

Abstract
Over 40 years ago, Francois Jacob proposed that levels of “integrons” explain how biological systems are constructed. Today, these networks of interactions between tissues, cells, organelles, metabolic pathways, genes, and individual molecules provide key insights into biology. We suggest that the wiring and interdependency between subsystems within a network are useful to understand the aging process. The breakdown of one subsystem (e.g. an organelle) can have ramifications for other interconnected subsystems, leading to the sequential collapse of subsystem functions. On the other hand, the interconnected nature of homeostatic wiring can provide organisms with the means of compensating for the decline of one subsystem. This occurs at multiple levels in an organism – e.g. between organelles or between tissues. We review recent data that highlight the importance of such interconnectivity/communication in the aging process; both in progressive decline and longevity assurance.
“As a net is made up by a series of knots, so everything in this world is connected by a series of knots. If anyone thinks that the mesh of a net is an independent, isolated thing, he is mistaken.
… and each mesh has its place and responsibilities in relation to other meshes.” Buddha
Introduction
Aging is one of the highest risk factors known for most human diseases, including cancer, neurodegeneration, diabetes, and metabolic syndrome. Given the importance of these diseases in the population, as well as other age-associated phenotypes that contribute to frailty as people age, there is a keen interest to define and understand the aging process. In recent decades there have been regular intervals of excitement that “the key” to unlocking our understanding of aging has been discovered. However the number of keys has been increasing with the time spent looking for them and rather than coming to a clearer understanding (unifying theory), the number of hypotheses to define the aging process and explain it has been increasing. While this may be perceived as confusing, more realistically it reflects that as a field, the study of aging is still early in its development. It is at a stage where the process of aging continues to be defined and discoveries continue to help us realize there is still more underlying biology to understand. As a consequence, what we learn from model systems are very important for helping us to define and understand the aging process.
We begin by considering recent developments in the aging field in light of a fundamental property of biological systems – interconnectivity [1]. All levels of interaction contribute to the ultimate phenotype of an organism: interactions between tissues, between cells, between organelles, between metabolic pathways, between genes, between individual molecules and hormones [e.g. [2]]. With the help of network analysis, the complexity of such interactions can be visualized – though admittedly, not always in a manner of easy comprehension – to develop new ideas and questions relevant to the aging process. If we consider organelles as one type of subsystem within a network, then when organelle A declines with age, a connection with organelle B may result in B’s decline as well (depicted in Figure 1). These age-relevant connections can come in two flavors. In one case, a normal functional connection may become broken as organelle A declines – e.g. A normally provides a biosynthetic product to B. Alternatively, a novel connection is created, with pathological consequences for B, as A declines. It is also possible that more than one subsystem may be sensitive to aging, albeit through distinct routes. How and whether these types of events occur, is critical to developing a better understanding about aging.
Figure 1. A cascade of interactions between organelles help define the aging process.
