

Malignant peripheral nerve sheath tumors are amongst the most challenging mesenchymal malignancies to treat. Their frequent association with a seemingly simple genetic aberration—the loss of the tumor suppressor gene neurofibromin—belies genomic complexity that has rendered effective therapy elusive to date. This review aims to detail elements of current optimal clinical management and summarize recent data on potential molecular drivers and targets, with a view to charting the course for future progress in patient care.
Keywords: Sarcoma, Malignant peripheral nerve sheath tumor, Neurofibromatosis type 1, Molecular targeted therapy, Clinical trials
Learning Objectives
Explain the characteristics and treatment of malignant peripheral nerve sheath tumors, both in relation to neurofibromatosis type I and otherwise.
Cite the unique challenges in optimal management of malignant peripheral nerve sheath tumors.
Appraise the large amount of new data surrounding the potential molecular drivers, possible targets for therapy in this disease.
Abstract
Malignant peripheral nerve sheath tumors (MPNST) are uncommon, biologically aggressive soft tissue sarcomas of neural origin that pose tremendous challenges to effective therapy. In 50% of cases, they occur in the context of neurofibromatosis type I, characterized by loss of function mutations to the tumor suppressor neurofibromin; the remainder arise sporadically or following radiation therapy. Prognosis is generally poor, with high rates of relapse following multimodality therapy in early disease, low response rates to cytotoxic chemotherapy in advanced disease, and propensity for rapid disease progression and high mortality. The last few years have seen an explosion in data surrounding the potential molecular drivers and targets for therapy above and beyond neurofibromin loss. These data span multiple nodes at various levels of cellular control, including major signal transduction pathways, angiogenesis, apoptosis, mitosis, and epigenetics. These include classical cancer-driving genetic aberrations such as TP53 and phosphatase and tensin homolog (PTEN) loss of function, and upregulation of mitogen-activated protein kinase (MAPK) and (mechanistic) target of rapamycin (TOR) pathways, as well as less ubiquitous molecular abnormalities involving inhibitors of apoptosis proteins, aurora kinases, and the Wingless/int (Wnt) signaling pathway. We review the current understanding of MPNST biology, current best practices of management, and recent research developments in this disease, with a view to informing future advancements in patient care.
Implications for Practice:
Malignant peripheral nerve sheath tumors (MPNST) are amongst the most challenging mesenchymal malignancies to treat. They affect young and middle-aged adults, tend towards early metastasis, and often demonstrate resistance to chemotherapy. Their frequent association with a seemingly simple genetic aberration—the loss of the tumor suppressor gene neurofibromin—belies a quite prolific genomic complexity that has rendered effective therapy elusive to date. This review aims to detail elements of current optimal clinical management and summarize recent data on potential molecular drivers and targets, with a view to charting the course for future progress in patient care.
Introduction
Malignant peripheral nerve sheath tumors (MPNST) are believed to derive from peripheral nerves or demonstrate peripheral nerve differentiation. More specifically, they are defined as nerve sheath tumors arising from a peripheral nerve, from a pre-existing peripheral nerve sheath tumor, or in the setting of neurofibromatosis type 1 (NF1) syndrome. In other contexts, the diagnosis can only be made in the presence of demonstrable features of Schwannian differentiation [1]. MPNST may arise from a precursor plexiform neurofibroma, a benign tumor characterized by differentiated Schwann cells embedded in a varied microenvironment comprising perineural-like cells, fibroblasts, vascular cells, and mast cells. In contrast, schwannomas, benign peripheral nerve sheath tumors comprised exclusively of Schwann cells, do not give rise to malignancies. The term MPNST does not include tumors arising from the epineurium or the vasculature of peripheral nerves, but has replaced previous less well-defined terms of malignant schwannoma, neurogenic sarcoma, and neurofibrosarcoma. MPNST should also be distinguished from the vanishingly uncommon malignant granular cell tumor, yet another aggressive soft tissue tumor of Schwannian origin, which is more frequently associated with small nerve twigs, rather than large nerve trunks [1].
Epidemiology, Etiology, and Pathogenesis
MPNSTs comprise ∼2% of all sarcomas, a small fraction of a group of cancers that affect 5 people per million per year [2]. Whereas MPNST may arise at any age with no gender predilection, it tends to present earlier in life than most other genomically complex sarcomas, which are generally more prevalent beyond the sixth decade. The median age for sporadic MPNST is between 30 and 60 years, and that for NF1-associated MPNST is between 20 and 40 years [3].
Half of MPNSTs are associated with neurofibromatosis type 1 (NF1), the autosomal dominant condition that, affecting 1 in 3000 live births, represents the most common human cancer genetic predisposition syndrome. NF1 is characterized by multiple areas of cutaneous hyperpigmentation, termed café-au-lait spots, and numerous neurofibromas, the slowly progressing, pathologically heterogeneous nerve sheath tumors first described by the German pathologist von Recklinghausen in the 1880s. This syndrome is almost completely penetrant, but demonstrates variable expressivity, a phenotypic heterogeneity that renders accurate prediction and early detection of clinical complications like malignant transformation challenging. Cutaneous neurofibromas, superficial lesions derived from small peripheral nerve branches, affect virtually all NF1 patients, are generally asymptomatic, and have very low malignant potential. In contrast, plexiform neurofibromas are found in less than half of NF1 patients, but may cause significantly greater morbidity. Many are deep seated, arising from and involving single or multiple branches of larger nerves or nerve plexuses, and may grow to be quite large. Plexiform neurofibromas are often associated with a wide range of symptoms related to mass effect and can transform into MPNSTs. Other clinical features of NF1 include axillary freckling, optic gliomas, iris hamartomas termed Lisch nodules, bone dysplasia, and family history of NF1 in a first-degree relative; NF1 is diagnosed when any two of these seven criteria are met. NF1 can also be associated with cardiovascular abnormalities, learning deficiencies, and the development of a variety of malignancies, including leukemia, gastrointestinal stromal tumor, and rhabdomyosarcoma.
