

Abstract
A large number of epidemiological studies have linked a common single nucleotide polymorphism (SNP) in the human p53 gene to risk for developing a variety of cancers. This SNP encodes either an arginine or proline at position 72 (R72P) of the p53 protein, which can alter the apoptotic activity of p53 via transcriptional and non-transcriptional mechanisms. This SNP has also been reported to modulate the development of human papilloma virus (HPV)-driven cancers through differential targeting of the p53 variant proteins by the E6 viral oncoprotein. Mouse models for the p53 R72P polymorphism have recently been developed but a role for this SNP in modifying cancer risk in response to viral and chemical carcinogens has yet to be established experimentally. Here we demonstrate that the p53 R72P polymorphism modulates the hyperprolferative, apoptotic and inflammatory phenotypes caused by expression of the HPV16 E6 and E7 oncoproteins. Moreover, the R72P SNP also modifies the carcinogenic response to the chemical carcinogen 4NQO, in the presence and absence of the HPV16 transgene. Our findings confirm several human epidemiological studies associating the codon 72 proline variant with increased risk for certain cancers but also suggest that there are tissue-specific differences in how the R72P polymorphism influences the response to environmental carcinogens.
Introduction
The tumor suppressor p53 is a key player in stress responses that preserve genomic stability by responding to a variety of insults, including DNA damage, hypoxia, metabolic stress, and oncogene activation. Through both transcriptional and non-transcriptional mechanisms p53 can regulate apoptosis, autophagy, cell cycle progression, senescence, DNA repair and metabolism. Malfunction of the p53 pathway is an almost universal hallmark of human cancers, although the relative importance of the different p53 activities for tumor suppression is a matter of debate [1,2]. A common polymorphism in the p53 gene results in a protein with either an arginine or proline at amino acid 72 [3,4]. Cell culture-based studies have demonstrated that the p53 codon 72 arginine (R72) protein variant has a greater ability to suppress oncogenic transformation and increased apoptotic activity compared to the codon 72 proline (P72) variant [3,5,6]. Differences in gene-specific transcriptional regulation between the p53 polymorphic variants as well as an enhanced capability of the R72 protein to localize to mitochondria may both contribute to this differential apoptotic capacity.
The p53 R72P polymorphism has been extensively evaluated as a risk modifier for a variety of cancers and other diseases. Epidemiological studies have often associated the presence of the P72 encoding gene variant to increased risk for some cancers. However, in most cases there are conflicting reports and meta-analyses combining studies on lung, breast, and gastric cancers do not support a link between the p53 R72P SNP and cancer risk [4,7-9]. On the other hand, meta-analyses of studies examining esophageal squamous cell carcinoma (SCC) support the idea that individuals with a P72 variant allele are at greater risk for developing that cancer [10,11]. A meta-analysis examining risk for cervical cancer also suggests an association with the p53 R72P SNP [12]. This possibility was first raised by Storey et al, who proposed that women with the p53 R72/R72 genotype were at increased risk for cervical cancer because the R72 protein variant is preferentially targeted for degradation by the HPV E6 oncoprotein [13]. It is now known that HPV infection is also associated with the development of head and neck cancers, particularly squamus cell carcinoma (SCC) of the oropharynx [14,15]. Moreover, a strong interaction between HPV seropositivity and the R72P SNP for risk of SCC of the oropharynx has been reported [16-18]. However, in the case of HPV-associated head and neck SCC it is the P72-encoding allele of p53 that is associated with increased cancer risk.
We and others have recently developed mouse models for the p53 R72P polymorphism as tools to study the function of this SNP in genetically controlled but physiological relevant settings [19,20]. Pairs of models generated through different approaches each demonstrate that both human p53 variants are functional in mice [19,20]. Moreover, significant differences between the p53 variants in their abilities to induce apoptosis could be observed in these mouse models. However, depending on the tissue, either the R72 or P72 variant had higher apoptotic activity [19,20]. In one study it was shown that despite a significant difference in the acute apoptotic response to ultraviolet (UV) radiation, there was no significant difference in UV radiation-induced skin carcinogenesis between R72/R72 and P72/P72 genotypes [20]. Another study using similar humanized knock-in mouse models found the R72P SNP made no significant difference in lymphoma development driven by an Eμ-myc transgene [19]. Thus, there is no experimental data, to date, to support a role for the R72P SNP in modulating cancer development using these mouse models.
Here we used human p53 exon 4 knock-in mouse models to examine how the R72P SNP interacts with relevant environmental insults to modify the development of cancers of the oral cavity and esophagus. We found that this polymorphism modulated the hyperproliferative, apoptotic, and inflammatory responses to transgenic expression of the HPV16 E6 and E7 oncoproteins. Treatment of these HPV16 transgenic mice with the carcinogen 4-nitroquinoline-1-oxide (4NQO) induced tumor development, which was modified by the R72P polymorphism. Moreover, the R72P SNP also affected the development of esophageal cancers in 4NQO treated knock-in mice lacking the HPV16 transgene. Our findings demonstrate that this functional SNP in p53 can modify the response to viral and chemical carcinogens resulting in tissue-specific differences in tumor susceptibility.
Materials and methods
Transgenic mice
Human p53 exon 4 knock-in mice encoding either arginine (p53R72/R72) or proline (p53P72/P72) at codon 72 have been described [20]. K14-HPV16 transgenic mice expressing the E6 and E7 oncoproteins under the keratin 14 promoter were obtained from the NCI mouse repository [21]. Experimental mice were generated by crossing human p53 exon 4 knock-in mice with K14-HPV16 transgenic mice. All mice were maintained on the FVB/N inbred genetic background. Genomic DNA isolated from tail snips was used for genotyping the p53 R72P polymorphism as described [20]. Genotyping for the HPV16 transgene was performed using the NCI repository protocol. All mouse manipulations were approved by the University of Texas MD Anderson Cancer Center (UTMDACC) Institutional Animal Care and Use Committee and followed national guidelines.
