

Summary
Serious viral infections are a common cause of morbidity and mortality after allogeneic stem cell transplantation. They occur in the majority of allograft recipients and are fatal in 17–20%. These severe infections may be prolonged or recurrent and add substantially to the cost, both human and financial, of the procedure. Many features of allogeneic stem cell transplantation contribute to this high rate of viral disease. The cytotoxic and immunosuppressive drugs administered pretransplant to eliminate the host hematopoietic/immune system and any associated malignancy, the delay in recapitulating immune ontogeny post‐transplant, the immunosuppressive drugs given to prevent graft versus host disease (GvHD), and the effects of GvHD itself, all serve to make stem cell transplant recipients vulnerable to disease from endogenous (latent) and exogenous (community) viruses, and to be incapable of controlling them as quickly and effectively as most normal individuals.
Keywords: T cells, immunotherapies, transplantation
This article is part of a series of reviews covering Transplantation appearing in Volume 258 of Immunological Reviews.
Introduction: viral disease after hematopoietic stem cell transplantation
Serious viral infections are a common cause of morbidity and mortality after allogeneic hematopoietic stem cell transplantation (HSCT). They occur in the majority of allograft recipients and are fatal in 17–20% 1, 2. These severe infections may be prolonged or recurrent and add substantially to the cost, both human and financial, of the procedure 3, 4, 5. Many features of allogeneic HSCT contribute to this high rate of viral disease. The cytotoxic and immunosuppressive drugs administered pretransplant to eliminate the host hematopoietic/immune system and any associated malignancy, the delay in recapitulating immune ontogeny post‐transplant, the potential for induction of immune tolerance/T‐cell anergy upon exposure on an immature system to an active viral infection, the immunosuppressive drugs given to prevent graft versus host disease (GvHD), and the effects of GvHD itself, all serve to make HSCT recipients vulnerable to disease from endogenous (latent) and exogenous (community) viruses, and to be incapable of controlling them as quickly and effectively as most normal individuals.
Over the past 20 years, great strides have been made in identifying and implementing effective therapeutic agents for the prevention and treatment of many of the serious virus infections that afflict HSCT recipients. By combining effective agents with rapid molecular analyses that can detect the early reactivation of latent infections, it has been possible to transform the outcome of patients with post‐transplant cytomegalovirus (CMV) and Epstein Barr virus (EBV). Agents such as ganciclovir and rituximab 6, 7, 8, respectively, can now be used to control CMV and EBV before they cause serious or fatal illness. Despite these and other advances in early detection and prompt treatment, post‐transplant viral illness remains a major threat. As might be expected, improvements in viral prophylaxis and treatment have encouraged clinicians to extend the range of patients to whom HSCT can be offered as a curative option for their disease. A high proportion of patients who are now transplanted lack siblings who are identical for human leukocyte antigens (HLAs) (major histocompatibility antigens), necessitating the use of alternative stem cell sources including HLA‐partially matched unrelated donors, haploidentical related donors or cord blood units. These patients may require donor T cells to be removed from the infused stem cell preparation or may need to receive more intensive or prolonged post transplant immunosuppression, increasing their susceptibility to viral disease and prolonging the duration of this enhanced susceptibility. Moreover, the considerable success of ganciclovir and rituximab against CMV and EBV disease has yet to be followed by the availability of additional agents that can as effectively and safely treat the many other virus infections to which transplant patients are susceptible (Table 1). Even when pharmacological therapies are available, these agents are costly, associated with significant toxicities, and may lead to the outgrowth of drug‐resistant mutants. Since none of these agents improve virus‐specific T‐cell immunity, the root cause of post‐transplant viral infection, these diseases frequently recur after termination of treatment. In this review, we describe the immunotherapeutic strategies that have been explored by our own and other groups to achieve safe, effective, and sustained antiviral prophylaxis and treatment following HSCT and discuss the success and limitations of this approach.
Table 1.
Emerging viral pathogens implicated in complications after hematopoietic stem cell transplantation (HSCT)
| Virus | Incidence after allogeneic HSCT |
|---|---|
| HHV6 | 33–48% 120, 121, 122, 123, 124, 125 |
| HHV7 | 10–57% 126, 127 |
| Parvovirus | Up to 30% 128, 129, 130, 131 |
| BK virus | 10–25% 132, 133, 134, 135, 136 |
| Coronavirus | 8.8% 137, 138, 139 |
| Parainfluenza | 4–7% 3, 140, 141, 142, 143 |
| Influenza | 1.3–2.6% 143, 144 |
| RSV | 1.8–6% 140, 143, 145 |
| Metapneumovirus | 5% 146, 147 |
| Bocavirus | 5% 148, 149 |
| KI virus/WU virus | 1–8% 148, 149, 150, 151 |
| JC virus | Approximately 1% 152, 153 |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Donor lymphocyte infusions (DLI)
The first adoptive T‐cell transfer studies after allogeneic HSCT were based on the premise that peripheral blood from the stem cell donor would contain T cells able to mediate anti‐tumor and antiviral activity in the HSCT recipient 9, 10, 11, 12, 13. The expectation was that these cells would expand in the recipient after adoptive transfer and provide protective immunity. In terms of antiviral activity, providing that the donor has had prior virus exposure, donor peripheral blood does indeed contain memory T cells specific for a broad range of viruses, which reflect the life‐time exposure of the host to an array of pathogens, and DLI has proved to be an effective treatment for both adenovirus (AdV) infections and EBV‐associated post‐transplant lymphoproliferative disease (PTLD) 14, 15, 16. Unfortunately, broader application is limited by the low frequency of virus‐specific T cells compared with the much higher frequency of alloreactive T cells that can cause GvHD, a particular concern in HLA mismatched recipients who are most at risk of viral illness. Up to 10% of circulating T cells may be alloreactive 16, 17, while the frequency of T cells with a given antiviral specificity may be many logs lower (Fig. 1). Even when DLI is used to successfully treat EBV infections, a high rate of GvHD is observed, and T cells reactive to many common community viruses such as parainfluenza, respiratory syncytial virus (RSV), and human metapneumovirus (hMPV) are frequently all but undetectable in the peripheral blood of the stem cell donor, making this approach too high risk to be widely used to prevent or treat associated viral disease post‐transplant.
Figure 1.
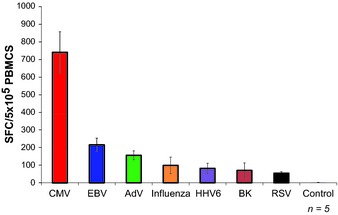
Frequency of virus‐specific T cells in peripheral blood. The frequency of circulating virus‐specific T cells directed against CMV (IE1 and pp65), EBV (LMP1, LMP2, EBNA1, EBNA3a, EBNA3b, EBNA3c, BZLF1), AdV (Hexon and Penton), Influenza (NP1 and MP1), HHV6 (U11, U14, and U90), BK virus (Large T and VP1), and RSV (N and F) was evaluated by IFNγ ELIspot. Results are presented as spot forming cells (SFC) per 5 × 105 PBMCs ± SEM (n = 5).
Reducing alloreactive T cells in DLI
To preserve the benefits and enhance the safety of DLI, techniques to selectively inactivate or deplete recipient‐specific alloreactive T cells have been evaluated.
