

Abstract
Background
2C-methyl-D-erythritol-2,4-cyclodiphosphate synthase (IspF) catalyzes the conversion of 4-diphosphocytidyl-2C-methyl-D-erythritol-2-phosphate to 2C-methyl-D-erythritol-2,4-cyclodiphosphate and cytidine monophosphate in production of isoprenoid-precursors via the methylerythritol phosphate biosynthetic pathway. IspF is found in the protozoan Plasmodium falciparum, a parasite that causes cerebral malaria, as well as in many Gram-negative bacteria such as Burkholderia cenocepacia. IspF represents a potential target for development of broad-spectrum antimicrobial drugs since it is proven or inferred as essential in these pathogens and absent from mammals. Structural studies of IspF from these two important yet distinct pathogens, and comparisons with orthologues have been carried out to generate reagents, to support and inform a structure-based approach to early stage drug discovery.
Results
Efficient recombinant protein production and crystallization protocols were developed, and high-resolution crystal structures of IspF from P. falciparum (Emphasis/Emphasis>IspF) and B. cenocepacia (BcIspF) in complex with cytidine nucleotides determined. Comparisons with orthologues, indicate a high degree of order and conservation in parts of the active site where Zn2+ is bound and where recognition of the cytidine moiety of substrate occurs. However, conformational flexibility is noted in that area of the active site responsible for binding the methylerythritol component of substrate. Unexpectedly, one structure of BcIspF revealed two molecules of cytidine monophosphate in the active site, and another identified citrate coordinating to the catalytic Zn2+. In both cases interactions with ligands appear to help order a flexible loop at one side of the active site. Difficulties were encountered when attempting to derive complex structures with other ligands.
Conclusions
High-resolution crystal structures of IspF from two important human pathogens have been obtained and compared to orthologues. The studies reveal new data on ligand binding, with citrate coordinating to the active site Zn2+ and when present in high concentrations cytidine monophosphate displays two binding modes in the active site. Ligand binding appears to order a part of the active site involved in substrate recognition. The high degree of structural conservation in and around the IspF active site suggests that any structural model might be suitable to support a program of structure-based drug discovery.
Keywords: Antimicrobial drug target, Isoprenoid biosynthesis, X-ray crystallography, Zn2+-dependent enzyme
Background
Isoprenoids contribute to a myriad of essential biological functions, for example as the prenyl groups that anchor proteins to membranes, as components of respiratory membranes, enzyme cofactors, and as scaffolds for the assembly of glycan structures [1-4]. Two distinct biosynthetic routes supply the universal isoprenoid-precursors isopentenyl pyrophosphate and dimethylallyl pyrophosphate. The mevalonate (MVA) pathway is present in animals, fungi and archaea while the 2C-methyl-D-erythritol-4-phosphate (MEP) pathway is widely distributed in bacteria, algae and several apicomplexan parasites, including Plasmodium species responsible for malaria [5,6]. Both MVA and MEP pathways occur in plants; the former operates in the cytoplasm and the latter in chloroplasts [7]. In most apicomplexan parasites the components of the MEP pathway are localized to an organelle known as the apicoplast, a relict chloroplast, which is also responsible for additional biosynthetic processes [8]. The organelle, which resembles a cyanobacterium is thought to have been acquired from an ancestral endosymbiotic event with an alga. All MEP pathway enzymes in P. falciparum are synthesized in the cytoplasm but they carry an apicoplast targeting sequence, which then drives localization [8]. The disruption of apicoplast replication results in parasites that are isopentenyl pyrophosphate auxotrophs [9].
Part of the interest in the MEP pathway concerns antimicrobial drug research and several observations support the idea that the pathway contains potential drug targets, enzymes whose inhibition may provide broad-spectrum antimicrobial activity [5,6]. We identified the Zn2+-dependent 2C-methyl-D-erythritol-2,4-cyclodiphosphate synthase [EC: 4.6.1.12] the fifth enzyme in the pathway as a target of interest. This enzyme, also known as IspF, catalyzes the conversion of 4-diphosphocytidyl-2C-methyl-D-erythritol-2-phosphate (CDP-MEP) to 2C-methyl-D-erythritol-2,4-cyclodiphosphate (MEcPP) and cytidine monophosphate (CMP, Figure 1). We considered a diverse set of criteria that have been established as key with respect to target assessment for early stage antimicrobial drug discovery [10]. These criteria include genetic and chemical validation of the target, and the availability of accurate structural information and appropriate reagents to guide the development of structure-activity relationships. Three primary factors were taken into consideration. First, genetic methods have identified that individual components of the pathway, including IspF are essential for bacterial survival. For example, the essentiality of the ispF gene for growth and survival of Bacillus subtilis, Escherichia coli and Mycobacterium tuberculosis is established [11-15]. Secondly, this pathway is absent from humans [5,6] potentially mitigating against toxicity issues. Thirdly, chemical validation of the pathway is provided by the antibacterial fosmidomycin, a potent inhibitor of 1-deoxy-D-xylulose 5-phosphate reductoisomerase, an enzyme that contributes to an early stage in the pathway. This last point regarding validation is reinforced by further work, which shows that fosmidomycin and derivatives are also efficacious in killing P. falciparum[16-18].
Figure 1.

Reaction catalyzed by IspF. A catalytic Zn2+ aligns and polarizes the β-phosphate to support nucleophilic attack by the 2-phosphate to produce MEcPP and CMP.
Previous studies have provided insight into aspects of IspF structure, specificity, and mechanism [19-22]. The structure of IspF has been derived from a dozen species in addition to the two new ones disclosed in this publication. More recently new IspF ligands including potent inhibitors have been identified [23-25], some of which have also been shown to inhibit the growth of P. falciparum within infected erythrocytes with respectable IC50 values of 1.4-1.6 μM [26]. These approaches have been complemented by NMR-based fragment screening and co-crystallization efforts targeting IspF from B. pseudomallei (BpIspF) [27].
Aims
We sought to further inform the assessment of IspF as a drug target with crystallographic studies of the enzyme from two serious human pathogens, the Gram-negative bacterium Burkholderia cenocepacia (BcIspF) and the parasitic protozoan Plasmodium falciparum (PfIspF).
Results and discussion
The structure of IspF
Efficient recombinant protein production systems, purification protocols and reproducible crystallization conditions have been established for BcIspF and PfIspF leading to yields of up to 25 mg L-1 and 2.5 mg L-1of E. coli culture respectively. High-resolution crystal structures of BcIspF in complex with CMP or citrate, and of PfIspF in complex with CDP, as well as an unliganded PfIspF structure have been determined. Crystallographic statistics are presented in Tables 1 and 2.
Table 1.