The genotypic hallmark of NF1 involves mutations to or other loss of the 350 kilobase gene NF1 on the long arm of chromosome 17, which encodes the tumor suppressor protein neurofibromin. This cytoplasmic protein possesses a guanosine triphosphatase (GTPase)-associated protein-related domain that inhibits the activity of the ras proto-oncogene by catalyzing the conversion of the active ras-GTP to its inactive GDP-bound conformation. Thus, NF1 inactivation leads to ras hyperactivity and consequent activation of multiple downstream survival and proliferative pathways, including the mitogen-activated protein kinase (MAPK), mammalian target of rapamycin (mTOR), and AKT (Mouse breed AK thymoma, also termed protein kinase B, or PKB) pathways. Biallelic loss of neurofibromin (one “hit” from the germline, and the second “hit” acquired somatically) resulting in ras activation is assumed to be directly responsible for the development of neurofibromas in NF1 syndrome. Yet the pathway to neurofibroma tumorigenesis is considerably more complex; NF1 mutations are likely necessary, but not sufficient to drive neoplastic change. Loss of heterozygosity at the NF1 locus has been demonstrated in human tissue samples and confirmed in mouse models as being permissive to neurofibroma formation [4]. However, murine studies have also revealed the importance of haploinsufficient NF1 mast cells to the promotion of inflammation and acceleration of tumor growth in plexiform neurofibromas [5]. The maturation, proliferation, and recruitment of these mast cells have been shown to be mediated by stem cell factor (SCF), the ligand for the KIT receptor tyrosine kinase (RTK) [6], suggesting SCF/KIT-dependent tumorigenic tumor-stromal interactions in plexiform neurofibromas, in addition to ras activation.
The subsequent molecular path from neurofibroma to MPNST in NF1 syndrome remains uncertain, although NF1 deficiency in and of itself is clearly insufficient, given that only approximately 10% of all NF1 patients eventually develop MPNST. The large number of molecular aberrations associated with MPNST in both preclinical and clinical studies across a variety of platforms is, on the other hand, not in doubt; MPNST is a genomically complex disease. Unsurprisingly, signal transduction pathways downstream of ras demonstrate evidence of increased activation. The MAPK pathway, mediated by the signal transduction kinases rapidly accelerated fibrosarcoma (RAF) MAPK/extracellular signal-regulated kinase (ERK) kinase (MEK), and ERK, demonstrates overexpression or upregulation of one or more of its elements in multiple studies of MPNST. For instance, phosphorylated MEK was overexpressed in >90% of MPNST tissues compared with 21% of benign neurofibromas in one study [7]. The phosphatidyl inositol 3 kinase-mouse strain AK thymoma-[mechanistic] target of rapamycin (PI3K/AKT/TOR) pathway, another major mitogenic signal transduction pathway downstream of ras, is also implicated in MPNST development. Expression of downstream targets of phosphorylated AKT and activated TOR assessed immunohistochemically on a tissue microarray was significantly higher in MPNSTs than neurofibromas, and the same pathways were found to be highly activated in MPNST cell lines [8]. Phosphatase and tensin homolog (PTEN), a key tumor suppressor regulating the PI3K/AKT/TOR pathway, has also been shown to be significantly downregulated in MPNST samples when compared with neurofibromas [9, 10]. Among the RTKs, the epidermal growth factor receptor (EGFR) has been prominently studied and associated with the development of MPNST. A recent study in a genetically engineered mouse model showed that EGFR overexpression was sufficient to transform neurofibroma into MPNST via Janus kinase 2/signal transducer and activator of transcription 3 (STAT3) activation [11]. The MNNG HOS transforming gene (MET) RTK is also of putative importance in MPNST pathogenesis; MET activation increased MPNST invasion, motility, and angiogenesis in an in vitro model, whereas MET knockdown markedly decreased tumor growth in severe combined immunodeficiency mice [12]. Finally, a recent study using both a murine forward genetic screen and tissue microarray studies of human tumor samples revealed evidence that activation of the canonical Wingless/int (Wnt) signaling pathway could induce transformation of immortalized Schwann cells, and that downregulation of this pathway was sufficient to reduce the tumorigenic phenotype of human MPNST cell lines [13].
As in the case of many malignancies, mutations in TP53 have been prominently associated with MPNST development; these mutations have been shown to result in MPNST development in several models, including elegant zebrafish models that have proved useful in modeling NF1-related tumors [14]. The actual incidence of TP53 aberrations in MPNST ranges widely across studies; a gene signature associated with TP53 inactivation was found in 16 of 20 tumor samples evaluated in one gene expression profiling study [15], whereas another study demonstrated TP53 mutations only in fewer than 25% of patient samples assessed [16].
Perhaps unexpectedly, a number of recent studies have demonstrated that there is an overall downregulation of genes in MPNST as compared with neurofibromas. In the gene expression study mentioned above, microarray analyses of global mRNA and microRNA (miRNA) expression profiles of both neurofibromas and MPNSTs revealed frequent loss of expression in the malignant transformation to MPNST [15]. Further studies have since corroborated these findings. Comparative methylome analyses showed that promoter hypermethylation may be responsible for much of this downregulation [17], whereas another study revealed the critical role of downregulation of further miRNA tumor suppressors, such as miR-29c, in the malignant progression of neurofibromas [18].
The molecular heterogeneity of MPNSTs goes beyond multiply activated pathways and widespread genomic aberrations. MPNSTs are known to have complex karyotypes, with an average of 18 aberrations per tumor based on metaphase comparative genomic hybridization analysis. Common karyotypic changes include gains from chromosome arms 7p, 8q, and 17q, and losses from 9p, 11q, 13q, and 17p [19]. MPNSTs demonstrate both inter- and intratumoral heterogeneity, and more than 70% of NF1-associated MPNSTs displayed intratumoral heterogeneity as shown by loss of heterozygosity analysis [20]. Even the canonical biallelic NF1 losses demonstrate significant heterogeneity; the spectrum of specific germline NF1 mutations in NF1 patients with MPNST has been shown to differ from both the somatic NF1 mutations in either sporadic or NF1-associated MPNST [21]. Finally, there are also data suggesting that the importance of neurofibromin loss in NF1-mutated MPNST may not be dependent on ras and MEK regulation, but rather mediated through the activation of alternative pathways like the bone morphogenetic protein 2 - small body size/mothers against decapentaplegic (BMP2-SMAD) pathway [22].
Although relatively similar molecular mechanisms are involved in the pathogenesis of sporadic MPNST, which comprise approximately 40% of all MPNST, there are some distinct differences between these tumors and the NF1-associated variety. Somatic NF1 mutations are far from universal in sporadic MPNST, affecting 41% of sporadic MPNSTs in one study [21]; the pathogenetic necessity of these mutations in this setting is thus unclear. In addition, TP53 mutations are more often associated with sporadic MPNST, whereas EGFR overexpression and Raf and PI3K/AKT pathway activation are more commonly seen in NF1-associated MPNST. In spite of these putative differences, no consistently distinctive gene expression profiles have been discovered for either subset to date [23].
The remaining 10% of MPNSTs arise secondary to previous irradiation and account for about 5% of radiotherapy (RT)-induced sarcomas, which arise most frequently in the setting of external beam RT for breast cancer or lymphoma. RT-induced sarcomas are known to have poorer outcomes compared with sporadic soft tissue sarcomas, independent of histologic type [24]. Inferior outcomes have been observed for RT-associated MPNST compared with NF1-associated or sporadic MPNST [24, 25].
