

Abstract
Diffuse large B cell lymphoma (DLBCL) is an aggressive form of non-Hodgkin lymphoma. While the initial treatment strategy is highly effective, relapse occurs in 40% of cases. Histone deacetylase inhibitors (HDACi) are a promising class of anti-cancer drugs but their single agent efficacy against relapsed DLBCL has been variable, ranging from few complete/partial responses to some stable disease. However, most patients showed no response to HDACi monotherapy for unknown reasons. Here we show that sensitivity and resistance to the hydroxamate HDACi, PXD101, can be modeled in DLBCL cell lines. Sensitivity is characterized by G2/M arrest and apoptosis and resistance by reversible G1 growth arrest. These responses to PXD101 are independent of several negative prognostic indicators such as DLBCL subtype, BCL2 and MYC co-expression, and p53 mutation, suggesting that HDACi might be used effectively against highly aggressive DLBCL tumors if they are combined with other therapeutics that overcome HDACi resistance. Our investigation of mechanisms underlying HDACi resistance showed that cyclin-dependent kinase inhibitors (CKIs), p21 and p27, are upregulated by PXD101 in a sustained fashion in resistant cell lines concomitant with decreased activity of the cyclin E/cdk2 complex and decreased Rb phosphorylation. PXD101 treatment results in increased association of CKI with the cyclin E/cdk2 complex in resistant cell lines but not in a sensitive line, indicating that the CKIs play a key role in G1 arrest. The results suggest several treatment strategies that might increase the efficacy of HDACi against aggressive DLBCL.
Keywords: histone deacetylase inhibitor, lymphoma, drug resistance, cell cycle, cell cycle inhibitors, MYC, BCL2
Introduction
The most commonly diagnosed non-Hodgkin lymphoma (NHL) is diffuse large B cell lymphoma (DLBCL). Molecular profiling studies have shown that DLBCL is a heterogeneous disease consisting of two predominant subtypes: germinal center B cell-like (GCB) and activated B cell-like (ABC).1 Overall, the current standard treatment, rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP), has prolonged survival in DLBCL patients.2,3 In general the GCB subtype is associated with a more favorable prognosis than the ABC subtype, yet significant fractions of patients with either subtype (30% GCB and 60% ABC) experience relapse within 3–5 years, which is associated with a high mortality rate.4 Much research effort has been directed toward characterization of tumor features that correlate with poor prognosis after R-CHOP therapy. Recent studies identified several indicators of inferior overall and progression-free survival after R-CHOP therapy including concurrent expression of MYC and BCL-25,6 or expression of mutant p53.7,8 This has initiated efforts to identify novel therapies that are targeted to patients with tumors such negative prognostic indicators.
Histone deacetylase inhibitors (HDACi) comprise a variety of chemicals that have shown promise in treatment of blood cancers. They are well-tolerated in clinical trials and show selectivity toward cancer cells relative to normal cells. These drugs target class I and II histone deacetylases (HDACs) with somewhat differing potencies and specificities.9 HDACs 1 and 2 are critical for B cell development and are required for proliferation induced by mitogens in mature B cells.10 Their expression in primary DLBCL samples of both ABC and GCB subtypes has been found to be elevated relative to normal lymphoid tissue.11,12 In preclinical experiments HDACi showed strong anti-proliferative and pro-apoptotic effects in a wide variety of cell types.9 Based on clinical trials, two HDACi, vorinostat and romidepsin, were approved to treat advanced cutaneous T cell lymphoma, a relatively rare form of NHL. Their efficacy as single agents against the much more commonly-occurring DLBCL has been highly variable, ranging from a few complete/partial responses to some instances of stable disease. However, the majority of DLBCL patients did not respond to HDACi alone.13-16
The clinical results suggest that many relapsed DLBCL tumors are resistant to the apoptotic effects of HDACi and have spurred research aimed at identifying other therapeutics that act synergistically with HDACi to bring about tumor regression.17,18 HDACi are known to downregulate both c-Myc (MYC)19,20 and BCL2 expression,21 suggesting that they could be effective against DLBCL positive for both proteins (double-positive). However, rational selection of synergistic therapeutics is challenging because the mechanistic bases of sensitivity and resistance to HDACi in DLBCL are unknown.
In the current study we documented the response of a variety of DLBCL cell lines to PXD101 (belinostat), a representative of the hydroxamate class of pan-HDACi. We observed growth inhibition in all cell lines which was due to either cytotoxicity or cell cycle arrest. The cytotoxic response was characterized by commitment to apoptosis that did not require prolonged exposure to the drug. The cytostatic response was characterized uniformly by reversible G1 arrest. We contend that these distinct responses model sensitivity and resistance to HDACi in DLBCL. We also establish that all of the DLBCL cell lines tested are double- positive, expressing both MYC and BCL2, indicating that they share characteristics of DLBCL tumors that are associated with poor prognosis after R-CHOP treatment. Our comparative analysis of the sensitive and resistant cell lines has revealed a key difference in regulation of cyclin E/cdk2 and the cyclin-dependent kinase inhibitors (CKI), p21 and p27 that is independent of p53 status and contributes to its effects on cell cycle progression. These findings suggest that HDACi may be used effectively against double-positive DLBCL in combination with other therapeutics that target cell cycle progression or arrest.
Results
Response to PXD101 in DLBCL cell lines
To characterize HDACi response in DLBCL, we performed a systematic examination of a variety of DLBCL cell lines representing both GCB and ABC subtypes exposed to PXD101, also known as belinostat. This drug has been evaluated in clinical trials16 and, like suberoylanilide hydroxamic acid (SAHA), is a member of the hydroxamic acid class of pan-HDACi. To document the growth effects of this drug we performed a dose response assessment using the MTS assay. We observed growth inhibition at 24 or 48 h treatment over a wide range of PXD101 doses and calculated IC50s for the 24 h treatment period. All cell lines were growth inhibited with IC50s in the submicromolar range (Table 1) with a 5-fold difference between the IC50s of the most and least sensitive lines, similar to observations made by Kalac et al.18 using a distinct set of DLBCL cell lines.
Table 1. IC50 concentrations for growth inhibition at 24 h PXD101 treatment as determined by MTS assay.
| Cell line | DLBCL subtype | t(14;18) status | IC50(μM) |
|---|---|---|---|
| SUDHL6 | GCB | Positive | 0.15 |
| OCI-Ly19 | GCB | Positive | 0.30 |
| SUDHL4 | GCB | Positive | 0.45 |
| DB | GCB | Positive | 0.77 |
| SUDHL8 | GCB | Negative | 0.40 |
| OCI-Ly3 | ABC | Negative | 0.63 |
| U2932 | ABC | Negative | 0.36 |
To examine the nature of the growth inhibition, we performed cell cycle analysis and apoptosis assays on a subset of these cell lines. Cells were treated with PXD101 for 0, 24, 48, or 72 h at the IC50 determined at 24 h treatment in the MTS assays. The Annexin V/propidium iodide (PI) assay was used to detect apoptosis and cell death. PXD101 induced significant levels of apoptosis in three GCB-type lines (DB, OCI-Ly19, and SUDHL6), as shown in Figure 1A, C, and E, as well as in the ABC-type OCI-Ly3 cells (not shown). Cell cycle analysis revealed that the cytotoxic response to PXD101 in DB, OCI-Ly19, and SUDHL6 is associated with accumulation of cells in the G2/M phase of the cell cycle by 24 h treatment (Fig. 1B, D, and F). In all three cell lines, the percentage of cells in G2/M declines progressively at 48 and 72 h and correlates with the onset of apoptosis, suggesting that the cells accumulate in G2/M and then undergo apoptosis.
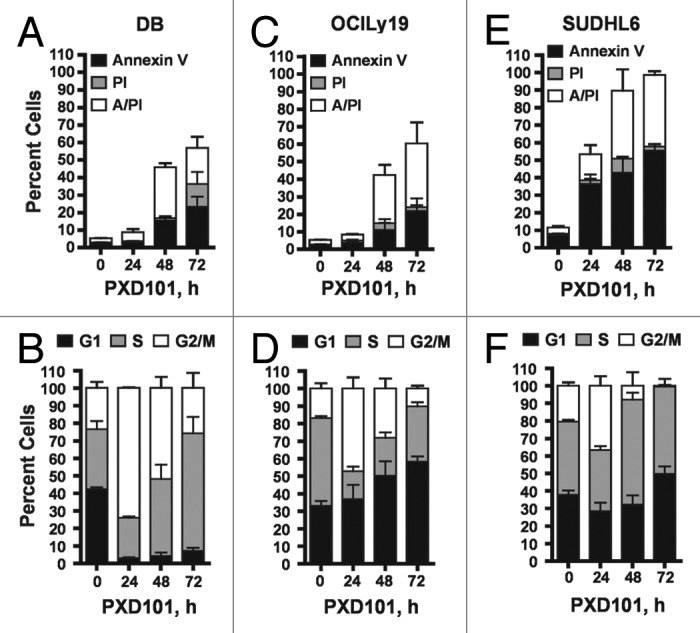
Figure 1. The cytotoxic response to PXD101. DB (A and B), OCI-Ly19 (C and D), and SUDHL6 (E and F) cells were treated with PXD101 at the IC50 concentrations determined for each as shown in Table 1. At 0, 24, 48, and 72 h treatment cells were harvested and subjected to Annexin V/PI assay (A, C, and E) or cell cycle analysis (B, D, and F). The graphs shown represent the results of 3–4 independent experiments. Error bars represent SEM.