Crystallographic statistics for Pf IspF
| Structure/PDB | Pf IspF:CDP/4C81 | Unliganded Pf IspF/4C82 |
|---|---|---|
| Space group |
H3 |
H3 |
| Unit cell dimensions |
|
|
|
a, b, c (Å) |
84.9, 84.9, 101.0 |
84.2, 84.2, 102.5 |
| Resolution range
a
(Å) |
41.63-1.60 (1.69-1.60) |
29.71-2.00 (2.11-2.00) |
| No. reflections |
181781 (16968) |
41802 (5780) |
| Unique reflections |
35607 (5194) |
18068 (2622) |
| Completeness (%) |
99.4 (99.3) |
98.6 (97.0) |
|
Rmerge
b
(%) |
9.3 (42.0) |
9.4 (48.2) |
| Multiplicity |
5.1 (3.3) |
2.3 (2.2) |
| < I/σ(I)> |
11.1 (2.6) |
5.3 (2.0) |
| Wilson B (Å2) |
24.4 |
32.0 |
|
Rwork
c
/Rfree
d
(%) |
14.4, 16.5 |
19.4, 22.6 |
| Number of residues/waters/ligands |
156, 85 waters, 1 CDP, 2 Zn2+, 3 Cl-, 1 SO42- |
156, 94 waters, 2 Zn2+, 1 SO42- |
| RMSD bond lengths (Å)/ |
0.005 |
0.019 |
| Bond angles (º) |
1.000 |
2.087 |
| Mean B-factors (Å2) |
|
|
| Overall |
22.9 |
37.6 |
| Main chain, side chain |
25.1, 33.8 |
34.2, 39.5 |
| Waters |
35.2 |
47.8 |
| Ligands |
34.9, 28.3 (0.6, 0.4 CDP) |
44.5 (Zn2+, SO42-) |
| 35.8 (Zn2+, Cl-, SO42-) | ||
| Ramachandran plot |
|
|
| Favoured regions (%) |
98.6 |
95.3 |
| Allowed regions (%) |
0.7 |
4.0 |
| Outliers | 1 outlier: Asp79 | 1 outlier: Asp79 |
aValues in parentheses refer to the highest resolution shell. bR merge = ∑ hkl ∑ i |I i (hkl) − < I(hkl) > |/∑ hkl ∑ i I i (hkl); where I i (hkl) is the intensity of the ith measurement of reflection hkl and < I(hkl) > is the mean value of I i (hkl) for all i measurements. cR work = ∑ hkl ‖F o | ‒ |F c ‖/∑|F o |, where F o is the observed structure factor and F c is the calculated structure factor. dR free is the same as R work except calculated with a subset, 5%, of data that are excluded from the refinement calculations.
Table 2.
Crystallographic statistics for Bc IspF
| Structure/PDB | Bc IspF:CMP/4C8G | Bc IspF:2CMP/4C8E | Bc IspF:citrate/4C8I |
|---|---|---|---|
| Space group |
C2 |
C2 |
C2 |
| Unit cell dimensions |
|
|
|
|
a, b, c (Å), |
97.2, 88.6, 72.4, |
97.9, 89.3, 73.0, |
131.7, 52.5, 72.1, |
| β |
103.9° |
104.2° |
94.6° |
| Resolution range (Å) |
29.84-2.00 (2.10-2.00) |
19.32-1.90 (2.00-1.90) |
40.90-2.00 (2.10-2.00) |
| No. reflections |
98451 (13335) |
164120 (24760) |
119715 (15957) |
| Unique reflections |
38409 (5579) |
47918 (6972) |
33460 (4696) |
| Completeness (%) |
96.0 (95.4) |
99.9 (100.0) |
99.4 (96.1) |
|
Rmerge (%) |
11.0 (32.2) |
13.8 (32.3) |
3.2 (7.8) |
| Multiplicity |
2.6 (2.4) |
3.4 (3.6) |
3.6 (3.4) |
| < I/σ(I)> |
5.9 (2.7) |
6.2 (3.1) |
23.9 (12.8) |
| Wilson B (Å2) |
16.5 |
16.3 |
18.7 |
|
Rwork/Rfree (%) |
23.7, 28.0 |
20.4, 25.1 |
15.6, 19.8 |
| Number of residues |
458, 173 waters, |
483, 420 waters, |
460, 320 waters, |
| Waters/ligands |
3 CMP, 3 Zn2+, 1 Mg2+, 1 PO43- |
6 CMP, 3 Zn2+, 1 SO42- |
3 citrates,3 Zn2+, 1 PO43- |
| 1di(hydroxyethyl)ether | |||
| RMSD bond lengths (Å)/ |
0.020 |
0.011 |
0.020 |
| bond angles (º) |
2.198 |
1.526 |
2.101 |
| Mean B-factors (Å2) |
|
|
|
| Overall |
14.5 |
17.2 |
18.9 |
| Main chain, side chain |
13.6, 15.3 |
18.7, 20.4 |
16.2, 20.1 |
| Waters |
16.5 |
29.9 |
26.7 |
| Ligands |
15.1 (CMP) 24.8 (Zn2+, Mg2+, PO43-) |
18.2 (CMP 1) |
23.4, 36.8, 43.1 (citrates) |
| 28.7 (CMP 2) |
26.1 (Zn2+, PO43-) |
||
| 25.0 (other ligands) | |||
| Ramachandran plot |
|
|
|
| Favoured regions (%) |
96.9 |
97.8 |
97.7 |
| Allowed regions (%) |
2.2 |
2.2 | 2.3 |
| Outliers | 4 outliers: Ser37A/C, Tyr29B, Gly17C |
The calculated masses of the BcIspF and PfIspF subunits are 19.3 kDa and 20.5 kDa respectively and the polypeptide folds into a single α/β domain that consists of a four-stranded mixed β-sheet on one side with three α-helices on the other (Figure 2). IspF forms a stable homotrimer as observed in size-exclusion chromatography, which returns an estimate of 60 kDa for each. In all IspF crystal structures three subunits form a compact bell-shaped assembly approximately 45 Å by 60 Å in the axial and equatorial dimensions respectively (Figure 2). A surface area approximately equivalent to that of one subunit is buried on oligomerization. The accessible surface area (ASA) of an IspF monomer averages out as approximately 8130 Å2 for BcIspF and 8680 Å2 for PfIspF. The ASA for the BcIspF trimer is 16080 Å2, and for the PfIspF trimer 18180 Å2. In the case of BcIspF the asymmetric unit consists of three subunits, chains A, B, C, which are related by three-fold non-crystallographic symmetry (NCS). NCS is high with least squares overlays of 161Cα positions in the range 0.2 - 0.3 Å. One subunit is present in the asymmetric unit of PfIspF and the trimer is generated by the symmetry operations (−x + y, -x, z) and (−y, x-y, z). A least squares superimposition of Cα positions from both PfIspF structures matches all 156 residues with an RMSD of 0.3 Å. Since there are no pronounced differences between the CDP bound and unliganded PfIspF structures only the former is discussed.
Figure 2.