Immunohistochemical staining for Ki67 and cleaved Lamin A
Tissue samples (skin, tongue and head) from six week old mice were fixed, paraffin embedded and immunohistochemically (IHC) stained for Ki67 (DAKO, Glostrup, Denmark) as a marker of proliferation and cleaved lamin A (Cell Signaling Technology, Danvers, MA) as a marker of apoptosis using protocols developed by UTMDACC Science Park Histology Facility Core. Esophageal tumor samples were stained similarly. Positive cells were counted using digital images captured with the Aperio ScanScope CS slide scanner and analyzed with the manufacturer's GENIE software (Aperio Technologies, Vista, CA).
Carcinogen treatment
Six week old mice were given the carcinogen 4-Nitroquinoline-1-oxide (4NQO) (Sigma, St Louis, MO) at a concentration of 10 μg/ml in drinking water. K14-HPV16 transgenic mice were treated for 3-5 weeks and non-transgenic, R72P knock-in mice were treated for 16 weeks. Mice were then returned to normal drinking water until 24 weeks from the start date of treatment.
Pathology
Mice were sacrificed when moribund or at the completion of the carcinogenesis study and subjected to complete necropsy with identification of gross lesions. Gross lesions and organs were collected, fixed in formalin for 24 h, then switched to 70% ethanol for storage. Tissue sections were embedded in paraffin, sectioned, and stained with hematoxylin and eosin (H&E) using standard techniques. Slides were submitted to the UTMDACC Mutant Mouse Pathology Facility Core for expert pathological analysis by a board-certified veterinary pathologist. Immediately after euthanasia, blood was collected from some mice by intracardial puncture. Blood was submitted to the UTMDACC Michael Keeling Center for Comparative Medicine and Research for routine clinical chemistry and hematology testing.
Inflammation was scored as 1 (minimal to mild), 2 (moderate), or 3 (marked) based on the total number of inflammatory cells present, including neutrophils, eosinophils, and mononuclear cells. The cell types comprising the inflammatory infiltrate varied from esophagus to esophagus and from region to region of the same esophagus. Inflammatory cells were located predominantly in the lamina propria/submucosa, and inflammation was generally more marked in and around tumors that in regions of the esophagus without tumors.
Primary keratinocyte cultures
Primary keratinocytes were isolated from newborn pups (0-3 days) as described [22]. Briefly, pups were washed in ethanol and cold-PBS, then skin was removed, washed with cold-PBS and trypsinized overnight at 4°C. The epidermis was carefully separated from the dermis, minced with scissors and filtered through a cell strainer. Cells were then suspended in Defined Keratinocyte-SFM (Invitrogen, CA) supplemented with antibiotic, 2.5% FBS and keratinocyte growth factor and seeded in 6 cm tissue culture dishes (one pup/dish). After 4 h incubation, media was replaced with serum free Defined Keratinocyte media with growth factor supplement. At 50-80% confluency, cells were harvested and processed for isolation of RNA or lysed to obtained cellular protein for immunoblotting.
Real-time quantitative RT-PCR
RNA was extracted from primary keratinocytes using RNeasy kit (Qiagen, Valencia). After reverse transcription, cDNAs were subjected to quantitative real-time PCR using the SYBR green gene expression assay (Applied biosystem). RNA levels were normalized to the endogenous control gene GAPDH.
Western blot analysis
Protein from primary keratinocyte lysate was quantified by bicinchoninic acid (BCA) colorimetric assay (Pierce, Rockford, IL). Equal quantities of protein (20 μg/lane) were run on SDS polyacrylamide gels and transferred onto polyvinylidene fluoride (PVDF) membranes. Immunoblotting was done using the following primary antibodies: p53 (Vector laboratories, Burlingame, CA); Bax, p21 and actin (Santa Cruz Biotechnology, Santa Cruz, CA).
Statistical Analysis
Statistical comparisons between specific groups were done by two tailed Student's t-test. Differences between groups with a p value of <0.05 were considered statistically significant. Where appropriate, the average values of indicated sample numbers are presented and expressed as mean ± SE.
Results
The p53 R72P polymorphism modulates the phenotype of K14-HPV16 transgenic mice
K14-HPV16 transgenic mice expressing the E6 and E7 oncoproteins [21] were crossed with p53R72/R72 and p53P72/P72 mice in which human exon 4 sequences replace mouse exon 4 sequences [20]. K14-HPV16 transgenic mice heterozygous for the p53 R72 and P72 alleles generated from that cross were further backcrossed to p53R72/R72 and p53P72/P72 mice to generate K14-HPV16 transgenic and non-transgenic mice homozygous for either the p53 R72 or P72 alleles. All mouse models used in this study were in the FVB/N strain background.
Expression of the HPV16 E6 oncogene was assessed in primary keratinocytes isolated from newborn pups of the various genotypes by real-time PCR (Figure 1A). As expected, E6 was expressed in the K14-HPV16 transgenic mice, but not the non-transgenic mice, and the R72P polymorphism did not affect the level of E6 expression. The expression of p53 mRNA was also similar among the different genotypes. Unexpectedly, the expression levels of several p53 target genes were unaffected by the K14-HPV16 transgene despite the expression of E6, which is known to promote p53 protein degradation (Figure 1A). Although there was a slight trend towards lower expression of several p53 target genes in P72/P72 mice compared to R72/R72 mice, this was only significant in the case of Puma in the K14-HPV16 transgenic mice. Western blot analysis showed similar levels of p53, Bax, and p21 protein expression in primary keratinocytes isolated from the knock-in models with and without the K14-HPV16 transgene (Figure 1B).