Inactivation of alloreactive T cells
Alloantigen‐specific T cells can be selectively anergized by exploiting the requirement of T cells to receive both an HLA‐restricted, antigen‐specific signal, and secondary co‐stimulatory signals if they are to become successfully activated and proliferate. One such costimulatory signal is delivered by the B7.1 and B7.2 molecules on antigen‐presenting cells (APCs) to the CD28 receptor on T cells, and blockade of this interaction using fusion proteins (such as CTLA4‐Ig) or monoclonal antibodies (such as anti‐B7.1 and ‐B7.2) during T‐cell receptor (TCR)‐APC engagement has been demonstrated to induce antigen‐specific hyporesponsiveness in T cells. Guinan et al. 18, 19 have successfully used both blockade strategies clinically. Two days prior to transplant they harvested haploidentical donor marrow, which was incubated with recipient‐derived mononuclear cells in the presence of either CTLA‐4–Ig or anti‐B7 monoclonal antibodies. These anergized T cells were then administered at the same time as the HSCT. In two pilot studies, they found that even when products contained large numbers of alloanergized donor T cells (range 7–129 × 106 CD3+ cells/kg) there was acceptable engraftment with less severe acute GvHD than historical control recipients of haploidentical stem cells replete with unmanipulated T cells. In addition, they reported that the treated subjects had more rapid immune reconstitution than historic controls, with detectable in vivo expansion of virus‐specific T cells 20.
Selective allodepletion ex vivo
Alternatively, alloreactive T cells may be physically removed from the donor graft prior to infusion. Several groups have co‐cultured donor T cells with recipient‐derived APCs of various origin including peripheral blood mononuclear cells (PBMCs), activated T cells 21, EBV‐transformed lymphoblastoid cell lines (EBV‐LCL) 22, 23, dendritic cells (DCs) 24, and fibroblasts 25. Any recipient‐reactive T cell within the donor cell population should be stimulated by the recipient APC, and subsequently upregulate activation markers such as CD25 23, 26, 27, 28, 29, CD69 30, CD71 24, 31, CD137 25, 32, and HLA‐DR and proliferate. These reactive cells can then be selectively eliminated by magnetic‐bead coupled monoclonal antibodies targeted to activation antigens or by immunotoxins, induction of apoptosis, or photodynamic purging 21, 23, 25, 26, 27, 32, 33, 34, 35, 36, 37, 38, 39.
CD25‐immunotoxin and photodynamic depletion‐based approaches have both been used clinically. Andre‐Schmutz et al. 40 used CD25 conjugated to the ricin α chain to allodeplete haploidentical (n = 13) or matched unrelated (n = 3) donor T cells that were then infused early (days 15–47) post‐transplant to a cohort of pediatric patients. Each patient received multiple infusions of 1–8 × 105 allodepleted cells/kg, with no GvHD prophylaxis. Even at the highest dose level, the allodepleted T cells were found to be safe, in contrast with earlier studies reporting GvHD ≥Grade II in 40% of patients after infusion of 1 × 105 unmanipulated donor T cells/kg. In addition, the investigators found evidence that the allodepleted cells retained a virus‐specific component. Two patients with EBV or CMV developed a high frequency of circulating virus‐specific T‐cell precursors postinfusion and both cleared their respective infections. Solomon et al. 27 applied this approach to adults, infusing 16 patients (median 65 years) with haploidentical T cells that were also allodepleted with an anti‐CD25 immunotoxin. This group of patients would have been considered to be at high risk of developing GvHD but in fact only eight patients developed this complication, which was mild to moderate (GvHD Grades I–II) and steroid responsive in six and severe only in two. The development of GvHD appeared to correlate with the efficiency of allodepletion. Our group also demonstrated that the infusion of CD25‐allodepleted cells to haploidentical HSCT recipients was safe and that a threshold of at least 1 × 105 allodepleted T cells/kg was required to accelerate antiviral T‐cell recovery 23, 41, 42.
More recently, Mielke et al. 43 used photodynamic purging to prepare allodepleted T cells for infusion. They used a photoactive rhodamine derivative, 4,5‐dibromorhodamine 123 (TH9402), which efficiently diffuses into both resting and activated T cells. While resting T cells can actively extrude the dye, it is selectively retained in the mitochondria of activated cells due to the inactivation of the multidrug transporter. These activated cells are then sensitive to visible light (514 nm), which results in mitochondrial oxidation and cell death. Taking advantage of this property, Mielke et al. 43 co‐cultured donor cells with activated recipient‐derived T cells followed by addition of TH9402 and exposure to visible light. Twenty‐four HLA‐identical sibling HSCT recipients received a dose of 5 × 106 photodepleted cells/kg on day 0. Unfortunately, the results were disappointing. Both acute and chronic GvHD were frequent, and infectious complications associated with viral (CMV, AdV, BK virus, RSV), bacterial and fungal pathogens were common and unexpectedly prolonged and severe so that the trial was halted. In this study, it was unclear whether severe GvHD was associated with variability in the infused photodepleted products.
Overall, the above studies demonstrated the feasibility of add‐back allodepleted T‐cell therapy. They also emphasized the importance of choosing the optimal recipient‐derived stimulator cell to achieve robust alloactivation, the impact that variability in the efficiency of allodepletion can have on in vivo safety, and the narrow and unpredictable therapeutic window between the desired antiviral activity and unwanted GvHD. Moreover, since T cells specific for the majority of viral pathogens circulate at frequencies that are lower than EBV and CMV (Fig. 1), it is likely that extremely high doses of allodepleted T cells would be needed to provide full spectrum anti‐viral protection and that, even after allodepletion, the cell doses required would likely exceed the GvHD threshold. Finally, the allodepletion approach, at present, can only reliably be used when donor and recipient are haploidentical. Where there are fewer HLA disparities or differences only at minor histocompatibility antigens (e.g. in an HLA matched sibling transplant), the degree of alloactivation produced in the mixed lymphocyte cultures is insufficient for consistent removal of all alloreactive T cells. Consequently, the ratio of viral reactive to alloreactive T cells is not sufficiently improved to obtain beneficial antiviral effects in the absence of increased GvHD.
Selective allodepletion in vivo
An alternative to ex vivo allodepletion is to administer donor T cells that incorporate a suicide or safety switch that can be activated only in the event of GvHD, allowing recipients to take full advantage of the antiviral benefits associated with donor T‐cell infusions. Moreover, if the suicide switch is functional only in activated cells, and the patient has GvHD but no viral infection, induction of suicide may deplete the alloreactive component while sparing virus‐reactive cells capable of responding to future virus reactivation or infection.
The most widely tested in vivo allodepletion approach uses the thymidine kinase gene from herpes simplex virus I (HSV‐tk) 44. TK expression in transgenic T cells catalyzes the phosphorylation of the non‐toxic prodrug ganciclovir into the active agent. After transformation into the final triphosphate form by cellular kinases, the drug acts as a GTP analog, thus inhibiting DNA chain elongation and killing dividing cells. Several phase I–II studies have shown that ganciclovir administration can be used to deplete transferred TK‐modified cells in vivo and no adverse events related to gene transfer have been reported 45, 46, 47, 48, 49, 50. However, induction of transgenic cell death may require many days and is usually incomplete, potentially delaying clinical benefit. In addition, since ganciclovir is required for cell elimination this precludes its use as an antiviral agent (e.g. for the treatment of CMV) in this highly susceptible patient population. Finally, the TK gene product can be immunogenic 51, 52. For example, the relatively immune competent patients post HLA‐identical HSCT can mount a TK‐directed CD8+ T‐cell response leading to the premature and unintentional elimination of infused cells 53, 54. Despite these potential limitations, phase I and II clinical studies have shown TK‐T cells can consistently benefit immune reconstitution and that GvHD can be controlled by ganciclovir administration so that the approach is now being evaluated in a multicenter, multi‐national phase III study that it is hoped will allow licensure of this important approach.