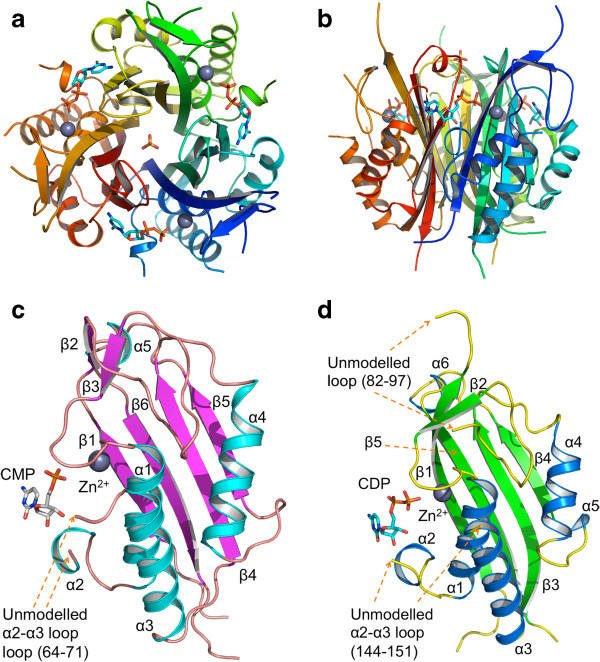
Overall structure of IspF. IspF forms a homotrimer. a) Axial view. b) Side view. The figure has been drawn using the coordinates for PfIspF in complex with CDP; a trimer has been generated from symmetry-related subunits. Subunits (displayed as ribbons) are colored blue, orange and green respectively. Zn2+ cations are displayed as grey spheres, sulfate and CDP are displayed as sticks. The secondary structure of PfIspF and BcIspF subunits is shown in c) and d) respectively, similarly orientated. The location of missing loops is highlighted with arrows.
Structural comparisons of IspF orthologues from the Protein Data Bank (PDB) were carried out using the DALI server [28]. Pairwise sequence identities range from 28 to 90%, Z scores from 18 to 31 and RMSD values from 0.3 to 2.5 Å. The apicoplast targeting sequence of Plasmodium spp. and the chloroplast targeting sequence of Arabidopsis thaliana IspF were excluded in the calculation of sequence identities. Using representative IspF structures from each of the twelve unique organisms in the PDB, an average RMSD of Cα positions was calculated against both proteins. The mean of the average RMSD was 1.5 Å and therefore the comparisons are indicative of an enzyme that has a highly conserved structure.
The two IspF variants studied here, with sequence identity of 38%, a DALI Z score 23 and RMSD value typically around 1.7 Å, can be taken to represent a comparison of structures that are amongst the most divergent. A structure-based sequence alignment of BcIspF with PfIspF is shown in Figure 3 and this, together with comparisons based on consideration of 854 IspF sequences, provides a context in which to discuss key features. PfIspF contains a 19 residue insertion at the N-terminus, Lys76 - Glu94, immediately after β1 that does not align with bacterial IspF sequences. Here, residues Tyr82 - Phe97 could not be modeled due to the lack of ordered electron density (Figure 2c).
Figure 3.

Structure-based sequence alignment of BcIspF and PfIspF. The alignment has been annotated with elements of secondary structure. Strictly conserved residues are highlighted in black. Three residues that coordinate Zn2+ are marked with a yellow star. Residues that bind CDP or CMP 1, are marked with a grey disk and those that bind CMP 2 with an orange triangle. Residues that bind MEcPP in EcIspF [20] are marked with a magenta triangle. A conserved glutamate that binds either Mg2+ or Mn2+ in EcIspF structures is highlighted with a cyan star. This figure was prepared using ALINE[29].
The most similar structures of each new IspF structure were noted. BcIspF aligns 151 residues with an RMSD of 0.4 Å to BpIspF (Z = 31, 90% sequence identity). PfIspF aligns 143Cα positions with an RMSD of 0.8 Å, and 151 residues with an RMSD of 1.2 Å with subunits of the enzyme from P. vivax (PvIspF, Z = 25, 66% sequence identity) and A. thaliana (AtIspF, Z = 25, 42% sequence identity) respectively. The PvIspF structure at 2.3 Å resolution was deposited in the PDB without an associated publication [PDB: 3B6N]. Although Zn2+ has been modeled at the active site (discussed below), the corresponding B-factor of 119 Å2 is 2.6 times greater than the average B-factor of the protein atoms, 46 Å2, and the side chain of a coordinating histidine, His170, is poorly oriented. We conclude that a water molecule may be a more likely occupant of this metal binding site. In contrast, in our structure of a Plasmodium IspF, which is at 1.60 Å resolution, the B-value for Zn2+ of 20 Å2 is lower than the average B-factor over all the protein atoms of 29 Å2.
The enzyme active site is located in a cleft at the interface of two subunits (Figure 2a) and here Zn2+ is coordinated by the side chains of two histidines and an aspartate [19]. The coordinating residues are Asp10, His12 and His44 in BcIspF, Asp71, His73 and His123 in PfIspF (Figures 3 and 4). These residues are strictly conserved in IspF. In the E. coli enzyme (EcIspF) Glu135, helps coordinate either a Mg2+ or Mn2+ together with the bridging phosphates of the substrate or CDP [19,20]. Although the residue is conserved as Glu137/216 (Figure 4) we do not observe any cation binding here. In 81% of IspF sequences a glutamate is observed at this position, and this is conservatively replaced by aspartate in a further 18%.
Figure 4.

Superposition of the active site in BcIspF and PfIspF. Residues that bind CMP or CDP are displayed as sticks. Sequence numbers for BcIspF are followed by those for PfIspF, and residues contributed from the adjacent subunit are marked with’. BcIspF and PfIspF are colored cyan and green respectively. Key interactions are detailed in the text.
The highly conserved histidine and serine combination (PfIspF: His115-Ser116, BcIspF: His36-Ser37, identical in over 96% IspF sequences) located on the loop that leads to α1, adjacent to the Zn2+ binding site (Figure 2c, d), display elevated B-factors in all structures and in one chain of the BcIspF:CMP complex the electron density was of insufficient quality to allow the region to be modelled, indicating a degree of flexibility. Electron density is absent or poorly defined for the loop linking α2 to α3 in many of the IspF structures. This segment occurs at one side of the active site (Asp144 - Asn151 in PfIspF) and (Phe63 - Ala73 in BcIspF). This α2 - α3 loop could be modeled in the BcIspF:2CMP complex although the residues have elevated B-factors (average 47 Å2), compared to the average B-value for the entire protein (19 Å2). The same loop was also modelled for chain A in the BcIspF-citrate complex. These observations are consistent with the inference that the α2 - α3 loop on one side of the active site is flexible [16,17].
A hydrophobic cavity occurs at the core of the IspF trimer. In BcIspF the cavity is lined by the side chains of Tyr9, Val11, Ile101 and Tyr141, and the floor of the cavity is formed by a ring of six hydrophobic residues, Val3 and Leu155 donated by all three subunits. Similarly in PfIspF side chains of Tyr70, Ile72, Ile180 and Val220 line this cavity, and the side chains of Phe230 form the floor of the cavity. Anions such as sulfate, phosphate or pyrophosphate bind at the entrance to this cavity in several IspF structures, for example phosphate is present in AtIspF [30]. In the structure of BcIspF crystallized with (NH4)2SO4 as a precipitant, a sulfate is bound by the side chain of Arg144. In PfIspF, a sulfate binds close to this site but dislodged such that anion coordination involves the main chain amides of nearby residues, Val220 from all three subunits, rather than a basic side chain. When BcIspF is crystallized in the presence of Na2HPO4, Tyr141 amide groups bind a phosphate in a similar fashion. Isoprenoid-species such as geranyl- or farnesyl-pyrophosphate have been observed in the central cavity of some IspF trimers and a possible role in feedback regulation has been suggested [31-33]. A molecule of di(hydroxyethyl)ether; a likely decomposition product or impurity of PEG 3350 used in crystallization, occupies the cavity in the BcIspF:2CMP structure (Table 2), possibly mimicking a prenyl chain. A solvated Mg2+ with octahedral coordination is present in the cavity of the BcIspF:CMP structure forming interactions similar to those observed in the structure of IspF from B. pseudomallei[27], data not shown.