Clinical Presentation and Diagnosis
As noted above, half of all MPNSTs arise in the context of NF1 syndrome, usually in association with pre-existing plexiform neurofibromas. The lifetime risk of developing MPNST in a patient with NF1 syndrome is 8%–13% [26]. Patients present with a rapidly enlarging mass that may be painful or cause local neurological symptoms such as weakness or paresthesias. The development of new, worsening, or persistent pain in the neurofibroma of a patient with NF1 is an important symptom that should always be conscientiously evaluated, even if there was already pre-existing chronic discomfort from the lesion. The most common sites of involvement include the nerve roots and bundles in the extremities and pelvis, particularly the sciatic nerve. In most instances, the size of the mass is greater than 5 cm at presentation (Fig. 1A, 1B), and up to 50% of patients present with metastatic disease, usually to the lung.
Figure 1.

Radiological findings in malignant peripheral nerve sheath tumors (MPNST). (A): 24-year-old male with neurofibromatosis type 1 presenting with large thigh mass. Contrast MRI revealed dominant mass within the left vastus medialis muscle with heterogeneous signal intensity and enhancement with areas of central necrosis. Biopsy confirmed MPNST, for which he underwent local resection and adjuvant RT. (B): 45-year-old male presenting with thoracic cord compression. Magnetic resonance imaging revealed a homogeneously enhancing mass within the right posterior mediastinum extending through the right T3-T4 neural foramen into the spinal canal with associated compression of the spinal cord. Biopsy confirmed MPNST; he underwent extensive resection and adjuvant radiotherapy.
Magnetic resonance imaging is the most useful imaging modality for characterizing the anatomical extent of the tumor for surgical planning. Fluorodeoxyglucose-positron emission tomography (FDG-PET) has been studied to evaluate the key clinical task of differentiating benign neurofibromas from MPNST in patients with NF1. One study demonstrated reliable and replicable differentiation with relatively high specificity [27]; this high degree of diagnostic accuracy with FDG-PET remains to be replicated in other series.
Histologic features of MPNST are rather nonspecific (Fig. 2A--2C). Generally, tumors are composed of monotonous spindle cells arranged in intersecting fascicles. Pleomorphic variants also exist. At low power, alternating hyper- and hypocellular areas may be present, often with hypercellular areas localized in close proximity to blood vessels. Compared with benign neurofibromas, MPNST usually demonstrate a marked increase in tumor cellularity, pleomorphism, and mitotic activity and show a more organized cellular growth pattern, with less extracellular matrix material. Occasionally, a spectrum of changes may be seen, ranging from atypical neurofibromas to high-grade MPNST. The former are associated with increased nuclear pleomorphism in the absence of mitotic activity or cellularity. A recent study revealed recurrent cyclin-dependent kinase 2A (CDKN2A) and CDKN2B deletions in atypical neurofibromas; aberrations at this gene locus were also found in MPNSTs evaluated, but not benign neurofibromas [28]. These data suggest atypical neurofibromas to be true premalignant lesions, pointing toward the need to consider pre-emptive therapeutic intervention when such neurofibromas are detected. Graded by either the National Cancer Institute or the Fédération Nationale des Centres de Lutte Contre le Cancer systems [1], the majority of MPNSTs will be intermediate to high grade. Heterologous elements, such as skeletal muscle, bone, cartilage, and blood vessels, are present in approximately 15% of tumors [1]. Heterologous elements may portend an even poorer prognosis; MPNSTs demonstrating skeletal muscle differentiation (malignant Triton tumors) are particularly aggressive and associated with poor prognosis. There is no pathognomonic molecular or immunohistochemical study for MPNST. S100 protein is weakly and patchily present in <50% of cases; strong diffuse staining nearly always excludes a diagnosis of MPNST. Thus, in the absence of a history of NF1 or gross or microscopic evidence of association of tumor with nerve sheath or neurofibroma, the most reliable method of diagnosis remains electron microscopy, which can identify ultrastructural features of Schwann cells.
Figure 2.
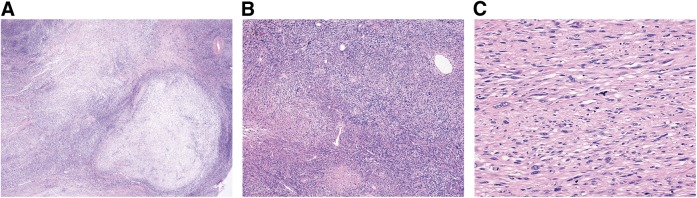
Histopathologic features of (A): Low power view (×20) of MPNST demonstrating variable hypo- and hypercellular areas. (B): Low power view (×40) showing more cellular MPNST with scattered pleomorphic cells. (C): Moderate power view (×100) showing hypercellular area with intersecting fascicles of monotonous spindle cells.
Prognostic Factors
In general, MPNST is known to have high metastatic potential and poor prognosis. Reported long-term outcomes vary widely across multiple series, with 5-year survival ranging between 15% and 50%. Most data on clinicopathologic factors are derived from several retrospective single institution studies analyzing between 100 and 200 patients each. Large tumor size at presentation (typically >5 cm) has been the most consistently determined adverse prognostic factor across all series [7, 25, 29]. Other reported factors include tumor grade, truncal location, surgical margin status, local recurrence, and heterologous rhabdomyoblastic differentiation. There remain no well-defined or widely reproducible molecular prognosticators. One series found an association with nuclear p53 expression with inferior outcome [7]; AKT and TOR pathway activation (identified immunohistochemically) were negatively prognostic in another study [30], whereas MET activation has also been suggested to portend inferior prognosis [12].
Large tumor size at presentation (typically >5 cm) has been the most consistently determined adverse prognostic factor across all series. Other reported factors include tumor grade, truncal location, surgical margin status, local recurrence, and heterologous rhabdomyoblastic differentiation.
The true prognostic impact of NF1 syndrome in MPNST remains somewhat in flux. Several large series report significantly worse outcomes for MPNST arising in the setting of NF1 compared with sporadic disease, with inferior responses to cytotoxic chemotherapy and 5-year survivals that are up to 50% worse [31–33]. A meta-analysis of several European studies, however, suggested that, whereas NF1 may have been negatively prognostic in studies before the year 2000, this effect was subsequently lost, possibly because of better overall surveillance and more rapid intervention at earlier stages of disease in patients with NF1 accruing from improvements in imaging and diagnostic techniques [34].
