

Abstract
Retinoblastoma is the most common intraocular malignancy of childhood. In developing countries, treatment is limited, long-term survival rates are low and current chemotherapy causes significant morbidity to pediatric patients and significantly limits dosing. Therefore there is an urgent need to identify new therapeutic strategies to improve the clinical outcome of patients with retinoblastoma. Here, we investigated the effects of two natural compounds okadaic acid (OKA) and parthenolide (PN) on human retinoblastoma Y79 cells. For the first time we showed that OKA/PN combination at subtoxic doses induces potent synergistic apoptotic effects accompanied by lowering in p-Akt levels, increasing in the stabilized forms of p53 and potent decrease in pS166-Mdm2. We also showed the key involvement of PTEN which, after OKA/PN treatment, potently increased before p53, thus suggesting that p53 activation was under PTEN action. Moreover, after PTEN-knockdown p-Akt/ pS166Mdm2 increased over basal levels and p53 significantly lowered, while OKA/PN treatment failed both to lower p-Akt and pS166-Mdm2 and to increase p53 below/over their basal levels respectively. OKA/PN treatment potently increased ROS levels whereas decreased those of GSH. Reducing cellular GSH by l-butathionine-[S,R]-sulfoximine treatment significantly anticipated the cytotoxic effect exerted by OKA/PN. Furthermore, the effects of OKA/PN treatment on both GSH content and cell viability were less pronounced in PTEN silenced cells than in control cells. The results provide strong suggestion for combining a treatment approach that targets the PTEN/Akt/Mdm2/p53 pathway.
Keywords: retinoblastoma, Y79 cells, synergistic apoptotic effects, oxidative stress, natural drugs, PTEN/Akt/Mdm2/p53 pathway, parthenolide, okadaic acid
Introduction
Retinoblastoma is a genetically determined tumor which represents the most common intraocular malignancy of infancy and early childhood.1 It represents the prototypic model for inherited cancers with the pivotal genetic event being the inactivation of both copies of RB1 gene at the chromosomal locus 13q14.2 This determines the inactivation or loss of the retinoblastoma protein (pRb) which, in normal conditions, targets E2F1 transcription factor blocking entry into the S phase of cell cycle.3 Retinoblastoma equally affects both genders and all races and is responsible for 1% of cancer deaths of children and 5% of blindness. Most children are diagnosed with retinoblastoma in the first or second year of life, although later presentation can occur, and very rare cases have even been reported in adults.4 The cells of origin are postulated to be retinal stem cells or retinal progenitor cells.5 The untreatment of retinoblastoma is always fatal and the patients die of intracranial extension and disseminated disease within two years. The first goal of treatment is survival, followed by globe salvage, then preservation of vision. In the last years repeated cycles of chemotherapy with carboplatin, vincristine and etoposide combined with cryotherapy and laser photocoagulation have improved the ocular salvage rate for children with bilateral retinoblastoma,6 and treatment in the early stages of disease holds a good prognosis for survival and salvage of visual function. In late stages, however, the prognosis for ocular function and even survival is jeopardized and children often experience recurrence after treatment. In addition, as ototoxicity is a possible side effect of carboplatin,7 the evaluation of hearing loss in patients who frequently have impaired vision is very important. Children with the inherited form have a high risk of developing other cancers later in life4 and very long-term follow-up of retinoblastoma patients revealed an emerging excess risk of mortality in hereditary retinoblastoma survivors. After 50 years of follow-up, hereditary retinoblastoma survivors, most of whom were treated with radiotherapy, had a 25.5% cumulative risk of dying from a subsequent malignant neoplasm compared with a 1.0% cumulative risk for nonhereditary retinoblastoma survivors, most of whom were treated surgically.8 The high mortality risks persisted for cancers of the bone, connective tissue, brain and other parts of the nervous system, and for melanoma.
In the past 15 years our laboratory was actively engaged in studying the cytotoxic effect of drugs in human retinoblastoma cells and their molecular mechanism of action. Our studies, performed in human Y79 retinoblastoma cells, have demonstrated that in these cells the lack of functional pRb is accompanied by upregulation of p53 and this makes Y79 cells prone to apoptosis.9 We have also shown that a number of drugs act as apoptotic inducers in Y79 cells through either extrinsic or intrinsic pathways, via p53-independent or -dependent mechanisms,10,11 and with some drugs inducing significant synergistic effects when employed in combination.12
Recently, our attention has been paid to the use of natural compounds that do not have cytotoxic effects on normal cells and exert potent cytotoxic effects on cancer cells. We have demonstrated that paclitaxel (PTX), a complex diterpene isolated from Taxus Brevifolia, whose target is the mitotic tubulin/microtubule system, is a potent inducer of apoptosis in human Y79 retinoblastoma cells;13 in addition, when subtoxic doses of PTX and β-lapachone (LPC)—a natural naphthoquinone compound extracted from the bark of the lapacho tree Tabebuia avellanedae—were combined, they potently synergized in inducing apoptosis.14
Here, we investigated the effects of parthenolide (PN), a sesquiterpene lactones derived from the leaves of feverfew (Tanacetum parthenium), and of okadaic acid (OKA), a lipophilic derivative of a C38 fatty acid produced by several marine dinoflagellates and isolated from the sponges Halichondria okadai and Halichondria melanodocia.
PN has attracted much attention because of its anti-inflammatory and anti-cancer activities.15-18 PN contains an α-methylene-γ-lactone ring and an epoxide moiety which are able to interact with nucleophilic sites of biologically important molecules thus modulating multiple targets and contributing to its various in vitro and in vivo effects.19
OKA acts by inhibiting serine/threonine protein phosphatases (PP1 and specially PP2A)20 and this results in the hyperphosphorylation of many cell proteins with the disregulation of a variety of cellular processes. Apoptosis has been widely reported as a relevant mechanism of OKA-mediated toxicity; however, molecular pathways in OKA-driven apoptosis still remain unclear.21
Here, we demonstrate for the first time that subtoxic doses of OKA/PN combinations are particularly effective in inducing synergistic apoptotic effects in human retinoblastoma Y79 cells. We also describe the possible molecular mechanism of this synergistic effect.