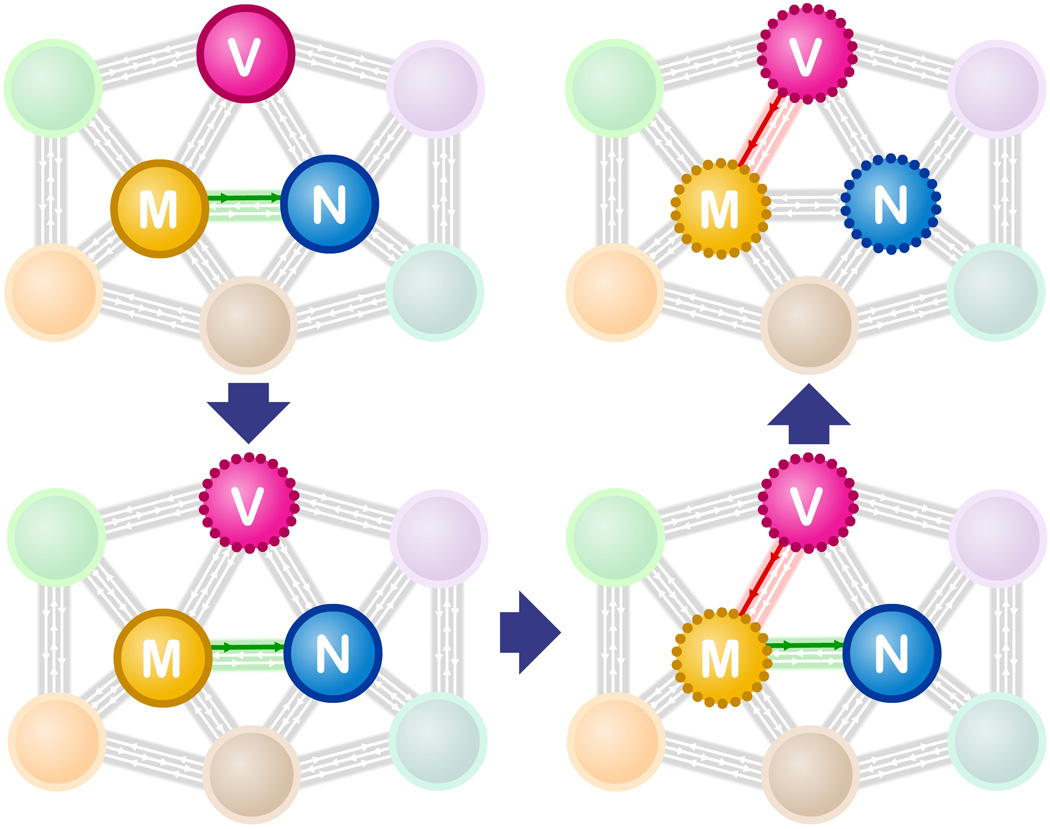
The myriad of connections between subsystems (colored spheres) in a cell are represented here as gray arrows. The vacuole (V), mitochondria (M) and nucleus (N) are represented in this network schematic. As described in the text, reduced vacuole function (indicated by a perforated circle – bottom left) early in a yeast cell’s life span, leads to an apparent pathological interaction (red arrow) with mitochondria that in turn leads to reduced mitochondrial function (bottom right). The dysfunctional mitochondria no longer produce sufficient iron-sulfur clusters (loss of green arrow), which are required for normal DNA replication & repair in the nucleus.
A second type of interaction important in the aging process is the response of a biological system to changes – i.e. the basis of homeostasis and biological anticipation. As an organism ages, what types of response or compensation occur, and what is the consequence of the response? For instance, is compensation “successful” and help to prevent dysfunction, or does compensation result in unintended (negative) consequences, throwing different, interconnected, subsystems into dysfunction?
Recent results illustrating the importance of such interconnectivity in the aging process are highlighted below.
An example of the aging process at the cellular level: inter-organelle dependency
An individual (mother) cell of the budding yeast, Saccharomyces cerevisiae, divides asymetrically and produces a finite number of daughter cells. This attribute has made it a model system for replicative cellular aging and has led the way for several discoveries that are shared with aging phenotypes in metazoa [3].
An example of subsystem dependency in the aging process is increased nuclear genome instability. In diploid lab strains carrying multiple heterozygous alleles, the frequency of loss-of-heterozygosity (LOH) increases 40–100 fold in the progeny of old mothers compared to progeny from young mothers [4]. The increase in LOH correlated with the daughters of old mother cells displaying mitochondrial dysfunction [5], including a reduced inner membrane potential (ΔΨ). The ΔΨ is required for many aspects of mitochondrial function, including import of nuclear encoded proteins such as those involved in iron-sulfur cluster (ISC) biosynthesis [6,7]. Indeed, a mitochondrial-related defect in ISC biosynthesis was shown to play a causal role in the nuclear genome instability of aging cells. This is consistent with ISCs being required cofactors in a number of enzymes involved in DNA replication and repair [8,9]. Thus several critical genome maintenance proteins may become dysfunctional as a result of the reduced levels of ISCs.