Treatment
In the setting of localized disease, as is the case with all soft tissue sarcomas, complete surgical extirpation with clear margins is the treatment of choice. Multiple retrospective datasets have shown the negative prognostic impact of involved margins and local recurrence. Crucially, even in large, nonextremity MPNSTs, as often arises in NF1, for which extensive gross total resections would be associated with significant morbidity, data exist to suggest the independent prognostic importance of complete surgical resection [35]. As in the case with most large (>5 cm) high-grade limb sarcomas, adjuvant radiation is advocated to reduce local recurrence. The risk-benefit profile of adjuvant radiation in patients with NF1 must be carefully discussed with all patients in view of the heightened risk of radiation-induced sarcomas. There are no randomized data examining adjuvant chemotherapy specifically in MPNST. In histologically unselected populations of soft tissue sarcomas, data exist from meta-analyses to suggest marginal survival benefit [36], implying that adjuvant chemotherapy remains a consideration for motivated patients. The recently reported SARC006 phase II trial conducted by the Sarcoma Alliance for Research (SARC) evaluated the role of chemotherapy with doxorubicin, ifosfamide, and etoposide in 48 locally advanced or metastatic MPNST patients. It revealed encouraging disease stabilization and responses accruing from neoadjuvant chemotherapy that rendered subsequent local therapy feasible in the majority of patients with localized disease [37]. In this study, there was also a suggestion that NF1-associated disease predicted for inferior responses to chemotherapy compared with sporadic disease. Although by no means definitive, these data suggest that chemotherapy may yet have a role in the multimodality treatment of selected MPNST patients with nonmetastatic disease.
In the setting of advanced or metastatic MPNST, outcomes are generally poor. Doxorubicin and ifosfamide are the most active agents in unselected soft tissue sarcomas, with a Response Evaluation Criteria in Solid Tumors (RECIST) response rate of approximately 25% for the combination. Responses are no better in MPNST, reported to be 21% in a multi-institution retrospective study pooling MPNST patients across multiple soft tissue sarcoma trials [38].
Previous gene expression analyses of MPNST tumor samples have identified topoisomerase II α as the most overexpressed gene in MPNSTs relative to benign neurofibroma [39]. There exist very limited data suggesting the possible greater activity of etoposide than doxorubicin in MPNST. A case series demonstrated significant responses to the combination of carboplatin and etoposide in two MPNST patients who were refractory to doxorubicin and ifosfamide [40]. In the earlier described SARC006 study of chemotherapy in MPNST, patients were given two cycles of doxorubicin combined with ifosfamide (IA), followed by two cycles of etoposide combined with ifosfamide (IE); more objective responses were obtained in both NF1-associated and sporadic MPNSTs following the administration of IE than IA [37].
Poor results with conventional agents have stimulated interest in exploring rationally developed targeted therapeutics in MPNST, building upon the large amount of molecular data surrounding MPNST pathogenesis amassed to date (Table 1). The family of ras proto-oncogenes aberrantly activated with loss of neurofibromin is an obvious, if to date elusive, target for many cancers, including MPNST. One strategy involves exploiting the need of ras to localize to the cytoplasmic surface of the cell membrane through farnesylation, in which a lipid group is attached to the ras protein posttranslationally. Interrupting this process, necessary for efficient signaling, can potentially abrogate ras activity. This can be achieved at the level of blocking lipid synthesis, through the use of 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMGCoA) reductase inhibitors (statins), or at the level of protein prenylation, using farnesyl transferase inhibitors. Clinical studies with these agents have not, however, realized this promise; a phase I trial of tipifarnib in children with NF1-associated plexiform neurofibromas and refractory solid tumors revealed no objective responses [41]. The MAPK pathway, activated immediately downstream of ras, and comprising the sequential phosphorylation of Raf family, MEK, and ERK kinases in a mitogenic cascade, would appear to be a rational pathway to target, and MEK inhibition has shown promising results in vitro [42]. However, a multicenter phase II trial evaluating sorafenib, a small-molecule oral tyrosine kinase inhibitor, revealed no objective RECIST responses in the 12 patients with MPNST [43], although some symptomatic relief was observed. A phase I study evaluating sorafenib in nine pediatric patients with NF1 and plexiform neurofibromas also revealed no responses [44].
Table 1.
Completed and ongoing clinical trials of targeted therapies in malignant peripheral nerve sheath tumor- or neurofibromatosis type 1-related plexiform neurofibromas
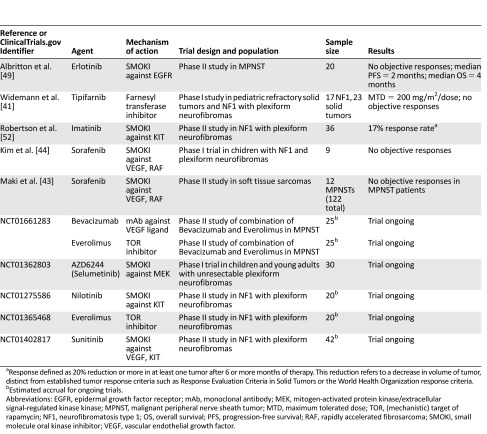
Another pathway closely linked with neurofibromin-deficient ras and most likely important in the pathogenesis of MPNSTs is PI3K/AKT/TOR. In vitro analyses revealed constitutive activation of TOR in NF1-deficient cells, an aberrant activation dependent on ras and PI3K [45]. Multiple studies have shown promising activity of TOR inhibitors against MPNST in preclinical studies in vitro and in vivo, either singly or in combination with other agents [8, 29, 45, 46]. Importantly, there are data to suggest the possible need to appropriately enrich populations for optimal therapeutic effect.
Expression level of PTEN, a tumor suppressor that negatively regulates the TOR pathway, has been suggested as a potential biomarker for TOR pathway blockade efficacy, although the data in support of this have been mixed. It may also be necessary to combine TOR inhibitors with other agents, to optimize therapeutic blockade of the pathway, in view of the complex and multifarious axes of control and feedback limiting the efficacy of single pathway blockade. For example, studies in xenograft models have revealed apoptotic resistance secondary to significant productive autophagy accompanying growth arrest of MPNST cells in response to PI3K/TOR inhibition; the addition of genetic and pharmacologic inhibition of autophagy reversed apoptotic resistance and induced significant cell death [47]. TOR inhibitors have also demonstrated synergism when combined with statins, inhibitors of ras production and activity, in models in vitro [10]. The addition of inhibitors of the molecular chaperone heat shock protein 90 (hsp90) to therapy with rapalogues is yet another promising strategy, mechanistically underpinned by this combination abrogating the cellular stress response machinery required to survive proteotoxic stress engendered by reactive oxygen species, thus leading to cell death [48]. In an elegant study, this combination demonstrated striking in vivo tumoricidal activity significantly surpassing the cytostatic activity seen with TOR inhibition alone [46]. A phase I/II trial of the hsp90 inhibitor ganetespib in combination with sirolimus in MPNST is currently being developed.