We have investigated an alternative safety‐switch in which we transduced allodepleted T cells with a retroviral vector encoding an inducible human caspase 9 (iC9) suicide gene and a selectable marker (truncated human CD19) to enable enrichment of the transduced cells 55, 56, 57. The iC9 gene product is activated by exposure to a small molecule chemical inducer of dimerization (CID) leading to rapid T‐cell death by triggering the intrinsic (mitochondrial) apoptosis pathway. We gave iC9‐expressing T cells to haploidentical pediatric HSCT recipients, and if the patients developed GvHD, we gave a single dose of the dimerizing drug AP1903. We found that CID treatment eliminated 90% of the infused transgenic cells within 30 min, with a further log depletion during the next 24 h 55. The patients' GvHD responded fully and did not recur even when the residual transgenic T cells re‐expanded. The recovering iC9 T cells, however, did retain antiviral activity, suggesting selective sparing of these cells over the more activated alloreactive iC9 T cells that had caused GvHD. We found no evidence of an immune response against the transgenic cells. The use of an otherwise bioinert small molecule to dimerize and activate iC9 allows the retention of important antiviral agents, including ganciclovir, for therapeutic use.
Direct enrichment of virus‐specific T cells
An alternative means of safely providing antiviral protection after HSCT relies on the direct isolation of virus‐specific T cells from donor peripheral blood for subsequent adoptive transfer. Peptide‐HLA multimers and cytokine‐secretion capture columns have both been adapted to serve this purpose. Multimer selection isolates T cells based on the ability of their antigen‐specific receptor (TCR) to bind to a complex of synthetic peptide‐loaded recombinant HLA molecules. While the approach is therefore independent of a defined phenotypic or functional characteristic, it requires prior knowledge of immunodominant epitopes and is restricted by HLA type. At present, multimers are most readily made with class I HLA antigens, which can select only CD8+ T cells and not the class II HLA‐restricted CD4+ T‐cell subset. This may limit the breadth and duration of any immune response following adoptive transfer. Even when class I HLA antigens are used, individual multimer complexes vary unpredictably in their stability and affinity for a given TCR, so that it is not possible at present to make effective multimers for every immunodominant epitope for each HLA class I polymorphism. In contrast, the cytokine capture approach selects T cells (both CD4+ and CD8+) based on their ability to secrete effector cytokines in response to viral antigen stimulation and is thus neither limited to specific peptides nor particular HLA types.
Selection of virus‐specific T cells by multimers
T cells detect antigens via TCR interaction with peptides bound to self‐HLA molecules presented on the surface of APCs. The TCR/peptide‐HLA interaction is very weak and typically lasts for no longer than a few seconds at physiological temperatures. Advances over the past 15 years, however, have produced multimeric forms of soluble peptide‐HLA molecules with enhanced avidity that can be utilized ex vivo to both visualize and select T cells that bear cognate TCRs. To date, two distinct multimer formats, tetramers, and pentamers, have been used to select T cells for adoptive transfer. Tetramers consist of four peptide‐loaded HLA molecules held in a tetrahedral configuration that typically allows only 3 HLA‐peptide complexes to bind to cognate TCRs (Fig. 2). The five HLA‐peptide groups of HLA‐pentamers, however, are arranged in a planar configuration so that all five complexes are available for TCR binding thereby increasing both the binding sensitivity and avidity.
Figure 2.
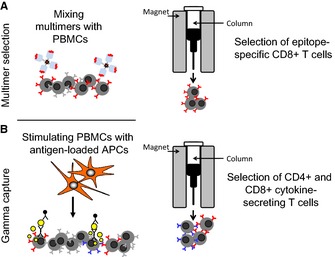
Rapid section of virus‐specific T cells. Peptide‐HLA multimers (A) and cytokine‐secretion capture (B) have both been used clinically to select virus‐specific T cells for adoptive transfer. Multimer selection isolates T cells based on the ability of their TCR to bind to a complex of synthetic peptide‐loaded recombinant HLA molecules, while the cytokine capture approach selects T cells (both CD4+ and CD8+) based on their ability to secrete effector cytokines in response to viral antigen stimulation.
Cobbold et al. 58 were the first to use multimer‐selected T cells clinically. They selected CMV‐specific T cells, since CD8+ T cells specific for this clinically important virus are found at high frequency in healthy seropositive individuals (typically 0.5–4% of the CD8+ T‐cell pool) which facilitates their direct isolation ex vivo 58. The investigators used a panel of CMV IE1 and pp65 tetramers to select specific T cells from nine SCT donors that were administered to six recipients of a matched related donor transplant and three recipients of matched unrelated transplants who had CMV reactivation. Although the infused product was composed exclusively of CD8+ T cells, the cells expanded by several logs in vivo, and in eight of nine cases CMV viral load became undetectable post transfer. No case of CMV disease was seen. Uhlin et al. 59 used pentamers to isolate T cells from the mother of a cord blood transplant recipient with rituximab‐resistant EBV‐PTLD. Based on shared haplotypes, available pentamers, and detectable specific populations in peripheral blood, the investigators used two HLA‐A2‐restricted epitopes from EBV BMLF1 (lytic) and LMP2 (latent) to select specific T cells for adoptive transfer. These directly selected cells expanded postinfusion and produced a complete clinical response. Although antiviral protection was non‐sustained, since EBV‐PTLD recurred at 12 months, the disease was eradicated by a second infusion of the same cells 59. The same group subsequently used multimer selection to isolate virus‐specific T cells either from cryopreserved donor material or from third party donor peripheral blood to treat eight patients with CMV (n = 6; HLA‐A2, B7 and B35 pp65 peptide‐loaded pentamers), AdV (n = 1; HLA‐A1 hexon‐derived peptide), or EBV (n = 1; HLA‐A2 LMP2‐ and BMLF1‐derived peptides) infections after allogeneic HSCT 60. Six of these eight patients showed a decrease in viral titers within 2 weeks of receiving the selected cells 60.
More recently, researchers have prepared streptamers for clinical application, which are multimers in which the binding between the complex of peptide‐loaded HLA molecules and the TCR of antigen‐specific T cells is reversible by addition of a competitor molecule that causes the streptamer to monomerize 61. The rapid release of the streptamer from the T cells means that the final infused product is indistinguishable from untreated T cells. As a consequence, the European Medicines Agency considers streptamer‐selected cells as ‘non‐advanced therapy medicinal products' (non‐ATMP), which require substantially less testing prior to clinical use than agents classified as ATMPs. Recently Schmitt et al. 62 reported the first clinical streptamer experience in two patients with refractory CMV. After a single infusion of 2.2 × 105 HLA‐B7+/CMV pp65‐specific CD8+ T cells/kg (purity of 97%), the frequency of CMV‐specific T cells in patient 1 increased from 0% to a maximum of 27.1% of all peripheral blood T cells. Patient 2 received a lower cell dose (0.37 × 105 HLA‐A24+/CMVpp65‐specific CD8+ T cells/kg) but nevertheless had a detectable increase of CMV‐specific CD8+ T cells, from 0.03% to 0.48%. These expanded cells were confirmed to be donor in origin by analysis of donor chimerism through single‐tandem repeats analysis, T‐cell receptor excision circles, and Vβ‐chain typing. Clinically, the T‐cell infusion resulted in CMV clearance without GvHD.