But what, then, caused a decline in the mitochondrial ΔΨ of aged mother cells? Here the power of prior knowledge from genome-wide genetic screens in young cells was leveraged to screen in aging cells for genes that delayed the onset of the age-associated mitochondrial defect. The genes identified led to the discovery that vacuole pH increased early in the lifespan of the yeast mother cell and that this change led to the reduced mitochondrial ΔΨ [10]. (The vacuole is the yeast lysosome and most acidic compartment of the cell.) The link between vacuolar pH increase and reduced mitochondrial membrane potential appears to be mediated by the vacuole’s reduced capacity to store neutral amino acids (import and storage requires an acidic vacuole). How reduction in neutral amino acid storage leads to a decline in mitochondrial membrane potential remains to be determined, but mitochondria-dependent catabolism of excess cytoplasmic amino acids may place a large burden on proton-dependent mitochondrial carrier proteins and, in the process, overwhelm mitochondrial ΔΨ.
When more than one aging phenotype occurs in the same cell: Are they connected?
Like vacuolar pH, another early asymmetry in age characteristics is the disproportionate accumulation of hydrogen peroxide in mother cells during cytokinesis - a phenomenon linked to an asymmetrical inheritance of pristine and active ROS defense systems [14]. Inactivation of peroxide reductases is one consequence of such a progressive accumulation of peroxides during aging in both yeast and rats [15–17] and that this inactivation impacts the rate of aging is evident by the fact that boosting the repair of damaged peroxiredoxins in yeast extends life span [16]. An obvious question emerging from such data is whether the repair of peroxiredoxin is bypassing or counteracting the detrimental effects of the early breakdown of vacuolar pH control and ISC defects, i.e. are the vacuolar/ICS and ROS/peroxiredoxin pathways of aging interconnected or independent?
Another canonical replicative aging phenotype in S. cerevisiae is the accumulation of extra chromosomal rDNA circles (ERCs) [18]. The yeast rDNA locus consists of 100–200 copies of a directly repeated unit containing a sequence that can act as an origin of replication. As a consequence of the structure of the rDNA locus, recombination can generate extrachromosomal circles. ERCs accumulate in mother cells with replicative age because they replicate and experience mother-biased segregation [19,20]. Although ERCs were hypothesized as a causal agent in replicative aging more than 15 years ago, the mechanism by which ERCs limit a mother cell’s life span is still a matter of conjecture, but recent data suggest that the abundance of ERCs in yeast mother cells may act as a sink for limiting replication factors [21]. Currently, ERC formation appears to be a stochastic event that is not obviously connected to the early changes in aging – vacuolar pH and hydrogen peroxide accumulation. Thus, does ERC formation represent another independent means of cell aging? If so, are different cells in a population aging via different means? Examination of terminal phenotypes of aged yeast cells, suggest that there are indeed different means to the end [22,23]. Lastly, if more than one subsystem decays via independent means in a cell, might they ultimately affect a common cellular subsystem that results in the cell’s demise? For instance, given that several DNA replication and repair factors are dependent upon ISCs, and that ERCs may be titrating away replication factors, could DNA replication completely “collapse” in some aging cells?