In contrast, PXD101 induced no significant apoptosis relative to vehicle-treated controls in SUDHL4 and SUDHL8 cells (Fig. 2A and C). In the ABC-type line U2932 a greater amount of apoptosis was observed involving about 25% of cells as shown in Figure 2E. Cell cycle analysis showed that PXD101 triggers a cytostatic response in these lines characterized by accumulation of cells in the G1 phase of the cell cycle (Fig. 2B, D, and F). At least 75% of cells accumulate in G1 by 24–48 h treatment which is largely maintained with further drug exposure, indicating that PXD101 induces G1 arrest in these cell lines.
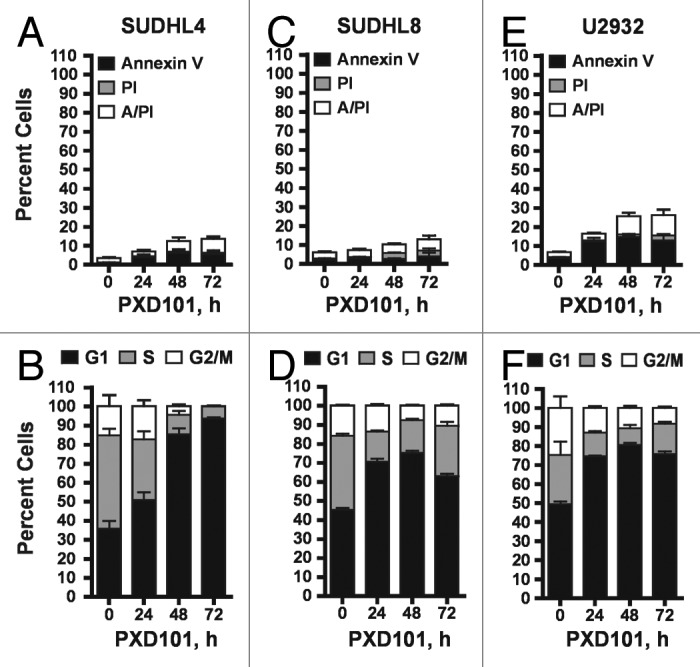
Figure 2. The cytostatic response to PXD101. SUDHL4 (A and B), SUDHL8 (C and D), and U2932 (E and F) cells were treated with PXD101 at the IC50 concentrations determined for each as shown in Table 1. At 0, 24, 48, and 72 h treatment cells were harvested and subjected to Annexin V/PI assay (A, C, and E) or cell cycle analysis (B, D, and F). The graphs shown represent the results of 3–4 independent experiments. Error bars represent SEM.
To investigate the relationship between the extent of drug exposure and the cellular response, we performed drug removal experiments. The cell lines which have a cytotoxic response to PXD101 were exposed to PXD101 for various times up to 24 h prior to drug removal. After 24 h further incubation in drug-free medium, they were examined for evidence of apoptosis (Fig. 3A and B). The results show that only 8–24 h exposure to PXD101 is sufficient to induce similar amounts of cell death in DB and OCI-Ly19 cells as observed with 48 h of continuous exposure (compare with Fig. 1A and C, 48 h). This indicates that the cells become committed to the apoptotic program within 24 h of treatment such that continued exposure to PXD101 is not required. Thus, we classify this group of cell lines as sensitive to HDACi.

Figure 3. Reversibility of PXD101 responses. (A and B) DB (A) or OCI-Ly19 (B) cells were treated at their respective IC50 concentrations for 0, 2, 4, 8, or 24 h after which the cells were washed and resuspended in fresh medium without drug. After 24 h further incubation, the cells were harvested and subjected to Annexin V/PI assay. (C–F) SUDHL4 (C), U2932 (D), and SUDHL8 (E) were treated with either DMSO or PXD101 (at IC50 concentrations) for 48 h. Cells were then washed and incubated in drug-free medium for a further 0, 24, 48, or 72 h. The cells were harvested and subjected to cell cycle analysis. The graphs shown represent the results of 3–4 independent experiments. Error bars represent SEM.
The cell lines which have a cytostatic response to PXD101 were treated for 48 h to allow for G1 arrest. Drug was then washed out and the cells were incubated with drug-free medium for an additional 48 h prior to cell cycle analysis. Figure 3C–E shows that the PXD101-induced G1 arrest is fully reversible. Within 48 h after drug removal, all of the cell lines revert to a cell cycle distribution that is indistinguishable from that of the controls treated with DMSO. Although PXD101 is anti-proliferative in these cell lines, the effect is not sustained once the drug is withdrawn. Therefore, we classify this phenotype as a form of drug resistance that could be analogous to non-response in the clinic.
Expression of of c-Myc (MYC) and BCL2 in PXD101-sensitive or -resistant cell lines
Expression of both MYC and BCL2 in DLBCL tumors has been associated with poor prognosis.5 Thus, we asked whether the MYC and BCL2 proteins are co-expressed in any of the DLBCL cell lines tested above. Equal amounts of multiple, independently-generated whole cell extracts from each cell line were run on an SDS-PAGE gel and subjected to immunoblotting with antibodies against MYC, BCL2, and GAPDH. Figure 4A shows that all cell lines tested co-express MYC and BCL2. We then considered whether overall levels of MYC or BCL2 in the various cell lines correlated with sensitivity or resistance to PXD101 because overexpression of BCL2 in Eμ-myc tumors in mice caused them to arrest in G1 in response to vorinostat treatment.22 The graphs in Figure 4B–D show levels of each protein in the various extracts expressed relative to levels of the same protein in one of the OCI-Ly19 extracts. In contrast to relatively even expression of GAPDH (Fig. 4D), the cell lines express varying levels of MYC and BCL2 (Fig. 4B and C). However, there is no clear relationship between relative levels of these two proteins and the cellular response to HDACi. In the case of MYC, the relative expression level was very similar in DB (PXD101-sensitive), SUDHL4, and U2932 (both PXD101-resistant) (Fig. 4A and B). For BCL2, levels in the PXD101-resistant cell lines were highly variable with SUDHL4 having the lowest levels of all 5 cell lines tested and U2932 having the highest (Fig. 4A and C). Interestingly, BCL2 from SUDHL8 cells reproducibly had a slower mobility in SDS-PAGE gels (Fig. 4A). It has been shown that phosphorylation or mutation of BCL2 can slow its mobility23 and mutations in BCL2 occur in 10–37% of GCB-type DLBCL tumors.24,25 Of all the cell lines shown, SUDHL8 had the lowest levels of BCL2 mRNA by far (Fig. S1A), consistent with the fact that it does not have the t(14;18) translocation that deregulates BCL2 transcription. Thus, because BCL2 protein levels in SUDHL8 cells are similar to those observed in other cell lines, its expression is likely determined by post-transcriptional mechanisms.

Figure 4. Sensitivity or resistance to PXD101 is not correlated with expression of MYC or BCL2. (A–C) Whole cell extracts were generated from the cell lines shown in 2–3 independent experiments. Equal amounts of protein were separated by SDS-PAGE and subjected to western transfer and immunoblotting with c-myc, BCL2, or GAPDH antibodies. All of the samples shown were run on the same SDS-PAGE gel and blotted simultaneously to accurately measure relative levels of each protein. The results of analysis are shown graphically for c-myc (B) and BCL2 (C). Levels of each protein are expressed relative to those in Exp. One of OCI-Ly19 cells. (D) Cells were treated with PXD101 for the times shown. Whole cell extracts were generated and equal amounts of protein were separated by SDS-PAGE, and subjected to Western transfer and immunoblotting with MYC, BCL2, or GAPDH antibodies. BCL2 shown for SUDHL8 correspond to the species with altered mobility as shown in (A). The results shown are representative of three independent experiments.
Regulation of BCL2 and MYC after drug exposure was also examined, as shown in Figure 4E. Over the 72 h treatment period, Bcl-2 levels are slightly upregulated in all the cell lines examined with the exception of DB. In contrast, MYC levels are uniformly downregulated in all the cell lines. Transcription of the MYC gene has been reported to be downregulated by HDACi in a variety of cell types. In fact MYC mRNA levels decrease by 50–60% in all the cell lines tested in the first 4–8 h of PXD101 treatment (Fig. S1C and D). However, in all lines but U2932, this is a transient repression; MYC mRNA levels return to baseline by 24–48 h treatment. This is not reflected by an increase in MYC protein levels indicating that PXD101 also represses MYC expression by post-transcriptional mechanisms. MYC protein can be acetylated at multiple sites26,27 but its functional impact is not clear. Acetylation of MYC has been reported to both prolong26,27 and decrease its half-life.28
Differential regulation of cell cycle regulatory proteins in PXD101-sensitive and -resistant DLBCL cell lines
Progression through G1 is unhindered in the PXD101-sensitive cell lines but blocked in the resistant lines. Therefore, we examined the expression and phosphorylation of proteins that regulate G1 progression. Inactivation of the retinoblastoma protein (Rb) through phosphorylation is a key event that allows G1 progression through the restriction point, and HDACi have been shown to cause decreased Rb phosphorylation in some cell types.29-32 Since phosphorylation of Rb slows its migration through SDS-PAGE gels, we first used an antibody against total Rb to determine whether PXD101 changes Rb mobility. While we found that U2932 cells do not express detectable levels of Rb protein, Figure 5A shows that Rb mobility increases with the length of PXD101 treatment in all the other cell lines, indicating a shift from hyperphosphorylated to hypophosphorylated Rb. This shift was confirmed with the use of antibodies against the latter. Figure 5C and D show the accumulation of hypophosphorylated Rb in SUDHL4 and SUDHL8 cells. Surprisingly, PXD101 significantly downregulated total Rb levels in all Rb-expressing cell lines (Fig. 5A). In contrast, Rb mRNA is not significantly downregulated in any of the cell lines with the exception of OCI-Ly19, where it decreases by about 40%. In DB and SUDHL4 cells Rb mRNA levels are upregulated by PXD101. This contrasts with an 80% decrease in total Rb protein, as shown for SUDHL4 and SUDHL8 cells (Fig. 5E and F). These results indicate that Rb levels are regulated by post-transcriptional mechanisms in response to PXD101 treatment. It is noteworthy that U2932 cells have Rb mRNA but little to no Rb protein, suggesting that the cells have at least one intact and actively-transcribed copy of the Rb gene. In fact Rb mRNA levels in U2932 cells are not significantly different from those measured in SUDHL8 (Fig. S1B). Altogether these observations imply that DLBCL cells have robust post-transcriptional mechanisms to regulate expression of Rb protein.

