE, van Meurs M, et al. Differential in situ cytokine profiles of Langerhans-like cells and T cells in Langerhans cell histiocytosis: Abundant expression of cytokines relevant to disease and treatment. Blood. 1999;94:4195–4201. [PubMed] [Google Scholar]
- 16.Lichtenstein L. Histiocytosis X; integration of eosinophilic granuloma of bone, Letterer-Siwe disease, and Schüller-Christian disease as related manifestations of a single nosologic entity. AMA Arch Pathol. 1953;56:84–102. [PubMed] [Google Scholar]
- 17.Caux C, Dezutter-Dambuyant C, Schmitt D, et al. GM-CSF and TNF-alpha cooperate in the generation of dendritic Langerhans cells. Nature. 1992;360:258–261. doi: 10.1038/360258a0. [DOI] [PubMed] [Google Scholar]
- 18.Manz MG, Miyamoto T, Akashi K, et al. Prospective isolation of human clonogenic common myeloid progenitors. Proc Natl Acad Sci USA. 2002;99:11872–11877. doi: 10.1073/pnas.172384399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Geissmann F, Manz MG, Jung S, et al. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327:656–661. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Merad M, Manz MG, Karsunky H, et al. Langerhans cells renew in the skin throughout life under steady-state conditions. Nat Immunol. 2002;3:1135–1141. doi: 10.1038/ni852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaplan DH, Jenison MC, Saeland S, et al. Epidermal Langerhans cell-deficient mice develop enhanced contact hypersensitivity. Immunity. 2005;23:611–620. doi: 10.1016/j.immuni.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 22.Chorro L, Sarde A, Li M, et al. Langerhans cell (LC) proliferation mediates neonatal development, homeostasis, and inflammation-associated expansion of the epidermal LC network. J Exp Med. 2009;206:3089–3100. doi: 10.1084/jem.20091586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ginhoux F, Merad M. Ontogeny and homeostasis of Langerhans cells. Immunol Cell Biol. 2010;88:387–392. doi: 10.1038/icb.2010.38. [DOI] [PubMed] [Google Scholar]
- 24.Chen W, Wang J, Wang E, et al. Detection of clonal lymphoid receptor gene rearrangements in Langerhans cell histiocytosis. Am J Surg Pathol. 2010;34:1049–1057. doi: 10.1097/PAS.0b013e3181e5341a. [DOI] [PubMed] [Google Scholar]
- 25.Doulatov S, Notta F, Eppert K, et al. Revised map of the human progenitor hierarchy shows the origin of macrophages and dendritic cells in early lymphoid development. Nat Immunol. 2010;11:585–593. doi: 10.1038/ni.1889. [DOI] [PubMed] [Google Scholar]
- 26.West DS, Dogan A, Quint PS, et al. Clonally related follicular lymphomas and Langerhans cell neoplasms: Expanding the spectrum of transdifferentiation. Am J Surg Pathol. 2013;37:978–986. doi: 10.1097/PAS.0b013e318283099f. [DOI] [PubMed] [Google Scholar]
- 27.Hunger RE, Sieling PA, Ochoa MT, et al. Langerhans cells utilize CD1a and Langerin to efficiently present nonpeptide antigens to T cells. J Clin Invest. 2004;113:701–708. doi: 10.1172/JCI19655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van der Vlist M, Geijtenbeek TB. Langerin functions as an antiviral receptor on Langerhans cells. Immunol Cell Biol. 2010;88:410–415. doi: 10.1038/icb.2010.32. [DOI] [PubMed] [Google Scholar]
- 29.Birbeck MS, Breathnach AS, Everall JD. An electron microscope study of basal melanocytes and high-level clear cells (Langerhans cells) in vitiligo. J Invest Dermatol. 1961;37:51. [Google Scholar]