Examples of organelle/tissue interconnectivity behind life span extension
While nuclear, vacuolar, cytoplasmic, and mitochondrial defects can cause a sequential degeneration of interconnected subsystems, some organelle dysfunctions can actually extend life span. Recent examples from yeast highlight that mitochondrial dysfunctions can trigger a “successful” nuclear response that delays aging. One such interconnected pathway, described as “mitochondrial back-signaling”, involves inter-organelle coordination of ribosomal biosynthesis between mitochondria and the nucleus [24]. Another link between inter-organelle signaling and aging is provided by data demonstrating that inactivating members of the mitochondrial translation control (MTC) module required for COB and COX translation causes a robust sirtuin-dependent increased genomic silencing and extension of replicative life span [25]. While the data on MTC-deficient cells has been interpreted as demonstrating that the nuclear silencing apparatus senses and responds to functional defects in mitochondria through PKA/TOR signaling [26] it is possible that the effects of MTC deficiency on aging and silencing are more direct and involves mitochondrial proteins with a dual role in translation and signaling [25].
Consistent with mitochondrial dysfunction triggering a survival response in yeast, it is becoming clear that an analogous system might be conserved in metazoa. Mutation or RNAi perturbation of mitochondrial electron transport chain (ETC) subunits can extend longevity in both worms and flies [27–30]. In worms, the survival response involves the unfolded protein response of the mitochondria (UPRmt) [31] and might also impinge upon ROS signaling. Additionally, stoichiometric imbalance of the ETC components, by reduction of mitochondrial translation via RNAi or pharmacological inhibition, can also increase lifespan and depends upon activation of the UPRmt [32]. This phenomenon might be conserved in vertebrates as indicated by the extreme longevity of mice displaying polymorphisms in loci of the mitochondrial ribosome [32].
With the findings that organelle dysfunction can lead to the onset of stress responsive pathways that promote survival in unicellular and multicellular organisms, a crucial question in the field has been to uncover whether these responses function only within a cell, or whether there is broad communication across cells. In regards to age-related tissue decline, if each cell independently determined its own stress responsiveness a model would emerge whereby a certain proportion of cells would have to become unresponsive to give rise to tissue decline. However, if stress responsiveness within each cell of a tissue was coordinated by a master cell type, a different model would emerge whereby the ability to communicate stress would be the major driving force behind age-related tissue decline. Each model need not be mutually exclusive.
Aging in particular presents a difficult challenge towards the homeostatic regulation of metabolism, and age is one of the greatest risk factors for a wide range of endocrine-based diseases, including obesity, atherosclerosis, and diabetes [33]. This challenge appears to be exacerbated by a severely diminished capacity in older organisms to appropriately regulate cellular stress response pathways. In the nematode C. elegans, for example, the ability to activate the cytosolic heat shock response and the UPRER is abrogated in older animals [34]. The failed induction of stress responses can subsequently affect overall survivorship of the organism: stress responsive transcription factors are required for the longevity caused by both reduced insulin/IGF1-like signaling (IIS) and dietary restriction (DR) [35–37] . Similarly, the UPRmt is required for the lifespan extension observed in nematodes with mutations in mitochondrial genes [31,32].
Importantly, UPRER, cytoplasmic heat shock, and mitochondrial stress appear capable of regulating stress in a cell non-autonomous or endocrine-like fashion (Figure 2). If just two out of the 302 neurons in C. elegans are ablated, for example, the whole organism loses the capacity to upregulate its heat shock response [38]. Neuronal induction of the UPRmt is also capable of activating the UPRmt in the distal cells [31]. Furthermore, neuronal induction of one branch of the UPRER, the ire1/xbp-1 branch, can activate the UPRER in distal, non-neuronal tissues involving a neuroendocrine mechanism [33]. These observations suggest that additional compartments within the cell might be capable of communicating to more distal tissues such that they can coordinately regulate their stress response networks.
Figure 2. Communication of proteostasis stress networks between cells and tissues is essential for metazoan aging.

Signaling between cells is part of the aging process. In C. elegans, mitochondrial proteotoxic stress in the nervous system leads to mitokines being sent to other cells. This signaling elicits a mitochondrial stress response in distal cells of this metazoan and essential for the increased longevity of animals with mitochondrial proteome imbalance. Similarly, this phenomenon has been observed for the IRE-1/XBP-1 endoplasmic reticulum stress response as well as the heat shock response, both of which can extend lifespan when ectopically induced.