Figure 1. Expression of HPV16 E6 and cellular genes in the p53 and RB/E2F pathways.
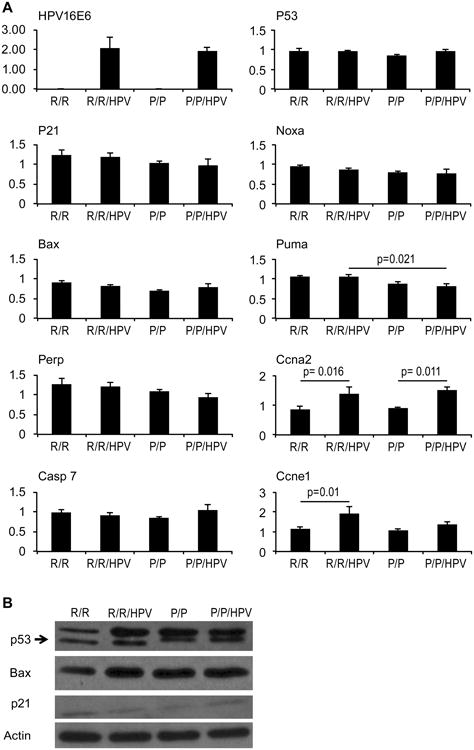
Primary mouse keratinocytes were isolated from 1-3 day old pups of the indicated genotypes. (A) Total RNA was isolated and subjected to quantitative real-time PCR for the indicated genes. The averages of triplicate assays are presented for each gene. Bars represent standard error and statistically significant differences (p<0.05) are indicated. (B) Western blot analysis of p53, Bax, p21, and actin was performed using whole cell extracts (20mg/lane) of primary keratinocytes isolated from the indicated genotypes.
Because the HPV E7 oncoprotein targets the RB tumor suppressor, several target genes downstream of the RB/E2F pathway were also examined in primary keratinocytes. Ccna2, encoding cyclin A2, and Ccne1, encoding cyclin E, were both expressed at higher levels in K14-HPV16 transgenic keratinocytes compared to non-transgenic keratinocytes (Figure 1A). In contrast, the pro-apoptotic E2F target gene caspase 7 was not elevated in transgenic cells. The R72P polymorphism did not significantly affect the expression of these E2F target genes in the presence or absence of the K14-HPV16 transgene.
At six weeks of age, several squamous epithelial tissues were taken for immunohistochemical (IHC) analysis, including the skin, oral mucosa, and tongue. In each of these tissues K14-HPV16 transgenic mice displayed hyperproliferation as measured by an increase in the number of cells staining positive for Ki67 (Figure 2A). In the epidermis, there was also a statistically significant difference between K14-HPV16;p53P72/P72 and K14-HPV16;p53R72/R72 mice, with the R72 variant associated with higher levels of Ki67 staining. A similar trend was also observed in the oral mucosa and tongue, although these differences did not reach statistical significance.
Figure 2. Modulation of proliferation and apoptosis by the R72P SNP in K14-HPV16 transgenic mice.
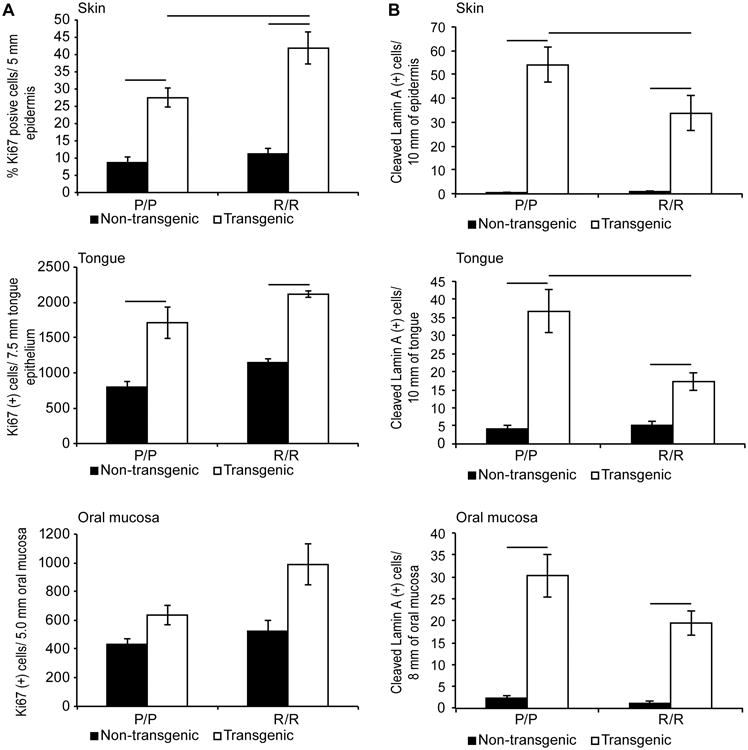
(A) Skin, oral mucosa and tongue tissues were IHC stained for Ki67 as a marker of proliferation. Results were quantified and the averages from six animals of each genotype are presented. B) The same tissues were IHC stained for cleaved lamin A as a marker of apoptosis. Results were quantified and the averages from six mice in each group are presented. Vertical bars indicate standard error and horizontal bars indicate statistically significant differences between groups (p<0.05).