Expression level of PTEN, a tumor suppressor that negatively regulates the TOR pathway, has been suggested as a potential biomarker for TOR pathway blockade efficacy, although the data in support of this have been mixed. It may also be necessary to combine TOR inhibitors with other agents, to optimize therapeutic blockade of the pathway, in view of the complex and multifarious axes of control and feedback limiting the efficacy of single pathway blockade.
Hyperactive or overexpressed RTKs are attractive therapeutic targets for small-molecule inhibitors and monoclonal antibodies and have been successfully targeted in several cancers with spectacular therapeutic effect over the last decade. Multiple RTKs have been shown to be dysregulated in MPNST in both the preclinical and clinical setting (Fig. 3). EGFR overexpression in MPNST has been reported in multiple molecular assays and has potential roles in malignant transformation of neurofibromas and invasion. Recently promising preclinical data have suggested the efficacy of pharmacological inhibition of the epidermal growth factor receptor-signal transducer and activator of transcription 3(EGFR-STAT3) pathway, in inhibiting MPNST transformation and tumorigenesis in xenograft models [11]. There has, however, been no clinical replication of these data to date; an early-phase clinical trial of erlotinib revealed no objective responses and was closed early [49].
Figure 3.
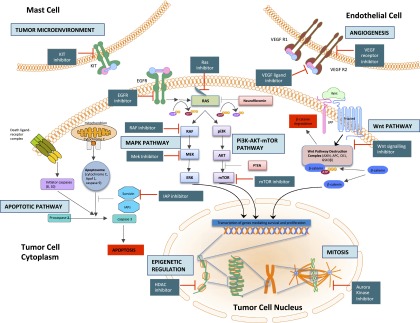
Pathways and potential targets in malignant peripheral nerve sheath tumors (MPNST). Depicted in this cartoon are the multiple nodes implicated in the pathogenesis of MPNST, involving intracellular signaling pathways, epigenetic regulation, mitosis, angiogenesis and interactions with the tumor environment. Alongside these nodes are indicated the currently available classes of pharmacological agents that can act upon them, thence potentially retarding tumor growth and bringing therapeutic benefit.
Abbreviations: EGFR, epidermal growth factor receptor; VEGF, vascular endothelial growth factor; VEGF R1/R2, vascular endothelial growth factor receptor 1/2; RAF, rapidly accelerated fibrosarcoma; MAPK, mitogen-activated protein kinase; MEK, mitogen-activated protein kinase/extracellular signal-regulated kinase kinase; IAP, inhibitor of apoptotic proteins; HDAC, Histone deacetylase; ERK, extracellular-signal-regulated kinase; PI3K, phosphatidyl inositol 3-kinase; AKT, mouse strain AK thymoma; TOR, (mechanistic) target of rapamycin; PTEN, phosphatase and tensin homolog.
The results of studies employing inhibitors of angiogenesis in sarcomas have been mixed. Phase II studies using both monoclonal antibodies and tyrosine kinase inhibitors directed against the vascular endothelial growth factor(VEGF) ligand or receptors have revealed modest benefit only, if any, and no discernible benefit specifically for MPNSTs [42, 50]. The recently reported PALETTE study, evaluating the multitargeted tyrosine kinase inhibitor pazopanib, with activity against VEGF and platelet-derived growth factor signaling, against placebo, revealed a 3-month benefit in progression-free survival in patients with nonadipocytic sarcomas [51], although no data on histologic subtype-specific results were provided beyond reporting data on leiomyosarcoma and synovial sarcoma. Further development of antiangiogenic therapy must aim to incorporate robust biomarkers of clinical efficacy, which have been to date elusive. In addition, single-agent antiangiogenic therapies are often associated with objective responses of less than 10%, as in the PALETTE trial; the addition of other agents as part of rationally designed combination therapies will thus need to be explored in a disease like MPNST, in which objective tumor responses are often important. A trial launched by SARC testing the combination of everolimus with bevacizumab in patients with refractory MPNST is currently recruiting patients and may shed further light on the therapeutic import of these pathways.
As earlier described, the SCF/KIT pathway appears to be important in the recruitment and maintenance of the microenvironment of plexiform neurofibromas. Following on from this mechanistic insight and encouraging single case responses, a phase II trial was undertaken to evaluate the activity of Imatinib in patients with NF1-associated plexiform neurofibromas [52]. Response was defined by a 20% or greater reduction in volume of at least one tumor after 6 or more months of therapy, and not by RECIST or World Health Organization response criteria. This was achieved in 26% of patients, with substantial interpatient and intertumor variability in tumor reduction, a heterogeneity that highlights the need for yet undefined biomarkers to select appropriate patient subgroups for optimal benefit.
Several other promising targets for therapy have emerged from recent preclinical studies. Histone deacetylase inhibitors (HDACi) have been found to induce cell death selectively in cells demonstrating enhanced ras signaling [53] as is the case in MPNST. Significant in vivo and in vitro activity in NF1-associated MPNSTs has been demonstrated independent of TP53 mutational status, and HDACi-induced autophagy is emerging as a pharmacologically surmountable source of resistance [54]. Survivin, the protein critically involved in several cellular processes, including survival, cellular division, and adaptation to stress, and whose expression is commonly found in transformed cells, has emerged as another promising target. Following the discovery of amplification of baculoviral inhibitor of apoptosis repeat-containing 5 (BIRC5), which encodes the antiapoptotic protein survivin, in human MPNST samples [55], survivin was found to be highly expressed in human MPNST cell lines. Survivin knockdown was associated with abrogation of MPNST cell growth, and targeting of survivin with a small-molecule inhibitor inhibited tumor growth and metastases in mouse xenograft models [56].
Recently, novel techniques for elucidation of key molecular drivers of MPNST have yielded some tantalizing data with regard to malignant transformation from neurofibromas and have suggested exciting new potential therapeutic targets. The Sleeping Beauty transposon-based somatic mutagenesis system in mice, in which a series of DNA-mobile elements is coupled with an enzyme that catalyzes the mobilization and reintegration of the mobile elements within the mouse genome, has gained prominence in recent years as a tool for cancer discovery, given its ability to rapidly induce cancer in transgenic mice as well as the ease of identification of the mutated genes [57]. Two publications highlighted the importance of specific genes and pathways in MPNST utilizing this system as a forward genetic screen. In the first, the importance of the canonical Wnt pathway to Schwann cell and neurofibroma progression to malignancy was demonstrated, with Wnt pathway targeting producing encouraging results in combination with TOR inhibition [13]. The second investigation using this platform reinforced the importance of known pathways such as PI3K/AKT/TOR and i/Wnt/β-catenin, in addition to identifying new proto-oncogenes involved in MPNST maintenance, such as forkhead box protein R2 (FOXR2) [58]. A ras-driven transcriptome analysis revealed dramatic overexpression and amplification of the downstream MAPK target aurora kinase A (AURKA) in MPNSTs but not neurofibromas, with inhibition of AURKA reducing cell survival in vitro and causing tumor stasis in xenograft models [59]. In spite of these encouraging discoveries, absence of significant clinical success with such novel agents as aurora kinase inhibitors underscores the need for rigorous clinical validation of these targets before significant progress can be claimed.