Is trans-cellular stress communication always good for the organism? Probably not: A growing body of evidence suggests that communication of stress from non-neuronal tissues can lead to cell non-autonomous tissue decline and decreased longevity. For example, inappropriate signaling of the UPRmt or UPRER from muscle cells in the worm results in decreased stress resistance and reduced longevity [31,33]. Additionally, excessive HSP90 in non-neuronal cells results in induction of the heat shock response in distal cells and a concomitant shortening of lifespan [39]. Finally, elegant work studying cellular senescence of the adipose tissue in mice has found that introduction of senescent cells into otherwise normal adipose reduces lipogenesis and adipose function through the action of senescent associated secreted factors [40].
Concluding remarks
The quality control systems of an organism’s organelles, tissues, and organs have traditionally been approached separately. Focusing instead on system interconnectivity, stress-sensitive nodes, and inter-organelle/tissue communication offers a different slant on damage propagation and the aging process. For example, part of the stochastic nature of survival may be understood in view of each subsystem displaying distinct predispositions to becoming non-functional in various individuals due to particular allelic makeups or to specific environmental and external factors – this could lead to waves of decline in different meshes of the network, each having a qualitatively different effect on survival/fitness. Furthermore, inter-organelle communications that produce successful compensatory responses, also depend on the spatial (tissue, organelle) origin of the signal. Thus, the node that fails first may have a greater impact on whether compensatory cross-talk responses will be successful or fail to bring the system back from the “tipping point”.
It is becoming increasingly clear that communication within cells and among cells aimed at restoring homeostasis under conditions of stress is a key feature of successful anti-aging programs. With the stochastic decline of survival with age, we must then begin to ask if there is a driving event to this stochasticity? Is it merely a failure of independent systems within a cell, among cells or could it be explained by the failure to communicate between cells? To date, at least three core aging pathways have been discovered that appear to transcend phyla: insulin/IGF1 signaling, the ETC pathway, and dietary restriction. Reduction in the activity of any one of these pathways results in lifespan extension and most have been shown to delay cell and tissue decline in one way or another. However, could these pathways also impinge on the efficiency of communication within cells and among cells? For example, it is possible that these pathways play a more prominent role in either propagating or perceiving signals of cellular stress, rather than increasing the robustness of “aging-sensitive” nodes in the homeostasis networks.
Finally, the concept of interconnectivity raises a number of obvious and intriguing questions. For example, what are the aging-sensitive nodes? Are these nodes conserved between cell types and species? What kinds of design principles make a node robust or sensitive to age-related changes? Do multiple aging pathways run in parallel due to the decline of multiple nodes or do such pathways converge and generate the same aging factors? How do different subsystems/meshes of the network cross-communicate with each other in a cell non-autonomous manner? Does such cross-talk also include decisions to destroy damaged subsystems, such as cells and organelles that have been pushed over the edge? We believe that these questions are worth pursuing in the quest for a better understanding of the aging process and how this process can be postponed.
Highlights.
Interconnectivity and homeostatic feedback are fundamental properties of biological systems.
Specific nodes in the interconnected network of an organism are more sensitive to change and thus mediate aging.
Interdependency between subsystems can lead to progressive decline by sequential collapse of homeostasis.