While these clinical successes hold promise, multimer selection of cells currently remains limited to patients who are infected with immunologically well‐characterized viruses (e.g. CMV or EBV) with defined epitopes, known HLA‐restricting elements, and who have donors with a relatively high frequency of circulating reactive T cells in their peripheral blood. Otherwise sufficiently large starting numbers of donor PBMCs may not be readily available (e.g. if the donor is unrelated to the recipient).
IFNγ‐capture
An alternative means by which virus‐specific T cells can be rapidly selected for adoptive transfer is based on their ability to produce and secrete effector cytokines. To date, only cells that secrete IFNγ in an antigen‐dependent manner have been used clinically. The IFNγ capture technique is based on the premise that antigen‐specific T cells are capable of secreting IFNγ following short‐term (12–16 h) antigen exposure, and these populations can be specifically captured by labeling cells with an anti‐IFNγ monoclonal antibody conjugated to a leukocyte‐specific (CD45) antibody followed by magnetic selection with anti‐IFNγ microbeads. Thus, unlike multimer selection this approach can be used to select both CD4+ and CD8+ antigen‐specific T cells in an HLA unrestricted manner.
Feuchtinger et al. 63 were the first to use this selection platform as a means of treating AdV infections in the post‐HSCT setting. AdV has a particularly high incidence after pediatric HSCT and remains a significant cause of morbidity and mortality in immunocompromised individuals, in whom it may produce pneumonia, hemorrhagic cystitis, nephritis, colitis, hepatitis, and encephalitis 64, 65. Several reports have shown that clearance of infection is associated with detection of AdV‐specific T cells 66, 67. Thus, in this study Feuchtinger et al. 63 stimulated donor PBMCs (matched related – n = 1, matched unrelated – n = 3, mismatched unrelated – n = 4, haploidentical – n = 1) with AdV lysate overnight, followed by magnetic selection of the IFNγ‐secreting population and then infused the cells into nine pediatric patients with systemic AdV infections (defined as persistent or recurrent AdV DNA in peripheral blood and/or stool after 2 weeks of antiviral chemotherapy and lacking AdV‐specific T cells). The infusions were found to be safe and despite the small numbers of infused polyclonal T cells (range 1.2 × 103–5 × 104 CD3+ T cells/kg: CD4+, mean 63%, CD8+, mean 29%), were associated with clinical benefit, since five of six evaluable patients showed a significant decrease in viral load in peripheral blood and stool with a corresponding increase in the frequency of AdV‐specific T cells in the blood. In a follow‐up study, the group applied this approach to treat chemorefractory CMV disease or reactivation 68. After short‐term (16 h) stimulation with the immunodominant CMV‐pp65 protein and capture of IFNγ‐secreting cells, a mean of 21.3 × 103 CD3+ T cells/kg were infused into 18 patients (matched related n = 1, matched unrelated n = 3, mismatched unrelated n = 3, haploidentical n = 11). The infusions were well‐tolerated with only a single case of GvHD reported. Fifteen of the 18 infused patients cleared their CMV viremia or had significant reduction (>1 log) of viral load. Peggs et al. 69 also reported the adoptive transfer of IFNγ‐captured CMV‐specific T cells for disease prophylaxis and pre‐emptive treatment. In their study, pp65 protein or peptide pools were used to stimulate T cells. Post‐selection a median of 2840 CD4+ and 630 CD8+ CMV‐specific T cells/kg were infused early (median day 35) post‐transplant and expansion of both populations was detected in vivo. However, acute GvHD was observed in eight of 18 patients (Grades I–III), two of whom required systemic steroid therapy; three patients developed extensive chronic GvHD.
IFNγ capture can also be used to obtain therapeutically effective EBV‐specific T cells. Moosmann et al. 70 selected IFNγ‐secreting cells following 12 h of exposure to 23 HLA class I and II‐associated peptides derived from 11 EBV antigens [five latent antigens (LMP2, EBNA1, EBNA3A, EBNA3B, EBNA3C), four immediate early/early antigens (BZLF1, BRLF1, BMLF1, BHRF1), and two late/structural antigens (BLLF1, BNRF1)] 70. The full set of 23 peptides was used as a generic stimulating pool for each donor, irrespective of the individual's HLA haplotype. The captured cells were given to six allogeneic HSCT recipients who had developed biopsy‐proven EBV‐PTLD that was progressing following 3–14 days conventional therapy (including reduction in immunosuppression and administration of rituximab or cidofovir). Three of the patients responded, but three, with multiorgan dysfunction at the time of T‐cell transfer, showed no/low EBV‐specific T‐cell activity postinfusion and all failed to respond. Icheva et al. 71 took a similar approach but focused on targeting the EBV EBNA1 antigen, based on its central role in maintaining viral persistence, its universal expression in EBV‐PTLD, and the fact that it has been shown to contain immunodominant CD4+ and CD8+ T‐cell epitopes. The investigators stimulated donor PBMCs either with whole EBNA1 protein or EBNA1 overlapping peptide pools, and gave IFNγ captured cells to 10 patients with EBV‐PTLD. No toxicities were observed, and eight of 10 patients showed in vivo expansion of EBNA1‐specific T cells, which was associated with a clinical and virologic response in seven, defined as a decrease of viral load >1 log and resolution of PTLD. In the two patients in whom in vivo T‐cell expansion was absent, no clinical improvement was observed.
Clinical trials of the IFNγ capture‐based approach have shown to be safe and to produce clinical activity for CMV, AdV, and EBV. The availability of ‘universal’ selection agents that can be used irrespective of HLA haplotypes, rather than individualized multimers made for each viral epitope and HLA polymorphism and the ability to select both CD4+ and CD8+ T cells rather than just class I HLA‐restricted CD8+ T cells are both potential advantages over multimer‐based selection techniques. Nevertheless, both γ‐capture and multimer selection techniques require large volumes of starting donor blood, and even then only small numbers of captured cells are obtained, limiting the crucial purity and potency testing that is required prior to patient transfer. As a consequence, both approaches are restricted to viruses with a high circulating precursor frequency.
Infusion of ex vivo expanded virus‐specific T cells
Immune recovery after HSCT can also be safely enhanced by infusing virus‐specific T cells (VSTs) that have been selectively amplified ex vivo using repetitive rounds of stimulation with APCs expressing target viral antigens. The in vitro stimulation with viral antigens selectively enriches virus‐reactive T cells and correspondingly dilutes alloreactive T cells. By this means even low frequency virus‐directed T cells can be obtained in substantial numbers from small quantities of peripheral blood, so that the strategy can produce VSTs directed to viruses for which the circulating T‐cell frequency is below the practicable threshold required for either multimer or IFNγ capture assays.
Ex vivo expansion of virus‐specific T cells requires the identification of immunodominant antigens in target viruses that induce protective VSTs as well as a delivery system to transfer the antigen to effective APCs. To avoid expanding alloreactive T cells, the APCs used to generate the VSTs must be autologous and express HLA molecules that present virus‐derived peptides. The APC must also express the costimulatory molecules required to support robust T‐cell activation and expansion. All reagents must also be compliant with current good manufacturing practices (cGMP) for cell therapies, which limits the use of some cell types, antigen sources, and other reagents. Finally, the time required to produce, test, and release the expanded product is significant. These many obstacles have limited broader application of the approach, but, as we describe below, all are being progressively surmounted.