- 30.Mizumoto N, Takashima A. CD1a and langerin: Acting as more than Langerhans cell markers. J Clin Invest. 2004;113:658–660. doi: 10.1172/JCI21140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Merad M, Ginhoux F, Collin M. Origin, homeostasis and function of Langerhans cells and other langerin-expressing dendritic cells. Nat Rev Immunol. 2008;8:935–947. doi: 10.1038/nri2455. [DOI] [PubMed] [Google Scholar]
- 32.Romani N, Clausen BE, Stoitzner P. Langerhans cells and more: Langerin-expressing dendritic cell subsets in the skin. Immunol Rev. 2010;234:120–141. doi: 10.1111/j.0105-2896.2009.00886.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Idoyaga J, Suda N, Suda K, et al. Antibody to Langerin/CD207 localizes large numbers of CD8alpha+ dendritic cells to the marginal zone of mouse spleen. Proc Natl Acad Sci USA. 2009;106:1524–1529. doi: 10.1073/pnas.0812247106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Geissmann F, Lepelletier Y, Fraitag S, et al. Differentiation of Langerhans cells in Langerhans cell histiocytosis. Blood. 2001;97:1241–1248. doi: 10.1182/blood.v97.5.1241. [DOI] [PubMed] [Google Scholar]
- 35.Park HJ, Jeon YK, Lee AH, et al. Use of the JL1 epitope, which encompasses the nonglycosylation site of CD43, as a marker of immature/neoplastic Langerhans cells. Am J Surg Pathol. 2012;36:1150–1157. doi: 10.1097/PAS.0b013e31825b9914. [DOI] [PubMed] [Google Scholar]
- 36.Yu RC, Chu C, Buluwela L, et al. Clonal proliferation of Langerhans cells in Langerhans cell histiocytosis. Lancet. 1994;343:767–768. doi: 10.1016/s0140-6736(94)91842-2. [DOI] [PubMed] [Google Scholar]
- 37.Willman CL, Busque L, Griffith BB, et al. Langerhans’-cell histiocytosis (histiocytosis X)—a clonal proliferative disease. N Engl J Med. 1994;331:154–160. doi: 10.1056/NEJM199407213310303. [DOI] [PubMed] [Google Scholar]
- 38.Betts DR, Leibundgut KE, Feldges A, et al. Cytogenetic abnormalities in Langerhans cell histiocytosis. Br J Cancer. 1998;77:552–555. doi: 10.1038/bjc.1998.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.da Costa CE, Szuhai K, van Eijk R, et al. No genomic aberrations in Langerhans cell histiocytosis as assessed by diverse molecular technologies. Genes Chromosomes Cancer. 2009;48:239–249. doi: 10.1002/gcc.20634. [DOI] [PubMed] [Google Scholar]
- 40.Murakami I, Gogusev J, Fournet JC, et al. Detection of molecular cytogenetic aberrations in Langerhans cell histiocytosis of bone. Hum Pathol. 2002;33:555–560. doi: 10.1053/hupa.2002.124035. [DOI] [PubMed] [Google Scholar]
- 41.Chikwava KR, Hunt JL, Mantha GS, et al. Analysis of loss of heterozygosity in single-system and multisystem Langerhans’ cell histiocytosis. Pediatr Dev Pathol. 2007;10:18–24. doi: 10.2350/06-02-0045.1. [DOI] [PubMed] [Google Scholar]
- 42.Yousem SA, Colby TV, Chen YY, et al. Pulmonary Langerhans’ cell histiocytosis: Molecular analysis of clonality. Am J Surg Pathol. 2001;25:630–636. doi: 10.1097/00000478-200105000-00010. [DOI] [PubMed] [Google Scholar]
- 43.Schönfeld N, Dirks K, Costabel U, et al. Wissenschaftliche Arbeitsgemeinschaft für die Therapie von Lungenkrankheiten A prospective clinical multicentre study on adult pulmonary Langerhans’ cell histiocytosis. Sarcoidosis Vasc Diffuse Lung Dis. 2012;29:132–138. [PubMed] [Google Scholar]
- 44.Francus T, Klein RF, Staiano-Coico L, et al. Effects of tobacco glycoprotein (TGP) on the immune system. II. TGP stimulates the proliferation of human T cells and the differentiation of human B cells into Ig secreting cells. J Immunol. 1988;140:1823–1829. [PubMed] [Google Scholar]