Results
Okadaic acid and parthenolide combination sinergistically induce cytotoxicity in Y79 cells
We first examined the survival rate of human retinoblastoma Y79 cells after treatment with OKA and PN alone. Monolayer cells were treated for various times (3–48 h) with various doses of OKA (5–50 nM) or PN (50 nM–2 µM) and viability was studied by MTT assay. As shown in Figure 1A, treatment with either OKA or PN determined a time- and dose-dependent loss of cell viability. At 48 h after treatment, 5–30 nM OKA determined only little effects (5–20% loss of cell viability); the effects significantly increased with 40 nM (35% loss of cell viability) and reached their maximum with 50 nM (70% loss of cell viability). Figure 1A also shows that similar loss of cell viability was induced by treatment with PN alone within the range of concentrations of 50–500 nM, 1 μM, and 2 μM. To evaluate whether OKA/PN combination synergistically induces cytotoxic effects, a dose- and time-dependent effect analysis according to Chou and Talalay method was performed.22 Three different suboptimal concentrations of OKA (10, 20, and 30 nM) and two suboptimal concentrations of PN (250 and 500 nM) were chosen and assessed in combination. Figure 1B shows the results obtained when Y79 cells were treated for various times (3–12 h) with OKA/PN combination at concentrations which changed in a fixed ratio. As Figure 1B shows, the decrease in cell survival was much greater with the combined treatment than with single-drug treatments. In Figure 1C analysis of the combination index (CI) with respect to the proportion of dead cells (fraction affected, Fa) showed values of CI considerably lesser than 1.0, indicating strong synergistic interactions between the two compounds. To useful continue our study, we established to employ, when not otherwise specified, the combined concentrations of 20 nM OKA and 500 nM PN (since then indicated as OKA/PN), for 6 h of treatment, a condition that, as shown in Figure 1B, resulted in not more than 50% loss of cell viability.
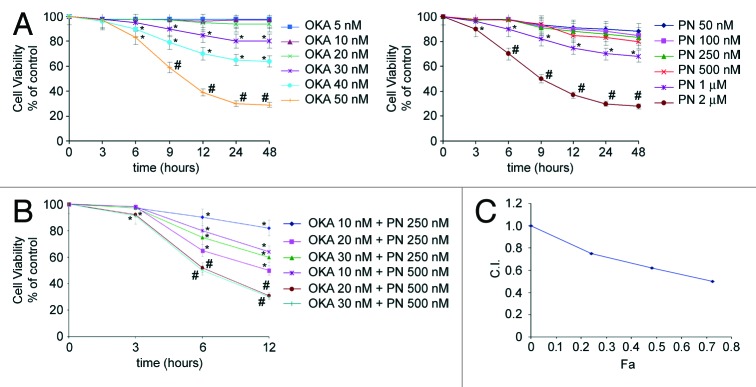
Figure 1. Synergistic interaction of OKA and PN in human retinoblastoma Y79 cells. The figure describes the time-and dose-dependent effects of OKA and PN treatment (A) and the effects of their combination (B) analyzed by MTT assay. Data are the mean ± SD of three independent experiments, each performed in triplicate, and expressed as percentage of the control. Data were considered significant at *P < 0.05 and highly significant at #P < 0.01 as compared with the control group. (C) Values of combination index (CI) were calculated in relation to the fraction affected (Fa). Values of CI less than 1.0 indicate synergistic interactions between the two drugs.
Treatment with OKA/PN induces apoptotic morphology
To elucidate the mechanism by which OKA/PN induced loss of cell viability in Y79 cells, we performed morphological analysis. In Figure 2A phase contrast (PC) microscopy shows that 6 h of OKA/PN treatment reduced cell number, also inducing dramatic modifications in cell shape, with rounding and loss of cell-cell contact. The effect seemed to be apoptotic as suggested by membrane blebbing, formation of apoptotic bodies and a tendency of cells to coat in the medium. Figure 2A shows that single-drug treatment did not change the number and the cell morphology. As chromatin condensation and nuclear fragmentation remain the hallmarks of apoptotic cells, apoptosis was assessed by nucleic acid staining. In Figure 2B Hoechst 33258 (H) staining shows that OKA/PN treatment induced typical apoptotic nuclei exhibiting highly fluorescent condensed chromatin, with bright blue spots, whereas control cells and cells treated with OKA or PN alone appeared normal, with nuclei staining weakly and homogeneously distributed. In Figure 2C the fluorescent dye technique by using A/E staining shows that OKA/PN treatment resulted in nuclear fragmentation with the coexistence of both, cells with nuclei presenting green bright spots (live-apoptotic cells), and cells with nuclei presenting red bright spots (dead-apoptotic cells). Control cells and cells treated with OKA or PN alone appeared normal, with uniform green nuclei.
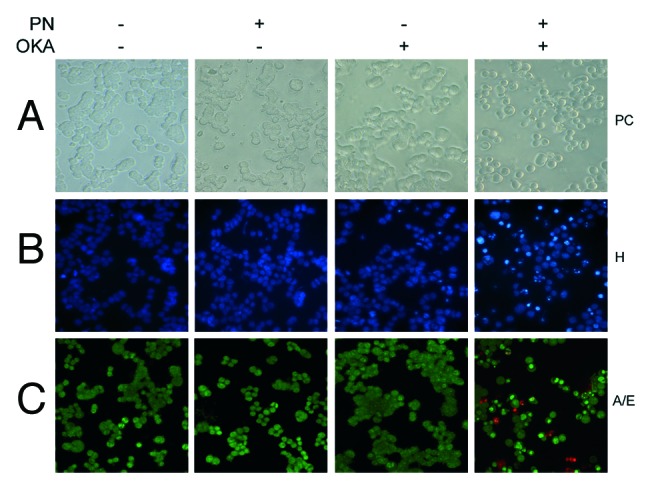
Figure 2. Apoptotic effects induced by OKA/PN combination in human retinoblastoma Y79 cells. The figure describes the effects OKA and PN treatment on cell morphology analyzed by (A) phase contrast (PC) microscopy, (B) fluorescence microscopy by Hoechst 33258 (H) staining, (C) fluorescence microscopy by acridine orange/ethidium bromide (A/E) staining. (Original magnification 400×.) Images are representative of at least four independent experiments.
Cytofluorimetric and western blot analyses confirm that OKA/PN treatment induces apoptosis
We performed cytofluorimetric analysis in the presence of propidium iodide, in order to quantify the percentage of cells in sub-G0-G1 phase. Bar histograms in Figure 3A show that at 3, 6, 12, and 24 h after treatment OKA/PN significantly induced cell accumulation in sub-G0/G1 phase. A small effect was already evident at 3 h after treatment (~8%), peaked at 6 h (~50%), was maximal at 12 h (62%), and did not change significantly thereafter. Neither OKA, nor PN alone induced sub-G0-G1 phase accumulation. In Figure 3B DNA content analysis describes as, at 6 h after OKA/PN treatment, cell cycle profile resulted in 50.3% accumulation of the cells in the sub-G0-G1 phase with concomitant loss from G0-G1 and G2-M phases. To further confirm that the sub G0/G1 peak was due to apoptosis and not necrosis, we performed Annexin V assay by flow cytometry. In Figure 3C cytofluorimetric analysis evidences that the cells accumulated in the sub-G0-G1 phase after OKA/PN treatments also contained a significant percentage of cells in early apoptosis. In Figure 3D the analysis describes the Annexin V/PI pattern at 6 h after OKA/PN treatment.