IHC staining for cleaved lamin A, a marker of apoptosis, was also performed in these same epithelial tissues. In each case positively staining cells were readily observed in tissues from K14-HPV16 transgenic mice while tissues from non-transgenic mice had very few positive cells (Figure 2B). Transgenic mice with the P72/P72 genotype displayed increased numbers of apoptotic cells compared to transgenic mice with the R72/R72 genotype although this was only statistically significant in the skin and tongue. This finding conflicts with reports suggesting that the R72 variant has greater apoptotic activity than the P72 variant [3,5,6].
The p53 R72P SNP modifies tumor development in HPV16 transgenic mice treated with the carcinogen 4NQO
To determine if the p53 R72P polymorphism would modulate the development of head and neck SCC, K14-HPV16 transgenic mice with either the p53 P72/P72 or R72/R72 genotypes were treated with the carcinogen 4NQO supplied in the drinking water. This model combining transgenic expression of E6 and E7 with 4NQO treatment has been successfully used to induce tumors in the oral cavity and upper digestive tract that resemble human cancers associated with HPV infection [23]. In the original protocol, mice were treated with 10 μg/ml of 4NQO in drinking water for 16 weeks [23]. However, we found that extended treatment of K14-HPV16 transgenic mice with this concentration of 4NQO was toxic. Thus, K14-HPV16 mice were treated with 4NQO for 3 weeks (group 1) or 4-5 weeks (group 2). Mice were then returned to normal drinking water and monitored for tumor development and health status. Mice were sacrificed when moribund or at 24 weeks from the start date of carcinogen treatment. In both groups of treated mice, K14-HPV16 transgenic mice with the R72/R72 genotype had increased survival compared to the P72/P72 genotype (Figure 3).
Figure 3. Survival of K14-HPV16 transgenic, p53 R72P polymorphic mice treated with 4NQO.
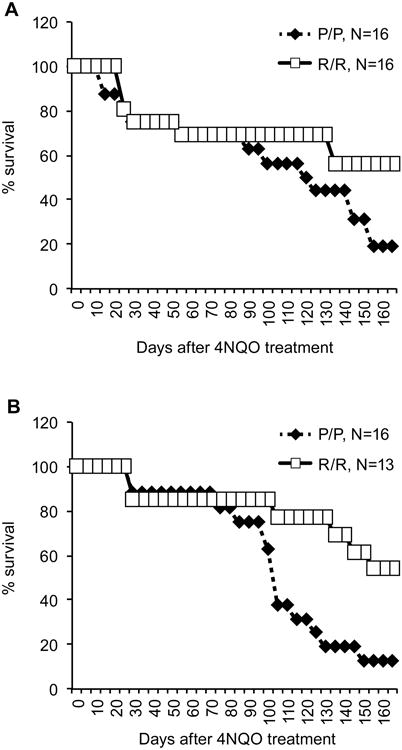
(A) Six week old K14-HPV16:p53R72/R72 and K14-HPV16:p53P72/P72 mice were given 10 μg/ml of 4NQO in drinking water for 3 weeks (20-24 days) and then returned to normal drinking water for the remainder of the 24 week study. Mice were monitored for health and any moribund mice were sacrificed before the end of the study. (B) A separate group of mice with the same genotypes as above were given 4NQO (10 μg/ml) in drinking water for 4 to 5 weeks and returned to normal drinking water for the remainder of the 24 week study. Survival was monitored as above.
Most K14-HPV16 transgenic mice treated with 4NQO developed at least one tumor in the oral mucosa, which includes the epithelium lining the cheeks, palate, and sublingual area (Figure 4A). Oral cavity tumors induced in non-transgenic mice and rats by oral exposure to 4NQO are predominately SCC of the tongue [24]. However, in the present studies, most tumors were SCC that arose from the gingiva adjacent to teeth (Figure 4B). A previous report documents the appearance of gingival squamous dysplasia in rats receiving oral 4NQO [25]. In our studies, it appears that the combination of the HPV16 transgene and 4NQO resulted in the development of frank SCC in the gingiva. The SCC that arose were well differentiated. They were characterized by extension of neoplastic epithelium below the neck of the tooth, loss of association between the epithelium and the root of the tooth, and the presence of a stromal response to the infiltrating epithelium. Invasiveness varied considerably, but most SCC were minimally to moderately invasive. The R72P polymorphism did not appear to alter tumor incidence or pathology in the oral mucosa.
Figure 4. The R72P polymorphism modulates the tumor spectrum of K14-HPV16 transgenic mice treated with 4NQO.