- 45.Youkeles LH, Grizzanti JN, Liao Z, et al. Decreased tobacco-glycoprotein-induced lymphocyte proliferation in vitro in pulmonary eosinophilic granuloma. Am J Respir Crit Care Med. 1995;151:145–150. doi: 10.1164/ajrccm.151.1.7812544. [DOI] [PubMed] [Google Scholar]
- 46.Nichols KE, Egeler RM, Perry VH, et al. Summary of the 12th Nikolas Symposium dendritic cell differentiation: Signals, signaling and functional consequences as clues to possible therapy. J Pediatr Hematol Oncol. 2003;25:193–197. doi: 10.1097/00043426-200303000-00003. [DOI] [PubMed] [Google Scholar]
- 47.Jeziorski E, Senechal B, Molina TJ, et al. Herpes-virus infection in patients with Langerhans cell histiocytosis: A case-controlled sero-epidemiological study, and in situ analysis. PLoS One. 2008;3:e3262. doi: 10.1371/journal.pone.0003262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.da Costa CE, Annels NE, Faaij CM, et al. Presence of osteoclast-like multinucleated giant cells in the bone and nonostotic lesions of Langerhans cell histiocytosis. J Exp Med. 2005;201:687–693. doi: 10.1084/jem.20041785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schouten B, Egeler RM, Leenen PJ, et al. Expression of cell cycle-related gene products in Langerhans cell histiocytosis. J Pediatr Hematol Oncol. 2002;24:727–732. doi: 10.1097/00043426-200212000-00009. [DOI] [PubMed] [Google Scholar]
- 50.Amir G, Weintraub M. Association of cell cycle-related gene products and NF-kappaB with clinical parameters in Langerhans cell histiocytosis. Pediatr Blood Cancer. 2008;50:304–307. doi: 10.1002/pbc.21198. [DOI] [PubMed] [Google Scholar]
- 51.Petersen BL, Rengtved P, Bank MI, et al. High expression of markers of apoptosis in Langerhans cell histiocytosis. Histopathology. 2003;42:186–193. doi: 10.1046/j.1365-2559.2003.01565.x. [DOI] [PubMed] [Google Scholar]
- 52.Bechan GI, Meeker AK, De Marzo AM, et al. Telomere length shortening in Langerhans cell histiocytosis. Br J Haematol. 2008;140:420–428. doi: 10.1111/j.1365-2141.2007.06904.x. [DOI] [PubMed] [Google Scholar]
- 53.da Costa CE, Egeler RM, Hoogeboom M, et al. Differences in telomerase expression by the CD1a+ cells in Langerhans cell histiocytosis reflect the diverse clinical presentation of the disease. J Pathol. 2007;212:188–197. doi: 10.1002/path.2167. [DOI] [PubMed] [Google Scholar]
- 54.de Graaf JH, Tamminga RY, Kamps WA, et al. Langerhans’ cell histiocytosis: Expression of leukocyte cellular adhesion molecules suggests abnormal homing and differentiation. Am J Pathol. 1994;144:466–472. [PMC free article] [PubMed] [Google Scholar]
- 55.de Graaf JH, Tamminga RY, Kamps WA, et al. Expression of cellular adhesion molecules in Langerhans cell histiocytosis and normal Langerhans cells. Am J Pathol. 1995;147:1161–1171. [PMC free article] [PubMed] [Google Scholar]
- 56.Geissmann F, Emile JF, Andry P, et al. Lack of expression of E-cadherin is associated with dissemination of Langerhans’ cell histiocytosis and poor outcome. J Pathol. 1997;181:301–304. doi: 10.1002/(SICI)1096-9896(199703)181:3<301::AID-PATH779>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 57.Sozzani S, Allavena P, D’Amico G, et al. Differential regulation of chemokine receptors during dendritic cell maturation: A model for their trafficking properties. J Immunol. 1998;161:1083–1086. [PubMed] [Google Scholar]