Figure 3. OKA/PN treatment induces apoptosis and activation of caspases in human retinoblastoma Y79 cells. (A) Graph summarizing percentages of cells in sub-G0-G1 phase, evaluated by flow cytometric analysis of propidium iodide DNA staining. Data are the mean ± SD of three independent experiments, each performed in triplicate. Data were considered significant at *P < 0.05 and highly significant at #P < 0.01 as compared with the control group. (B) Cell cycle profile at 6 h after OKA, PN, and OKA/PN treatment. (C) Graph summarizing percentages of early apoptotic cells (Annexin V+/PI−) measured by flow cytometric analysis of Annexin V labeling. Data are the mean ± SD of three independent experiments, each performed in triplicate. Data were considered significant at #P < 0.01 as compared with the control group. (D) Typical contour plots at 6 h after OKA, PN and OKA/PN treatment. (E) Western blot analysis of apoptotic markers. Actin was used as the loading control. Images are representative of at least four independent experiments.
We also investigated whether during the 3–12 h of OKA/PN treatment initiator and executioner caspases resulted activated. No activations were seen at 3 h after treatment either with single-drug or with OKA/PN treatment (not shown). At 6 h after OKA/PN treatment (Fig. 3E), a marked activation of initiator caspase 8 was evidenced as indicated by the increase in the active fragment level, with a small reduction in that of procaspase form. Treatment with OKA or PN alone also activated initiator caspase 8 but at a lesser degree than OKA/PN combination. Moreover, OKA/PN treatment activated the executioner caspase 3, caused the cleavage of Lamin B and reduced PARP level (Fig. 3E). At 12 h after OKA/PN treatment the levels of the analyzed proteins resulted similar to those described at 6 h after treatment (not shown).
OKA/PN treatment potently increases ROS levels and lowers GSH content
Recent studies have demonstrated that parthenolide induces apoptosis via multiple mechanisms including generation of reactive oxygen species (ROS).23 We were interested in evaluating whether apoptosis induced in Y79 cells by OKA/PN treatment even involved oxidative stress. To evaluate such an involvement, we measured intracellular free radical levels at various time-intervals (3–12 h) after drugs treatment, using the cell permeant dye H2-DCFDA. In Figure 4A measurements of H2-DCFDA fluorescence show that OKA/PN generated an increase in intracellular ROS which was already evident at 3 h after treatment, exhibited its maximum level at 6 h and progressively lowered at 9 and 12 h. Figure 4B shows the shift of fluorescence intensity observed at 6 h after OKA/PN treatment indicating the potent increase in ROS levels. As Figure 4B shows, neither OKA nor PN alone increased ROS levels. To determine whether ROS increase was accompanied by glutathione (GSH) consumption, we measured cellular GSH content using a colorimetric assay kit. The Figure 4C shows that OKA/PN treatment strongly reduced GSH content, with the concomitant increase in ROS level described in Figure 4A. Indeed, GSH content reached its lower level after 6 h treatment with OKA/PN and progressively recovered thereafter. After single-drug treatment only a small GSH decrease was measured. To verify whether GSH lowering could be responsible for the loss of cell viability induced by OKA/PN treatment, Y79 cells were pre-treated (4 h) with a non-toxic dose (100 μM) of the glutathione depleting agent l-butathionine-[S,R]-sulfoximine (BSO), a compound that, by inhibiting GSH synthesis, reduces cellular GSH level. Thereafter, the cells were treated for 3 h with the drugs and cell viability by MTT assay was measured. As shown in Figure 4D, in comparison with unpretreated cells, in BSO-pre-treated cells, 3 h of OKA/PN treatment resulted in a potent loss (−35%) of cell viability, whereas single-drug treatments did not determine reduction of cell viability. This suggests that the GSH decrease promoted by BSO anticipated OKA/PN time-dependent effects and described in Figure 1B. Overall, the results suggested that in Y79 cells GSH could be a critical factor in the control of cell survival/death.

Figure 4. OKA/PN induces ROS generation and depletion of intracellular GSH. (A) Graph shows ROS generation measured by flow cytometry as described in materials and methods. Data are the mean ± SD of three independent experiments, each performed in triplicate. Data were considered significant at *P < 0.05 and highly significant at #P < 0.01 as compared with the control group. (B) The figure shows the shift of fluorescence intensity, observed at 6 h after OKA/PN treatment and indicating a potent increase in ROS level. (C) The graph shows the induced depletion of intracellular GSH measured by the colorimetric assay reported in materials and methods. Data are the mean ± SD of three independent experiments, each performed in triplicate. Data were considered significant at *P < 0.05 and highly significant at #P < 0.01 as compared with the control group. (D) The figure shows the effect of pre-treatment with BSO on sensitivity to OKA/PN. Data are the mean ± SD of three independent experiments, each performed in triplicate. Data were considered significant at *P < 0.05 as compared with the control group.
OKA/PN treatment induces phospho-Akt and phospho-Mdm2 lowering, p53 increase, stabilization and activation, with PTEN being the key player
Here we evaluated whether PTEN (phosphatase and tensin homolog deleted on chromosome 10), a tumor suppressor recently involved in the regulation of cell fate, in particular senescence and apoptosis,24 is involved in the effects induced by OKA/PN treatment. We also analyzed the status of the serine-threonine kinase Akt, as it is known that PTEN specifically dephosphorylate phosphatidylinositol-3,4,5-trisphosphate (PIP3), thus antagonizing phosphatidylinositol 3-kinase (PI3K) activity and negatively regulating Akt, its downstream target. In Figure 5A western blot analysis shows that Y79 cells expressed significant basal levels of PTEN which markedly increased at 6 h after OKA/PN treatment. Figure 5A also shows that OKA/PN treatment did not modify Akt levels, whereas it markedly decreased the levels of p-Akt, its active form. No effects were determined by treatment with single-drugs.