Epstein Barr virus
EBV is a ubiquitous human herpesvirus, and over 95% of the adult population is seropositive, indicating a previous infection 72, 73. After primary infection, the virus persists in latent form in B lymphocytes and epithelial cells in the nasopharynx, and periodic replicative reactivations in B cells are tightly controlled by a very strong viral antigen‐specific T‐cell response so that up to 1–2% of circulating T cells in a normal EBV seropositive individual may be specific for EBV 74, 75, 76. After transplant when T‐cell immunity is impaired, EBV‐infected B cells that would normally be controlled by an effective EBV‐specific cytotoxic T‐cell response can outgrow, resulting in EBV‐PTLD. The risk of this complication after HSCT ranges from 1% to 20% with risk factors including the use of a T‐cell‐depleted stem cell product, a higher degree of HLA‐mismatch between donor and recipient, and the use of anti‐thymocyte globulin (ATG) in reduced intensity transplant‐conditioning regimens. PTLD developing after HSCT is usually derived from donor B cells but can be of recipient origin, particularly in patients who receive reduced‐intensity conditioning regimens that included ATG or alemtuzimab (Campath), or who receive cord blood transplants.
The outgrowing tumor cells in EBV‐PTLD are highly immunogenic and express all nine latent EBV antigens. Since almost all transplant donors are EBV seropositive with a high frequency of circulating EBV specific precursors, EBV‐PTLD is a good target disease to test the activity of ex vivo expanded virus antigen specific T cells. In the early 1990s, we were using selective T‐cell depletion with monoclonal antibodies to reduce the risk of GVHD in patients receiving transplants from matched unrelated donors or mismatched family members and about 10% of these patients developed EBV PTLD 77. We therefore devised a strategy to prevent and treat this complication by infusing EBV‐specific T cells generated by stimulating donor mononuclear cells with an irradiated EBV transformed donor LCL 78. LCLs could be easily generated from any donor by incubation with a laboratory strain of EBV and were excellent APCs that had the same pattern of viral gene expression as the outgrowing tumor cells. EBV‐specific T‐cell lines grown after stimulation with LCLs were polyclonal, containing both CD4+ and CD8+ T‐cells that recognized multiple latent and early lytic cycle EBV antigens.
We have reviewed our experience administering EBV‐specific T cells generated with this methodology as prophylaxis to over 100 high risk patients and as treatment for 13 patients with established PTLD (Tables 2 and 3). Of the patients who received the VSTs as prophylaxis, none developed PTLD compared with an 11.5% incidence in controls 22, 79, 80, 81. Thirteen patients received EBV‐specific T cells for active disease, and 11 achieved sustained complete remissions. In the first 26 patients, the T‐cell lines were genetically marked with a retroviral vector encoding the neomycin resistance gene, which enabled us to track the cells and show they could expand by up to 3–4 logs and persist for up to 9 years 22. This long‐term persistence may have been bolstered by infusion into lymphopenic recipients, in whom homeostatic mechanisms promoted lymphoid expansion and by continued T‐cell stimulation by periodic EBV reactivations. Animal studies also suggest that the presence of CD4+ T cells in the infused line is important for persistence and access to the memory pool. The infusions were well‐tolerated, although in patients with bulky disease, we observed reversible inflammatory reactions at disease sites due to infiltration with activated T cells. Of note, the infused cells were not alloreactive and there was no de novo GVHD after infusion 82.
Table 2.
Outcome of prophylaxis with donor virus‐specific T cells (VSTs) in our studies
| Specificity | Virus | Patients | No viral infection | Failure | References |
|---|---|---|---|---|---|
| Monovirus VSTs with prolonged culture | |||||
| LCL‐induced EBV VSTs | EBV | 105 | 105 | 0 |
Rooney et al. 80 Heslop et al. 79 Rooney et al. 81 Heslop et al. 22 |
| Bivirus VSTs with prolonged culture | |||||
| AD5/35 induced EBV/ADV CTLs | EBV | 13 | 13 | 0 | Leen et al. 101 |
| AdV | 12 | 12 | 0 | ||
| Trivirus VSTs with prolonged culture | |||||
| AD5/35 pp65 induced CMV/ADV/EBV VSTs | EBV | 20 | 20 | 0 | Leen et al. 102 |
| AdV | 20 | 20 | 0 | ||
| CMV | 14 | 14 | 0 | ||
| Trivirus VSTs with rapid culture | |||||
| Plasmid‐induced CMV/ADV/EBV VSTs | EBV | 9 | 9 | 0 | Gerdemann et al. 112 |
| AdV | 7 | 7 | 0 | ||
| CMV | 6 | 6 | 0 | ||
| Total | EBV | 147 | 147 | 0 | |
| AdV | 39 | 39 | 0 | ||
| CMV | 20 | 20 | 0 | ||
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Table 3.
Outcome of treatment with donor virus‐specific T cells (VSTs) in our studies
| Specificity | Virus | Patients | Partial or complete response | Failure | References |
|---|---|---|---|---|---|
| Monovirus VSTs with prolonged culture | |||||
| LCL‐induced EBV VSTs | EBV | 13 | 11 | 2 |
Rooney et al. 80 Heslop et al. 79 Rooney et al. 81 Heslop et al. 22 |
| Bivirus VSTs with prolonged culture | |||||
| AD5/35 induced EBV/ADV VSTs | EBV | 1 | 1 | 0 | Leen et al. 101 |
| AdV | 2 | 1 | 1 | ||
| Trivirus VSTs with prolonged culture | |||||
| AD5/35 pp65 induced CMV/ADV/EBV VSTs | EBV | 6 | 6 | 0 | Leen et al. 102 |
| AdV | 6 | 5 | 1 | ||
| CMV | 11 | 10 | 1 | ||
| Trivirus VSTs with rapid culture | |||||
| Plasmid induced CMV/ADV/EBV VSTs | EBV | 2 | 2 | 0 | Gerdemann et al. 112 |
| AdV | 4 | 4 | 0 | ||
| CMV | 5 | 4 | 1 | ||
| Total | EBV | 22 | 20 | 2 | |
| AdV | 12 | 10 | 2 | ||
| CMV | 16 | 14 | 2 | ||
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Similar response rates were seen in another study from Memorial Sloan Kettering Cancer Center (MSKCC), where Doubrovina et al. 83 treated 14 patients with active PTLD with donor‐derived EBV‐specific T cells and showed complete responses in 10. The T cells given to the three patients who failed to respond recognized the LCLs transformed with the laboratory strain B95‐8 used to stimulate them but not the strain of EBV expressed by the patients' tumors. Similarly, in one of the patients in our study who had an initial response then progressed, we found expression of an EBV variant with a deletion in the immunodominant epitopes recognized by the infused line 84. The infused line was able to target the parental tumor cells but the tumor cells with the deletion subsequently grew out. In another patient in the MSKCC series the donor VST line was skewed in its response to EBV antigens by an HLA antigen A1101, which was present only in the donor and the tumor was derived from recipient cells. The patient subsequently responded to a third party line that was matched at fewer antigens but had strong EBV activity through a shared antigen 83. This experience emphasizes the importance of ascertaining whether PTLD is of donor or recipient origin and of infusing a line with antiviral activity through a shared antigen.