Complexes with CMP and CDP
Residues from both subunits interact with and position cytidine nucleotides in the active site. These compounds constitute fragments of the substrate and provide clues about aspects of molecular recognition. The structure-based sequence alignment (Figure 3) and overlay of the active sites of BcIspF:CMP and PfIspF:CDP complexes (Figure 4) indicate that the key enzyme-ligand interactions and aspects of the structure that are implicated in substrate specificity and the mechanism are highly conserved. Note that in the PfIspF:CDP complex two conformers of CDP are present that differ in the orientation of the β-phosphate group. These refined satisfactorily at occupancy levels of 0.6 and 0.4.
In BcIspF, carbonyl groups from Ala102 and cis-Pro105 accept hydrogen bonds donated by cytosine N4, and the amide groups of Leu107 and Ala108 donate hydrogen bonds to cytosine N3 and O2 respectively. The ribose O2’ forms a hydrogen bond with Asp58 from the adjacent subunit while the amide of Gly60, also from the adjacent subunit, hydrogen bonds to the ribose O3’. Thr135 amide and side chain hydroxyl groups form hydrogen bonds to the α-phosphate. Lys134 NZ interacts with the CMP α-phosphate, and may contribute to catalysis by helping to position the transition-state intermediate or, in conjunction with Zn2+, to align the substrate for nucleophilic attack. This interaction in BcIspF is reminiscent of that previously reported for the active site Lys132 in Thermus thermophilus IspF [34], except that in that case the lysine interacts with the β-phosphate. The side chain of Lys106 is positioned over the cytosine and contributes van der Waals contacts to stabilize the ligand pose. For clarity this residue is omitted in Figure 4.
We analysed the levels of conservation of these eight IspF residues detailed as interacting with CMP or CDP. The residues for which the side chain is key, the glycine and cis-proline are highly conserved in IspF, at greater than 95%. Where main chain functional groups are used the nature of the side chain is less important and the level of conservation drops to 37% for the equivalent of Leu107 to 62% for Ala102 in BcIspF. Lys134 is anomalous. This is only lysine in 20% of the sequences and is more often a threonine, 76% of sequences. Presumably the reduction in size allows space that a water molecule could occupy to help facilitate substrate binding.
Surprisingly, a second molecule of CMP was observed in the active site when BcIspF was co-crystallized in the presence of 10 mM ligand. This second CMP is likely due to the high concentration used and represents an artifact of crystallization, unlikely to have physiological significance. This new cytidine-binding site is referred to as position 2 and residues that bind this ligand are from the same subunit that binds the active site Zn2+, indeed the CMP 2 phosphate coordinates the metal ion (Figure 5). The binding of CMP 2 within the three active sites of the asymmetric unit is very similar, for example the main chain conformation of the α2-α3 loop and the positions of Leu78 and His36 interacting with van der Waals forces on either side of the pyrimidine and helping to position it is essentially the same. In addition the position of Phe63 that helps to position Leu78 is also conserved. Phe63 is strictly conserved in 96% of IspF sequences, Leu78 in 77%. However, there are differences in the orientation of two side chains that change the detail of the hydrogen bonding interactions with CMP 2. In active site A (Figure 6) two solvent mediated links are noted involving the carbonyl groups of Ser64, Phe70 and Ala73 and cytosine O2 and N3. A direct hydrogen bond is formed between N4 and the Ala73 carbonyl. The Phe70 and Ala73 carbonyl groups position a water molecule that binds cytosine N3 and the Ala73 carbonyl also interacts directly with cytosine N4. The carboxylate of Asp65 binds to the ribose O2’. In the other active sites (not shown), different rotamers of Ser37 place the side chain OG to accept a hydrogen bond from N4 but for Asp65 the rotamer orientations results in functional groups too distant for an interaction with the ribose. Overlay with the EcIspF-product complex shows that CMP 2 binds in the same location as MEcPP [20] and indeed exploits interactions with the same residues, the majority of which are conserved (Figures 3 and 7). Specifically, in common with observations regarding recognition of the pyrimidine moiety of substrates we note that residues that use side chain functional groups to interact with ligands in the MEcPP binding site are well conserved, those that use main chain groups less so. Ser37 is strictly conserved and Asp65 occurs in about 70% of IspF sequences but Ser64, Phe70 and Ala73 are strictly conserved in 4%, 43% and 58% respectively. The latter two residues are not conserved in PfIspF, being replaced by asparagine and lysine respectively (Figure 3).
Figure 5.

Binding pose of two cytidine nucleotides in BcIspF. Hydrogen bonding interactions between BcIspF, water molecules and both CMP molecules in one active site are depicted with black dashed lines. Residues that contribute to binding interactions only via the main chain are depicted without side chain atoms. Solvent molecules are depicted as red spheres. Components of one subunit are shown with C atoms yellow and for the other subunit, cyan. Electron density is displayed for CMP 2 as green chicken-wire corresponding to an |Fo-Fc| map (omit map) contoured at 2.5 σ.
Figure 6.

Key residues and interactions to bind CMP 2 in BcIspF. The color scheme is the same as used in Figure 5.
Figure 7.
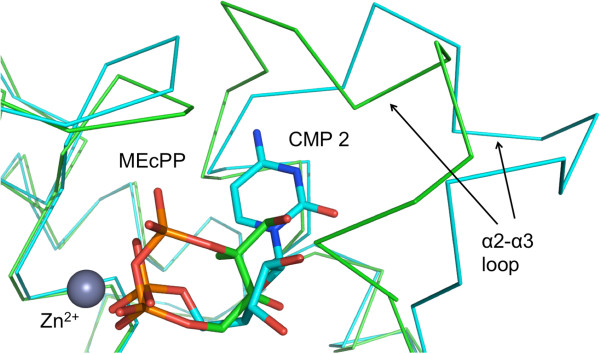
CMP 2 occupies the same site as MEcPP. An overlay of the EcIspF product complex and BcIspF:CMP 2. CMP 2 binds in a similar site as MEcPP and also coordinates Zn2+. The α2-α3 loop at one side of the binding site (EcIspF: Phe61-Ser73; BcIspF: Phe63-Ser75) adopts a different conformation.
The binding pose of CMP 2 offers clues about molecular features that might be considered for ligand design targeting this area of the active site. Potential ligands could, for example, aim to extend out to recreate the hydrogen bonding interactions displayed by the two ordered water molecules shown in Figure 6. It might also be useful to test placement of a basic component to interact with the acidic Asp65.