- 58.Sallusto F, Schaerli P, Loetscher P, et al. Rapid and coordinated switch in chemokine receptor expression during dendritic cell maturation. Eur J Immunol. 1998;28:2760–2769. doi: 10.1002/(SICI)1521-4141(199809)28:09<2760::AID-IMMU2760>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 59.Dieu MC, Vanbervliet B, Vicari A, et al. Selective recruitment of immature and mature dendritic cells by distinct chemokines expressed in different anatomic sites. J Exp Med. 1998;188:373–386. doi: 10.1084/jem.188.2.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fleming MD, Pinkus JL, Fournier MV, et al. Coincident expression of the chemokine receptors CCR6 and CCR7 by pathologic Langerhans cells in Langerhans cell histiocytosis. Blood. 2003;101:2473–2475. doi: 10.1182/blood.V101.7.2473. [DOI] [PubMed] [Google Scholar]
- 61.Annels NE, Da Costa CE, Prins FA, et al. Aberrant chemokine receptor expression and chemokine production by Langerhans cells underlies the pathogenesis of Langerhans cell histiocytosis. J Exp Med. 2003;197:1385–1390. doi: 10.1084/jem.20030137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nichols KE, Arceci RJ. BRAF, a piece of the LCH puzzle. Blood. 2010;116:1825–1827. doi: 10.1182/blood-2010-06-289934. [DOI] [PubMed] [Google Scholar]
- 63.Tadmor T, Tiacci E, Falini B, et al. The BRAF-V600E mutation in hematological malignancies: A new player in hairy cell leukemia and Langerhans cell histiocytosis. Leuk Lymphoma. 2012;53:2339–2340. doi: 10.3109/10428194.2012.706289. [DOI] [PubMed] [Google Scholar]
- 64.Sahm F, Capper D, Preusser M, et al. BRAFV600E mutant protein is expressed in cells of variable maturation in Langerhans cell histiocytosis. Blood. 2012;120:e28–e34. doi: 10.1182/blood-2012-06-429597. [DOI] [PubMed] [Google Scholar]
- 65.Bates SV, Lakshmanan A, Green AL, et al. BRAF V600E-positive multisite Langerhans cell histiocytosis in a preterm neonate. AJP Rep. 2013;3:63–66. doi: 10.1055/s-0033-1338168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yousem SA, Dacic S, Nikiforov YE, et al. Pulmonary Langerhans cell histiocytosis: Profiling of multifocal tumors using next-generation sequencing identifies concordant occurrence of BRAF V600E mutations. Chest. 2013;143:1679–1684. doi: 10.1378/chest.12-1917. [DOI] [PubMed] [Google Scholar]
- 67.Haroche J, Charlotte F, Arnaud L, et al. High prevalence of BRAF V600E mutations in Erdheim-Chester disease but not in other non-Langerhans cell histiocytoses. Blood. 2012;120:2700–2703. doi: 10.1182/blood-2012-05-430140. [DOI] [PubMed] [Google Scholar]
- 68.Haroche J, Cohen-Aubart F, Emile JF, et al. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim-Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood. 2013;121:1495–1500. doi: 10.1182/blood-2012-07-446286. [DOI] [PubMed] [Google Scholar]
- 69.Paraiso KH, Fedorenko IV, Cantini LP, et al. Recovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. Br J Cancer. 2010;102:1724–1730. doi: 10.1038/sj.bjc.6605714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Diamond EL, Abdel-Wahab O, Pentsova E, et al. Detection of an NRAS mutation in Erdheim-Chester disease. Blood. 2013;122:1089–1091. doi: 10.1182/blood-2013-02-482984. [DOI] [PubMed] [Google Scholar]
- 71.Favara BE, Steele A. Langerhans cell histiocytosis of lymph nodes: A morphological assessment of 43 biopsies. Pediatr Pathol Lab Med. 1997;17:769–787. [PubMed] [Google Scholar]