The results of these and other studies confirmed that donor‐derived EBV‐specific T‐cell therapy using T‐cell lines generated using LCLs as stimulator cells is safe and effective as prophylaxis and treatment for PTLD after HSCT 85, 86, 87, 88. While the strategy has been implemented at multiple institutions, a limitation is the time taken to generate the LCL line (around 4 weeks) and then the VST line (3–6 weeks). Furthermore, as illustrated above, the use of a laboratory strain of EBV to generate the LCL lines may stimulate a line that does not recognize all EBV strains and makes moving the approach to licensing studies more difficult.
Cytomegalovirus
CMV is a latent β‐herpesvirus that in immunocompetent individuals usually causes asymptomatic infection. However, reactivation of CMV in immunocompromised individuals can result in significant morbidity and mortality, with clinical manifestations including interstitial pneumonitis, gastroenteritis, fevers, hepatitis, encephalitis, and retinitis 8, 89. In the immunocompetent host, CMV‐specific CD4+ and CD8+ T cells play an important role in immune protection from both primary infection and subsequent reactivations. Therefore, a number of groups have investigated whether the adoptive transfer of in vitro expanded donor‐derived VSTs can provide protection during the immunodeficient phase that follows HSCT.
In pioneering work, Walter et al. 90 used fibroblasts infected with the AD169 strain of CMV to stimulate donor T cells followed by limiting‐dilution cloning to isolate cytolytic CD8+ CMV‐directed T cells for adoptive transfer. Cells were administered prophylactically to 14 recipients of HLA‐matched transplants at weekly intervals in doses escalating from 3.3 × 106 to 1 × 109/kg from 30 to 40 days post‐transplant. While the majority of patients lacked any CMV‐specific T‐cell activity prior to infusion, such activity was subsequently detected in all recipients. TCR clonotyping studies showed that the transferred cells persisted for at least 8 weeks but progressively declined in patients who did not develop a concomitant endogenous CMV‐specific CD4+ T‐cell response. This observation highlights the importance of CD4+ helper T cells in sustaining antiviral activity in vivo. Neither CMV viremia nor disease developed in any of the treated patients. More recently, the same group performed a phase II study in which donor‐derived CD8+ and CD4+ T‐cell clones were combined and infused prophylactically to 35 CMV seropositive HSCT recipients. The infusions were safe and immediately augmented CMV‐specific T‐cell immunity. Perruccio et al. 91 also prepared T‐cell clones using donor PBMCs loaded with CMV lysate as a stimulus. In this study, the investigators exclusively generated CD4+ CMV‐specific T‐cell clones, which were infused to 25 haploidentical HSCT recipients at doses ranging from 105 to 3 × 106 cells/kg. Again the cells proved to be safe in vivo and infusion accelerated the recovery of endogenous CD8+ CMV‐directed T cells and controlled CMV antigenemia 91.
As an alternative to infusing clones with a limited diversity of antiviral specificity, other groups have used polyclonal T cells directed to a broader array of CMV antigens and epitopes to prevent and treat CMV infections post‐transplant. For example, Einsele et al. 92 generated CMV‐directed lines using donor PBMCs loaded with CMV lysate as a stimulus. The resultant VSTs contained both CD4+ and CD8+ T cells, which were infused to eight matched or mismatched related HSCT recipients who lacked endogenous CMV‐directed immunity and who were suffering from drug‐resistant CMV viremia (defined as the presence of CMV DNA in the peripheral blood after a minimum of 4 weeks of antiviral chemotherapy). Infusions of 1 × 107 cells/m2 were safe and resulted in a significant viral load reduction in seven evaluable patients, which was durable in five 92. In the two patients with the highest viral loads, however, the antiviral effect was only transient. One of these patients responded to a second VST infusion, whereas the other succumbed to fatal CMV encephalitis after refusing a second dose of cells. Similar successful outcomes were reported by Peggs et al. 93, who used DCs loaded with inactivated CMV antigen to stimulate PBMCs from allogeneic HSCT donors. Sixteen patients were treated with 1 × 105 VSTs/kg at a median of 36 days post‐transplant. The infusions were safe and reconstituted sufficient antiviral immunity to enable additional antiviral drugs to be avoided for eight patients. Finally, Gottlieb et al. reported a phase II study in which CMV‐VSTs were generated using either peptide‐loaded DCs or DCs transduced with an adenoviral vector encoding the full length pp65 antigen. A total of 50 patients received a single dose of 2 × 107 VSTs/m2 from day 28 post‐transplant, and their outcomes were compared with a group of contemporaneous controls. There was no difference in acute or chronic GvHD between the groups and overall and progression‐free survival was similar. However, there was a reduction in the percentage of patients who required CMV‐directed antiviral therapy (17% versus 36%) and in the total number of treatment days in the VST cohort (3.4 days versus 8.9 days) 94, 95, 96. Thus, VSTs can be of direct benefit in preventing/treating CMV reactivations, reducing the requirement for antiviral therapy with a corresponding reduction in drug costs and drug and disease‐associated morbidity.
To increase the practicality of using adoptively transferred T cells and to extend the application to additional viruses, we have developed protocols for the generation of VSTs with simultaneous activity against multiple agents.
Multivirus VSTs
To generate VSTs directed to the most common post‐transplant serious viral infections (CMV, AdV, and EBV), we prepared APCs consisting of activated monocytes and EBV‐LCLs transduced with a clinical grade adenoviral vector, either encoding the immunodominant CMV‐pp65 antigen for the generation of trivirus VSTs from CMV seropositive donors 97, 98, or modified with a null vector for the generation of bivirus‐directed VSTs from CMV seronegative donors 99, 100, 101. Thus, the EBV‐LCL presents viral antigens derived from the transforming EBV, the transducing adenoviral vector, and the encoded CMV pp65 transgene. When used to stimulate donor PBMCs, these ‘double’ or ‘triple‐positive’ LCLs consistently reactivated polyclonal VSTs with activity against the target viruses in a single culture. We administered small numbers (5 × 106–1.5 × 108 cells/m2) of these in vitro expanded donor‐derived bivirus or trivirus VSTs to recipients (age range 1–63 years) of haploidentical, HLA‐matched related or unrelated donor transplants, none of whom developed GVHD. Postinfusion, we observed a detectable increase in the precursor frequency of T cells directed against both EBV and CMV in peripheral blood, but circulating AdV‐specific T cells increased only in those recipients who also had evidence of an AdV infection, demonstrating the importance of in vivo restimulation by viral antigens to support the expansion of the infused cells. We also saw evidence of activity against all three viruses in vivo 101, 102. Ten of 11 patients with CMV reactivation (defined as increased CMV DNA or CMV antigen positive) had a complete or partial response (defined as >50% reduction in the viral load) to VSTs, with a coincident increase in CMV‐specific T cells. Similarly, seven patients with evidence of EBV reactivation (including 1 with EBV‐PTLD) also responded to VSTs, with a marked expansion of EBV‐reactive T cells. Finally, six or eight recipients with AdV infections (defined as culture positive in their respiratory tract, blood, or stool) or disease at the time of VST infusion had a marked reduction in their adenoviral load concomitant with a rise in the frequency of their circulating AdV‐specific T cells. This included one patient with progressive adenoviral pneumonia requiring maximal ventilatory support who had a complete response to VSTs with clearance of AdV from his respiratory tract. All results are summarized in Tables 2 and 3. It is also noteworthy that neither bivirus nor trivirus VST recipients developed de novo AdV infections post‐infusion, in contrast with an incidence of 68% in similar patients who did not receive VSTs 67, suggesting that the infused cells may survive at levels below the threshold for detection and protect patients long term, perhaps by entering the memory pool. This possibility is supported by our ability to detect AdV‐reactive cells after a single ex vivo restimulation of blood T cells with adenoviral antigens 99, 100, 103. Hence, broad spectrum antiviral protection and treatment can be provided from a single infusion of cells and small numbers of T cells can provide long‐term antiviral protection.