BcIspF-citrate complex
Attempts to produce unliganded BcIspF crystals for ligand or fragment soaking experiments initially appeared to be successful and highly ordered samples were obtained. However, citrate was present in the best crystallization conditions and the electron density maps were consistent with this molecule coordinating the metal ion and therefore complicating ligand-binding studies. Two binding modes were observed. Citrate was well defined in one active site with an average B-factor of about 23 Å2, but less so in the other two where it was modeled with occupancy 0.75 and the average B-factors are approximately 37 and 43 Å2. For comparison, the average B-factor for protein atoms in this structure is approximately 19 Å2. The best-defined citrate (Figure 8) binds to the active site Zn2+via both carboxylate and hydroxyl groups and this is also observed at one other site. Here the citrate forms hydrogen-bonding interactions with the main chain amides of His36 and Ser37, and side chains of Asp65 and Lys134. The interaction involving Asp65 is likely a consequence of the low pH at which crystals were obtained. Additional solvent mediated interactions to link citrate to the protein also occur (not shown). The normally flexible and poorly ordered α2-α3 loop appears to lock down over the bound citrate adopting a configuration similar to that observed in the EcIspF-MEcPP complex [20]. In the other citrate-binding pose (not shown) only a carboxylate coordinates to Zn2+ but hydrogen bonds are also formed exploiting the same functional groups on the protein, the amides of His36 and Ser37, Ser37 OG, and Lys134 NZ. It is clear from these results, that any attempts to investigate ligand binding at the Zn2+ site will require crystallization conditions that do not include citrate.
Figure 8.
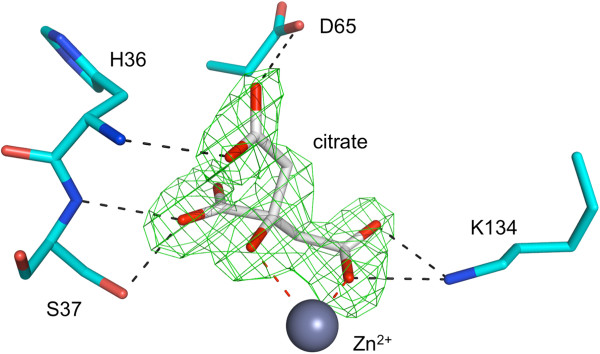
Citrate binding to Zn2+ in BcIspF. A well-resolved citrate binds to Zn2+. An omit map is displayed for citrate in green chicken wire contoured at 3 σ. Residues are shown as sticks and a grey sphere represents Zn2+. Interactions are marked with dashed lines. Solvent mediated interactions are not shown.
Co-crystallization with potential Zn2+ coordinating ligands
A screen of Zn2+-binding ligands previously identified L-tryptophan hydroxamate as a ligand for IspF suggesting that the presence of such a group may anchor ligands in the active site [25]. Subsequent studies with BpIspF reported a number of metal ion coordinating ligands [27]. Crystallization trials of PfIspF and BcIspF in the presence of several such Zn2+ coordinating ligands (e.g. fosmidomycin, benzohydroxamic acid, acetohydroxamic acid and pyrithione), testing a range of concentrations together with the absence or presence of CMP were carried out. Citrate was not present in these crystallization trials. Numerous datasets have been measured and analyzed but we have not observed any evidence of selected ligands at the Zn2+ site. In one case a 2.0 Å resolution dataset was obtained from BcIspF co-crystallized with 10 mM pyrithione. It quickly became evident that neither Zn2+ nor pyrithione were present in the structure (not shown) and this suggests that the ligand may have extracted Zn2+ from the active site.
Conclusions
The structures of IspF from a bacterial (BcIspF) and a protozoan (PfIspF) pathogen have been determined and compared to orthologues. A high degree of structural similarity in and around the active sites is noted. These proteins have been assessed for suitability as platforms for structure-based drug discovery. The cytidine-binding pocket is relatively rigid in IspF, whereas the pocket that binds the methylerythritol-phosphate portion of the substrate is relatively flexible. Numerous attempts to obtain the structure of protein-ligand complexes failed suggesting that the crystallization conditions may need to be adjusted or alternatives found. We note that in each case high ionic strength was necessary to obtain well-ordered crystals. Successful co-crystallization with small molecule ligands has been reported for EcIspF and BpIspF [24,25,27] and given the high degree of structural similarity in the IspF active site it might be prudent to use those enzymes for early stage drug discovery studies. Previously [26], AtIspF was used as a surrogate for PfIspF due to difficulties in working with the later. We also noted problems with the solubility of PfIspF and subsequently identified conditions to circumvent this issue. However, given the high level of conservation we have described then any IspF might be considered a suitable surrogate for the apicomplexan enzymes. The unexpected observations that a second molecule of CMP can occupy the active site as well as the presence of citrate interacting with Zn2+ in BcIspF are noteworthy. These ligands provide templates for ligand design addressing metal ion coordination and interactions in the MEcPP-binding pocket.
Methods
Protein production
The gene encoding BcIspF was amplified from B. cenocepacia genomic DNA (strain J2315) by PCR using 5′- CAT ATG GAC TTC AGA ATC GGA CAA GG −3′ and 5′- GGA CCT CAG CCG CCC TGC TTC ACC −3′ as the forward and reverse primers respectively (Thermo Scientific). The PCR product was ligated into TOPO-Blunt-II (Invitrogen) then subcloned into a modified pET15b vector (Novagen), which produces a histidine-tagged protein with a Tobacco Etch Virus (TEV) protease site. A single nucleotide error in the forward primer gave a single amino acid mutation F3V. The gene encoding IspF from P. falciparum strain 3D7, [GenBank:XP_001349603], was synthesized (Genscript) with codons optimized for recombinant expression in E. coli. The sequence encoding residues 1–59, a predicted apicoplast targeting sequence, was excluded and the codon for Cys60 was replaced with one for serine. This synthetic gene was sub-cloned into the same modified pET15b vector as Bc ispF. Experimental procedures for recombinant protein production using E. coli BL-21 (DE3) Gold following published methods [19].
Purification of PfIspF
PfIspF precipitates at temperatures below 18°C therefore all procedures were performed at room temperature unless otherwise stated. Cells were harvested by centrifugation at 3000 g for 30 minutes at 18°C and re-suspended in 100 mM KCl, 100 mM L-arginine, 50 mM CHES, pH 9.5 (buffer A). DNaseI and a protease inhibitor tablet (EDTA-free, Roche) were added prior to cell lysis by passage through a French pressure cell at 16000 psi. Cell lysate was clarified by ultracentrifugation at 40000 g for 30 minutes at 18°C (Avanti centrifuge, Beckmann) and the supernatant was syringe filtered (Sartorius).
Lysate was loaded onto a nickel ion affinity chromatography column (5 mL HisTrap HP, GE Healthcare) using an FPLC system (Äkta Explorer, GE Healthcare). A linear concentration gradient of imidazole was applied with elution of PfIspF occurring at 200 mM mM imidazole. The fractions containing IspF were pooled and dialyzed against buffer A. The affinity tag (His-tag) was cleaved with His-tagged TEV protease at 20°C for 16 hours. Protease-treated sample was re-applied to a nickel ion affinity chromatography column, and the cleaved protein eluted. Fractions were analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and those containing PfIspF were pooled. The sample was further purified on a size-exclusion chromatography column (Superdex 200 26/60, GE Healthcare) equilibrated with buffer A. This column had been calibrated with molecular weight standards (Bio-Rad). The level of PfIspF purity was confirmed by SDS-PAGE and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Protein concentration was estimated spectrophotometrically using a theoretical extinction coefficient, ϵ: 11920 M-1 cm-1 at 280 nm, calculated using ProtParam[35].