- 72.de Graaf JH, Tamminga RY, Dam-Meiring A, et al. The presence of cytokines in Langerhans’ cell histiocytosis. J Pathol. 1996;180:400–406. doi: 10.1002/(SICI)1096-9896(199612)180:4<400::AID-PATH701>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 73.Kannourakis G, Abbas A. The role of cytokines in the pathogenesis of Langerhans cell histiocytosis. Br J Cancer Suppl. 1994;23:S37–S40. [PMC free article] [PubMed] [Google Scholar]
- 74.Brown RE. Angiotensin-converting enzyme, transforming growth factor beta(1), and interleukin 11 in the osteolytic lesions of Langerhans cell histiocytosis. Arch Pathol Lab Med. 2000;124:1287–1290. doi: 10.5858/2000-124-1287-ACETGF. [DOI] [PubMed] [Google Scholar]
- 75.Andersson By U, Tani E, Andersson U, et al. Tumor necrosis factor, interleukin 11, and leukemia inhibitory factor produced by Langerhans cells in Langerhans cell histiocytosis. J Pediatr Hematol Oncol. 2004;26:706–711. doi: 10.1097/00043426-200411000-00004. [DOI] [PubMed] [Google Scholar]
- 76.Oh Y, Oh I, Morimoto J, et al. Osteopontin has a crucial role in osteoclast-like multinucleated giant cell formation. J Cell Biochem. 2013 doi: 10.1002/jcb.24695. [E-pub ahead of print] [DOI] [PubMed] [Google Scholar]
- 77.Vignery A. Macrophage fusion: Are somatic and cancer cells possible partners? Trends Cell Biol. 2005;15:188–193. doi: 10.1016/j.tcb.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 78.Cives M, Ciavarella S, Dammacco F, et al. Cell fusion in myeloma marrow microenvironment: Role in tumor progression. Crit Rev Oncog. 2013;18:75–95. doi: 10.1615/critrevoncog.v18.i1-2.50. [DOI] [PubMed] [Google Scholar]
- 79.Tucci M, Ciavarella S, Strippoli S, et al. Immature dendritic cells from patients with multiple myeloma are prone to osteoclast differentiation in vitro. Exp Hematol. 2011;39:773–783, e1. doi: 10.1016/j.exphem.2011.04.006. [DOI] [PubMed] [Google Scholar]
- 80.Cives M, Simone V, Brunetti O, et al. Novel lenalidomide-based combinations for treatment of multiple myeloma. Crit Rev Oncol Hematol. 2013;85:9–20. doi: 10.1016/j.critrevonc.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 81.Coury F, Annels N, Rivollier A, et al. Langerhans cell histiocytosis reveals a new IL-17A-dependent pathway of dendritic cell fusion. Nat Med. 2008;14:81–87. doi: 10.1038/nm1694. [DOI] [PubMed] [Google Scholar]
- 82.Allen CE, McClain KL. Interleukin-17A is not expressed by CD207(+) cells in Langerhans cell histiocytosis lesions. Nat Med. 2009;15:483–484; author reply 484–485. doi: 10.1038/nm0509-483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Peters TL, McClain KL, Allen CE. Neither IL-17A mRNA nor IL-17A protein are detectable in Langerhans cell histiocytosis lesions. Mol Ther. 2011;19:1433–1439. doi: 10.1038/mt.2011.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Murakami I, Morimoto A, Oka T, et al. IL-17A receptor expression differs between subclasses of Langerhans cell histiocytosis, which might settle the IL-17A controversy. Virchows Arch. 2013;462:219–228. doi: 10.1007/s00428-012-1360-6. [DOI] [PubMed] [Google Scholar]