Figure 5. PXD101 treatment induces loss of Rb protein and Rb phosphorylation. (A and B) The cell lines shown were treated with PXD101 for up to 72 h. (A) Whole cell extracts were subjected to western blotting with antibodies against total Rb protein or α-tubulin. (B) Total RNA was extracted from cells and used to measure levels of Rb mRNA by RT-qPCR. (C and D) Whole cell extracts from PXD101-treated SUDHL4 (C) or SUDHL8 (D) cells were subjected to western blotting with antibodies against Rb phosphorylated at either Ser780 or Ser795, hypophosphorylated Rb, or α-tubulin. (E and F) Levels of total Rb, pRb Ser780, and pRb Ser795 were quantitated from non-saturated images and normalized to levels of α-tubulin for SUDHL4 (E) and SUDHL8 (F) cells. Normalized values from each timepoint of PXD101 treatment are expressed as fractions or multiples of the normalized value from untreated cells for each individual experiment. All of the results shown are representative of 2–4 independent experiments.
Phosphorylation of Rb is performed by the G1-specific cyclin-dependent kinase complexes, cyclin D/cdk4/6 and cyclin E/cdk2. Rb serine 780 (S780) is specifically targeted by the cyclin D-containing complexes.33,34 Figure 5C and D show that phosphorylation of serine 780 decreases dramatically in SUDHL4 and SUDHL8 cells over the course of PXD101 treatment. This could be due to the loss of Rb protein but quantitative analysis shows that S780 phosphorylation falls off more quickly than total Rb (Fig. 5E and F) suggesting a decrease in cyclin D complex activity. This could be due to decreased expression of cyclin D; HDACi have been shown to significantly downregulate levels of cyclin D1 and D2 in other cell types.31,35 Through PCR, we determined that DB, OCI-Ly19, SUDHL4, SUDHL8, and U2932 cells express cyclin D3 mRNA only; U2932 also has cyclin D1 but not cyclin D2 mRNA (not shown). We measured D cyclin levels in these cell lines by immunoblot (Fig. S2) and did not observe uniform downregulation of cyclin D3 across the five cell lines. In fact, there was no correlation with its pattern of regulation by PXD101 and cellular response to the drug. In U2932 cells we found that cyclin D1 was expressed at very low levels which were moderately decreased in response to PXD101. Altogether the results suggest that downregulation of D cyclin levels is not a key mechanism by which PXD101 controls G1 progression in DLBCL cells.
Serine 795 in Rb (S795) can be phosphorylated by either cyclin D- or cyclin E-containing complexes.33,34 S795 phosphorylation in SUDHL4 and SUHDL8 cells was also dramatically decreased by PXD101 treatment at a rate faster than the loss of total Rb (Fig. 5E and F), suggesting that activity of the cyclin E/cdk2 complex may decline. This complex is a major regulator of G1 progression; its activation allows cells to pass the restriction point in G1 and commit to DNA replication and mitosis. Therefore, we performed immunoprecipitation (IP)-kinase assays with cyclin E antibody and histone H1 as substrate. The results show significant decreases in roscovitine-sensitive kinase activity by 24 h of PXD101 treatment in all three resistant cell lines (Fig. 6A and B). By 48 h, when G1 arrest is achieved, kinase activity declined by at least 75% relative to untreated cells (Fig. 6B). In contrast, cyclin E/cdk2 activity increases significantly in DB cells by 24 h treatment. We did not measure activity at 48 h due to high levels of apoptosis.
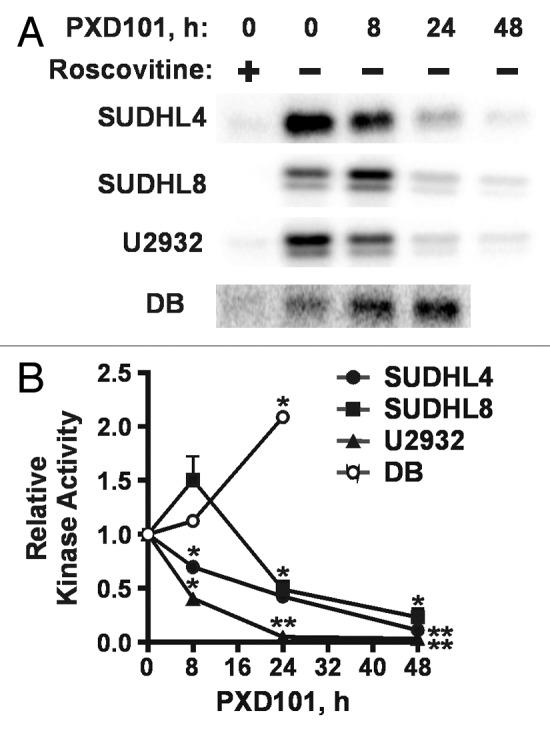
Figure 6. Differential effects of PXD101 on activity of cyclin E/cdk2. Whole cell extracts from cells treated with PXD101 for 0, 8, 24, and 48 h were used to perform immunoprecipitation with cyclin E antibody. (A) Bound fractions were subjected to kinase assay with purified histone H1 as substrate. Roscovitine was added to one sample to ensure that the labeling detected was due to the activity of cyclin-dependent kinases. Following SDS-PAGE, gels were dried and exposed to phosphorimaging screens to visualize radiolabeled protein. Representative results from 3 independent experiments in each cell line. (B) Graphical summary of results from 3–4 independent replicates of IP-kinase assays. (*P < 0.05, **P < 0.01).
The activity of the cyclin E/cdk2 complex is inhibited by association with the CIP/KIP family of cyclin-dependent kinase inhibitors (CKI), including p21, p27, and p57.36 HDACi have been shown to upregulate the expression of p21 in a wide variety of cell types, and there are several reports of increased p57 expression.35,37 Therefore, we treated cells for up to 72 h and monitored levels of the CKI by western blot. Levels of p57 were unchanged in all of the cell lines (not shown), but p21 and p27 levels were increased by PXD101 (Fig. 7A and B). Interestingly, in the cell lines sensitive to PXD101 p21 was only transiently upregulated (Fig. 7B), peaking at 6–8 h treatment in OCI-Ly19 and at 24 h in DB before returning to baseline levels. Levels of p27 were unaffected by PXD101 in OCI-Ly19 cells while in DB cells p27 levels increased at 72 h treatment, after the onset of apoptosis (Fig. 1A). In contrast, p21 was upregulated in a sustained fashion in the PXD101-resistant cell lines (Fig. 7A). p21 levels were increased by 6 h in SUDHL4 and U2932 cells and elevated levels were maintained out to 72 h. SUDHL8 cells do not express detectable levels of p21. Levels of p27 increased significantly in all the PXD101-resistant cell lines by 24 h treatment.

Figure 7. PXD101 effects on expression of cyclin-dependent kinase inhibitors (CKI) p21 and p27 and modification of p53. (A and B) The cell lines shown were treated with PXD101 for 0, 2, 4, 8, 24, 48, and 72 h. Whole cell extracts were generated and subjected to western blotting with antibodies against p21, p27, hsp90, α-tubulin, or GAPDH. PXD101-resistant cell lines are shown in (A) while PXD101-sensitive cell lines are shown in (B). (C and D) Cells were treated with PXD101 (P), doxorubicin (2 μM) (D), or Nutlin-3A (10 or 20 μM) (N) for 8 or 24 h. Whole cell extracts from treated and untreated cells were generated and subjected to western blotting with the antibodies indicated. PXD101-resistant cell lines are shown in (C) while PXD101-sensitive cell lines are shown in (D). The blots shown are representative of 3 independent experiments.
Upregulation of p21 by HDACi has been found in other cell types to be either p53-independent31,38,39 or p53-dependent.40 While HDACi are not directly genotoxic, they can induce a DNA damage response in some cell types by multiple mechanisms (reviewed in ref. 41). To determine whether PXD101 activates p53 in any of the DLBCL cell lines, we evaluated changes in p53 levels and modification status and assayed p53 function. To examine p53 modifications we treated cells with PXD101, doxorubicin, or Nutlin-3A. Doxorubicin is a known genotoxic agent that induces p53 signaling while Nutlin-3A, which disrupts the p53-MDM2 interaction,42 is a non-genotoxic inducer of p53. A recent report showed that the HDACi, romidepsin, induced p53 phosphorylation at Thr18,40 which is an indicator of an activated DNA damage response.43 However, as shown in Figure 7C and D, PXD101 does not increase Thr18 phosphorylation in any of the DLBCL cell lines tested. As expected, doxorubicin causes a robust increase in Thr18 phosphorylation while Nutlin-3A has little to no effect. PXD101 also does not increase p53 acetylation with the exception of DB cells where we observed a small, but significant increase in p53 acetylation. This does not strictly correlate with PXD101 sensitivity because p53 acetylation did not change in OCI-Ly19 cells (Fig. 7D). In contrast, doxorubicin treatment resulted in clear increases in p53 acetylation in all cell lines, while Nutlin3A upregulated it in SUDHL4, DB, and OCI-ly19 cells. Total p53 levels either did not change (SUDHL8, DB) or declined (SUDHL4, U2932, and OCI-ly19) in response to PXD101 treatment, but generally increased in response to doxorubicin treatment. We assayed p53 function by measuring expression of its downstream target, PUMA. Figure S4 shows that PUMA levels increase in response to doxorubicin, as expected, but decrease in response to PXD101 treatment in SUDHL4 and OCI-Ly19 cells, both of which express wt p53 (Table S1). Altogether the data indicate that p53 is not activated by PXD101 in these DLBCL cell lines and thus, p21 upregulation is p53-independent.