Conclusion
Biologically, MPNSTs present a conundrum—although in most cases associated with a seemingly simple genetic aberration (the loss of neurofibromin on chromosome 17), they simultaneously display genomic complexity more commonly associated with sarcomas that arise in older patients, a factor that no doubt contributes to the difficulties to date faced in treating these sarcomas that are often intransigent to conventional therapy.
We are in need of a better understanding of the biology of, and refining clinical biomarkers for transformation of benign neurofibromas into MPNSTs in the setting of NF1. The explosion of molecular and preclinical data in recent years gives hope that we may be starting to gain some traction over this complexity. In particular, targeting of both the TOR and hsp90 pathways appears to be one strategy of considerable promise based on existing data; results of early clinical trials employing this approach are eagerly awaited, as are those of other combinations involving TOR inhibitors. The complexity of ras signaling is an obvious target but has proved a nettlesome signaling pathway to target to date, although downstream elements such as MEK may be more easily inhibited. Focus on novel proapoptotic agents such as survivin inhibitors is another broad approach to cancer that may be beneficial in this aneuploid group of cancers. Continued efforts should be undertaken to better understand tumor resistance to systemic therapy, crosstalk and alternative activation pathways, so that effective early clinical studies can be designed employing rational combinations of targeted agents.
In this rare and diagnostically challenging disease, efforts already underway must aspire toward innovative trial designs that maximize patient resources, buttressed by multicenter collaborations to improve the quality and quantity of meaningful translational and clinical data. With these efforts, the significant challenges to improve care for patients with this form of soft tissue sarcoma can be met.
This article is available for continuing medical education credit at CME.TheOncologist.com.
Author Contributions
Conception/Design: Robert G. Maki, Mohamad Farid, Angela Cioffi
Provision of study material or patients: Elizabeth G. Demicco, Linda Ahn, Pamela R. Merola
Collection and/or assembly of data: Mohamad Farid, Elizabeth G. Demicco, Roberto Garcia
Data analysis and interpretation: Robert G. Maki, Mohamad Farid, Elizabeth G. Demicco, Angela Cioffi
Manuscript writing: Robert G. Maki, Mohamad Farid, Elizabeth G. Demicco
Final approval of manuscript: Robert G. Maki, Mohamad Farid, Elizabeth G. Demicco, Roberto Garcia, Linda Ahn, Pamela R. Merola, Angela Cioffi
Disclosures
Robert G. Maki: Bayer Healthcare Pharmaceuticals (C/A, H, RF). The other authors indicated no financial relationships.
Section Editor: Jaap Verweij: None
Reviewer “A”: None
Reviewer “B”: Novartis, Bayer, Eisai, Pfizer, PharmaMar, GlaxoSmithKline (RF); GlaxoSmithKline (H)
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1.Fletcher CDM, Bridge JA, Hogendoorn PCW. et al. WHO Classification of Tumours of Soft Tissue and Bone. Lyon, France: IARC; 2013. [Google Scholar]
- 2.Ng VY, Scharschmidt TJ, Mayerson JL, et al. Incidence and survival in sarcoma in the United States: A focus on musculoskeletal lesions. Anticancer Res. 2013;33:2597–2604. [PubMed] [Google Scholar]
- 3.Widemann BC. Current status of sporadic and neurofibromatosis type 1-associated malignant peripheral nerve sheath tumors. Curr Oncol Rep. 2009;11:322–328. doi: 10.1007/s11912-009-0045-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cichowski K, Shih TS, Schmitt E, et al. Mouse models of tumor development in neurofibromatosis type 1. Science. 1999;286:2172–2176. doi: 10.1126/science.286.5447.2172. [DOI] [PubMed] [Google Scholar]
- 5.Zhu Y, Ghosh P, Charnay P, et al. Neurofibromas in NF1: Schwann cell origin and role of tumor environment. Science. 2002;296:920–922. doi: 10.1126/science.1068452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Staser K, Yang FC, Clapp DW. Pathogenesis of plexiform neurofibroma: Tumor-stromal/hematopoietic interactions in tumor progression. Annu Rev Pathol. 2012;7:469–495. doi: 10.1146/annurev-pathol-011811-132441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zou C, Smith KD, Liu J, et al. Clinical, pathological, and molecular variables predictive of malignant peripheral nerve sheath tumor outcome. Ann Surg. 2009;249:1014–1022. doi: 10.1097/SLA.0b013e3181a77e9a. [DOI] [PubMed] [Google Scholar]
- 8.Zou CY, Smith KD, Zhu QS, et al. Dual targeting of AKT and mammalian target of rapamycin: A potential therapeutic approach for malignant peripheral nerve sheath tumor. Mol Cancer Ther. 2009;8:1157–1168. doi: 10.1158/1535-7163.MCT-08-1008. [DOI] [PubMed] [Google Scholar]
- 9.Gregorian C, Nakashima J, Dry SM, et al. PTEN dosage is essential for neurofibroma development and malignant transformation. Proc Natl Acad Sci USA. 2009;106:19479–19484. doi: 10.1073/pnas.0910398106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bradtmöller M, Hartmann C, Zietsch J, et al. Impaired Pten expression in human malignant peripheral nerve sheath tumours. PLoS One. 2012;7:e47595. doi: 10.1371/journal.pone.0047595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu J, Patmore DM, Jousma E et al. EGFR-STAT3 signaling promotes formation of malignant peripheral nerve sheath tumors. Oncogene 2013 [E-pub ahead of print]. [DOI] [PMC free article] [PubMed]