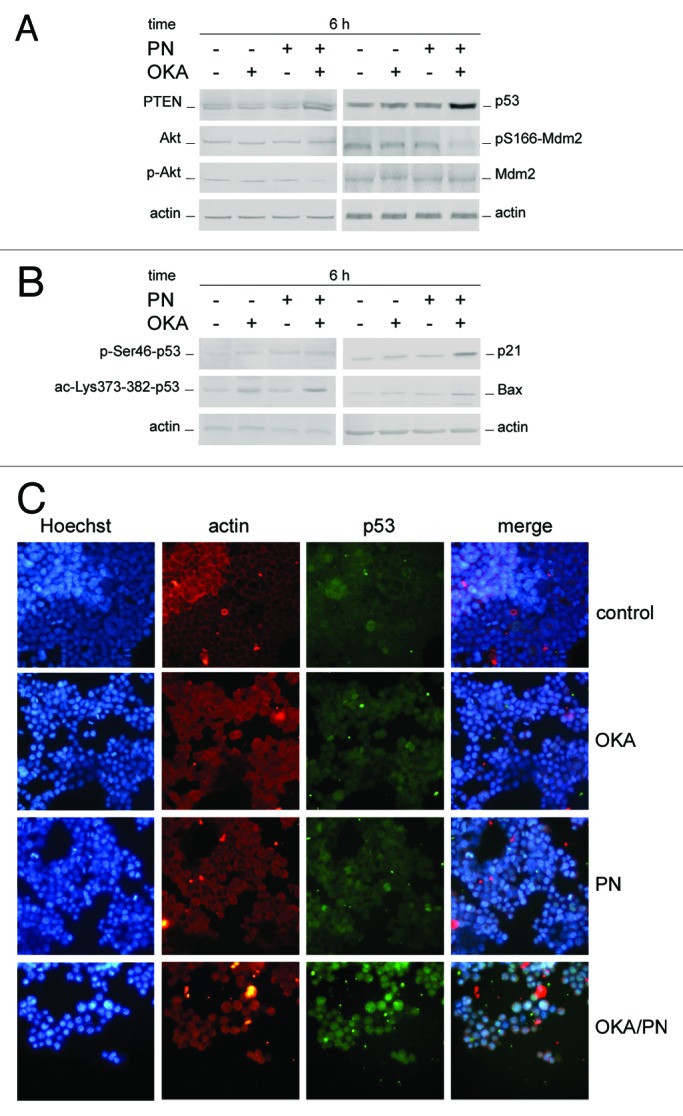
Figure 5. Effects of OKA/PN treatment on PTEN/Akt/Mdm2/p53 pathway. (A and B) describe western blotting analyses after 6 h of drugs treatment. Actin was used as the loading control. Images are representative of at least four independent experiments. (C) The effect of 6 h of drugs treatment on p53 localization by fluorescence microscopy. Cells were triple stained with (1) Hoechst 33258 dye (blue), to localize the nucleus; (2) rhodamine-conjugated secondary antibody (red) to localize cytosolic actin; (3) FITC-conjugated secondary antibody (green) to localize cytosolic/nuclear p53. The right panels show the merge of the three dyes. (Original magnification 400×.) Images are representative of at least four independent experiments.
Knowing that PI3K–Akt signaling can control p53 functions by regulating Mdm2 localization,25 we examined the effects of OKA/PN treatment on the levels of both p53 and pS166-Mdm2. As Figure 5A shows at 6 h after OKA/PN treatment p53 potently increased and pS166-Mdm2 potently decreased, while Mdm2 did not change. Little effects were seen with single-drug treatments. We then decided to verify both the status of p53 and its cell localization. In Figure 5B western blot analyses of phosphorylated (p) and acetylated (ac) p53 forms show that 6 h of OKA/PN treatment markedly increased the basal levels of p-Ser46-p53 and ac-Lys-373/382-p53. The results suggested that these effects could be responsible for p53 stabilization and its trapping in the nucleus. The figure also shows that OKA/PN treatment potently increased the levels of both p21WAF1 and bax, thus suggesting that p53 is transcriptionally active. Also single-drug treatment increased the levels of these proteins, but these effects were much lesser strong than with combined treatment.
In Figure 5C immunofluorescence analysis of p53 intracellular localization shows that 6 h of OKA/PN treatment induced a potent increase in p53 levels with its massive nuclear accumulation, as highlighted by merged image. Only small effects on p53 were observed with single-drug treatment.
Aimed at identifying the molecular player responsible for the effects activated by OKA/PN treatment, we analyzed the effects induced by the drugs at early times of treatment (30–120 min). In Figure S1 western blot analysis shows that PTEN levels markedly increased, after OKA/PN treatment, long before than p53, with PTEN increasing being accompanied by potent lowering in p-Akt and pS166-Mdm2 levels.
Silencing PTEN gene subverts the levels of the molecular players which control apoptosis
To establish whether PTEN may be the key player in the activation of the apoptotic pathway, we have depleted PTEN gene by small-interfering-RNA (siRNA).
First, we checked the effects of PTEN-targeting siRNA on the expression of endogenous PTEN in Y79 cells. Cells transfected with scrambled siRNA (Scr-siRNA) were employed as control. In Figure 6A western blot analysis shows that, after 24–72 h of PTEN-siRNA transfection, PTEN levels potently lowered. The effects were observed at 24 h after transfection and peaked at 48 h. Thereafter, PTEN levels went up markedly, so that at 72 h after transfection we only observed a small decrease in its level. This suggested that, at that time, the temporary transcriptional silencing was in rapid recovery. Overall, the results suggested that the optimal silencing efficiency was reached at 48 h after transfection. The knockdown of PTEN protein was specific, as no PTEN protein reduction was observed in cells transfected with Scr-siRNA. Therefore, either Scr-siRNA or PTEN-siRNA cells transfected, at 48 h after transfection, and untransfected cells were treated with the drugs for 6 h. Figure 6B shows that in Scr-siRNA transfected cells, OKA/PN treatment determined the same effects induced in untransfected cells (i.e., a potent increase in PTEN levels) instead, in PTEN-siRNA transfected cells, OKA/PN treatment failed to raise the low levels of PTEN determined by its silencing. Figure 6B also shows that, similarly to the effect determined on PTEN levels, OKA/PN failed to raise the low levels of p53 determined by PTEN silencing. With regard to the effects of OKA/PN on the levels of p-Akt and pS166-Mdm2, the results show that, in comparison with untransfected and Scr-siRNA transfected cells—where OKA/PN treatment potently lowered their levels—in PTEN-siRNA transfected cells OKA/PN treatment was unable to reduce p-Akt and pS166-Mdm2 levels. We also studied the effect of OKA/PN treatment on GSH content and on cell viability. As Figure 6C shows, in PTEN-siRNA cells at 6 h after OKA/PN treatment were observed only modest effects on either GSH content or cell viability. Instead, at 12 h GSH levels decreased by about 25% and cell viability decreased by about 38%. Overall, the findings suggest that PTEN can be the key player that orchestrates the molecular events involved in the activation of the apoptotic pathway.

Figure 6. Effects of silencing PTEN gene on the molecular players which control apoptosis. Western blot analysis of (A) knockdown efficiency of PTEN protein, and (B) of 6 h of drugs treatment on both untransfected cells and PTEN-siRNA or scrambled siRNA (Scr-siRNA) transfected cells. Actin was used as the loading control. Images are representative of at least four independent experiments. (C) Effects of 6 and 12 h of OKA/PN treatment on both GSH content (left) and cell viability (right) under the conditions described in (B). Data are the mean ± SD of three independent experiments, each performed in triplicate. Data were considered significant at *P < 0.05 and highly significant at #P < 0.01 as compared with Scr-siRNA transfected cells.
Discussion
Retinoblastoma is considered a curable cancer in the developed world, yet it can cause significant morbidity and, rarely, mortality.26 However, 80% of all the pediatric malignancies occurs in developing countries, where treatment is limited and long-term survival rates are low. In addition, existing therapies for retinoblastoma (i.e., chemotherapy and focal therapies) result in unfavorable outcomes at both the systemic and local levels.27,28 Vincristine, etoposide, and carboplatin—the agents commonly used systemically in the treatment of retinoblastoma—determine side effects (abdominal pain, nausea, vomiting, and myelosuppression) which cause significant morbidity to pediatric patients and significantly limit dosing.29 Thus, the identification of both new drugs and new molecular targets is necessary to facilitate the development of novel therapeutics.