To determine whether the sustained upregulation of p21 and p27 observed in PXD101-resistant cell lines resulted in increased association with the cyclin E/cdk2 complex, we performed co-IPs in parallel with the IP-kinase assays shown in Figure 6 to measure the presence of the CKI in the complex. In the SUDHL4 and U2932 cell lines, we observed increased levels of p21 in the cyclin E/cdk2 complex at 24 and 48 h PXD101 treatment (Fig. 8). Accordingly, in SUDHL8 cells, p27 association with the complex increased by 24 h and was highest at 48 h treatment. In contrast, the association of p21 with cyclin E/cdk2 in DB cells did not change over 24 h of treatment in spite of the fact that p21 levels are significantly elevated 24 h after PXD101 exposure. It is noteworthy that the amounts of cyclin E and cdk2 immunoprecipitated do not decrease over the course of the treatment period in any of the cell lines. Altogether the results strongly indicate that sustained upregulation of the CKIs plays a role in the PXD101-induced G1 arrest through association with cyclin E/cdk2 and subsequent inhibition of its kinase activity.
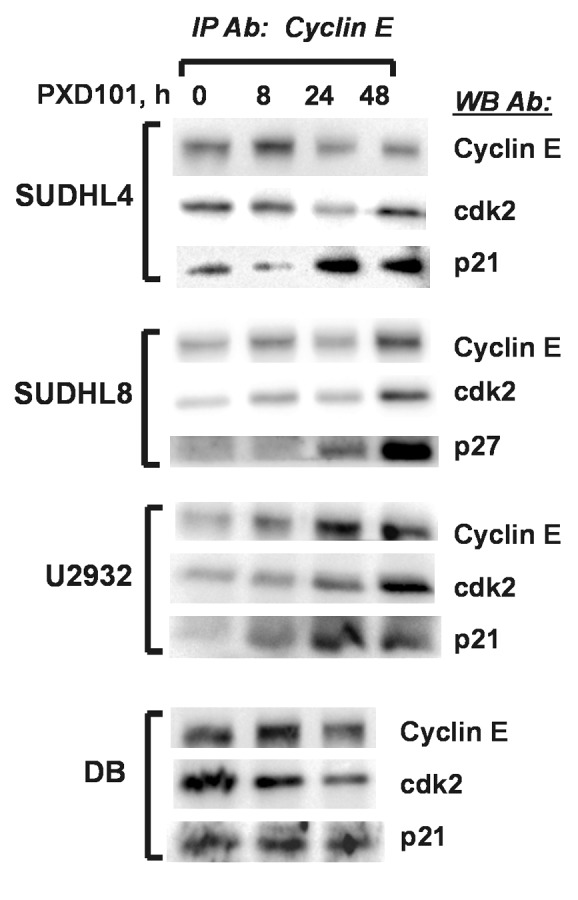
Figure 8. PXD101 induces association of CKI with cyclin E/cdk2 in resistant DLBCL cell lines. Whole cell extracts from cells treated with PXD101 for 0, 8, 24, and 48 h were used to perform immunoprecipitation with cyclin E antibody. Bound fractions were subjected to western blotting with antibodies against cyclin E and cdk2, and either p21 or p27. The results shown are representative of 3–4 independent experiments.
Discussion
In clinical trials evaluating HDACi as single agents, their action against relapsed DLBCL was found to be variable and mostly ineffective. However, since HDACi are generally well-tolerated and show selectivity toward tumor cells, there is interest in combining them with other therapeutics to increase effectiveness against DLBCL.17,18,44 Rational selection of companion drugs is challenging because the mechanistic bases of resistance to HDACi exhibited by DLBCL tumors are unknown and no models have been developed to study them. With the current study we have progressed toward defining the determinants of resistance by establishing that resistance and sensitivity to a prototypical hydroxamate HDACi can be modeled in DLBCL cell lines which express genes significantly associated with poor prognosis in DLBCL patients. We then used this model system to reveal key differences in the response of sensitive and resistant cell lines to HDACi that impact cell cycle progression and may modulate cellular sensitivity to the pro-apoptotic effects of these drugs in DLBCL.
A variety of DLBCL cell lines have been found to be growth-inhibited by HDACi,17,18,45-47 but it is not always clear whether this is due to cytotoxicity or to cell cycle arrest. We found that DLBCL cell lines did not respond uniformly to treatment with PXD101, having either a cytotoxic or cytostatic response. These responses did not correlate with DLBCL subtype (GCB vs. ABC) or the presence of the chromosomal translocation which deregulates BCL2 expression. In addition, no correlation was observed with co-expression of MYC and BCL2 proteins, relative basal levels of these proteins, or their regulation by PXD101.
The cytostatic response to PXD101 was characterized uniformly by G1 arrest with little apoptosis. Because we found that it was fully reversible if PXD101 was removed, we contend that it is a form of resistance. In the clinic HDACi are generally administered in cycles with patients being exposed to a single dose for several days at a time followed by a period of recovery prior to the next cycle.15,16 Thus, during the time of treatment, resistant tumor cells may cease proliferating through G1 arrest and then re-enter the cell cycle once treatment has stopped. This idea is supported by a recent study using the Eμ-myc model of B cell lymphoma which concluded that the pan-HDACi vorinostat was most effective at prolonging survival in mice with Eμ-myc tumors when it induced tumor cells to undergo apoptosis.22 Overexpression of Bcl-2 or Bcl-XL in these tumors caused G1 cell cycle arrest in response to vorinostat treatment rather than apoptosis. In mice with such tumors vorinostat did not prolong survival. Although we did not find a correlation between PXD101 resistance and BCL2 expression in the DLBCL cell lines, these results strongly indicate that G1 cell cycle arrest can be a form of resistance to HDACi in B cell lymphomas.
HDACi-induced G1 arrest has been observed in other cell types and is generally thought to be protective against the apoptotic effects of these drugs.48 Interestingly, the mechanisms by which HDACi have been reported to induce G1 arrest appear to be cell type-specific. In some cell types upregulation of CKI (p21, p57, and p16)10,30,49,50 or downregulation of cyclin D131,51,52 have been shown to mediate HDACi-induced G1 arrest while in others the arrest is p21- and p53-independent.53 HDACi have also been shown to induce senescence.53,54 In the DLBCL cell lines tested, analysis of proteins that are key regulators of G1 progression showed that PXD101 had dramatic effects on Rb protein. Levels of total Rb were greatly decreased in response to PXD101 treatment in both sensitive and resistant cell lines by post-transcriptional mechanisms. To our knowledge this has not been documented in other cell types. Rb can be acetylated55,56 but its functions may be context-specific. Rb acetylation is required for differentiation in muscle- and keratinocyte-derived cell lines but has no impact on its cell cycle functions.56,57 Rb acetylation has also been shown to inhibit its phosphorylation55,58 and interaction with E2F1 in response to DNA damage signaling.58 Its role in regulating Rb turnover, if any, is unknown.
In agreement with reports of HDACi effects in other cell types,29-32 PXD101 induced dramatic loss of Rb phosphorylation in the Rb-expressing DLBCL cell lines tested. However, decreased Rb phosphorylation may have distinct causes and functions in the sensitive and resistant cell lines. Caspase-induced Rb degradation has been found to make an important contribution to the induction of apoptosis under certain circumstances including exposure of cells to cytotoxic drugs.59 Rb dephosphorylation by protein phosphatase 1 (PP1) in response to pro-apoptotic stimuli precedes the onset of apoptosis and has the effect of making Rb a more efficient substrate of caspase-3.60,61 Importantly, this loss of Rb phosphorylation occurred with no decrease in the activity of cyclin E- or A-cdk complexes. This is consistent with our analysis of DB cells, in which Rb phosphorylation decreases over a time frame in which cyclin E/cdk2 activity increases (Figs. 5A and 6B). Thus, in the cell lines which have a cytotoxic response to PXD101, loss of Rb phosphorylation may be due to dephosphorylation in preparation for programmed cell death.