- 12.Torres KE, Zhu QS, Bill K, et al. Activated MET is a molecular prognosticator and potential therapeutic target for malignant peripheral nerve sheath tumors. Clin Cancer Res. 2011;17:3943–3955. doi: 10.1158/1078-0432.CCR-11-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Watson AL, Rahrmann EP, Moriarity BS, et al. Canonical Wnt/β-catenin signaling drives human schwann cell transformation, progression, and tumor maintenance. Cancer Discov. 2013;3:674–689. doi: 10.1158/2159-8290.CD-13-0081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berghmans S, Murphey RD, Wienholds E, et al. tp53 mutant zebrafish develop malignant peripheral nerve sheath tumors. Proc Natl Acad Sci USA. 2005;102:407–412. doi: 10.1073/pnas.0406252102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Subramanian S, Thayanithy V, West RB, et al. Genome-wide transcriptome analyses reveal p53 inactivation mediated loss of miR-34a expression in malignant peripheral nerve sheath tumours. J Pathol. 2010;220:58–70. doi: 10.1002/path.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Verdijk RM, den Bakker MA, Dubbink HJ, et al. TP53 mutation analysis of malignant peripheral nerve sheath tumors. J Neuropathol Exp Neurol. 2010;69:16–26. doi: 10.1097/NEN.0b013e3181c55d55. [DOI] [PubMed] [Google Scholar]
- 17.Feber A, Wilson GA, Zhang L, et al. Comparative methylome analysis of benign and malignant peripheral nerve sheath tumors. Genome Res. 2011;21:515–524. doi: 10.1101/gr.109678.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Presneau N, Eskandarpour M, Shemais T, et al. MicroRNA profiling of peripheral nerve sheath tumours identifies miR-29c as a tumour suppressor gene involved in tumour progression. Br J Cancer. 2013;108:964–972. doi: 10.1038/bjc.2012.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brekke HR, Ribeiro FR, Kolberg M, et al. Genomic changes in chromosomes 10, 16, and X in malignant peripheral nerve sheath tumors identify a high-risk patient group. J Clin Oncol. 2010;28:1573–1582. doi: 10.1200/JCO.2009.24.8989. [DOI] [PubMed] [Google Scholar]
- 20.Thomas L, Mautner VF, Cooper DN, et al. Molecular heterogeneity in malignant peripheral nerve sheath tumors associated with neurofibromatosis type 1. Hum Genomics. 2012;6:18. doi: 10.1186/1479-7364-6-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bottillo I, Ahlquist T, Brekke H, et al. Germline and somatic NF1 mutations in sporadic and NF1-associated malignant peripheral nerve sheath tumours. J Pathol. 2009;217:693–701. doi: 10.1002/path.2494. [DOI] [PubMed] [Google Scholar]
- 22.Sun D, Haddad R, Kraniak JM, et al. RAS/MEK-independent gene expression reveals BMP2-related malignant phenotypes in the Nf1-deficient MPNST. Mol Cancer Res. 2013;11:616–627. doi: 10.1158/1541-7786.MCR-12-0593. [DOI] [PubMed] [Google Scholar]
- 23.Katz D, Lazar A, Lev D. Malignant peripheral nerve sheath tumour (MPNST): The clinical implications of cellular signalling pathways. Expert Rev Mol Med. 2009;11:e30. doi: 10.1017/S1462399409001227. [DOI] [PubMed] [Google Scholar]
- 24.Gladdy RA, Qin LX, Moraco N, et al. Do radiation-associated soft tissue sarcomas have the same prognosis as sporadic soft tissue sarcomas? J Clin Oncol. 2010;28:2064–2069. doi: 10.1200/JCO.2009.25.1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.LaFemina J, Qin LX, Moraco NH, et al. Oncologic outcomes of sporadic, neurofibromatosis-associated, and radiation-induced malignant peripheral nerve sheath tumors. Ann Surg Oncol. 2013;20:66–72. doi: 10.1245/s10434-012-2573-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Evans DG, Baser ME, McGaughran J, et al. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet. 2002;39:311–314. doi: 10.1136/jmg.39.5.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferner RE, Golding JF, Smith M, et al. [18F]2-fluoro-2-deoxy-D-glucose positron emission tomography (FDG PET) as a diagnostic tool for neurofibromatosis 1 (NF1) associated malignant peripheral nerve sheath tumours (MPNSTs): A long-term clinical study. Ann Oncol. 2008;19:390–394. doi: 10.1093/annonc/mdm450. [DOI] [PubMed] [Google Scholar]
- 28.Beert E, Brems H, Daniëls B, et al. Atypical neurofibromas in neurofibromatosis type 1 are premalignant tumors. Genes Chromosomes Cancer. 2011;50:1021–1032. doi: 10.1002/gcc.20921. [DOI] [PubMed] [Google Scholar]
- 29.Stucky CC, Johnson KN, Gray RJ, et al. Malignant peripheral nerve sheath tumors (MPNST): The Mayo Clinic experience. Ann Surg Oncol. 2012;19:878–885. doi: 10.1245/s10434-011-1978-7. [DOI] [PubMed] [Google Scholar]
- 30.Endo M, Yamamoto H, Setsu N, et al. Prognostic significance of AKT/mTOR and MAPK pathways and antitumor effect of mTOR inhibitor in NF1-related and sporadic malignant peripheral nerve sheath tumors. Clin Cancer Res. 2013;19:450–461. doi: 10.1158/1078-0432.CCR-12-1067. [DOI] [PubMed] [Google Scholar]
- 31.Porter DE, Prasad V, Foster L, et al. Survival in malignant peripheral nerve sheath tumours: A comparison between sporadic and neurofibromatosis type 1-associated tumours. Sarcoma. 2009;2009:756395. doi: 10.1155/2009/756395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ferrari A, Miceli R, Rey A, et al. Non-metastatic unresected paediatric non-rhabdomyosarcoma soft tissue sarcomas: Results of a pooled analysis from United States and European groups. Eur J Cancer. 2011;47:724–731. doi: 10.1016/j.ejca.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carli M, Ferrari A, Mattke A, et al. Pediatric malignant peripheral nerve sheath tumor: The Italian and German soft tissue sarcoma cooperative group. J Clin Oncol. 2005;23:8422–8430. doi: 10.1200/JCO.2005.01.4886. [DOI] [PubMed] [Google Scholar]
- 34.Kolberg M, Høland M, Agesen TH, et al. Survival meta-analyses for >1800 malignant peripheral nerve sheath tumor patients with and without neurofibromatosis type 1. Neuro-oncol. 2013;15:135–147. doi: 10.1093/neuonc/nos287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dunn GP, Spiliopoulos K, Plotkin SR, et al. Role of resection of malignant peripheral nerve sheath tumors in patients with neurofibromatosis type 1. J Neurosurg. 2013;118:142–148. doi: 10.3171/2012.9.JNS101610. [DOI] [PubMed] [Google Scholar]
- 36.Pervaiz N, Colterjohn N, Farrokhyar F, et al. A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft-tissue sarcoma. Cancer. 2008;113:573–581. doi: 10.1002/cncr.23592. [DOI] [PubMed] [Google Scholar]
- 37.Widemann BC, Reinke D, Helman L, et al. SARC006: Phase II trial of chemotherapy in sporadic and neurofibromatosis type 1 (NF1)-associated high-grade malignant peripheral nerve sheath tumors (MPNSTs) J Clin Oncol. 31, 2013 (suppl; abstr 10522) [Google Scholar]