Manufacturing limitations for VSTs
As described above, the administration of ex vivo activated and expanded antigen‐specific VSTs with single or multivirus specificity has a proven clinical track record of safety and efficacy in the immunocompromised host that spans almost 20 years. Unfortunately, however, until recently the approach has proved difficult to scale for broad application. Manufacturing of VSTs is technically demanding and prolonged; for trivirus VSTs, for example, the generation of the EBV‐LCL requires 4–6 weeks followed by an additional 4–6 weeks for VST activation and expansion, and a further 1–2 weeks for identity, sterility, and potency testing. In addition, manufacturing must be performed in a facility approved to make cells that meet cGMP standards, and such facilities are expensive to build and maintain. In addition, the costs of manufacturing and release testing live EBV virus (B95‐8 strain) and clinical grade viral vectors are also high. Finally, all these processes demand high skill and substantial time commitment from manufacturing technologists. Fortunately, it has now become possible to address many of these issues of cost, complexity, and scalability.
Reducing the VST manufacturing cost
To reduce costs and alleviate concerns associated with the use of live virus/viral vectors to manufacture VSTs, we have investigated alternative sources of antigen with which to stimulate donor PBMCs. We replaced our adenovector and EBV‐LCLs by generating DNA plasmids encoding immunogenic antigens from EBV (EBNA1, LMP2, BZLF1), CMV (IE1, pp65), and AdV (Hexon, Penton), which could be introduced into APCs, such as DCs, using the clinically acceptable AMAXA nucleofection system. In preclinical studies, we achieved high level transgene expression following nucleofection of DCs with good viability during the period of T‐cell activation and the reproducible generation of VSTs that were phenotypically similar to our conventionally generated VSTs (mix of CD4+ and CD8+ T cells, a proportion of which retained expression of the central memory marker CD62L), and that contained virus‐specific T cells at frequencies that were comparable or superior to conventional products 104. Clinical grade plasmid‐DNA can be rapidly and cost‐effectively produced in scalable quantities with excellent long‐term stability. Since plasmids are non‐infectious, non‐replicative, and integrate poorly into the transfected cell genome, this approach also alleviates safety concerns. Thus, transition to such an antigen source would substantially lower the production costs of VSTs.
More recently we evaluated direct stimulation of donor PBMCs with commercially available GMP‐grade peptide mixtures (pepmixes). These consist of 15mer peptide libraries derived from viral antigens that overlap by 11 amino acids. Since each peptide is 15 amino acids in length, the pepmixes cover all possible CD8+ and the majority of CD4+ epitopes 105. These 15mer‐produced VSTs were phenotypically and functionally equivalent to conventionally generated VSTs. This refinement eliminates the need for DCs or other specialized APCs and for virus/viral vectors for stimulation, resulting in a >50% reduction in manufacturing costs.
Reducing VST manufacturing complexity
Conventional VSTs are made by sequential expansion steps in traditional 24‐well tissue‐culture plates, with weekly restimulation and regular splitting of the cultured cells. This system is extremely labor intensive, highly skilled, and essentially unscalable. The frequent media changes required to optimize nutrient and cytokine levels and remove waste products make contamination a constant risk and still allow T cells to undergo significant cell death. Efforts to substitute the closed and scalable bioreactor systems that are widely used in other types of clinical cell culture systems failed to produce VSTs with the same anti‐viral specificity and effector function. To address this logistic issue, we collaborated with Wilson Wolf Manufacturing to develop a new disposable gas permeable rapid expansion device (G‐Rex), which dramatically reduces T‐cell apoptosis during culture, resulting in more efficient in vitro expansion. In this static device, gas exchange (O2 in and CO2 out) occurs across a gas permeable silicon membrane at the base, allowing for the initial input media volume to be increased, which in turn increases the available nutrients and dilutes waste products without the need for culture agitation, frequent culture feeding, or continuous medium perfusion 106, 107, 108. These optimized growth conditions allow for higher antigen‐specific T‐cell densities per unit surface area to be achieved (8–10 × 106 per cm2 in the G‐Rex compared with approximately 1.5 × 106 per cm2 in wells), and incorporation of this platform substantially simplifies VST production by minimizing the number and complexity of cell manipulations without affecting the phenotype or function of the generated T‐cell lines 109.
Reducing antigenic competition
Until recently, a maximum of three viruses could be successfully targeted in a single VST. Efforts to make VSTs that were consistently specific for additional viruses had foundered on the problem of antigenic competition. This phenomenon occurs due to competition between virus‐derived peptides to become bound to HLA molecules expressed on shared APCs and the physical constraints on the capacity of APCs to simultaneously engage T cells with different specificities if these are present at substantially different frequencies. Indeed, even the conventional trivirus (EBV+CMV+AdV)‐directed VSTs described above are dominated by CMV‐specific T cells at the expense of the EBV and AdV‐specific components, so that both the breadth of AdV epitopes recognized and the magnitude of the response to them is less in the trivirus products than in bivirus products generated using EBV‐LCLs transduced with an adenoviral vector lacking the CMV‐pp65 transgene 100. This antigenic competition and the resultant production of lines with restricted viral specificities precludes the generation of potent, broad‐spectrum effector T cells that would be effective against the complete array of pathogens present in HSCT recipients.
Our conventional VST manufacturing process did not incorporate exogenous cytokines, which we discovered adversely affected VST proliferation and survival in vitro, increasing their susceptibility to activation‐induced cell death (AICD), and contributing to a restricted repertoire of epitope recognition. Thus, we increased the range of viral antigens that could be recognized by a single VST line and thereby mitigated the impact of antigenic competition. We supplemented cultures of peptide‐pulsed PBMCs with cytokines shown to support T‐cell proliferation in vitro and in vivo (IL‐2, IL‐15), as well as cytokine combinations (IL‐4+IL‐7) that support retention of a central memory phenotype, and promote activated T‐cell survival by upregulating anti‐apoptotic molecules 110, 111. When lines were supplemented with IL‐4+IL‐7, we observed expansion and survival of both CD4+ and CD8+ virus‐specific T cells within the PBMCs, which recognized multiple viral epitopes and killed virus‐infected targets 105. The induced cells were Th1‐polarized despite exposure to IL‐4, a prototypic Th2 cytokine. Importantly, these VST cultures lacked alloreactive T cells, even when the VSTs had been produced by only a single in vitro stimulation with viral peptides. By this approach, we were able to alleviate antigenic competition both within the APC and between T cells.