In the cell lines which undergo a cytostatic response to PXD101, we observe rapid and dramatic loss of Rb phosphorylation at both S780 and S795 (Fig. 5E and F). Since levels of the D cyclins are not strongly affected by PXD101 treatment (Fig. S2), the rapid decline in S780 phosphorylation is likely due to impaired activity of cyclin D complexes in resistant cells. Rb phosphorylation decreased by 80% over 48 h of PXD101 treatment in SUDHL4 and SUDHL8 cells (Fig. 5E and F), consistent with a decrease in cyclin E/cdk2 activity of a similar magnitude in all of the PXD101-resistant cell lines (Fig. 6B). Since this is the same time period over which G1 arrest is established (Fig. 2), we conclude that impaired activity of both cyclin D- and cyclin E-containing cdk complexes plays a key role in causing G1 arrest. In SUDHL4 and SUDHL8 cells this dual impairment results in the accumulation of hypophosphorylated, active Rb. In U2932 cells, which lack Rb expression, other substrates of these cdk complexes may contribute to the establishment of G1 arrest. It is noteworthy that PXD101 induces a higher amount of apoptosis in U2932 cells than in the SUDHL4 or SUDHL8 cells. One potential explanation is that the lack of Rb expression makes for a less efficient G1 arrest, thereby allowing a small fraction of cells to escape G1 and proceed to G2/M, where HDACi are known to induce mitotic aberrations that cause apoptosis.62
The CKIs p21 and p27 can directly interact with cyclin D and cyclin E-containing cdk complexes to regulate their activity. The functional effect of p21 or p27 association with cyclin D-containing complexes is complicated; their interaction can be inhibitory or stimulatory to complex activity depending on variables such as stoichiometry or phosphorylation.63-65 However, their association with the cyclin E/cdk2 complex is only inhibitory. Consistent with other reports,35,37 we found that PXD101 treatment strongly induced p21 expression within 24 h in all the cell lines except SUDHL8, which have no detectable expression of the protein. In addition, the p21 upregulation was p53-independent because we found little evidence of p53 activation by PXD101 in any of the cell lines. One major difference between the sensitive and resistant cell lines was in the kinetics of PXD101-induced p21 expression. In the sensitive lines, expression of p21 protein was transient but in the PXD101-resistant lines it was sustained. Studies of the mechanism of p21 upregulation by HDACi have focused exclusively on the initial activation of transcription (reviewed in ref. 66) so it is unclear why p21 mRNA levels decline in the PXD101-sensitive cell lines. Surprisingly, p27 expression was induced in all the resistant lines by 24 h PXD101 treatment. In concordance with the PXD101-induced loss of cyclin E/cdk2 activity in resistant cell lines, increased association of p21 (SUDHL4 and U2932) or p27 (SUDHL8) with this complex was detectable by 24 h treatment. Importantly, p21 association with the cyclin E/cdk2 complex in the PXD101-sensitive line, DB, was unaffected by PXD101 treatment over a 24 h period, consistent with both the lack of cyclin E/cdk2 inhibition and G1 arrest in DB cells. Altogether we contend that the sustained upregulation of p21 and/or p27 leads to increased association with the cyclin E/cdk2 complex to inhibit it from promoting G1 progression in PXD101-resistant DLBCL cell lines.
The cytotoxic response to PXD101 was characterized by G2/M arrest followed by apoptosis and did not require continuous exposure to drug. Other cancer cell lines have been shown to undergo G2/M arrest in response to HDACi.62 It has been shown that HDACi can induce the spindle assembly checkpoint to cause an M phase arrest. However, in the continued presence of the drugs, the checkpoint fails to be maintained and the cells exit mitosis without properly partitioning their genomes (see ref. 67 and references therein). This is thought to be a major mechanism by which HDACi cause cytotoxicity. It is noteworthy that the PXD101-resistant cell lines do not arrest in G2/M. Expression of wild-type (wt) p53 has been shown to sensitize cells to paclitaxel, a drug which, like HDACi, can induce arrest in G2/M through activation of the spindle assembly checkpoint and cause subsequent apoptosis.68 Thus, the mutational status of p53 could distinguish between cell lines that are sensitive or resistant to PXD101. However, this does not appear to be the case because PXD101-sensitive cell lines differ in their p53 status (Table S1). OCI-Ly19 cells have wt p53 while DB cells express mutant p53.69 The PXD101-resistant cells are also distributed between wt and mutant p53. Expression profiling studies are underway to identify gene expression patterns linked to G2/M arrest and apoptosis in an effort to understand the underlying mechanisms. In addition, genes uniquely expressed in the sensitive cell lines may serve as predictive biomarkers of HDACi sensitivity in DLBCL. Currently only one such marker has been identified in CTCL70; more are needed.
There are several markers that predict poor prognosis in DLBCL patients after treatment with R-CHOP, including tumors of the ABC subtype, the presence of the t(14;18) BCL2 translocation (specifically in tumors of GCB origin),71 co-expression of MYC and BCL2,5,6 and the expression of mutant p53.7,8 It is significant that PXD101 is effective against cell lines with translocated BCL2 and co-expression of MYC and BCL2 proteins (double-positive), regardless of their p53 status. This strongly suggests that HDACi have excellent potential to be used effectively against relapsed DLBCL in combination with other therapeutics that circumvent mechanisms of HDACi resistance.
Since our study strongly indicates that sustained upregulation of p21 or p27 leads to G1 arrest and PXD101 resistance, HDACi might be combined effectively with therapeutics that target p21 or p27 expression in DLBCL. In fact, the cdk inhibitor, flavopiridol, has been found to prevent HDACi-induced p21 expression and promote cell death in leukemia cell lines.72 In addition, HDACi might also be combined successfully with chemicals that induce the spindle assembly checkpoint, such as vincristine or paclitaxel.73 Because they do not arrest in G2/M it appears that PXD101 does not induce this checkpoint in the resistant cells.74,75 However, in combination with drugs that can, PXD101 may cause the failure of the checkpoint and induce apoptosis.76 The model system we have developed will be used to test these drug combinations as well as to further define mechanisms of sensitivity and resistance that may reveal additional therapeutics which could be used effectively with HDACi against aggressive DLBCL tumors.
Materials and Methods
Cell lines and reagents
Seven DLBCL-derived cell lines were cultured in RPMI medium supplemented with1 10% FBS and gentamicin (SUDHL4, SUDHL6, and U2932),2 15% FBS, 1mM sodium pyruvate, and gentamicin (DB and OCILY3),3 10% FBS, 1 mM sodium pyruvate and gentamicin (OCILY19),4 or 10% FBS, 1 mM glutamine, and gentamicin (SUDHL8). All cell lines were maintained at 37 °C in a humidified atmosphere containing 5% CO2. Prior to every experiment cells were seeded at 2 × 105 to 4 × 105cells/ml with a 50/50 mixture of conditioned and fresh medium.
PXD101 was obtained from the Cancer Therapy Evaluation Program (National Cancer Institute). Antibodies against Bcl-2 (50E3), MYC (D84C12), cyclin D3 (DCS22), cyclin D1 (92G2), p21, p27, p57, Rb (4H1), phospho-Rb (Ser 780), phospho-Rb (Ser 795), cyclin E (HE12), phospho-p53 (Thr18), and α-tubulin were obtained from Cell Signaling Technology, Inc. Underphosphorylated Rb antibody was purchased from BD Biosciences. GAPDH (FL-335), cdk2, cyclin E (C-19), p53 (DO-1), and acetylated p53 (Lys382) antibodies were acquired from Santa Cruz Biotechnology, Inc. PUMA antibody was purchased from Abcam.
Cell growth, cell cycle, and cell death assays
Cell growth was measured by MTS assay according to manufacturer’s specifications (Cell titer 96® Aqueous One Solution, Promega). Cells were seeded in 96-well plates and treated in triplicate with a wide range of PXD101 doses. IC50s for growth inhibition were calculated using curve-fitting software (GraphPad Prism). The IC50 concentrations determined at 24 h PXD101 treatment for each cell line were used in all subsequent experiments.
Apoptosis and cell cycle phase distribution were estimated from flow cytometric data using ModFit LT 3.0 (Verity Software House). Cells were treated either with DMSO or PXD101. After treatment, cells were washed once with cold 1X Dulbecco’s modified PBS (D-PBS); 1 × 106 cells were used to measure Annexin V positive cells according to manufacturer’s specifications (Annexin V-FITC kit, ENZO Life Sciences). For analysis of cell cycle distribution, cells were fixed with 70% ethanol, centrifuged at 500× g for 15 min. Ethanol was removed and 0.5 ml of cold PBS, 20 μl of propidium iodide and 12.5 μl of RNase A (20 mg/ml) were added. Samples were incubated at 37 °C for 30 min prior to flow cytometry.
Western blotting
After treatment, cells were washed once with cold D-PBS. Cell pellets were flash-frozen in liquid nitrogen and kept at −80 °C until use. Whole cell extracts were generated by resuspending cell pellets in RIPA buffer (150 mM sodium chloride, 1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 50 mM Tris, pH 8.0, protease inhibitor cocktail (Roche) and phosphatase inhibitors [10 mM NaF, 25 mM β-glycerophosphate]). Resuspended pellets were transferred into Qiashredder columns (Qiagen) and centrifuged at 12 000× g for 5 min. Supernatants were collected and protein concentration was determined by Bradford method. For immunoblotting 100 μg of protein from each sample were separated by SDS-PAGE gel electrophoresis. Resolved proteins were transferred to 0.45 μm nitrocellulose membranes and subjected to immunoblotting.
RNA analysis
Total RNA was isolated from DLBCL cells treated with PXD101 or vehicle (DMSO) by the following method. Cells were washed once with D-PBS, pelleted, and mixed with Trizol (Invitrogen). Chloroform was added and the mixtures were vortexed for 15 s. Separation of aqueous and organic phases was achieved by pre-incubation at room temperature for 3 min followed by centrifugation at 12 000 × g. The aqueous phase was transferred to a clean tube and slowly mixed with an equal volume of 70% ethanol. Samples were then applied to spin-columns and RNA was isolated according to manufacturer’s instructions (Nucleospin II, Clontech).
Synthesis of cDNA was performed using the iScript kit (BioRad). Measurement of gene expression was performed using quantitative PCR with 2× SyBr green buffer (Bioline) and gene-specific primers as per manufacturer’s instructions in a StepOnePlus instrument (ABI/Life Technologies). Gene-specific primer pairs were derived from PrimerBank (Harvard, http://pga.mgh.harvard.edu/primerbank/) and purchased from Sigma-Aldrich. Primer sequences are as follows: BCL2 forward, 5′-GAACTGGGGG AGGATTGTGG-3′, reverse, 5′-CCGGTTCAGG TACTCAGTCA; MYC forward, 5′-CCACAGCAAA CCTCCTCACA G-3′, reverse, 5′-GCAGGATAGT CCTTCCGAGT G-3′; Rb forward, 5′-TTGGATCACA GCGATACAAA CTT-3′, reverse, 5′-AGCGCACGCC AATAAAGACA T-3′; p21 forward, 5′-GTCACTGTCT TGTACCCTTG TG-3′, reverse, 5′-CGGCGTTTGG AGTGGTAGAA A-3′; GAPDH forward, 5′-CATGAGAAGT ATGACAACAG CCT-3′, reverse, 5′-AGTCCTTCCA CGATACCAAA GT-3′. For drug treatment studies the relative concentrations of mRNAs were calculated by the ΔΔCt method using GAPDH and HPRT1 as reference housekeeping genes. Relative expression of a particular gene between cell lines was also calculated using the ΔΔCt method except that one replicate from one cell line was used for normalization purposes (ΔCt).