The search for drugs that are non-cytotoxic to normal cells and can effectively target cancer cells has led to the discovery of the potent anticancer activity of some natural products which are chemopreventive or therapeutic against cancers.30 One major goal of anticancer drug discovery is to develop innovative therapies that exhibit a real improvement in effectiveness and/or tolerability. To found natural drug combinations which can kill cancer cells by synergistically acting at subtoxic doses, may be a good goal. Recently, we have demonstrated that, in Y79 cells, PTX/LPC combination at subtoxic doses potently induces apoptosis accompanied by potent lowering in proteins inhibitor of apoptosis and by activation of Bid and caspases 3 and 6 with lamin B and PARP breakdown.14 We have also shown that Y79 cells contain constitutively activated Akt which counteracts apoptotic signals by forming a cytosolic complex with p53 and pS166-Mdm2. It is known that pS166-Mdm2 can enter the nucleus where it can induce p53 export, thus trapping it in the cytosolic complex which drives its degradation by ubiquitin–proteasome system. Our previous results showed that PTX/LPC treatment induced a weakness of Akt-Mdm2-p53 complex by strongly lowering pS166-Mdm2 levels and this increased nuclear p53 levels. Overall, our results suggested that p-Akt lowering was at the root of the apoptotic action exerted by PTX/LPC combination and provided strong validation for a treatment approach that targets inhibitor of apoptosis proteins and survival signals.
Constitutive Akt activation is related to a more aggressive phenotype in various human cancers31 and it contributes to cancer progression via pleiotropic effects, including suppression of apoptosis and modulation of cell-cycle regulation. Akt plays a key role in mitogenic pathway controlled by PI3K. Under normal PI3K/Akt signaling, PI3K is recruited to the cell surface through binding to a receptor tyrosine kinase (RTK) and phosphorylates PIP2 to generate PIP3, a critical second messenger which leads to the activation of Akt.32 Akt is crucial in downstream signaling of multiple RTK of pathogenic importance in cancer development,33 in particular in the mitogenic signal activated by IGF-1R/IRS-1 combination.34,35 We have previously shown that in Y79 cells IGF-1/IGF-1R mediates an autocrine growth mechanism which induces the tyrosine phosphorylation of IRS-1 and we suggested that this autocrine loop can be the leading cause of the uncontrolled proliferation of human retinoblastoma Y79 cells.36
Most highly malignant human cancers have dysregulated PI3K/Akt signaling cascades which lead to abnormally increased cell proliferation.33 Here, we show for the first time that in human retinoblastoma Y79 cells combination of OKA with PN at subtoxic doses, induces a potent synergistic apoptotic effect. Our results demonstrate that, likely to PTX/LPC combination, OKA/PN combination induces apoptosis by lowering p-Akt levels and favoring p53 stabilization through a potent decrease in the level of pS166-Mdm2. However, here we also show the key involvement of PTEN which potently increases after OKA/PN treatment. Unlike most of the protein tyrosine phosphatases, PTEN preferentially dephosphorylates phosphoinositide substrates by converting PIP3 to PIP2.37 In such a manner, PTEN functions as a tumor suppressor by antagonizing PI3K/Akt signaling pathway. However, PTEN may have additional phosphatase-independent activities and can also play other critical roles in multiple aspects of cancer development.38 PTEN is the second most frequently mutated gene in human cancer after p53,39 and PTEN loss is observed in many malignant tumors where it is often a marker for advanced neoplastic disease.40,41 Upon PTEN loss, excessive accumulation of PIP3 at the plasma membrane recruits and activates Akt, potently driving cell proliferation and apoptosis resistance.42 Conversely, treatment of cancer cells with drugs which induce PTEN upregulation determines potent apoptotic effects.43 Apoptosis is an active process which can be triggered in tumor cells by a number of different stimuli, among which considerable evidence implicates oxidative stress as a mediator.44 To this regard, it has been suggested that programmed cell death has evolved as a mechanism to eliminate cells which become producers of large amount of ROS.45 The biological activity of PN is thought to be mediated through its α-methylene-γ-lactone moiety, which can react with nucleophiles, especially with cysteine thiol groups. By its chemical properties PN modifies cellular redox state and induces cell death.31 Even OKA is a strong inducer of oxidative stress, in particular on neuronal cells.46 We have already shown that in Y79 cells apoptosis can be mediated by oxidative stress.16 Here we demonstrate that apoptosis induced by OKA/PN treatment is accompanied by a potent increase in ROS level with a simultaneous decreases in that of GSH; the results also show that reducing cellular GSH levels by glutathione depleting agent BSO, significantly anticipates the time of cytotoxic effect exerted by OKA/PN. Indeed, in cells pretreated with BSO, OKA/PN treatment markedly induced loss in cell viability after 3 h of treatment, an event that in non-pretreated cells was determined after 6 h of treatment. Overall, the results suggest that Y79 cells could be prone to oxidative stress.
In this study we show a molecular scenario in which PTEN plays a key role in OKA/PN-induced effects. Indeed, PTEN induction and p-Akt lowering were already evident after 30 min of OKA/PN treatment, significantly before p53 increase. This suggests that p53 activation could be under the action of PTEN as also evidenced by knockdown of PTEN where pAkt/pS166-Mdm2 increased over basal levels, while p53 significantly lowered. In addition, in PTEN-silenced cells OKA/PN treatment was unable to lower both p-Akt and pS166-Mdm2 below basal levels and to increase p53 over basal level. Regarding to cell viability, oxidative stress and GSH content, we observed that, respect to control cells, in PTEN-silenced cells OKA/PN treatment induced a lesser-reduction of cell viability, a lesser production of ROS and a lower decrease in GSH content.
To our knowledge our study report for the first time both a synergistic apoptotic action between OKA and PN and the involvement of PTEN as key player in the apoptotic mechanism in human retinoblastoma Y79 cells. The results provide strong suggestion for a combining treatment approach that targets the PTEN/Akt/Mdm2/p53 pathway.
Materials and Methods
Cell culture
Human retinoblastoma Y79 cells—purchased from Interlab Cell Line Collection—were cultured in suspension in T-75 flask in RPMI 1640, supplemented with 15% (v/v) heat-inactivated fetal bovine serum, 2 mM l-glutamine and antibiotics (50 U/ml penicillin–50 μg/ml streptomycin) (Euroclone) in a humidified atmosphere of 5% CO2 in air at 37 °C. Media were replaced every 2–3 d. For monolayer cultures Y79 cells were seeded into 96-well plates (5 × 104 cells/well) or 6-well plates (1.5 × 106 cells/well), precoated with 5 μg/cm2 poly-d-lysine (Sigma-Aldrich). Cells were treated with OKA and/or PN for the time indicated in the results and, then were removed by pipetting up and down and pelleted at 1000× g. OKA and PN were supplied by Sigma-Aldrich, prepared as a stock solution (621 μM and 40 mM respectively) in dimethylsulfoxide, protected from light, preserved at −20 °C and diluted to working concentration with culture medium. Final concentrations of DMSO never exceeded 0.04%.