Cell communication between subsystems can compensate for age-associated decline and may extend life span.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Jacob F. La logique du vivant: une histoire de l’hérédité. Gallimard, Paris: 1970. [Google Scholar]
- 2.Soltow QA, Jones DP, Promislow DEL. A network perspective on metabolism and aging. Integr. Comp. Biol. 2010;50:844–854. doi: 10.1093/icb/icq094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Steinkraus KA, Kaeberlein M, Kennedy BK. Replicative aging in yeast: the means to the end. Annu Rev Cell Dev Biol. 2008;24:29–54. doi: 10.1146/annurev.cellbio.23.090506.123509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McMurray MA, Gottschling DE. An age-induced switch to a hyper-recombinational state. Science. 2003;301:1908–1911. doi: 10.1126/science.1087706. [DOI] [PubMed] [Google Scholar]
- 5.Veatch JR, McMurray MA, Nelson ZW, Gottschling DE. Mitochondrial dysfunction leads to nuclear genome instability via an iron-sulfur cluster defect. Cell. 2009;137:1247–1258. doi: 10.1016/j.cell.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neupert W, Herrmann JM. Translocation of proteins into mitochondria. Annu Rev Biochem. 2007;76:723–749. doi: 10.1146/annurev.biochem.76.052705.163409. [DOI] [PubMed] [Google Scholar]
- 7.Lill R, Hoffmann B, Molik S, Pierik AJ, Rietzschel N, Stehling O, Uzarska MA, Webert H, Wilbrecht C, Mühlenhoff U. The role of mitochondria in cellular iron-sulfur protein biogenesis and iron metabolism. Biochim Biophys Acta. 2012 doi: 10.1016/j.bbamcr.2012.05.009. [DOI] [PubMed] [Google Scholar]
- 8.Netz DJA, Stith CM, Stümpfig M, Köpf G, Vogel D, Genau HM, Stodola JL, Lill R, Burgers PMJ, Pierik AJ. Eukaryotic DNA polymerases require an iron-sulfur cluster for the formation of active complexes. Nat Chem Biol. 2012;8:125–132. doi: 10.1038/nchembio.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.White MF, Dillingham MS. Iron-sulphur clusters in nucleic acid processing enzymes. Curr. Opin. Struct. Biol. 2012;22:94–100. doi: 10.1016/j.sbi.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 10. Hughes AL, Gottschling DE. An early age increase in vacuolar pH limits mitochondrial function and lifespan in yeast. Nature. 2012;492:261–265. doi: 10.1038/nature11654. Evidence for a causal link between dysfunction of one organelle leading to dysfunction in another organelle with age; changes in vacuolar acidity lead to mitochondrial dysfunction. In addition, this link appears to be initiated by a defect in amino acid storage in the vacuole.
- 11.Aguilaniu H, Gustafsson L, Rigoulet M, Nyström T. Asymmetric inheritance of oxidatively damaged proteins during cytokinesis in Saccharomyces cerevisiae: a Sir2p dependent mechanism. Science. 2003;299:1751–1753. doi: 10.1126/science.1080418. [DOI] [PubMed] [Google Scholar]
- 12.Erjavek N, Larsson L, Grantham J, Nyström T. Accelerated aging and failure to segregate damaged proteins in Sir2 mutants can be suppressed by overproducing the protein aggregation-remodeling factor Hsp104p. Genes Dev. 2007;21:2410–2421. doi: 10.1101/gad.439307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu B, Larsson L, Caballero A, Hao X, Öling D, Grantham J, Nyström T. The Polarisome is required for segregation and retrograde transport of protein aggregates. Cell. 2010;140:257–267. doi: 10.1016/j.cell.2009.12.031. [DOI] [PubMed] [Google Scholar]
- 14.Erjavek N, Nyström T. Sir2p-dependent cytoskeleton formation and mitotic segregation of damaged proteins – a process regulating the antioxidant capacity of yeast daughter cells. Proc Natl Acad Sci USA. 2007;26:10877–10881. doi: 10.1073/pnas.0701634104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Musicco C, Capelli V, Pesce V, Timperio AM, Calvani M, Mosconi L, Zolla L, Cantatore P, Gadaleta MN. Accumulation of overoxidized peroxiredoxin III in aged rat liver mitochondria. Biochim Biophys Acta. 2009;1787:890–896. doi: 10.1016/j.bbabio.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 16. Molin M, Yang J, Hanzén S, Toledano MB, Labarre J, Nyström T. Life span extension and H(2)O(2) resistance elicited by caloric restriction require the peroxiredoxin Tsa1 in Saccharomyces cerevisiae. Mol Cell. 2011;43:823–833. doi: 10.1016/j.molcel.2011.07.027. This study demonstrated that caloric restriction requires the presence of the peroxredoxin Tsa1, and its repair enzyme Srx1, to extend the lifespan of yeast mother cells. It also showed that ectopically boosting the repair of hyperoxidized Tsa1 is sufficient to extend life span in the absence of caloric restriction.