Immunoprecipitation and kinase assay
DLBCL cells were treated with PXD101 or vehicle (DMSO). After treatment, cells were washed once with cold D-PBS. Washed cell pellets were flash-frozen in liquid nitrogen and kept at −80 °C until use. Whole cell extracts were generated by resuspending cell pellets in lysis buffer (50 mM HEPES pH 7.3, 250 mM NaCl, 1 mM EDTA, 2.5 mM EGTA, 0.1% Tween-20, 10% glycerol, 1 mM DTT, protease inhibitors [1× Complete, Roche] and phosphatase inhibitors [5 M NaF, 25 mM β-glycerophosphate, and 0.1 mM sodium orthovanadate]). After 30 min incubation on ice, cell debris was removed by centrifugation and protein concentration was determined by Bradford assay. For immunoprecipitations, 300 μg (kinase assays) or 700 μg (co-immunoprecipitations) extract protein was diluted to 0.5 ml in wash buffer C (50 mM Hepes pH 7.3, 1 mM DTT, protease and phosphatase inhibitors). Samples were pre-cleared for 1 h at 4 °C with Gammabind G sepharose (GE Healthcare Life Sciences) and exposed to either 2 μg anti-cyclin E (C-19, Santa Cruz Biotechnology) or 2 μg anti-GFP (3E6, Invitrogen) along with Gammabind G sepharose for 3 h at 4 °C with rotation. Beads were pelleted by gentle centrifugation and washed as follows: 1× with wash buffer A (50 mM Hepes pH 7.3, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 1.0% Triton X-100, 10% glycerol, 1 mM DTT, protease and phosphatase inhibitors), 2× with wash buffer B (50 mM Hepes pH 7.3, 1 M NaCl, 1.5 mM MgCl2, 1 mM EGTA, 1.0% Triton X-100, 10% glycerol, 1 mM DTT, protease and phosphatase inhibitors), and 3× with wash buffer C.
For kinase assays washed immunoprecipitates were suspended in kinase buffer (50 mM Hepes pH 7.3, 1 mM DTT, 10 mM MgCl2, 10 μM ATP, phosphatase inhibitors) containing 5 μg purified Histone H1 (SignalChem) and 10 μCi 32P-γ-ATP (Amersham). Roscovitine at a final concentration of 5 μM was added to an immunoprecipitate derived from untreated cells to ensure that the activity measured was derived from a cyclin-dependent kinase. Samples were then incubated at 30 °C for 30 min with vortexing every 5 min. Reactions were terminated by adding 5× SDS-PAGE buffer (0.2 M Tris, pH 6.8, 10% SDS, 50% glycerol, 0.25 M DTT, 0.01% Bromophenol Blue). After SDS-PAGE, gels were dried for 1 h at 80 °C under vacuum. Radioactive labeling was visualized using a Pharos Fx Plus Molecular Imager (BioRad) and quantitated using ImageLab software (BioRad).
For co-immunoprecipitations, washed immunoprecipitates were resuspended in 2× SDS-PAGE buffer (80 mM Tris pH 6.8, 4% SDS, 20% glycerol, 0.1 M DTT, 0.004% Bromophenol Blue) and heated to 95 °C for 5 min. Supernatants were resolved with SDS-PAGE. Following western transfer, membranes were incubated with antibodies against the following: cyclin E (HE12), cdk2, and p21 or p27.
Supplementary Material
Acknowledgments
The authors would like to acknowledge the staff of the Flow Cytometry Shared Service at the Arizona Cancer Center for services rendered. We also thank Dr Tracy Brooks (U Mississippi) for assistance with MTS assays and analysis of MYC expression. We are grateful to members of the Lymphoma Research Group at the UA for helpful suggestions and critique during the progress of the study. This work was supported by a pilot project grant from the Southwest Environmental Health Sciences Center (ES006694, PI S Lau) to CLS, an award from the Arizona Biomedical Research Commission to CLS, and a career development award and developmental project award to CLS (Lymphoma SPORE [1 P50 CA-130805-05, PI RI Fisher]).
Glossary
Abbreviations:
- DLBCL
diffuse large B cell lymphoma
- HDAC
histone deacetylase
- HDACi
histone deacetylase inhibitor
- GCB
germinal center B cell-like
- ABC
activated B cell-like
- NHL
non-Hodgkin lymphoma
- BCL2
B cell CLL/lymphoma 2
- MYC
c-myc
- Rb
retinoblastoma protein
- cdk2
cyclin-dependent kinase 2
- CKI
cyclin-dependent kinase inhibitor
- SAHA
suberoylanilide hydroxamic acid
Disclosure of Potential Conflicts of Interest
CLS and LMR receive financial support for basic research studies from Spectrum Pharmaceuticals which holds the commercial license for belinostat (PXD101). However, none of this money was used to support the work reported in the accompanying manuscript. Spectrum Pharmaceuticals had no influence on the study goals, execution, or preparation of the manuscript nor did they provide the drug used in the study.
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/25941
References
- 1.Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–11. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 2.Coiffier B, Thieblemont C, Van Den Neste E, Lepeu G, Plantier I, Castaigne S, Lefort S, Marit G, Macro M, Sebban C, et al. Long-term outcome of patients in the LNH-98.5 trial, the first randomized study comparing rituximab-CHOP to standard CHOP chemotherapy in DLBCL patients: a study by the Groupe d’Etudes des Lymphomes de l’Adulte. Blood. 2010;116:2040–5. doi: 10.1182/blood-2010-03-276246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feugier P, Van Hoof A, Sebban C, Solal-Celigny P, Bouabdallah R, Fermé C, Christian B, Lepage E, Tilly H, Morschhauser F, et al. Long-term results of the R-CHOP study in the treatment of elderly patients with diffuse large B-cell lymphoma: a study by the Groupe d’Etude des Lymphomes de l’Adulte. J Clin Oncol. 2005;23:4117–26. doi: 10.1200/JCO.2005.09.131. [DOI] [PubMed] [Google Scholar]
- 4.Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI, Gascoyne RD, Muller-Hermelink HK, Smeland EB, Giltnane JM, et al. Lymphoma/Leukemia Molecular Profiling Project The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med. 2002;346:1937–47. doi: 10.1056/NEJMoa012914. [DOI] [PubMed] [Google Scholar]
- 5.Johnson NA, Slack GW, Savage KJ, Connors JM, Ben-Neriah S, Rogic S, Scott DW, Tan KL, Steidl C, Sehn LH, et al. Concurrent expression of MYC and BCL2 in diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol. 2012;30:3452–9. doi: 10.1200/JCO.2011.41.0985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hu S, Xu-Monette ZY, Tzankov A, Green T, Wu L, Balasubramanyam A, Liu WM, Visco C, Li Y, Miranda RN, et al. MYC/BCL2 protein coexpression contributes to the inferior survival of activated B-cell subtype of diffuse large B-cell lymphoma and demonstrates high-risk gene expression signatures: a report from The International DLBCL Rituximab-CHOP Consortium Program. Blood. 2013;121:4021–31, quiz 4250. doi: 10.1182/blood-2012-10-460063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stefancikova L, Moulis M, Fabian P, Vasova I, Zedek F, Ravcukova B, Muzik J, Kuglik P, Vranova V, Falkova I, et al. Prognostic impact of p53 aberrations for R-CHOP-treated patients with diffuse large B-cell lymphoma. Int J Oncol. 2011;39:1413–20. doi: 10.3892/ijo.2011.1170. [DOI] [PubMed] [Google Scholar]
- 8.Xu-Monette ZY, Wu L, Visco C, Tai YC, Tzankov A, Liu WM, Montes-Moreno S, Dybkaer K, Chiu A, Orazi A, et al. Mutational profile and prognostic significance of TP53 in diffuse large B-cell lymphoma patients treated with R-CHOP: report from an International DLBCL Rituximab-CHOP Consortium Program Study. Blood. 2012;120:3986–96. doi: 10.1182/blood-2012-05-433334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene. 2007;26:5541–52. doi: 10.1038/sj.onc.1210620. [DOI] [PubMed] [Google Scholar]
- 10.Yamaguchi T, Cubizolles F, Zhang Y, Reichert N, Kohler H, Seiser C, Matthias P. Histone deacetylases 1 and 2 act in concert to promote the G1-to-S progression. Genes Dev. 2010;24:455–69. doi: 10.1101/gad.552310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marquard L, Poulsen CB, Gjerdrum LM, de Nully Brown P, Christensen IJ, Jensen PB, Sehested M, Johansen P, Ralfkiaer E. Histone deacetylase 1, 2, 6 and acetylated histone H4 in B- and T-cell lymphomas. Histopathology. 2009;54:688–98. doi: 10.1111/j.1365-2559.2009.03290.x. [DOI] [PubMed] [Google Scholar]
- 12.Gloghini A, Buglio D, Khaskhely NM, Georgakis G, Orlowski RZ, Neelapu SS, Carbone A, Younes A. Expression of histone deacetylases in lymphoma: implication for the development of selective inhibitors. Br J Haematol. 2009;147:515–25. doi: 10.1111/j.1365-2141.2009.07887.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kelly WK, O’Connor OA, Krug LM, Chiao JH, Heaney M, Curley T, MacGregore-Cortelli B, Tong W, Secrist JP, Schwartz L, et al. Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. J Clin Oncol. 2005;23:3923–31. doi: 10.1200/JCO.2005.14.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’Connor OA, Heaney ML, Schwartz L, Richardson S, Willim R, MacGregor-Cortelli B, Curly T, Moskowitz C, Portlock C, Horwitz S, et al. Clinical experience with intravenous and oral formulations of the novel histone deacetylase inhibitor suberoylanilide hydroxamic acid in patients with advanced hematologic malignancies. J Clin Oncol. 2006;24:166–73. doi: 10.1200/JCO.2005.01.9679. [DOI] [PubMed] [Google Scholar]