Cell viability
For these studies the cells were seeded into 96-well plates as above reported. Twenty-four hours after seeding, cells were treated with drugs for the time indicated in the Results. Cell viability was determined by the MTT colorimetric assay.47 MTT is reduced to purple formazan in the mitochondria of living cells. The absorbance of the formazan was measured at 570 nm, with 630 nm as a reference wavelength using an automatic ELISA plate reader (OPSYS MR, Dynex Technologies). After drugs treatment, cell survival was estimated as a percentage of the value of the vehicle-treated control. In some experiments trypan blue was used to count viable cells with comparable results.
Apoptotic morphology
Apoptotic morphology was detected by either Hoechst 33258 fluorescent staining (Sigma-Aldrich) or by acridine orange and ethidium bromide (A/E) (Sigma-Aldrich), the fluorescent DNA-binding dyes. For these studies, after seeding into 96-well plates, Y79 cells were treated with OKA and PN individually and in combination for the indicated times at 37 °C. Thereafter, the incubation medium was removed and cells were washed with phosphate-buffered saline (PBS). For Hoechst 33258 staining, the cells were fixed with methanol/acid acetic (3:1) for 10 min at room temperature and after washing with PBS, the cells were incubated with Hoechst 33258 (5 μg/ml) for 10 min at room temperature. For A/E staining, the cells were treated with a solution composed of A/E (100 μg/ml PBS of each dye). Samples were examined on a Leica DM IRB inverted microscope (Leica Microsystems Srl) equipped with fluorescence optics and suitable filters for DAPI, FITC and rhodamine detection; images were photographed and captured by a computer-imaging system (Leica DC300F camera and Adobe Photoshop for image analysis).
Immunofluorescence analysis to evaluate cytoplasmic–nuclear shuttling of p53
Y79 cells were grown in 6-well plates, after treatments the cells were washed with PBS, fixed with 3.7% formaldehyde and permeabilized with 0.1% Triton X-100 (Sigma) in PBS. All antibodies were diluted in PBS + 1% BSA + 0.05% NaN3 (Sigma-Aldrich). The cells were incubated overnight at 4°C with a mixture of rabbit monoclonal antibody against actin (diluted 1:2000; Sigma) and mouse monoclonal antibody against p53 (diluited 1:500; Santa Cruz Biotechnology Inc.). The cells were then washed in PBS, incubated with Cy3-conjugated donkey anti-rabbit IgG (H+L) secondary antibody (diluted 1:100; Jackson ImmunoResearch Laboratories) for 1 h at room temperature and then with Cy2-conjugated donkey anti-mouse IgG (H+L) secondary antibody (diluted 1:100; Jackson ImmunoResearch Laboratories) for another hour. Nuclei were visualized after staining with Hoechst 33258 (5 μg/ml) at room temperature for 10 min. Samples were examined on a Leica DM IRB inverted microscope, as above reported.
Flow cytometry analysis
Apoptosis was also studied by flow cytometry of either DNA content or Annexin V labeling. For DNA staining, cell suspensions were centrifuged, washed 3 times with PBS and resuspended at 1 × 106 cells/ml in PBS. Cells were mixed with cold absolute ethanol and stored for 1 h at 4 °C. After centrifugation, cells were rinsed 3 times in PBS and the pellet was suspended in 1 ml of propidium iodide (PI) staining solution (3.8 mM sodium citrate, 25 μg/ml PI, 10 μg/ml RNase A; Sigma-Aldrich) and kept in the dark at 4°C for 3 h prior to flow cytometry analysis. The proportion of cells giving fluorescence in the sub-G0/G1 peak of cell cycle was taken as a measure of apoptosis.
For Annexin V labeling, cell suspensions were centrifuged, washed 3 times with PBS and resuspended in 1X Annexin V binding buffer (BD Biosciences PharMingen) at a concentration of 1 × 106 cells/ml. One hundred microliters of cell suspension was then incubated with 5 μl of Annexin V-FITC (BD Biosciences PharMingen) and 5 μl of PI for 15 min at a room temperature in the dark. Double labeled with annexin V and PI allows a distinction of early apoptotic (annexin V+/PI−) and late apoptotic/necrotic (annexin V+/PI+) cells.
Flow cytometry analyses were performed by a COULTER EPICS XL flow cytometer (Beckman Coulter Srl) equipped with a single Argon ion laser (emission wavelength of 488 nm) and Expo 32 software. The green fluorescence was measured in the FL1 channel using a 515 nm BP filter and the red fluorescence was measured in the FL3 channel using a 620 nm BP filter. At least 1 × 104 cells per sample were analyzed and data were stored in list mode files.
Evaluation of ROS generation
Intracellular ROS production was determined by detecting the fluorescent intensity of the fluoroprobe 5-(and6)-carboxy-2’, 7’-dichlorodihydrofluorescein diacetate (H2-DCFDA) (Molecular Probes). Y79 cells grown in 6-well plates at the density of 1.0 × 106/well were treated with drugs. Thereafter, the cells were detached and collected by centrifugation into a 15 ml Falcon tube. The dissociated cells were resuspended in 0.5 ml of pre-warmed PBS containing H2-DCFDA (50 μM) and allowed to recover for 30 min in the darkness.
Flow cytometry analysis was performed by a COULTER EPICS XL flow cytometer, as above reported. The amount of ROS production was considered to be directly proportional to the green fluorescence intensity.
Measurement of intracellular glutathione (GSH)
The intracellular GSH concentration was measured using a commercial assay, provided by OXIS Research. After treatment, the cells (5 × 105/well) were collected, and centrifuged at 1000× g for 8 min. The pellets were washed in PBS and resuspended in 50 μl metaphosphoric acid (5 g/100 ml distilled water). GSH content was measured as described by manufacturer's instructions.
Then the samples were read at 400 nm. The absorbance was determined by an automatic ELISA plate reader. The final content of intracellular GSH was expressed as nanomoles of GSH per 106 cells.
Cell transfection of siRNA PTEN
Y79 cells were seeded (5 × 105 cells/well) in 6-well plates and cultured in RPMI 1640 medium, supplemented with 15% FBS, for 24 h to reach approximately 60–80% confluence before transfection. Specific siRNAs directed against PTEN, obtained by Santa Cruz Biotechnology as a pool of double-stranded RNA oligonucleotides, were transfected for 5 h into the cells at a final concentration of 50 nM, in the presence of 5 μl Lipofectamine 2000 (Invitrogen) in a final volume of 1 ml serum-antibiotic free RPMI 1640. At the end the reaction was stopped replacing the culture medium with RPMI 1640 +15% FBS. Cells were examined for PTEN downregulation and other properties 24–72 h after transfection. siRNA, consisting in a scramble sequence, was used as a negative control.