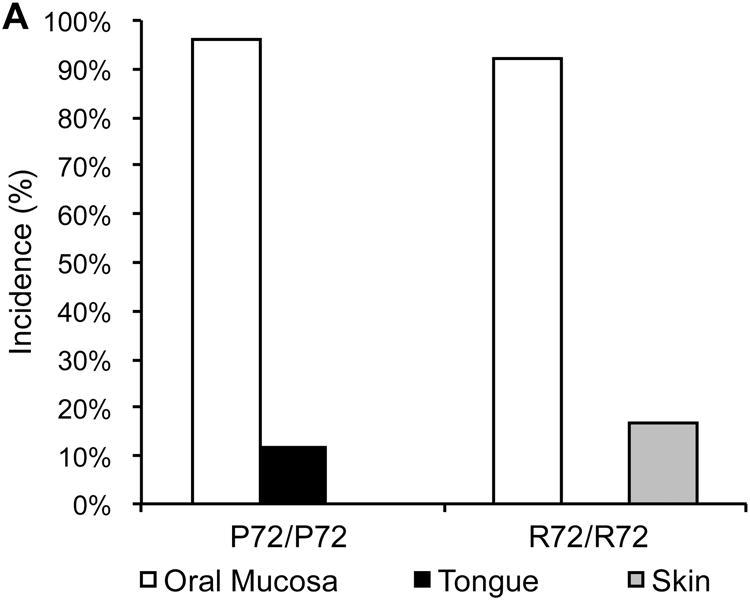
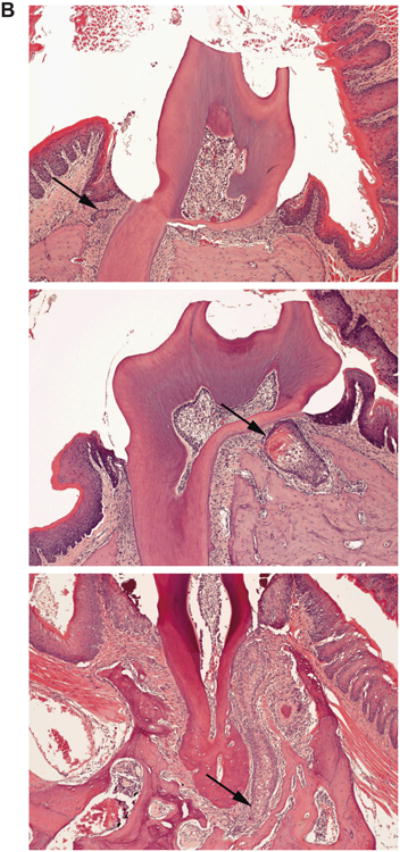
A) Tumor incidence is presented for the oral mucosa, tongue and skin of K14-HPV16 transgenic mice with either the p53R72/R72 (N=25) or p53P72/P72 (N=24) genotypes following treatment with 4NQO. B) K14-HPV16 transgenic mice treated with 4NAO developed SCC of the gingiva. SCC ranged from minimally invasive (top), to moderately invasive (middle) to markedly invasive (bottom). Arrows indicate islands and lobules of infiltrating neoplastic epithelium.
Although there was no difference between R72/R72 and P72/P72 mice in SCC of the oral mucosa, there were differences in two other cancers from the K14-HPV16 transgenic mice treated with 4NQO. Three out of 25 transgenic mice with the R72/R72 genotype developed SCC of the tongue while no transgenic mice with the P72/P72 genotype (0/24) developed SCC at this site (Figure 4A). On the other hand, four out of 24 transgenic mice with the P72/P72 genotype developed SCC of the skin, while no skin cancer was observed in transgenic mice with the R72/R72 genotype (0/25). This suggests that the R72P SNP may differentially modulate tumor development in the skin and tongue.
The R72P polymorphism affects markers of inflammation
A previous report analyzing other knock-in mouse models humanized for the R72P polymorphism found that the P72 variant of p53 interacted more efficiently with the NF-kB transcription factor than the R72 variant [19]. This differential association with NF-kB was associated with increased expression of inflammatory genes and an enhanced inflammatory response to lipopolysaccharide (LPS) in mice expressing the p53 P72 variant. To determine if inflammation in the K14-HPV16 transgenic mice differed between p53 genotypes, serum chemistry and complete blood counts were performed on experimental animals from each group. Transgenic mice with the P72/P72 genotype had significantly higher levels of total protein and gamma globulin compared to transgenic mice with the R72/R72 genotype (Figure 5A). On the other hand, transgenic mice with the P72/P72 genotype had lower levels of albumin resulting in a dramatic difference in the albumin/globin ratio between the two genotypes (Figure 5A).
Figure 5. The p53 P72 variant is associated with an enhanced inflammatory response in K14-HPV16 transgenic mice treated with 4NQO.
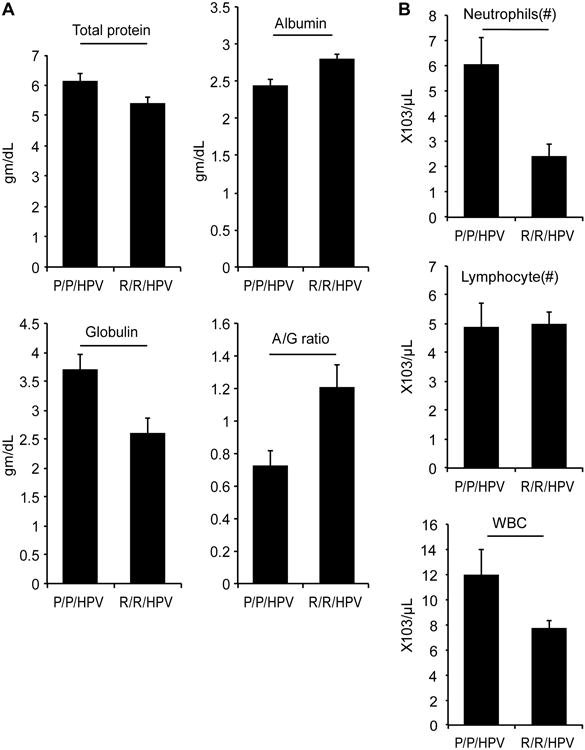
(A) The levels of total protein, albumin and gamma globulin were measured in the serum of K14-HPV16 transgenic mice expressing either the p53 P72 or R72 variants. The averages from 12 (P72/P72) or 15 (R72/R72) mice are presented. (B) Cell counts were performed on blood samples from the same mice as above. The averages for total numbers of white blood cells, neutrophils and lymphocytes are presented. Statistically significant differences are indicated by horizontal bars (p<0.05).