- 15.Crump M, Coiffier B, Jacobsen ED, Sun L, Ricker JL, Xie H, Frankel SR, Randolph SS, Cheson BD. Phase II trial of oral vorinostat (suberoylanilide hydroxamic acid) in relapsed diffuse large-B-cell lymphoma. Ann Oncol. 2008;19:964–9. doi: 10.1093/annonc/mdn031. [DOI] [PubMed] [Google Scholar]
- 16.Gimsing P, Hansen M, Knudsen LM, Knoblauch P, Christensen IJ, Ooi CE, Buhl-Jensen P. A phase I clinical trial of the histone deacetylase inhibitor belinostat in patients with advanced hematological neoplasia. Eur J Haematol. 2008;81:170–6. doi: 10.1111/j.1600-0609.2008.01102.x. [DOI] [PubMed] [Google Scholar]
- 17.Dasmahapatra G, Lembersky D, Kramer L, Fisher RI, Friedberg J, Dent P, Grant S. The pan-HDAC inhibitor vorinostat potentiates the activity of the proteasome inhibitor carfilzomib in human DLBCL cells in vitro and in vivo. Blood. 2010;115:4478–87. doi: 10.1182/blood-2009-12-257261. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 18.Kalac M, Scotto L, Marchi E, Amengual J, Seshan VE, Bhagat G, Ulahannan N, Leshchenko VV, Temkin AM, Parekh S, et al. HDAC inhibitors and decitabine are highly synergistic and associated with unique gene-expression and epigenetic profiles in models of DLBCL. Blood. 2011;118:5506–16. doi: 10.1182/blood-2011-02-336891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Polack A, Eick D, Koch E, Bornkamm GW. Truncation does not abrogate transcriptional downregulation of the c-myc gene by sodium butyrate in Burkitt’s lymphoma cells. EMBO J. 1987;6:2959–64. doi: 10.1002/j.1460-2075.1987.tb02601.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Lint C, Emiliani S, Verdin E. The expression of a small fraction of cellular genes is changed in response to histone hyperacetylation. Gene Expr. 1996;5:245–53. [PMC free article] [PubMed] [Google Scholar]
- 21.Duan H, Heckman CA, Boxer LM. Histone deacetylase inhibitors down-regulate bcl-2 expression and induce apoptosis in t(14;18) lymphomas. Mol Cell Biol. 2005;25:1608–19. doi: 10.1128/MCB.25.5.1608-1619.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lindemann RK, Newbold A, Whitecross KF, Cluse LA, Frew AJ, Ellis L, Williams S, Wiegmans AP, Dear AE, Scott CL, et al. Analysis of the apoptotic and therapeutic activities of histone deacetylase inhibitors by using a mouse model of B cell lymphoma. Proc Natl Acad Sci U S A. 2007;104:8071–6. doi: 10.1073/pnas.0702294104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deng X, Gao F, Flagg T, May WS., Jr. Mono- and multisite phosphorylation enhances Bcl2’s antiapoptotic function and inhibition of cell cycle entry functions. Proc Natl Acad Sci U S A. 2004;101:153–8. doi: 10.1073/pnas.2533920100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pasqualucci L, Trifonov V, Fabbri G, Ma J, Rossi D, Chiarenza A, Wells VA, Grunn A, Messina M, Elliot O, et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat Genet. 2011;43:830–7. doi: 10.1038/ng.892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schuetz JM, Johnson NA, Morin RD, Scott DW, Tan K, Ben-Nierah S, Boyle M, Slack GW, Marra MA, Connors JM, et al. BCL2 mutations in diffuse large B-cell lymphoma. Leukemia. 2012;26:1383–90. doi: 10.1038/leu.2011.378. [DOI] [PubMed] [Google Scholar]
- 26.Vervoorts J, Lüscher-Firzlaff JM, Rottmann S, Lilischkis R, Walsemann G, Dohmann K, Austen M, Lüscher B. Stimulation of c-MYC transcriptional activity and acetylation by recruitment of the cofactor CBP. EMBO Rep. 2003;4:484–90. doi: 10.1038/sj.embor.embor821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patel JH, Du Y, Ard PG, Phillips C, Carella B, Chen CJ, Rakowski C, Chatterjee C, Lieberman PM, Lane WS, et al. The c-MYC oncoprotein is a substrate of the acetyltransferases hGCN5/PCAF and TIP60. Mol Cell Biol. 2004;24:10826–34. doi: 10.1128/MCB.24.24.10826-10834.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Faiola F, Liu X, Lo S, Pan S, Zhang K, Lymar E, Farina A, Martinez E. Dual regulation of c-Myc by p300 via acetylation-dependent control of Myc protein turnover and coactivation of Myc-induced transcription. Mol Cell Biol. 2005;25:10220–34. doi: 10.1128/MCB.25.23.10220-10234.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mitsiades N, Mitsiades CS, Richardson PG, McMullan C, Poulaki V, Fanourakis G, Schlossman R, Chauhan D, Munshi NC, Hideshima T, et al. Molecular sequelae of histone deacetylase inhibition in human malignant B cells. Blood. 2003;101:4055–62. doi: 10.1182/blood-2002-11-3514. [DOI] [PubMed] [Google Scholar]
- 30.Vaziri C, Stice L, Faller DV. Butyrate-induced G1 arrest results from p21-independent disruption of retinoblastoma protein-mediated signals. Cell Growth Differ. 1998;9:465–74. [PubMed] [Google Scholar]
- 31.Sandor V, Senderowicz A, Mertins S, Sackett D, Sausville E, Blagosklonny MV, Bates SE. P21-dependent g(1)arrest with downregulation of cyclin D1 and upregulation of cyclin E by the histone deacetylase inhibitor FR901228. Br J Cancer. 2000;83:817–25. doi: 10.1054/bjoc.2000.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rosato RR, Almenara JA, Grant S. The histone deacetylase inhibitor MS-275 promotes differentiation or apoptosis in human leukemia cells through a process regulated by generation of reactive oxygen species and induction of p21CIP1/WAF1 1. Cancer Res. 2003;63:3637–45. [PubMed] [Google Scholar]
- 33.Zarkowska T, Mittnacht S. Differential phosphorylation of the retinoblastoma protein by G1/S cyclin-dependent kinases. J Biol Chem. 1997;272:12738–46. doi: 10.1074/jbc.272.19.12738. [DOI] [PubMed] [Google Scholar]
- 34.Kitagawa M, Higashi H, Jung HK, Suzuki-Takahashi I, Ikeda M, Tamai K, Kato J, Segawa K, Yoshida E, Nishimura S, et al. The consensus motif for phosphorylation by cyclin D1-Cdk4 is different from that for phosphorylation by cyclin A/E-Cdk2. EMBO J. 1996;15:7060–9. [PMC free article] [PubMed] [Google Scholar]
- 35.Seo JS, Cho NY, Kim HR, Tsurumi T, Jang YS, Lee WK, Lee SK. Cell cycle arrest and lytic induction of EBV-transformed B lymphoblastoid cells by a histone deacetylase inhibitor, Trichostatin A. Oncol Rep. 2008;19:93–8. [PubMed] [Google Scholar]
- 36.Besson A, Dowdy SF, Roberts JM. CDK inhibitors: cell cycle regulators and beyond. Dev Cell. 2008;14:159–69. doi: 10.1016/j.devcel.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 37.Shin JY, Kim HS, Park J, Park JB, Lee JY. Mechanism for inactivation of the KIP family cyclin-dependent kinase inhibitor genes in gastric cancer cells. Cancer Res. 2000;60:262–5. [PubMed] [Google Scholar]
- 38.Huang L, Sowa Y, Sakai T, Pardee AB. Activation of the p21WAF1/CIP1 promoter independent of p53 by the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) through the Sp1 sites. Oncogene. 2000;19:5712–9. doi: 10.1038/sj.onc.1203963. [DOI] [PubMed] [Google Scholar]
- 39.Nakano K, Mizuno T, Sowa Y, Orita T, Yoshino T, Okuyama Y, Fujita T, Ohtani-Fujita N, Matsukawa Y, Tokino T, et al. Butyrate activates the WAF1/Cip1 gene promoter through Sp1 sites in a p53-negative human colon cancer cell line. J Biol Chem. 1997;272:22199–206. doi: 10.1074/jbc.272.35.22199. [DOI] [PubMed] [Google Scholar]
- 40.Wang H, Zhou W, Zheng Z, Zhang P, Tu B, He Q, Zhu WG. The HDAC inhibitor depsipeptide transactivates the p53/p21 pathway by inducing DNA damage. DNA Repair (Amst) 2012;11:146–56. doi: 10.1016/j.dnarep.2011.10.014. [DOI] [PubMed] [Google Scholar]
- 41.Eot-Houllier G, Fulcrand G, Magnaghi-Jaulin L, Jaulin C. Histone deacetylase inhibitors and genomic instability. Cancer Lett. 2009;274:169–76. doi: 10.1016/j.canlet.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 42.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–8. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 43.Jenkins LM, Durell SR, Mazur SJ, Appella E. p53 N-terminal phosphorylation: a defining layer of complex regulation. Carcinogenesis. 2012;33:1441–9. doi: 10.1093/carcin/bgs145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bots M, Johnstone RW. Rational combinations using HDAC inhibitors. Clin Cancer Res. 2009;15:3970–7. doi: 10.1158/1078-0432.CCR-08-2786. [DOI] [PubMed] [Google Scholar]