Western blot analysis
For these studies, after treatment, cells were washed in PBS and lysed for 30 min at 4 °C in ice-cold lysis buffer (1% NP40, 0.1% SDS, and 0.5% sodium deoxycolate in PBS) containing protease inhibitor cocktail. Then, cells were sonicated three times for 10 s. Equal amounts of protein samples (40 μg/lane) were subjected to SDS-PAGE and then transferred to a nitrocellulose membrane (Bio-Rad Laboratories Srl) for detection with specific antibodies anti-phospho Mdm2 (Ser166), anti-phospho p53 (Ser46) (diluted 1:500; Cell Signaling), anti-acetyl p53 (Lys 373–382) (diluited 1:500; Millipore Corporation). Unless otherwise specified all antibodies (diluted 1:1000) were purchased from Santa Cruz Biotechnology. The secondary horseradish peroxidase-labeled goat anti-mouse and goat anti-rabbit antibodies were from Amersham Life Science Inc. Immunoreactive signals were detected using enhanced chemiluminescence (ECL) reagents (Bio-Rad). The correct protein loading was confirmed by stripping the immunoblot and reprobing with primary antibody for actin (diluted 1:500; Sigma). Protein concentration was assayed using Bradford method (Bio-Rad Laboratories).
Statistical analysis
Data, represented as mean ± S.D., were analyzed using the 2-tailed Student t test. Differences were considered significant when P < 0.05.
Supplementary Material
Acknowledgments
This study was partially supported by grants from Italian Ministry of Education, University and Research (MIUR) ex-60%, 2007; MIUR; contract number 867/ 06/ 07/2011; MIUR; contract number 2223/12/19/2011; MIUR-PRIN; contract number 144/01/26/2012.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/25944
References
- 1.Aerts I, Lumbroso-Le Rouic L, Gauthier-Villars M, Brisse H, Doz F, Desjardins L. Retinoblastoma. Orphanet J Rare Dis. 2006;1:31. doi: 10.1186/1750-1172-1-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goodrich DW. The retinoblastoma tumor-suppressor gene, the exception that proves the rule. Oncogene. 2006;25:5233–43. doi: 10.1038/sj.onc.1209616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Giacinti C, Giordano A. RB and cell cycle progression. Oncogene. 2006;25:5220–7. doi: 10.1038/sj.onc.1209615. [DOI] [PubMed] [Google Scholar]
- 4.Melamud A, Palekar R, Singh A. Retinoblastoma. Am Fam Physician. 2006;73:1039–44. [PubMed] [Google Scholar]
- 5.Zhong X, Li Y, Peng F, Huang B, Lin J, Zhang W, Zheng J, Jiang R, Song G, Ge J. Identification of tumorigenic retinal stem-like cells in human solid retinoblastomas. Int J Cancer. 2007;121:2125–31. doi: 10.1002/ijc.22880. [DOI] [PubMed] [Google Scholar]
- 6.Antoneli CB, Ribeiro KC, Steinhorst F, Novaes PE, Chojniak MM, Malogolowkin M. Treatment of retinoblastoma patients with chemoreduction plus local therapy: experience of the AC Camargo Hospital, Brazil. J Pediatr Hematol Oncol. 2006;28:342–5. doi: 10.1097/00043426-200606000-00004. [DOI] [PubMed] [Google Scholar]
- 7.Jehanne M, Mercier G, Doz F. Monitoring of ototoxicity in young children receiving carboplatin for retinoblastoma. Pediatr Blood Cancer. 2009;53:1162. doi: 10.1002/pbc.22124. [DOI] [PubMed] [Google Scholar]
- 8.Marees T, van Leeuwen FE, de Boer MR, Imhof SM, Ringens PJ, Moll AC. Cancer mortality in long-term survivors of retinoblastoma. Eur J Cancer. 2009;45:3245–53. doi: 10.1016/j.ejca.2009.05.011. [DOI] [PubMed] [Google Scholar]
- 9.Giuliano M, Lauricella M, Vassallo E, Carabillò M, Vento R, Tesoriere G. Induction of apoptosis in human retinoblastoma cells by topoisomerase inhibitors. Invest Ophthalmol Vis Sci. 1998;39:1300–11. [PubMed] [Google Scholar]
- 10.Vento R, Giuliano M, Lauricella M, Carabillò M, Di Liberto D, Tesoriere G. Induction of programmed cell death in human retinoblastoma Y79 cells by C2-ceramide. Mol Cell Biochem. 1998;185:7–15. doi: 10.1023/A:1006836428202. [DOI] [PubMed] [Google Scholar]
- 11.Vento R, D’Alessandro N, Giuliano M, Lauricella M, Carabillò M, Tesoriere G. Induction of apoptosis by arachidonic acid in human retinoblastoma Y79 cells: involvement of oxidative stress. Exp Eye Res. 2000;70:503–17. doi: 10.1006/exer.1998.0810. [DOI] [PubMed] [Google Scholar]
- 12.Giuliano M, Lauricella M, Calvaruso G, Carabillò M, Emanuele S, Vento R, Tesoriere G. The apoptotic effects and synergistic interaction of sodium butyrate and MG132 in human retinoblastoma Y79 cells. Cancer Res. 1999;59:5586–95. [PubMed] [Google Scholar]
- 13.Drago-Ferrante R, Santulli A, Di Fiore R, Giuliano M, Calvaruso G, Tesoriere G, Vento R. Low doses of paclitaxel potently induce apoptosis in human retinoblastoma Y79 cells by up-regulating E2F1. Int J Oncol. 2008;33:677–87. [PubMed] [Google Scholar]
- 14.D’Anneo A, Augello G, Santulli A, Giuliano M, di Fiore R, Messina C, Tesoriere G, Vento R. Paclitaxel and beta-lapachone synergistically induce apoptosis in human retinoblastoma Y79 cells by downregulating the levels of phospho-Akt. J Cell Physiol. 2010;222:433–43. doi: 10.1002/jcp.21983. [DOI] [PubMed] [Google Scholar]
- 15.Pajak B, Gajkowska B, Orzechowski A. Molecular basis of parthenolide-dependent proapoptotic activity in cancer cells. Folia Histochem Cytobiol. 2008;46:129–35. doi: 10.2478/v10042-008-0019-2. [DOI] [PubMed] [Google Scholar]
- 16.Kawasaki BT, Hurt EM, Kalathur M, Duhagon MA, Milner JA, Kim YS, Farrar WL. Effects of the sesquiterpene lactone parthenolide on prostate tumor-initiating cells: An integrated molecular profiling approach. Prostate. 2009;69:827–37. doi: 10.1002/pros.20931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guzman ML, Rossi RM, Karnischky L, Li X, Peterson DR, Howard DS, Jordan CT. The sesquiterpene lactone parthenolide induces apoptosis of human acute myelogenous leukemia stem and progenitor cells. Blood. 2005;105:4163–9. doi: 10.1182/blood-2004-10-4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carlisi D, D’Anneo A, Angileri L, Lauricella M, Emanuele S, Santulli A, Vento R, Tesoriere G. Parthenolide sensitizes hepatocellular carcinoma cells to TRAIL by inducing the expression of death receptors through inhibition of STAT3 activation. J Cell Physiol. 2011;226:1632–41. doi: 10.1002/jcp.22494. [DOI] [PubMed] [Google Scholar]