- 85.Hogarty MD. IL-17A in LCH: Systemic biomarker, local factor, or none of the above? Mol Ther. 2011;19:1405–1406. doi: 10.1038/mt.2011.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kukreja A, Radfar S, Sun BH, et al. Dominant role of CD47-thrombospondin-1 interactions in myeloma-induced fusion of human dendritic cells: Implications for bone disease. Blood. 2009;114:3413–3421. doi: 10.1182/blood-2009-03-211920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tucci M, Stucci S, Strippoli S, et al. Dendritic cells and malignant plasma cells: An alliance in multiple myeloma tumor progression? The Oncologist. 2011;16:1040–1048. doi: 10.1634/theoncologist.2010-0327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tucci M, Stucci S, Savonarola A, et al. Immature dendritic cells in multiple myeloma are prone to osteoclast-like differentiation through interleukin-17A stimulation. Br J Haematol. 2013;161:821–831. doi: 10.1111/bjh.12333. [DOI] [PubMed] [Google Scholar]
- 89.Olsson Åkefeldt S, Ismail MB, Valentin H, et al. Targeting BCL2 family in human myeloid dendritic cells: a challenge to cure diseases with chronic inflammations associated with bone loss. Clin Dev Immunol. 2013;701305 doi: 10.1155/2013/701305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Olsson Åkefeldt S, Maisse C, Belot A, et al. Chemoresistance of human monocyte-derived dendritic cells is regulated by IL-17A. PLoS One. 2013;8:e56865. doi: 10.1371/journal.pone.0056865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Alexandrescu S, Tatevian N, Czerniak BA, et al. Morphoproteomics provides support for TGF-β pathway signaling in the osteoclastogenesis and immune dysregulation of osteolytic Langerhans cell histiocytosis. Int J Clin Exp Pathol. 2012;5:503–511. [PMC free article] [PubMed] [Google Scholar]
- 92.Ohnishi K, Komohara Y, Sakashita N, et al. Macrophages in Langerhans cell histiocytosis are differentiated toward M2 phenotype: Their possible involvement in pathological processes. Pathol Int. 2010;60:27–34. doi: 10.1111/j.1440-1827.2009.02472.x. [DOI] [PubMed] [Google Scholar]
- 93.Frick JS, Grünebach F, Autenrieth IB. Immunomodulation by semi-mature dendritic cells: A novel role of Toll-like receptors and interleukin-6. Int J Med Microbiol. 2010;300:19–24. doi: 10.1016/j.ijmm.2009.08.010. [DOI] [PubMed] [Google Scholar]
- 94.Senechal B, Elain G, Jeziorski E, et al. Expansion of regulatory T cells in patients with Langerhans cell histiocytosis. PLoS Med. 2007;4:e253. doi: 10.1371/journal.pmed.0040253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Egeler RM, Favara BE, Laman JD, et al. Abundant expression of CD40 and CD40-ligand (CD154) in paediatric Langerhans cell histiocytosis lesions. Eur J Cancer. 2000;36:2105–2110. doi: 10.1016/s0959-8049(00)00296-3. [DOI] [PubMed] [Google Scholar]
- 96.McClain KL, Laud P, Wu WS, et al. Langerhans cell histiocytosis patients have HLA Cw7 and DR4 types associated with specific clinical presentations and no increased frequency in polymorphisms of the tumor necrosis factor alpha promoter. Med Pediatr Oncol. 2003;41:502–507. doi: 10.1002/mpo.10366. [DOI] [PubMed] [Google Scholar]
- 97.Bernstrand C, Carstensen H, Jakobsen B, et al. Immunogenetic heterogeneity in single-system and multisystem Langerhans cell histiocytosis. Pediatr Res. 2003;54:30–36. doi: 10.1203/01.PDR.0000069844.50684.7D. [DOI] [PubMed] [Google Scholar]
- 98.Minkov M, Pötschger U, Grois N, et al. Bone marrow assessment in Langerhans cell histiocytosis. Pediatr Blood Cancer. 2007;49:694–698. doi: 10.1002/pbc.21227. [DOI] [PubMed] [Google Scholar]
- 99.Galluzzo ML, Braier J, Rosenzweig SD, et al. Bone marrow findings at diagnosis in patients with multisystem Langerhans cell histiocytosis. Pediatr Dev Pathol. 2010;13:101–106. doi: 10.2350/09-05-0651-OA.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Caselli D, Aricò M, EBMT Paediatric Working Party The role of BMT in childhood histiocytoses. Bone Marrow Transplant. 2008;41(suppl 2):S8–S13. doi: 10.1038/bmt.2008.46. [DOI] [PubMed] [Google Scholar]