- 17.Nyström T, Yang J, Molin M. Peroxiredoxins, gerontogenes linking aging to genome instability and cancer. Genes Dev. 2012;26:2001–2008. doi: 10.1101/gad.200006.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sinclair DA, Guarente L. Extrachromosomal rDNA circles--a cause of aging in yeast. Cell. 1997;91:1033–1042. doi: 10.1016/s0092-8674(00)80493-6. [DOI] [PubMed] [Google Scholar]
- 19.Shcheprova Z, Baldi S, Frei SB, Gonnet G, Barral Y. A mechanism for asymmetric segregation of age during yeast budding. Nature. 2008;454:728–734. doi: 10.1038/nature07212. [DOI] [PubMed] [Google Scholar]
- 20.Gehlen LR, Nagai S, Shimada K, Meister P, Taddei A, Gasser SM. Nuclear geometry and rapid mitosis ensure asymmetric episome segregation in yeast. Curr Biol. 2011;21:25–33. doi: 10.1016/j.cub.2010.12.016. [DOI] [PubMed] [Google Scholar]
- 21. Kwan EX, Foss EJ, Tsuchiyama S, Alvino GM, Kruglyak L, Kaeberlein M, Raghuraman MK, Brewer BJ, Kennedy BK, Bedalov A. A Natural Polymorphism in rDNA Replication Origins Links Origin Activation with Calorie Restriction and Lifespan. PLoS Genet. 2013;9:e1003329. doi: 10.1371/journal.pgen.1003329. A quantitative trait locus (QTL) is identified, and then verified, as a major contributor to explain the difference between two strains with different lifespans.
- 22.Xie Z, Zhang Y, Zou K, Brandman O, Luo C, Ouyang Q, Li H. Molecular phenotyping of aging in single yeast cells using a novel microfluidic device. Aging Cell. 2012;11:599–606. doi: 10.1111/j.1474-9726.2012.00821.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee SS, Avalos Vizcarra I, Huberts DHEW, Lee LP, Heinemann M. Whole lifespan microscopic observation of budding yeast aging through a microfluidic dissection platform. Proc Natl Acad Sci USA. 2012;109:4916–4920. doi: 10.1073/pnas.1113505109. These two papers allow individual yeast cells to be monitored microscopically throughout their entire lifespan.