- 38.Kroep JR, Ouali M, Gelderblom H, et al. First-line chemotherapy for malignant peripheral nerve sheath tumor (MPNST) versus other histological soft tissue sarcoma subtypes and as a prognostic factor for MPNST: An EORTC soft tissue and bone sarcoma group study. Ann Oncol. 2011;22:207–214. doi: 10.1093/annonc/mdq338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Skotheim RI, Kallioniemi A, Bjerkhagen B, et al. Topoisomerase-II alpha is upregulated in malignant peripheral nerve sheath tumors and associated with clinical outcome. J Clin Oncol. 2003;21:4586–4591. doi: 10.1200/JCO.2003.07.067. [DOI] [PubMed] [Google Scholar]
- 40.Steins MB, Serve H, Zühlsdorf M, et al. Carboplatin/etoposide induces remission of metastasised malignant peripheral nerve tumours (malignant schwannoma) refractory to first-line therapy. Oncol Rep. 2002;9:627–630. [PubMed] [Google Scholar]
- 41.Widemann BC, Salzer WL, Arceci RJ, et al. Phase I trial and pharmacokinetic study of the farnesyltransferase inhibitor tipifarnib in children with refractory solid tumors or neurofibromatosis type I and plexiform neurofibromas. J Clin Oncol. 2006;24:507–516. doi: 10.1200/JCO.2005.03.8638. [DOI] [PubMed] [Google Scholar]
- 42.Mattingly RR, Kraniak JM, Dilworth JT, et al. The mitogen-activated protein kinase/extracellular signal-regulated kinase kinase inhibitor PD184352 (CI-1040) selectively induces apoptosis in malignant schwannoma cell lines. J Pharmacol Exp Ther. 2006;316:456–465. doi: 10.1124/jpet.105.091454. [DOI] [PubMed] [Google Scholar]
- 43.Maki RG, D’Adamo DR, Keohan ML, et al. Phase II study of sorafenib in patients with metastatic or recurrent sarcomas. J Clin Oncol. 2009;27:3133–3140. doi: 10.1200/JCO.2008.20.4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim A, Dombi E, Tepas K, et al. Phase I trial and pharmacokinetic study of sorafenib in children with neurofibromatosis type I and plexiform neurofibromas. Pediatr Blood Cancer. 2013;60:396–401. doi: 10.1002/pbc.24281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johannessen CM, Reczek EE, James MF, et al. The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proc Natl Acad Sci USA. 2005;102:8573–8578. doi: 10.1073/pnas.0503224102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Johansson G, Mahller YY, Collins MH, et al. Effective in vivo targeting of the mammalian target of rapamycin pathway in malignant peripheral nerve sheath tumors. Mol Cancer Ther. 2008;7:1237–1245. doi: 10.1158/1535-7163.MCT-07-2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ghadimi MP, Lopez G, Torres KE, et al. Targeting the PI3K/mTOR axis, alone and in combination with autophagy blockade, for the treatment of malignant peripheral nerve sheath tumors. Mol Cancer Ther. 2012;11:1758–1769. doi: 10.1158/1535-7163.MCT-12-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.De Raedt T, Walton Z, Yecies JL, et al. Exploiting cancer cell vulnerabilities to develop a combination therapy for ras-driven tumors. Cancer Cell. 2011;20:400–413. doi: 10.1016/j.ccr.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Albritton KH, Rankin C, Coffin CM et al. Phase II study of erlotinib in metastatic or unresectable malignant peripheral nerve sheath tumors (MPNST). J Clin Oncol (Meeting Abstracts) June 2006;24 no. 18_suppl 9518. [Google Scholar]
- 50.D’Adamo DR, Anderson SE, Albritton K, et al. Phase II study of doxorubicin and bevacizumab for patients with metastatic soft-tissue sarcomas. J Clin Oncol. 2005;23:7135–7142. doi: 10.1200/JCO.2005.16.139. [DOI] [PubMed] [Google Scholar]
- 51.van der Graaf WT, Blay JY, Chawla SP, et al. EORTC Soft Tissue and Bone Sarcoma Group. PALETTE Study Group Pazopanib for metastatic soft-tissue sarcoma (PALETTE): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2012;379:1879–1886. doi: 10.1016/S0140-6736(12)60651-5. [DOI] [PubMed] [Google Scholar]
- 52.Robertson KA, Nalepa G, Yang FC, et al. Imatinib mesylate for plexiform neurofibromas in patients with neurofibromatosis type 1: A phase 2 trial. Lancet Oncol. 2012;13:1218–1224. doi: 10.1016/S1470-2045(12)70414-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Klampfer L, Huang J, Shirasawa S, et al. Histone deacetylase inhibitors induce cell death selectively in cells that harbor activated kRasV12: The role of signal transducers and activators of transcription 1 and p21. Cancer Res. 2007;67:8477–8485. doi: 10.1158/0008-5472.CAN-07-0210. [DOI] [PubMed] [Google Scholar]
- 54.Lopez G, Torres K, Liu J, et al. Autophagic survival in resistance to histone deacetylase inhibitors: Novel strategies to treat malignant peripheral nerve sheath tumors. Cancer Res. 2011;71:185–196. doi: 10.1158/0008-5472.CAN-10-2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Storlazzi CT, Brekke HR, Mandahl N, et al. Identification of a novel amplicon at distal 17q containing the BIRC5/SURVIVIN gene in malignant peripheral nerve sheath tumours. J Pathol. 2006;209:492–500. doi: 10.1002/path.1998. [DOI] [PubMed] [Google Scholar]
- 56.Ghadimi MP, Young ED, Belousov R, et al. Survivin is a viable target for the treatment of malignant peripheral nerve sheath tumors. Clin Cancer Res. 2012;18:2545–2557. doi: 10.1158/1078-0432.CCR-11-2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Howell VM. Sleeping beauty—a mouse model for all cancers? Cancer Lett. 2012;317:1–8. doi: 10.1016/j.canlet.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 58.Rahrmann EP, Watson AL, Keng VW, et al. Forward genetic screen for malignant peripheral nerve sheath tumor formation identifies new genes and pathways driving tumorigenesis. Nat Genet. 2013;45:756–766. doi: 10.1038/ng.2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Patel AV, Eaves D, Jessen WJ, et al. Ras-driven transcriptome analysis identifies aurora kinase A as a potential malignant peripheral nerve sheath tumor therapeutic target. Clin Cancer Res. 2012;18:5020–5030. doi: 10.1158/1078-0432.CCR-12-1072. [DOI] [PMC free article] [PubMed] [Google Scholar]