- 101.Steiner M, Matthes-Martin S, Attarbaschi A, et al. Importance of allogeneic T-cells for disease control after stem cell transplantation for high-risk Langerhans cell histiocytosis. Haematologica. 2007;92:e3–e4. doi: 10.3324/haematol.10993. [DOI] [PubMed] [Google Scholar]
- 102.Emile JF, Tartour E, Brugières L, et al. Detection of GM-CSF in the sera of children with Langerhans’ cell histiocytosis. Pediatr Allergy Immunol. 1994;5:162–163. doi: 10.1111/j.1399-3038.1994.tb00232.x. [DOI] [PubMed] [Google Scholar]
- 103.Emile JF, Peuchmaur M, Fraitag S, et al. Immunohistochemical detection of granulocyte/macrophage colony-stimulating factor in Langerhans’ cell histiocytosis. Histopathology. 1993;23:327–332. doi: 10.1111/j.1365-2559.1993.tb01215.x. [DOI] [PubMed] [Google Scholar]
- 104.Emile JF, Fraitag S, Andry P, et al. Expression of GM-CSF receptor by Langerhans’ cell histiocytosis cells. Virchows Arch. 1995;427:125–129. doi: 10.1007/BF00196516. [DOI] [PubMed] [Google Scholar]
- 105.Rolland A, Guyon L, Gill M, et al. Increased blood myeloid dendritic cells and dendritic cell-poietins in Langerhans cell histiocytosis. J Immunol. 2005;174:3067–3071. doi: 10.4049/jimmunol.174.5.3067. [DOI] [PubMed] [Google Scholar]
- 106.Misery L, Rougier N, Crestani B, et al. Presence of circulating abnormal CD34+ progenitors in adult Langerhans cell histiocytosis. Clin Exp Immunol. 1999;117:177–182. doi: 10.1046/j.1365-2249.1999.00950.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Leenen PJ, Egeler RM. Langerhans’ cell histiocytosis is caused by dysregulation of the E-cadherin-beta-catenin cascade: A hypothesis. Immunol Cell Biol. 1999;77:460–467. doi: 10.1046/j.1440-1711.1999.00856.x. [DOI] [PubMed] [Google Scholar]
- 108.Hutter C, Kauer M, Simonitsch-Klupp I, et al. Notch is active in Langerhans cell histiocytosis and confers pathognomonic features on dendritic cells. Blood. 2012;120:5199–5208. doi: 10.1182/blood-2012-02-410241. [DOI] [PubMed] [Google Scholar]
- 109.Gadner H, Minkov M, Grois N, et al. Histiocyte Society Therapy prolongation improves outcome in multisystem Langerhans cell histiocytosis. Blood. 2013;121:5006–5014. doi: 10.1182/blood-2012-09-455774. [DOI] [PubMed] [Google Scholar]
- 110.Girschikofsky M, Arico M, Castillo D, et al. Management of adult patients with Langerhans cell histiocytosis: Recommendations from an expert panel on behalf of Euro-Histio-Net. Orphanet J Rare Dis. 2013;8:72. doi: 10.1186/1750-1172-8-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Adam Z, Szturz P, Vaníček J, et al. Cladribine (2-chlorodeoxyadenosine) in frontline chemotherapy for adult Langerhans cell histiocytosis: A single-center study of seven cases. Acta Oncol. 2013;52:994–1001. doi: 10.3109/0284186X.2012.716164. [DOI] [PubMed] [Google Scholar]
- 112.Simko SJ, Tran HD, Jones J, et al. Clofarabine salvage therapy in refractory multifocal histiocytic disorders, including Langerhans cell histiocytosis, juvenile xanthogranuloma and Rosai-Dorfman disease. Pediatr Blood Cancer. 2013 doi: 10.1002/pbc.24772. [E-pub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Szturz P, Adam Z, Rehák Z, et al. Lenalidomide proved effective in multisystem Langerhans cell histiocytosis. Acta Oncol. 2012;51:412–415. doi: 10.3109/0284186X.2011.631581. [DOI] [PubMed] [Google Scholar]