Significantly more circulating white blood cells (WBC) were found in K14-HPV16 transgenic mice with the P72/P72 genotype compared to the R72/R72 genotype. This leukophilia was due to higher absolute numbers of neutrophils as no difference in the absolute numbers of lymphocytes was observed between genotypes (Figure 5B). Increased neutrophil numbers and gamma globulin levels in P72/P72 transgenic mice provide additional experimental evidence that the p53 P72 variant is associated with an enhanced inflammatory response, which may contribute to differences in tumorigenesis in certain settings.
Mice expressing the p53 P72 variant are more susceptible to esophageal carcinogenesis following long-term 4NQO treatment
Only two esophageal tumors were observed K14-HPV16 transgenic mice treated with 4NQO, one with the p53 R72/R72 genotype and one with the P72/P72 genotype (data not shown). To further explore a potential role for the R72P SNP in modulating esophageal carcinogenesis, p53R/72/R72 and p53P72/P72 mice without the K14-HPV16 transgene were treated with 10 μg/ml of 4NQO supplied in the drinking water for 16 weeks. These knock-in mice were then returned to normal drinking water for an additional eight weeks. Long-term treatment with 4NQO in the drinking water is known to induce esophageal tumors in wild type mice [26]. While SCC of the oral mucosa, tongue, and skin were rare in this cohort of mice, esophageal tumors were relatively common with some mice developing multiple tumors. In agreement with human epidemiological studies associating the p53 P72 allele with increased risk for esophageal cancer [10,11], P72/P72 knock-in mice had a significantly higher tumor incidence and increased tumor multiplicity compared to R72/R72 knock-in mice (Figure 6A and B).
Figure 6. p53P72/P72 knock-in mice are more susceptible to 4NQO-induced esophageal carcinogenesis compared to p53R72/R72 mice.
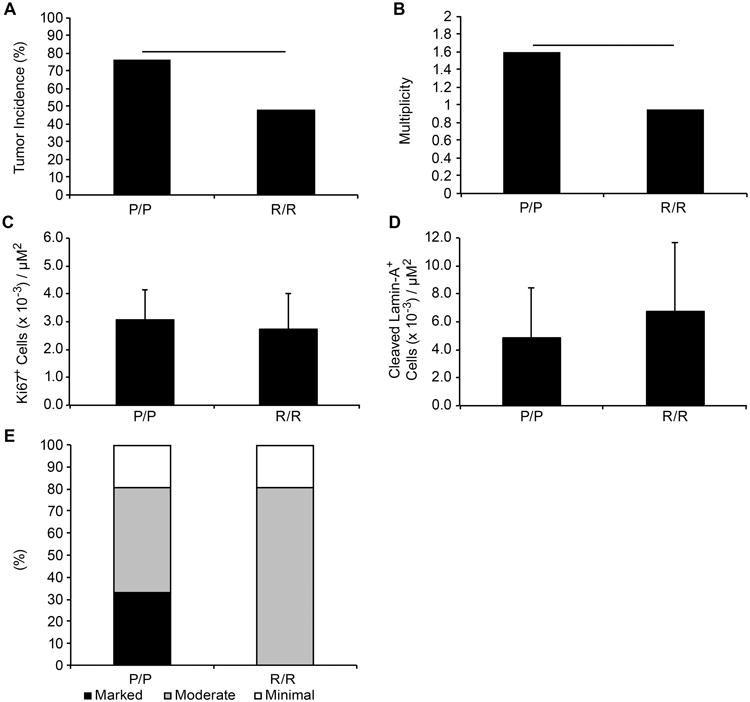
Mice encoding the p53 P72 (N=17) or R72 (N=21) variant were treated with 4NQO in drinking water (10 μg/ml) for sixteen weeks and returned to normal drinking water for eight weeks prior to sacrifice. Esophageal tumors were scored and the tumor incidence (A) and multiplicity (number of tumors per mouse) (B) is presented. Statistically significant differences are indicated by horizontal bars (p<0.05). Sections of esophageal tumors from p53R72/R72 and p53P72/P72 mice were stained for Ki67 (C) or cleaved lamin A (D). Slides were counted using the Aperio ScanScope CS and GENIE software. The average number of positive cells per area of tumor for each genotype is presented. (E) Esophageal tumors from the above mice were examined microscopically for inflammatory cell infiltrate and scored as mild, moderate or marked based on morphology and number of inflammatory cells in epithelial regions.
Proliferation in the esophageal tumors was similar between R72/R72 and P72/P72 mice as measured by the number of Ki67-positive cells per field (Figure 6C). Likewise, no significant difference in the number of apoptotic cells, indicated by staining for cleaved lamin A, was observed between genotypes (Figure 6D). Esophageal tumors from these mice were also scored for inflammation based on the level of cellular infiltrates. The character of the inflammatory infiltrates varied in different esophagi and from region to region in the same esophagus. Inflammatory cells were located predominately in the submucosal region and inflammation was more evident in and around tumors than in regions of the esophagus without tumors. Consistent with the blood and serum chemistry results from the K14-HPV16 experimental mice, a third of the p53P72/P72 knock-in mice treated with 4NQO for 16 weeks had marked inflammation of esophageal tissue while p53R72/R72 knock-in mice had only moderate or mild inflammation (Figure 6E).
Discussion
In these studies we have used humanized p53 exon 4 knock-in mouse models to explore how the R72P SNP impacts tumor development in the oral cavity and upper digestive tract. The carcinogen 4NQO supplied in the drinking water was used in these studies because it mimics the DNA damage and mutagenesis caused by similar carcinogens in cooked food and tobacco smoke [24,27]. Moreover, 4NQO carcinogenesis produces a spectrum of oral cavity and esophageal lesions that parallel the progression and histological features of human cancers. Using this well-established model we found that mice expressing the p53 P72 variant were more susceptible to esophageal SCC compared to mice encoding the R72 variant. This finding is consistent with several epidemiological studies associating the P72 allele with increased risk for esophageal SCC [10,11] and demonstrates that human cancer susceptibility due to this common polymorphism can be modeled in mice.