- 45.Gupta M, Ansell SM, Novak AJ, Kumar S, Kaufmann SH, Witzig TE. Inhibition of histone deacetylase overcomes rapamycin-mediated resistance in diffuse large B-cell lymphoma by inhibiting Akt signaling through mTORC2. Blood. 2009;114:2926–35. doi: 10.1182/blood-2009-05-220889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bodo J, Sedlak J, Maciejewski JP, Almasan A, Hsi ED. HDAC inhibitors potentiate the apoptotic effect of enzastaurin in lymphoma cells. Apoptosis. 2011;16:914–23. doi: 10.1007/s10495-011-0617-x. [DOI] [PubMed] [Google Scholar]
- 47.Cerchietti LC, Hatzi K, Caldas-Lopes E, Yang SN, Figueroa ME, Morin RD, Hirst M, Mendez L, Shaknovich R, Cole PA, et al. BCL6 repression of EP300 in human diffuse large B cell lymphoma cells provides a basis for rational combinatorial therapy. J Clin Invest. 2010;120:4569–82. doi: 10.1172/JCI42869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Robey RW, Chakraborty AR, Basseville A, Luchenko V, Bahr J, Zhan Z, Bates SE. Histone deacetylase inhibitors: emerging mechanisms of resistance. Mol Pharm. 2011;8:2021–31. doi: 10.1021/mp200329f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zupkovitz G, Grausenburger R, Brunmeir R, Senese S, Tischler J, Jurkin J, Rembold M, Meunier D, Egger G, Lagger S, et al. The cyclin-dependent kinase inhibitor p21 is a crucial target for histone deacetylase 1 as a regulator of cellular proliferation. Mol Cell Biol. 2010;30:1171–81. doi: 10.1128/MCB.01500-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Munro J, Barr NI, Ireland H, Morrison V, Parkinson EK. Histone deacetylase inhibitors induce a senescence-like state in human cells by a p16-dependent mechanism that is independent of a mitotic clock. Exp Cell Res. 2004;295:525–38. doi: 10.1016/j.yexcr.2004.01.017. [DOI] [PubMed] [Google Scholar]
- 51.Alao JP, Stavropoulou AV, Lam EW, Coombes RC, Vigushin DM. Histone deacetylase inhibitor, trichostatin A induces ubiquitin-dependent cyclin D1 degradation in MCF-7 breast cancer cells. Mol Cancer. 2006;5:8. doi: 10.1186/1476-4598-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hu J, Colburn NH. Histone deacetylase inhibition down-regulates cyclin D1 transcription by inhibiting nuclear factor-kappaB/p65 DNA binding. Mol Cancer Res. 2005;3:100–9. doi: 10.1158/1541-7786.MCR-04-0070. [DOI] [PubMed] [Google Scholar]
- 53.Wilting RH, Yanover E, Heideman MR, Jacobs H, Horner J, van der Torre J, DePinho RA, Dannenberg JH. Overlapping functions of Hdac1 and Hdac2 in cell cycle regulation and haematopoiesis. EMBO J. 2010;29:2586–97. doi: 10.1038/emboj.2010.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ogryzko VV, Hirai TH, Russanova VR, Barbie DA, Howard BH. Human fibroblast commitment to a senescence-like state in response to histone deacetylase inhibitors is cell cycle dependent. Mol Cell Biol. 1996;16:5210–8. doi: 10.1128/mcb.16.9.5210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chan HM, Krstic-Demonacos M, Smith L, Demonacos C, La Thangue NB. Acetylation control of the retinoblastoma tumour-suppressor protein. Nat Cell Biol. 2001;3:667–74. doi: 10.1038/35083062. [DOI] [PubMed] [Google Scholar]
- 56.Nguyen DX, Baglia LA, Huang SM, Baker CM, McCance DJ. Acetylation regulates the differentiation-specific functions of the retinoblastoma protein. EMBO J. 2004;23:1609–18. doi: 10.1038/sj.emboj.7600176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pickard A, Wong PP, McCance DJ. Acetylation of Rb by PCAF is required for nuclear localization and keratinocyte differentiation. J Cell Sci. 2010;123:3718–26. doi: 10.1242/jcs.068924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Markham D, Munro S, Soloway J, O’Connor DP, La Thangue NB. DNA-damage-responsive acetylation of pRb regulates binding to E2F-1. EMBO Rep. 2006;7:192–8. doi: 10.1038/sj.embor.7400591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chau BN, Wang JY. Coordinated regulation of life and death by RB. Nat Rev Cancer. 2003;3:130–8. doi: 10.1038/nrc993. [DOI] [PubMed] [Google Scholar]
- 60.Dou QP, An B, Will PL. Induction of a retinoblastoma phosphatase activity by anticancer drugs accompanies p53-independent G1 arrest and apoptosis. Proc Natl Acad Sci U S A. 1995;92:9019–23. doi: 10.1073/pnas.92.20.9019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang RH, Liu CW, Avramis VI, Berndt N. Protein phosphatase 1alpha-mediated stimulation of apoptosis is associated with dephosphorylation of the retinoblastoma protein. Oncogene. 2001;20:6111–22. doi: 10.1038/sj.onc.1204829. [DOI] [PubMed] [Google Scholar]
- 62.Schrump DS. Cytotoxicity mediated by histone deacetylase inhibitors in cancer cells: mechanisms and potential clinical implications. Clin Cancer Res. 2009;15:3947–57. doi: 10.1158/1078-0432.CCR-08-2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.LaBaer J, Garrett MD, Stevenson LF, Slingerland JM, Sandhu C, Chou HS, Fattaey A, Harlow E. New functional activities for the p21 family of CDK inhibitors. Genes Dev. 1997;11:847–62. doi: 10.1101/gad.11.7.847. [DOI] [PubMed] [Google Scholar]
- 64.Zhang H, Hannon GJ, Beach D. p21-containing cyclin kinases exist in both active and inactive states. Genes Dev. 1994;8:1750–8. doi: 10.1101/gad.8.15.1750. [DOI] [PubMed] [Google Scholar]
- 65.Ray A, James MK, Larochelle S, Fisher RP, Blain SW. p27Kip1 inhibits cyclin D-cyclin-dependent kinase 4 by two independent modes. Mol Cell Biol. 2009;29:986–99. doi: 10.1128/MCB.00898-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ocker M, Schneider-Stock R. Histone deacetylase inhibitors: signalling towards p21cip1/waf1. Int J Biochem Cell Biol. 2007;39:1367–74. doi: 10.1016/j.biocel.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 67.Gabrielli B, Chia K, Warrener R. Finally, how histone deacetylase inhibitors disrupt mitosis! Cell Cycle. 2011;10:2658–61. doi: 10.4161/cc.10.16.16953. [DOI] [PubMed] [Google Scholar]
- 68.Wahl AF, Donaldson KL, Fairchild C, Lee FY, Foster SA, Demers GW, Galloway DA. Loss of normal p53 function confers sensitization to Taxol by increasing G2/M arrest and apoptosis. Nat Med. 1996;2:72–9. doi: 10.1038/nm0196-72. [DOI] [PubMed] [Google Scholar]
- 69.Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, Johnson NA, Severson TM, Chiu R, Field M, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476:298–303. doi: 10.1038/nature10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Khan O, Fotheringham S, Wood V, Stimson L, Zhang C, Pezzella F, Duvic M, Kerr DJ, La Thangue NB. HR23B is a biomarker for tumor sensitivity to HDAC inhibitor-based therapy. Proc Natl Acad Sci U S A. 2010;107:6532–7. doi: 10.1073/pnas.0913912107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Visco C, Tzankov A, Xu-Monette ZY, Miranda RN, Tai YC, Li Y, Liu WM, d’Amore ES, Li Y, Montes-Moreno S, et al. Patients with diffuse large B-cell lymphoma of germinal center origin with BCL2 translocations have poor outcome, irrespective of MYC status: a report from an International DLBCL rituximab-CHOP Consortium Program Study. Haematologica. 2013;98:255–63. doi: 10.3324/haematol.2012.066209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dasmahapatra G, Almenara JA, Grant S. Flavopiridol and histone deacetylase inhibitors promote mitochondrial injury and cell death in human leukemia cells that overexpress Bcl-2. Mol Pharmacol. 2006;69:288–98. doi: 10.1124/mol.105.016154. [DOI] [PubMed] [Google Scholar]
- 73.Farrugia MM, Duan LJ, Reis MD, Ngan BY, Berinstein NL. Alterations of the p53 tumor suppressor gene in diffuse large cell lymphomas with translocations of the c-MYC and BCL-2 proto-oncogenes. Blood. 1994;83:191–8. [PubMed] [Google Scholar]
- 74.Hasegawa H, Yamada Y, Iha H, Tsukasaki K, Nagai K, Atogami S, Sugahara K, Tsuruda K, Ishizaki A, Kamihira S. Activation of p53 by Nutlin-3a, an antagonist of MDM2, induces apoptosis and cellular senescence in adult T-cell leukemia cells. Leukemia. 2009;23:2090–101. doi: 10.1038/leu.2009.171. [DOI] [PubMed] [Google Scholar]
- 75.Li CC, O’Connell CD, Beckwith M, Longo DL. Detection of p53 mutations in B cell non-Hodgkin’s lymphoma cell lines. Leukemia. 1995;9:650–5. [PubMed] [Google Scholar]
- 76.Amini RM, Berglund M, Rosenquist R, Von Heideman A, Lagercrantz S, Thunberg U, Bergh J, Sundström C, Glimelius B, Enblad G. A novel B-cell line (U-2932) established from a patient with diffuse large B-cell lymphoma following Hodgkin lymphoma. Leuk Lymphoma. 2002;43:2179–89. doi: 10.1080/1042819021000032917. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.