- 114.McClain KL, Kozinetz CA. A phase II trial using thalidomide for Langerhans cell histiocytosis. Pediatr Blood Cancer. 2007;48:44–49. doi: 10.1002/pbc.20578. [DOI] [PubMed] [Google Scholar]
- 115.Henter JI, Karlén J, Calming U, et al. Successful treatment of Langerhans’-cell histiocytosis with etanercept. N Engl J Med. 2001;345:1577–1578. doi: 10.1056/NEJM200111223452118. [DOI] [PubMed] [Google Scholar]
- 116.Chohan G, Barnett Y, Gibson J, et al. Langerhans cell histiocytosis with refractory central nervous system involvement responsive to infliximab. J Neurol Neurosurg Psychiatry. 2012;83:573–575. doi: 10.1136/jnnp-2011-300575. [DOI] [PubMed] [Google Scholar]
- 117.Rodríguez HN, García I, Alba A, et al. Infliximab-induced reactivated Langerhan’s cell histiocytosis in a patient with ulcerative colitis. Inflamm Bowel Dis. 2009;15:1286–1287. doi: 10.1002/ibd.20796. [DOI] [PubMed] [Google Scholar]
- 118.Jordan MB, McClain KL, Yan X, et al. Anti-CD52 antibody, alemtuzumab, binds to Langerhans cells in Langerhans cell histiocytosis. Pediatr Blood Cancer. 2005;44:251–254. doi: 10.1002/pbc.20181. [DOI] [PubMed] [Google Scholar]
- 119.Janku F, Amin HM, Yang D, et al. Response of histiocytoses to imatinib mesylate: Fire to ashes. J Clin Oncol. 2010;28:e633–e636. doi: 10.1200/JCO.2010.29.9073. [DOI] [PubMed] [Google Scholar]
- 120.Caponetti GC, Miranda RN, Althof PA, et al. Immunohistochemical and molecular cytogenetic evaluation of potential targets for tyrosine kinase inhibitors in Langerhans cell histiocytosis. Hum Pathol. 2012;43:2223–2228. doi: 10.1016/j.humpath.2012.03.014. [DOI] [PubMed] [Google Scholar]
- 121.Bechan GI, Lee DW, Zajonc DM, et al. Phage display generation of a novel human anti-CD1A monoclonal antibody with potent cytolytic activity. Br J Haematol. 2012;159:299–310. doi: 10.1111/bjh.12033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rust R, Kluiver J, Visser L, et al. Gene expression analysis of dendritic/Langerhans cells and Langerhans cell histiocytosis. J Pathol. 2006;209:474–483. doi: 10.1002/path.2003. [DOI] [PubMed] [Google Scholar]
- 123.Kis-Toth K, Bacskai I, Gogolak P, et al. Monocyte-derived dendritic cell subpopulations use different types of matrix metalloproteinases inhibited by GM6001. Immunobiology. 2013;218:1361–1369. doi: 10.1016/j.imbio.2013.06.012. [DOI] [PubMed] [Google Scholar]
- 124.Greshock J, Nathanson K, Medina A, et al. Distinct patterns of DNA copy number alterations associate with BRAF mutations in melanomas and melanoma-derived cell lines. Genes Chromosomes Cancer. 2009;48:419–428. doi: 10.1002/gcc.20651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Dhomen N, Reis-Filho JS, da Rocha Dias S, et al. Oncogenic Braf induces melanocyte senescence and melanoma in mice. Cancer Cell. 2009;15:294–303. doi: 10.1016/j.ccr.2009.02.022. [DOI] [PubMed] [Google Scholar]
- 126.Michaloglou C, Vredeveld LC, Soengas MS, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–724. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- 127.van der Pluijm G. Epithelial plasticity, cancer stem cells and bone metastasis formation. Bone. 2011;48:37–43. doi: 10.1016/j.bone.2010.07.023. [DOI] [PubMed] [Google Scholar]
- 128.Berres ML, Allen CE, Merad M. Pathological consequence of misguided dendritic cell differentiation in histiocytic diseases. Adv Immunol. 2013;120:127–161. doi: 10.1016/B978-0-12-417028-5.00005-3. [DOI] [PMC free article] [PubMed] [Google Scholar]