Purification of BcIspF
BcIspF was purified in similar fashion to PfIspF except that a different buffer, (100 mM NaCl, 100 mM Tris–HCl, pH 7.5, buffer B) was used. Removal of the His-tag reduced the solubility of the protein significantly. Once this problem was identified the tag was left in place. The temperature sensitivity observed for PfIspF was not seen for BcIspF and samples were kept either on ice or at 4°C throughout. BcIspF concentration was measured using the Bradford assay (Thermo Scientific).
Screening for crystallization conditions
An automated liquid-handling system (Phoenix, Art Robbins) was used for screening crystallization conditions in combination with commercially available screens. All samples were filtered through a 0.1 μm PVDF filter (Ultrafree, Millipore) immediately prior to screening. Sitting-drops were formed by mixing 100 nL sample with either 100 or 200 nL of reservoir. Reservoir volumes were 60 μl. Conditions identified from the screens were subsequently scaled up and optimized.
Crystallization of a PfIspF:CDP complex and unliganded PfIspF
PfIspF was concentrated to 6 mg mL-1 using a 10 kDa cutoff centrifugal filter (Vivaspin 20, Sartorius) to provide a stock solution for crystallization. The sample was supplemented with MgCl2 (2 mM) and CDP disodium salt (2 mM, Sigma-Aldrich). Hanging drops were prepared consisting of 2 μl sample mixed with 2 μl of reservoir (1.8-2.5 M (NH4)2SO4, 5 mM ZnCl2 and 100 mM Bis-Tris, pH 5.5). Diffusion against 1000 μl of reservoir led to the formation of small prismatic crystals (approximate dimensions 0.15 × 0.15 × 0.15 mm) within 2–4 days. Isomorphous crystals were also obtained with 10 mM fosmidomycin present in the crystallization drop. Crystals were transferred to drops containing saturated sucrose solution, for ~5 seconds before flash cooling in liquid nitrogen.
Crystallization of BcIspF complexes
BcIspF was concentrated to 5 mg mL-1 in buffer B, similar to PfIspF and the solution adjusted to include 2 mM MgCl2 and 20 mM CMP disodium salt (Sigma-Aldrich). A second sample was prepared at 10 mg mL-1 in buffer C (100 mM NaCl, 2 mM MgCl2, 100 mM sodium formate, pH 5.0) to which CMP (2 mM) was also added.
The BcIspF CMP 2 structure was obtained from crystals grown by mixing 2 μL of the sample in buffer B with reservoir (2 μL) containing 25% w/v PEG 3350, 2.2 M (NH4)2SO4, 0.1 M sodium formate, pH 5.0, and 1% v/v dioxane. Orthorhombic blocks reached maximum size within ten days. A crystal of approximate dimensions 0.15 × 0.10 × 0.10 mm was used for data collection. Buffer B supplemented with 20% v/v glycerol was used as a cryoprotectant.
The second sample gave the BcIspF CMP structure. This was crystallized by mixing 2 μL with 4 μL reservoir containing 5% w/v PEG 1000, 33% v/v ethanol, 0.1 M Na2HPO4, pH 4.0, and 2% v/v dioxane. A crystal of approximate dimensions 0.25 × 0.10 × 0.10 mm grew over six days and was used for data collection. A mixture of buffer C with reservoir (1:2), replacing PEG 1000 with PEG 4000 at 25% w/v, was used as a cryoprotectant.
The BcIspF:citrate structure was derived from protein concentrated to 10 mg mL-1 in buffer D (100 mM NaCl, 100 mM sodium formate, pH 5.0), and crystallized by mixing (1.5 μL) with a reservoir (3 μL) containing 5% w/v PEG 1000, 36% v/v ethanol, 0.1 M Na2HPO4 and 0.1 M citric acid, pH 4.2. A different crystal form to that previously found, grew within 4 days to approximate dimensions 0.4 × 0.1 × 0.1 mm. A mixture of sample buffer with reservoir (1:2), replacing PEG 1000 with PEG 4000 at 25% w/v, was used as a cryoprotectant. Datasets for the BcIspF complex crystals were collected on the in-house X-ray system as described for PfIspF.
Crystals were screened in-house using a rotating-anode X-ray source (MicroMax 007 HF, Rigaku) equipped with a dual image plate detector (RAXIS IV++). Diffraction data from the PfIspF:CDP complex were collected on beamline ID29 at the European Synchrotron Radiation Facility (ESRF, Grenoble) at −170°C using an ADSC Quantum 315r CCD detector. Data from the crystals grown in the presence of fosmidomycin were collected in-house. Subsequently it was determined that fosmidomycin was not ordered and this is therefore considered to be an unliganded PfIspF structure. Datasets for the BcIspF complex crystals were all collected on the in-house X-ray system described above.
Data processing, structure solution and refinement
Diffraction data were indexed and integrated using MOSFLM[36] or XDS[37] and scaled with SCALA[38] with 5% of data flagged for Rfree calculations. The Rfree flag was maintained for isomorphous structures. Initial structures were solved by molecular replacement using PHASER[39]. Different search models, edited to remove all non-protein atoms, were used for these calculations. The coordinates of E. coli IspF [PDB:1GX1] with ~33% sequence identity, [19] and B. pseudomallei [PDB:3F0G] 63% identity, [27] were used to determine the PfIspF:CDP and BcIspF:2CMP structures respectively. B-factors of the replacement models were adjusted to match the Wilson B-factor derived from scaling of the data. Subsequently these parent structures were used as the starting points for analyses of the other structures. Crystals of PfIspF display differing degrees of twinning as recognised in SCALA and a twin fraction of 0.14 was applied to the CDP complex.
Refinement calculations were performed with REFMAC5[40] and the resulting models together with electron density and difference density maps were inspected in COOT, which also allowed for model adjustment to improve the fit to density [41]. Water molecules, ions, ligands and conformational rotamers were added to the models as the refinements progressed. In the case of citrate molecules binding to BcIspF, one site was well ordered but the other two sites displayed more diffuse electron density and a decision was taken to set the occupancy to 0.75. These then refined satisfactorily with no residual features in the resulting difference density maps. Strict NCS restraints were employed in the early stages of refinement and then removed towards the latter stages. To confirm the presence of ligand molecules in the final models, |Fo-Fc| electron density difference maps, referred to as ‘omit maps’, were prepared by excluding ligands from Fc calculation, several cycles of refinment in REFMAC5 and inspection of the maps in COOT. The validation tools in COOT and the MolProbity server [42] were used to assess model geometry. Comparisons with structures in the PDB were performed using the DALI server [28]. The Protein Interfaces, Surfaces & Assemblies server, [43] was used to calculate surface and interface areas and figures were prepared with PyMOL[44].
Sequence analyses
A search in UniProt for IspF sequences using the E.C. number [4.6.1.12] returned 2458 entries. The list was filtered down to 2229 by excluding the bifunctional IspDF examples and fragmentary protein sequences. Sequences with greater than or equal to 90% identity were clustered and a single representative sequence identified for each cluster, reducing the list to 876 sequences. These were inspected and a decision taken to remove a number of sequences that still appeared to be fragmentary. A final set of 854 IspF sequences were submitted to the European Bioinformatics Institute-ClustalOmega multiple sequence alignment server [45]. The alignment was displayed in Jalview[46] and percentage conservation scores were manually calculated for all residues marked in Figure 3.