- 19.Koprowska K, Czyz M. [Molecular mechanisms of parthenolide’s action: Old drug with a new face] Postepy Hig Med Dosw (Online) 2010;64:100–14. [PubMed] [Google Scholar]
- 20.Bialojan C, Takai A. Inhibitory effect of a marine-sponge toxin, okadaic acid, on protein phosphatases. Specificity and kinetics. Biochem J. 1988;256:283–90. doi: 10.1042/bj2560283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rossini GP, Sgarbi N, Malaguti C. The toxic responses induced by okadaic acid involve processing of multiple caspase isoforms. Toxicon. 2001;39:763–70. doi: 10.1016/S0041-0101(00)00202-6. [DOI] [PubMed] [Google Scholar]
- 22.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 23.Kim YR, Eom JI, Kim SJ, Jeung HK, Cheong JW, Kim JS, Min YH. Myeloperoxidase expression as a potential determinant of parthenolide-induced apoptosis in leukemia bulk and leukemia stem cells. J Pharmacol Exp Ther. 2010;335:389–400. doi: 10.1124/jpet.110.169367. [DOI] [PubMed] [Google Scholar]
- 24.Kitagishi Y, Matsuda S. Redox regulation of tumor suppressor PTEN in cancer and aging (Review) Int J Mol Med. 2013;31:511–5. doi: 10.3892/ijmm.2013.1235. [Review] [DOI] [PubMed] [Google Scholar]
- 25.Chang CJ, Freeman DJ, Wu H. PTEN regulates Mdm2 expression through the P1 promoter. J Biol Chem. 2004;279:29841–8. doi: 10.1074/jbc.M401488200. [DOI] [PubMed] [Google Scholar]
- 26.Boubacar T, Fatou S, Fousseyni T, Mariam S, Fatoumata DT, Toumani S, Abdoul-Aziz D, Marouf KM. A 30-month prospective study on the treatment of retinoblastoma in the Gabriel Toure Teaching Hospital, Bamako, Mali. Br J Ophthalmol. 2010;94:467–9. doi: 10.1136/bjo.2009.159699. [DOI] [PubMed] [Google Scholar]
- 27.Laurie NA, Shih CS, Dyer MA. Targeting MDM2 and MDMX in retinoblastoma. Curr Cancer Drug Targets. 2007;7:689–95. doi: 10.2174/156800907782418266. [DOI] [PubMed] [Google Scholar]
- 28.Boutrid H, Jockovich ME, Murray TG, Piña Y, Feuer WJ, Lampidis TJ, Cebulla CM. Targeting hypoxia, a novel treatment for advanced retinoblastoma. Invest Ophthalmol Vis Sci. 2008;49:2799–805. doi: 10.1167/iovs.08-1751. [DOI] [PubMed] [Google Scholar]
- 29.Rizzuti AE, Dunkel IJ, Abramson DH. The adverse events of chemotherapy for retinoblastoma: what are they? Do we know? Arch Ophthalmol. 2008;126:862–5. doi: 10.1001/archopht.126.6.862. [DOI] [PubMed] [Google Scholar]
- 30.Bhanot A, Sharma R, Noolvi MN. Natural sources as potential anti-cancer agents: A review. Intl J Phytomed. 2011;3:9–26. [Google Scholar]
- 31.Nicholson KM, Anderson NG. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal. 2002;14:381–95. doi: 10.1016/S0898-6568(01)00271-6. [DOI] [PubMed] [Google Scholar]
- 32.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–7. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 33.Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497–510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Valentinis B, Baserga R. IGF-I receptor signalling in transformation and differentiation. Mol Pathol. 2001;54:133–7. doi: 10.1136/mp.54.3.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Metz HE, Houghton AM. Insulin receptor substrate regulation of phosphoinositide 3-kinase. Clin Cancer Res. 2011;17:206–11. doi: 10.1158/1078-0432.CCR-10-0434. [DOI] [PubMed] [Google Scholar]
- 36.Giuliano M, Vento R, Lauricella M, Calvaruso G, Carabillo M, Tesoriere G. Role of insulin-like growth factors in autocrine growth of human retinoblastoma Y79 cells. Eur J Biochem. 1996;236:523–32. doi: 10.1111/j.1432-1033.1996.00523.x. [DOI] [PubMed] [Google Scholar]
- 37.Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–8. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 38.Chalhoub N, Baker SJ. PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol. 2009;4:127–50. doi: 10.1146/annurev.pathol.4.110807.092311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yin Y, Shen WH. PTEN: a new guardian of the genome. Oncogene. 2008;27:5443–53. doi: 10.1038/onc.2008.241. [DOI] [PubMed] [Google Scholar]
- 40.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–7. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 41.Li L, Ross AH. Why is PTEN an important tumor suppressor? J Cell Biochem. 2007;102:1368–74. doi: 10.1002/jcb.21593. [DOI] [PubMed] [Google Scholar]
- 42.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–74. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu B, Wang X, Chi ZF, Hu R, Zhang R, Yang W, Liu ZG. Ursolic acid-induced apoptosis in K562 cells involving upregulation of PTEN gene expression and inactivation of the PI3K/Akt pathway. Arch Pharm Res. 2012;35:543–8. doi: 10.1007/s12272-012-0318-1. [DOI] [PubMed] [Google Scholar]
- 44.Jin Z, El-Deiry WS. Overview of cell death signaling pathways. Cancer Biol Ther. 2005;4:139–63. doi: 10.4161/cbt.4.2.1508. [DOI] [PubMed] [Google Scholar]
- 45.Skulachev VP. Why are mitochondria involved in apoptosis? Permeability transition pores and apoptosis as selective mechanisms to eliminate superoxide-producing mitochondria and cell. FEBS Lett. 1996;397:7–10. doi: 10.1016/0014-5793(96)00989-1. [DOI] [PubMed] [Google Scholar]
- 46.Valdiglesias V, Laffon B, Pásaro E, Cemeli E, Anderson D, Méndez J. Induction of oxidative DNA damage by the marine toxin okadaic acid depends on human cell type. Toxicon. 2011;57:882–8. doi: 10.1016/j.toxicon.2011.03.005. [DOI] [PubMed] [Google Scholar]
- 47.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol. 1983;32:671–6. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.