Emerging data implicate infection with oncogenic HPV subtypes with the development of head and neck cancers [15]. It was also suggested that the two p53 protein variants are differentially targeted for degradation by the HPV E6 oncoprotein [13]. To determine if the R72P SNP modifies the oncogenic effects of HPV, K14-HPV16 transgenic mice were crossed to the p53 exon 4 knock-in models. We found that the R72P polymorphism made little difference in the levels of p53 or the expression of p53 target genes. Nonetheless, the hyperproliferative and apoptotic responses to the HPV16 transgene were modulated by the R72P SNP.
Interestingly, the R72P SNP appeared to differentially modify cancer susceptibility in different tissues. The incidence of oral mucosa cancer was not significantly different between genotypes since almost all K14-HPV16 transgenic mice treated with 4NQO developed these lesions regardless of SNP status. However, K14-HPV16;p53P72/P72 mice displayed decreased survival compared to K14-HPV16;p53R72/R72 mice when treated with 4NQO, suggesting that the polymorphism may influence time to tumor onset. Moreover, only P72/P72 transgenic mice developed SCC of the tongue (3 of 25) in this study while only R72/R72 transgenic mice developed SCC of the skin (4 of 24). This finding is consistent with previous epidemiological studies showing that the R72P polymorphism impacts cancer risk in a tissue-specific manner [4,11]. Future studies should address whether the R72P SNP differentially modulates risk for SCC of the tongue and skin in humans, particularly those cancers associated with HPV.
It has been assumed that differences in the apoptotic potential between the two p53 variants underlies differences in cancer risk. This may be true for some cancers but recent mouse model data indicate that other p53 activities, including the maintenance of genome integrity and the regulation of cell metabolism, may be more important for tumor suppression than the induction of apoptosis [1,2]. In addition, the p53 R72P SNP may also modify the inflammatory response associated with tumor development. We observe increased serum gamma globulin and absolute numbers of circulating neutrophils in K14-HVP16 transgenic mice with the P72/P72 genotype compared to transgenic mice with the R72/R72 genotype. Espophageal tumors from P72/P72 mice treated long-term with 4NQO also displayed increased inflammatory infiltrates compared to tumors from R72/R72 mice. This suggests that differential regulation of the inflammatory response by the p53 polymorphic variants may also contribute to differences in cancer susceptibility and progression of at least some tumor types.
Acknowledgments
We thank Jennifer Smith, Pamela Blau and Carlos Perez for technical assistance, Kevin Lin for statistical analysis, Becky Brooks for preparation of the manuscript, Chris Brown for graphics and Fernando Benavides, Lezlee Coghlan, Dale Weiss and coworkers for animal care. This research was performed in partial fulfillment of the requirements for the PhD degree from The University of Texas Graduate School of Biomedical Sciences at Houston; University of Texas MD Anderson Cancer Center, Houston, TX. This research is supported in part by grants from the National Institutes of Health ES007784 to D.G.J., through MD Anderson's Cancer Center Support Grant CA016672, National Institute of Environmental Health Sciences Center Grant P30ES007784 and, by a BOYSCAST Fellowship to J.S. from the Department of Science and Technology, Government of India.
References
- 1.Li T, Kon N, Jiang L, et al. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell. 2012;149(6):1269–1283. doi: 10.1016/j.cell.2012.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu G, Parant JM, Lang G, et al. Chromosome stability, in the absence of apoptosis, is critical for suppression of tumorigenesis in Trp53 mutant mice. Nat Genet. 2004;36(1):63–68. doi: 10.1038/ng1282. [DOI] [PubMed] [Google Scholar]
- 3.Thomas M, Kalita A, Labrecque S, Pim D, Banks L, Matlashewski G. Two polymorphic variants of wild-type p53 differ biochemically and biologically. Mol Cell Biol. 1999;19(2):1092–1100. doi: 10.1128/mcb.19.2.1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Whibley C, Pharoah PD, Hollstein M. p53 polymorphisms: cancer implications. Nat Rev Cancer. 2009;9(2):95–107. doi: 10.1038/nrc2584. [DOI] [PubMed] [Google Scholar]
- 5.Bergamaschi D, Samuels Y, Sullivan A, et al. iASPP preferentially binds p53 proline-rich region and modulates apoptotic function of codon 72-polymorphic p53. Nat Genet. 2006;38(10):1133–1141. doi: 10.1038/ng1879. [DOI] [PubMed] [Google Scholar]
- 6.Dumont P, Leu JI, Della Pietra AC, 3rd, George DL, Murphy M. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat Genet. 2003;33(3):357–365. doi: 10.1038/ng1093. [DOI] [PubMed] [Google Scholar]
- 7.Commonly studied single-nucleotide polymorphisms and breast cancer: results from the Breast Cancer Association Consortium. J Natl Cancer Inst. 2006;98(19):1382–1396. doi: 10.1093/jnci/djj374. [DOI] [PubMed] [Google Scholar]