Abbreviations
CDP: Cytidine 5′-diphosphate; CDP-MEP: 4-diphosphocytidyl-2C-methyl-D-erythritol-2-phosphate; CMP: Cytidine 5′-monophosphate; IspF: 2C-methyl-D-erythritol-2,4-cyclodiphosphate synthase; MEcPP: 2C-methyl-D-erythritol-2,4-cyclodiphosphate; MEP: 2C-methyl-D-erythritol-4-phosphate; MVA: Mevalonate; NCS: Non-crystallographic symmetry; RMSD: Root-mean-standard deviation; SDS-PAGE: Sodium dodecyl sulfate polyacrylamide gel electrophoresis.
Competing interests
The authors declare they have no competing interests.
Authors’ contributions
Conceived and designed the experiments: PEFO’R JK-T AD WNH. Performed the experiments: PEFO’R JK-T PKF AD. Analysed the data: PEFO’R JK-T PKF WNH. Wrote the paper: PEFO’R JK-T WNH. All authors read and approved the final manuscript.
Contributor Information
Patrick EF O’Rourke, Email: p.orourke@dundee.ac.uk.
Justyna Kalinowska-Tłuścik, Email: kalinows@chemia.uj.edu.pl.
Paul K Fyfe, Email: p.k.fyfe@dundee.ac.uk.
Alice Dawson, Email: a.x.dawson@dundee.ac.uk.
William N Hunter, Email: w.n.hunter@dundee.ac.uk.
Acknowledgments
We thank Scott Cameron for assistance and staff at the ESRF for access to facilities and excellent support. Coordinates and structure factors are deposited in the PDB under accession codes: 4C81, 4C82, 4C8E, 4C8G, and 4C8I. Funded by the Wellcome Trust [grants 082596, 094090, 100476], the European Commission [the Aeropath project], and a Biotechnology and Biological Sciences Research Council - Pfizer studentship.
References
- Hunter WN. The non-mevalonate pathway of isoprenoid precursor biosynthesis. J Biol Chem. 2007;282:21573–21577. doi: 10.1074/jbc.R700005200. [DOI] [PubMed] [Google Scholar]
- Zhao L, Chang WC, Xiao Y, Liu HW, Liu P. Methylerythritol phosphate pathway of isoprenoid biosynthesis. Annu Rev Biochem. 2013;82:497–530. doi: 10.1146/annurev-biochem-052010-100934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohmer M, Knani M, Simonin P, Sutter B, Sahm H. Isoprenoid biosynthesis in bacteria: a novel pathway for the early steps leading to isopentenyl diphosphate. Biochem J. 1993;295:517–524. doi: 10.1042/bj2950517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohdich F, Kis K, Bacher A, Eisenreich W. The non-mevalonate pathway of isoprenoids: genes, enzymes and intermediates. Curr Opin Chem Biol. 2001;5:535–540. doi: 10.1016/S1367-5931(00)00240-4. [DOI] [PubMed] [Google Scholar]
- Hunter WN. Isoprenoid precursor biosynthesis offers potential targets for drug discovery against diseases caused by apicomplexan parasites. Curr Top Med Chem. 2011;11:2048–2059. doi: 10.2174/156802611796575867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohdich F, Bacher A, Eisenreich W. Isoprenoid biosynthetic pathways as anti-infective drug targets. Biochem Soc Trans. 2005;33:785–791. doi: 10.1042/BST0330785. [DOI] [PubMed] [Google Scholar]
- Lichtenthaler HK. The 1-deoxy-D-xylulose-5-phosphate pathway of isoprenoid biosynthesis in plants. Annu Rev Plant Physiol Plant Mol Biol. 1999;50:47–65. doi: 10.1146/annurev.arplant.50.1.47. [DOI] [PubMed] [Google Scholar]
- van Dooren GG, Striepen B. The algal past and parasite present of the apicoplast. Annu Rev Microbiol. 2013;67:271–289. doi: 10.1146/annurev-micro-092412-155741. [DOI] [PubMed] [Google Scholar]
- Yeh E, DeRisi JL. Chemical rescue of malaria parasites lacking an apicoplast defines organelle function in blood-stage Plasmodium falciparum. PLoS Biol. 2011;9:1–10. doi: 10.1371/journal.pbio.1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frearson JA, Wyatt PG, Gilbert IH, Fairlamb AH. Target assessment for antiparasitic drug discovery. Trends Parasitol. 2007;23:589–595. doi: 10.1016/j.pt.2007.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buetow L, Brown AC, Parish T, Hunter WN. The structure of Mycobacteria 2C-methyl-D-erythritol-2,4-cyclodiphosphate synthase, an essential enzyme, provides a platform for drug discovery. BMC Struct Biol. 2007;7:68. doi: 10.1186/1472-6807-7-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell TL, Brown ED. Characterization of the depletion of 2-C-methyl-D-erythritol-2,4-cyclodiphosphate synthase in Escherichia coli and Bacillus subtilis. J Bacteriol. 2002;184:5609–5618. doi: 10.1128/JB.184.20.5609-5618.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos N, Rodríguez-Concepción M, Sauret-Güeto S, Gallego F, Lois L, Boronat A. Escherichia coli engineered to synthesize isopentenyl diphosphate and dimethylallyl diphosphate from mevalonate: a novel system for the genetic analysis of the 2-C-methyl-D-erythritol 4-phosphate pathway for isoprenoid biosynthesis. Biochem J. 2001;353:59–67. [PMC free article] [PubMed] [Google Scholar]
- Freiberg C, Wieland B, Spaltmann F, Ehlert K, Brotz H, Labischinski H. Identification of novel essential Escherichia coli genes conserved among pathogenic bacteria. J Mol Microbiol Biotechnol. 2001;3:483–489. [PubMed] [Google Scholar]
- Sauret-Güeto S, Ramos-Valdivia A, Ibáñez E, Boronat A, Rodríguez-Concepción M. Identification of lethal mutations in Escherichia coli genes encoding enzymes of the methylerythritol phosphate pathway. Biochem Biophys Res Commun. 2003;307:408–415. doi: 10.1016/S0006-291X(03)01211-7. [DOI] [PubMed] [Google Scholar]
- Shigi Y. Inhibition of bacterial isoprenoid synthesis by fosmidomycin, a phosphonic acid-containing antibiotic. J Antimicrob Chemother. 1989;24:131–145. doi: 10.1093/jac/24.2.131. [DOI] [PubMed] [Google Scholar]
- Okuhara M, Kuroda Y, Goto T, Okamoto M, Terano H, Kohsaka M, Aoki H, Imanaka H. Studies on new phosphonic acid antibiotics I. FR-900098, isolation and characterization. J Antibiot. 1980;33:13–17. doi: 10.7164/antibiotics.33.13. [DOI] [PubMed] [Google Scholar]
- Jomaa H, Wiesner J, Sanderbrand S, Altincicek B, Wiedemeyer C, Hintz M, Türbachova I, Eberl M, Zeidler J, Lichtenthaler HK, Soldati D, Beck E. Inhibitors of the nonmevalonate pathway of isoprenoid biosynthesis as antimalarial drugs. Science. 1999;285:1573–1576. doi: 10.1126/science.285.5433.1573. [DOI] [PubMed] [Google Scholar]