- 24.Heeren G, Rinnerthaler M, Laun P, von Seyerl P, Kössler S, Klinger H, Hager M, Bogengruber E, Jarolim S, Simon-Nobbe B, et al. The mitochondrial ribosomal protein of the large subunit, Afo1p, determines cellular longevity through mitochondrial back-signaling via TOR1. Aging. 2009;13:622–636. doi: 10.18632/aging.100065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Caballero A, Ugidos A, Liu B, Öling D, Molin M, Kvint K, Hao X, Mignat C, Nachin L, Molin M, Nyström T. Absence of mitochondrial translation control proteins extend life span by activating sirtuin-dependent silencing. Mol Cell. 2011;42:390–400. doi: 10.1016/j.molcel.2011.03.021. [DOI] [PubMed] [Google Scholar]
- 26.Chen XJ. The search for nonconventional mitochondrial determinants of aging. Mol Cell. 2011;42:271–273. doi: 10.1016/j.molcel.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Copeland JM, Cho J, Lo T, Jr, Hur JH, Bahadorani S, Arabyan T, Rabie J, Soh J, Walker DW. Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Curr Biol. 2009;19:1591–1598. doi: 10.1016/j.cub.2009.08.016. [DOI] [PubMed] [Google Scholar]
- 28.Dillin A, Hsu AL, Arantes-Oliveira N, Lehrer-Graiwer J, Hsin H, Fraser AG, Kamath RS, Ahringer J, Kenyon C. Rates of behavior and aging specified by mitochondrial function during development. Science. 2002;298:2398–2401. doi: 10.1126/science.1077780. [DOI] [PubMed] [Google Scholar]
- 29.Lee SS, Lee RY, Fraser AG, Kamath RS, Ahringer J, Ruvkun G. A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat Genet. 2003;33:40–48. doi: 10.1038/ng1056. [DOI] [PubMed] [Google Scholar]
- 30.Feng J, Bussière F, Hekimi S. Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans. Dev Cell. 2001;1:633–644. doi: 10.1016/s1534-5807(01)00071-5. [DOI] [PubMed] [Google Scholar]
- 31. Durieux J, Wolff S, Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011;144:79–91. doi: 10.1016/j.cell.2010.12.016. Not every mitochondria contributes to the aging process equally, but rather key mitochondria from the nervous system coordinate stress resistance and longevity in the worm. Signaling from the nervous system to the periphery in response to neuronal mitochondrial proteotoxic stress via a secreted signal, termed a mitokine.
- 32. Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, Williams RW, Auwerx J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. 2013;497:451–457. doi: 10.1038/nature12188. Using recombinant inbred lines of mice loci linked to extended were uncovered to mediate expression of the mitochondrial ribosome (MRP). Genetic and pharmacological manipulation of the MRP resulted in increase lifespan in the worm and induction of the mitochondrial UPR in both worms and mice.
- 33. Taylor RC, Dillin A. XBP-1 Is a Cell-Nonautonomous Regulator of Stress Resistance and Longevity. Cell. 2013;153:1435–1447. doi: 10.1016/j.cell.2013.05.042. Induction of the ER UPR in the nervous system can send a cell non-autonomous signal via neurosecretion to distal cells to specifically increase ER stress resistance and longevity. This signal is distinct from the mitokine.
- 34.Ben-Zvi A, Miller EA, Morimoto RI. Collapse of proteostasis represents an early molecular event in Caenorhabditis elegans aging. Proc Natl Acad Sci USA. 2009;106:14914–14919. doi: 10.1073/pnas.0902882106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Steinkraus KA, Smith ED, Davis C, Carr D, Pendergrass WR, Sutphin GL, Kennedy BK, Kaeberlein M. Dietary restriction suppresses proteotoxicity and enhances longevity by an hsf-1-dependent mechanism in Caenorhabditis elegans. Aging Cell. 2008;7:394–404. doi: 10.1111/j.1474-9726.2008.00385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ogg S, Paradis S, Gottlieb S, Patterson GI, Lee L, Tissenbaum HA, Ruvkun G. The Fork head transcription factor DAF 16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature. 1997;389:994–999. doi: 10.1038/40194. [DOI] [PubMed] [Google Scholar]
- 37.Lin K, Dorman JB, Rodan A, Kenyon C. daf-16: An HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science. 1997;278:1319–1322. doi: 10.1126/science.278.5341.1319. [DOI] [PubMed] [Google Scholar]
- 38.Prahlad V, Cornelius T, Morimoto RI. Regulation of the cellular heat shock response in Caenorhabditis elegans by thermosensory neurons. Science. 2008;320:811–814. doi: 10.1126/science.1156093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van Oosten-Hawle P, Porter RS, Morimoto RI. Regulation of organismal proteostasis by transcellular chaperone signaling. Cell. 2013;153:1366–1378. doi: 10.1016/j.cell.2013.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232–236. doi: 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]