- 8.Matakidou A, Eisen T, Houlston RS. TP53 polymorphisms and lung cancer risk: a systematic review and meta-analysis. Mutagenesis. 2003;18(4):377–385. doi: 10.1093/mutage/geg008. [DOI] [PubMed] [Google Scholar]
- 9.Zhou Y, Li N, Zhuang W, et al. P53 codon 72 polymorphism and gastric cancer: a meta-analysis of the literature. International journal of cancer Journal international du cancer. 2007;121(7):1481–1486. doi: 10.1002/ijc.22833. [DOI] [PubMed] [Google Scholar]
- 10.Jiang DK, Yao L, Wang WZ, et al. TP53 Arg72Pro polymorphism is associated with esophageal cancer risk: a meta-analysis. World journal of gastroenterology: WJG. 2011;17(9):1227–1233. doi: 10.3748/wjg.v17.i9.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang B, Wang D, Zhang D, et al. Pro variant of TP53 Arg72Pro contributes to esophageal squamous cell carcinoma risk: evidence from a meta-analysis. European journal of cancer prevention: the official journal of the European Cancer Prevention Organisation. 2010;19(4):299–307. doi: 10.1097/CEJ.0b013e32833964bc. [DOI] [PubMed] [Google Scholar]
- 12.Jee SH, Won SY, Yun JE, Lee JE, Park JS, Ji SS. Polymorphism p53 codon-72 and invasive cervical cancer: a meta-analysis. International journal of gynaecology and obstetrics: the official organ of the International Federation of Gynaecology and Obstetrics. 2004;85(3):301–308. doi: 10.1016/j.ijgo.2003.08.017. [DOI] [PubMed] [Google Scholar]
- 13.Storey A, Thomas M, Kalita A, et al. Role of a p53 polymorphism in the development of human papillomavirus-associated cancer. Nature. 1998;393(6682):229–234. doi: 10.1038/30400. [DOI] [PubMed] [Google Scholar]
- 14.Dahlstrom KR, Adler-Storthz K, Etzel CJ, et al. Human papillomavirus type 16 infection and squamous cell carcinoma of the head and neck in never-smokers: a matched pair analysis. Clinical cancer research: an official journal of the American Association for Cancer Research. 2003;9(7):2620–2626. [PubMed] [Google Scholar]
- 15.Li G, Sturgis EM. The role of human papillomavirus in squamous carcinoma of the head and neck. Curr Oncol Rep. 2006;8(2):130–139. doi: 10.1007/s11912-006-0048-y. [DOI] [PubMed] [Google Scholar]
- 16.Chen X, Sturgis EM, El-Naggar AK, Wei Q, Li G. Combined effects of the p53 codon 72 and p73 G4C14-to-A4T14 polymorphisms on the risk of HPV16-associated oral cancer in never-smokers. Carcinogenesis. 2008;29(11):2120–2125. doi: 10.1093/carcin/bgn191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ji X, Neumann AS, Sturgis EM, et al. p53 codon 72 polymorphism associated with risk of human papillomavirus-associated squamous cell carcinoma of the oropharynx in never-smokers. Carcinogenesis. 2008;29(4):875–879. doi: 10.1093/carcin/bgn039. [DOI] [PubMed] [Google Scholar]
- 18.Perrone F, Mariani L, Pastore E, et al. p53 codon 72 polymorphisms in human papillomavirus-negative and human papillomavirus-positive squamous cell carcinomas of the oropharynx. Cancer. 2007;109(12):2461–2465. doi: 10.1002/cncr.22702. [DOI] [PubMed] [Google Scholar]
- 19.Frank AK, Leu JI, Zhou Y, et al. The codon 72 polymorphism of p53 regulates interaction with NF-{kappa}B and transactivation of genes involved in immunity and inflammation. Mol Cell Biol. 2011;31(6):1201–1213. doi: 10.1128/MCB.01136-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu F, Dolle ME, Berton TR, et al. Mouse models for the p53 R72P polymorphism mimic human phenotypes. Cancer Res. 2010;70(14):5851–5859. doi: 10.1158/0008-5472.CAN-09-4646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arbeit JM, Munger K, Howley PM, Hanahan D. Progressive squamous epithelial neoplasia in K14-human papillomavirus type 16 transgenic mice. J Virol. 1994;68(7):4358–4368. doi: 10.1128/jvi.68.7.4358-4368.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pierce AM, Gimenez-Conti IB, Schneider-Broussard R, Martinez LA, Conti CJ, Johnson DG. Increased E2F1 activity induces skin tumors in mice heterozygous and nullizygous for p53. Proc Natl Acad Sci USA. 1998;95:8858–8863. doi: 10.1073/pnas.95.15.8858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Strati K, Pitot HC, Lambert PF. Identification of biomarkers that distinguish human papillomavirus (HPV)-positive versus HPV-negative head and neck cancers in a mouse model. Proc Natl Acad Sci U S A. 2006;103(38):14152–14157. doi: 10.1073/pnas.0606698103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanojia D, Vaidya MM. 4-nitroquinoline-1-oxide induced experimental oral carcinogenesis. Oral Oncol. 2006;42(7):655–667. doi: 10.1016/j.oraloncology.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 25.Ribeiro DA, Salvadori DM. Gingival changes in wistar rats after oral treatment with 4-nitroquinoline 1-oxide. Eur J Dent. 2007;1(3):152–157. [PMC free article] [PubMed] [Google Scholar]
- 26.Tang XH, Knudsen B, Bemis D, Tickoo S, Gudas LJ. Oral cavity and esophageal carcinogenesis modeled in carcinogen-treated mice. Clin Cancer Res. 2004;10(1 Pt 1):301–313. doi: 10.1158/1078-0432.ccr-0999-3. [DOI] [PubMed] [Google Scholar]
- 27.Vered M, Yarom N, Dayan D. 4NQO oral carcinogenesis: animal models, molecular markers and future expectations. Oral Oncol. 2005;41(4):337–339. doi: 10.1016/j.oraloncology.2004.07.005. [DOI] [PubMed] [Google Scholar]