- Kemp LE, Bond CS, Hunter WN. Structure of 2C-methyl-D-erythritol 2,4-cyclodiphosphate synthase: an essential enzyme for isoprenoid biosynthesis and target for antimicrobial drug development. Proc Natl Acad Sci. 2002;99:6591–6596. doi: 10.1073/pnas.102679799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbacher S, Kaiser J, Wungsintaweekul J, Hecht S, Eisenreich W, Gerhardt S, Bacher A, Rohdich F. Structure of 2C-methyl-D-erythritol-2,4-cyclodiphosphate synthase involved in mevalonate-independent biosynthesis of isoprenoids. J Mol Biol. 2002;316:79–88. doi: 10.1006/jmbi.2001.5341. [DOI] [PubMed] [Google Scholar]
- Gabrielsen M, Bond CS, Hallyburton I, Hecht S, Bacher A, Eisenreich W, Rohdich F, Hunter WN. Hexameric assembly of the bifunctional methylerythritol 2,4-cyclodiphosphate synthase and protein-protein associations in the deoxy-xylulose-dependent pathway of isoprenoid precursor biosynthesis. J Biol Chem. 2004;279:52753–52761. doi: 10.1074/jbc.M408895200. [DOI] [PubMed] [Google Scholar]
- Gabrielsen M, Rohdich F, Eisenreich W, Gräwert T, Hecht S, Bacher A, Hunter WN. Biosynthesis of isoprenoids: a bifunctional IspDF enzyme from Campylobacter jejuni. Eur J Biochem. 2004;271:3028–3035. doi: 10.1111/j.1432-1033.2004.04234.x. [DOI] [PubMed] [Google Scholar]
- Baumgartner C, Eberle C, Diederich F, Lauw S, Rohdich F, Eisenreich W, Bacher A. Structure-based design and synthesis of the first weak non-phosphate inhibitors for IspF, an enzyme in the non-mevalonate pathway of isoprenoid biosynthesis. Helv Chim Acta. 2007;90:1043–1068. doi: 10.1002/hlca.200790105. [DOI] [Google Scholar]
- Crane CM, Kaiser J, Ramsden NL, Lauw S, Rohdich F, Eisenreich W, Hunter WN, Bacher A, Diederich F. Fluorescent inhibitors for IspF, an enzyme in the non-mevalonate pathway for isoprenoid biosynthesis and a potential target antimalarial therapy. Angew Chem Int Ed. 2006;45:1069–1074. doi: 10.1002/anie.200503003. [DOI] [PubMed] [Google Scholar]
- Ramsden NL, Buetow L, Dawson A, Kemp LA, Ulaganathan V, Brenk R, Klebe G, Hunter WN. A structure-based approach to ligand discovery for 2C-methyl-D-erythritol-2,4-cyclodiphosphate synthase: A target for antimicrobial therapy. J Med Chem. 2009;52:2531–2542. doi: 10.1021/jm801475n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geist JG, Lauw S, Illarionova V, Illarionov B, Fischer M, Gräwert T, Rohdich F, Eisenreich W, Kaiser J, Groll M, Scheurer C, Wittlin S, Alonso-Gómez JL, Schweizer WB, Bacher A, Diederich F. Thiazolopyrimidine inhibitors of 2-methylerythritol 2,4-cyclodiphosphate synthase (IspF) from Mycobacterium tuberculosis and Plasmodium falciparum. ChemMedChem. 2010;5:1092–1101. doi: 10.1002/cmdc.201000083. [DOI] [PubMed] [Google Scholar]
- Begley DW, Hartley RC, Davies DR, Edwards TE, Leonard JT, Abendroth J, Burris CA, Bhandari J, Myler PJ, Staker BL, Stewart LJ. Leveraging structure determination with fragment screening for infectious disease drug targets: MECP synthase from Burkholderia pseudomallei. J Struct Funct Genomics. 2011;12:63–76. doi: 10.1007/s10969-011-9102-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L, Rosenström P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–W549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond CS, Schüttelkopf AW. ALINE: a WYSIWYG protein-sequence alignment editor for publication-quality alignments. Acta Cryst D. 2009;65:510–512. doi: 10.1107/S0907444909007835. [DOI] [PubMed] [Google Scholar]
- Calisto BM, Perez-Gil J, Bergua M, Querol-Audi J, Fita I, Imperial S. Biosynthesis of isoprenoids in plants: structure of the 2C-methyl-D-erithrytol 2,4-cyclodiphosphate synthase from Arabidopsis thaliana. Comparison with the bacterial enzymes. Protein Sci. 2007;16:2082–2088. doi: 10.1110/ps.072972807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp LE, Alphey MS, Bond CS, Ferguson MA, Hecht S, Bacher A, Eisenreich W, Rohdich F, Hunter WN. The identification of isoprenoids that bind in the intersubunit cavity of Escherichia coli 2C-methyl-D-erythritol-2,4-cyclodiphosphate synthase by complementary biophysical methods. Acta Cryst D. 2005;61:45–52. doi: 10.1107/S0907444904025971. [DOI] [PubMed] [Google Scholar]
- Ni S, Robinson H, Marsing GC, Bussiere DE, Kennedy MA. Structure of 2C-methyl-D-erythritol-2,4-cyclodiphosphate synthase from Shewanella oneidensis at 1.6 Å: identification of farnesyl pyrophosphate trapped in a hydrophobic cavity. Acta Cryst D. 2004;60:1949–1957. doi: 10.1107/S0907444904021791. [DOI] [PubMed] [Google Scholar]
- Sgraja T, Kemp LE, Ramsden N, Hunter WN. A double mutation of Escherichia coli 2C-methyl-D-erythritol-2,4-cyclodiphosphate synthase disrupts six hydrogen bonds with, yet fails to prevent binding of, an isoprenoid diphosphate. Acta Cryst F. 2005;61:625–629. doi: 10.1107/S1744309105018762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishida H, Wada T, Unzai S, Kuzuyama T, Takagi M, Terada T, Shirouzu M, Yokoyama S, Tame JRH, Park S. Structure and catalytic mechanism of 2-C-methyl-D-erythritol 2,4-cyclodiphosphate (MECDP) synthase, an enzyme in the non-mevalonate pathway of isoprenoid synthesis. Acta Cryst D. 2003;59:23–31. doi: 10.1107/S0907444902017705. [DOI] [PubMed] [Google Scholar]
- Gasteiger E, Hoogland C, Gattiker A, Duvand S, Wilkins MR, Appel RD, Bairoch A. Protein identification and analysis tools on the ExPASy server. Totowa, NJ, USA: Humana Press, The Proteomics Protocols Handbook; 2005. pp. 571–607. [Google Scholar]
- Battye TGG, Kontogiannis L, Johnson O, Powell HR, Leslie AGW. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Cryst D. 2011;67:271–281. doi: 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Cryst D. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. Scaling and assessment of data quality. Acta Cryst D. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Skubák P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. REFMAC5 for the refinement of macromolecular crystal structures. Acta Cryst D. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Cryst D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Cryst D. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- De Lano WL. The PyMOL Molecular Graphics System. Schrödinger, LLC; 2002. [Google Scholar]
- Sievers F, Wilm A, Dineen DG, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins D. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview Version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]