Clinical results using rapidly generated VSTs
To learn whether our manufacturing modifications produced clinically active VSTs that performed as well as or better than conventionally generated VSTs, we initiated two clinical studies. In the first clinical trial, we assessed the safety and potency of trivirus VSTs, produced using plasmid‐nucleofected DCs as APCs followed by expansion for 10 days in the G‐Rex device in media supplemented with IL‐4+IL‐7. These rapidly generated trivirus VSTs were infused to 10 allogeneic HSCT recipients (five haploidentical, four matched unrelated donor, one mismatched unrelated donor, and one matched related donor), all of whom had active infections with one or more of our target viruses (3 CMV, 2 AdV, 2 EBV, 2 EBV+AdV, and 2 CMV+AdV). Each patient received 0.5–2 × 107 cells/m2 from day 27 post‐HSCT. One patient developed a mild and localized skin rash postinfusion, but this patient also had an intercurrent BK infection and had presented with a similar rash during an earlier episode of BK reactivation. No other infusion‐related toxicities were noted. Overall, our clinical response rate was equivalent to that achieved in our previous trials of trivirus‐directed T cells generated using EBV and AdV vectors for manufacturing, with 90% of subjects with active AdV, CMV, and/or EBV infections having a measurable increase in circulating T cells directed against the infecting virus(es), and clearance of both single and dual viral reactivation/disease. Importantly, the lines in this study were generated without exposure to biohazards (live virus/viral vectors) and were manufactured in only 17 days (versus 8–12 weeks for ‘conventional’ VSTs) and from 60 ml of peripheral blood without evident loss of efficacy 112.
More recently, we have initiated a clinical trial based around a further manufacturing simplification, in which donor PBMCs are stimulated by pepmix combinations spanning 12 immunogenic antigens from five clinically relevant viruses (EBV, CMV, AdV, BK, HHV6) followed by expansion for 10 days in the G‐Rex device using media supplemented with IL‐4+IL‐7 112, 113. To date, we have infused these pentavalent (p)VSTs to 10 allogeneic HSCT recipients at doses ranging from 5 × 106 to 2 × 107/m2 with no immediate infusional toxicities, and no de novo acute GvHD. This study is still ongoing, but to date the infused pVSTs have successfully controlled active infections associated with all five of the target viruses. Thus, it appears that rapidly produced (10 days) VSTs targeting five clinically relevant pathogens that are generated from 20 ml of peripheral blood can produce clinical benefit, and we are currently exploring the extension of this platform to include additional clinically relevant viruses.
The manufacturing modifications described above should help VST therapies become a standard of care for transplant recipients by facilitating adoption of the approach in sponsored late phase clinical studies.
Extending VST therapy to recipients irrespective of donor availability/immunity
Despite the promising results of VST therapy in the allogeneic HSCT setting, it may be more challenging to translate this therapeutic modality to recipients of grafts from seronegative donors such as those derived from cord blood, an increasingly important alternative source for stem cells. Two approaches may be used to serve such individuals.
VSTs from cord blood donors
Generation of a virus‐specific T‐cell product from cord blood for infusion is complicated by the low numbers of available cells, their naive phenotype, and the associated low frequency of virus‐reactive T cells. Hence, the generation of VSTs in vitro requires the use of donor DCs as professional APCs and the addition of enhancing cytokines IL‐12, IL‐15, and IL‐7. Whether such cells will have the same in vivo persistence and antiviral activity as VSTs generated from seropositive donor peripheral blood remains to be seen 114. At last report (Hanley et al., American Society of Hematology annual meeting, 2012), seven patients received between 5 × 106 and 2.5 × 107 cells/m2 on days 63–146 after cord blood transplant, with no infusion‐related toxicities or subsequent GvHD. Five patients had no initial infection or reactivation episodes. Two patients had evidence of CMV or EBV reactivation, both of which were controlled, although for CMV two VST infusions were required. Thus, early reports indicate that the infusion of these cord blood derived antigen‐specific T cells can support immune reconstitution in vivo.
Third party VST banks
Despite the clinical benefit associated with VST therapy and the ease of manufacturing afforded by our production refinements, the need to generate specific VSTs for each individual patient renders this approach impractical for widespread or urgent use, or when the donor lacks viral immunity (as for cord blood transplant described above). To circumvent this requirement, investigators have prepared and administered banks of closely HLA‐matched VSTs that are ‘off the shelf’ products and so are available for immediate use. Administration of such products is, however, associated with some concerns. Most lines administered will be mismatched at one or more HLA‐loci, which may either increase GvHD postinfusion due to alloreactivity of the line or reduce in vivo persistence and antiviral benefit, due to the alloreactivity of the recipient. Despite these concerns, a number of studies have shown that the approach is feasible, safe, and effective, with reports of a high level of clinical responses. For example, Haque et al. 115, 116 used third party EBV‐specific VSTs to treat PTLD after solid organ or HSCT transplantation. In this study, patients received four doses of 2 × 106 VSTs/kg at weekly intervals and the lines were selected for matching by low resolution typing and screened for high level killing of donor EBV‐LCLs and low level killing of patient‐derived PHA blasts. No patient developed organ rejection or GVHD, and the study showed response rates of 64% and 52% at 5 weeks and 6 months respectively. The degree of HLA matching between the line and the recipient ranged from 2/6 to 5/6 HLA antigens, and at 6 months, there was a statistically significant trend toward a better outcome with closer matching 115, 116. In a second report, two solid organ transplant recipients with CNS lymphoma received closely matched EBV‐specific T cells leading to complete resolution of brain lesions due to EBV‐LPD 117. Similarly, the Memorial Sloan Kettering group reported that third party EBV‐specific VSTs produced complete responses in four of five patients with EBV PTLD after HSCT (including two cord blood transplant recipients) 83, 118. More recently, our group applied this approach to treat patients with refractory CMV, AdV, or EBV infections 119. In this multicenter study, we established a bank of 32 trivirus‐directed VSTs using conventional manufacturing approaches, 18 of which were administered to 50 recipients of allogeneic HSCTs [marrow (n = 24), peripheral blood (n = 33), or cord blood (n = 12 single and 13 double units)]. Our bank was of sufficient size for us to identify a suitable line for 90% of the patients screened for participation. Lines for infusion were chosen based on the presence of activity against the infecting virus through a shared HLA allele(s), which requires a comprehensive analysis of viral epitopes in the VSTs and the identification of the HLA restricting elements associated with each. Once activity against the infecting virus through a shared HLA allele(s) was confirmed, further selection of the best line was based on the overall degree of HLA match. Of the 50 patients who were treated, 74% had a complete or partial response (74% for CMV, 78% for AdV and 67% for EBV), the majority of which were durable (Table 4). In vivo T‐cell persistence, monitored using deep sequencing analysis, indicated that donor VST‐derived TCR sequences became apparent concomitant with a reduction in viral titers and persisted 4–12 weeks postinfusion. Although long‐term (beyond 3 months post‐infusion) analysis was not performed the expectation is that the third party cells were eliminated concomitant with endogenous immune recovery. Indeed, of the 50 patients treated, 14 required between 2 and 6 VSTs to sustain clinical benefit. However, we saw no significant de novo GvHD, and only one episode of secondary stem‐cell graft failure (in a patient with relapse). Thus, this study, like the other applications of banked VSTs, observed no associated increase in toxicity.
Table 4.
Outcome of treatment with third party virus‐specific T cells (VSTs) in our studies
| Specificity | Virus | Patients | Complete or partial response | Failure | References |
|---|---|---|---|---|---|
| Trivirus VSTs with prolonged culture | |||||
| AD5/35 pp65 induced CMV/AdV/EBV VSTs | EBV | 9 | 6 | 3 | Leen et al. 119 |
| AdV | 18 | 14 | 4 | ||
| CMV | 23 | 17 | 6 | ||
| Total | 50 | 37 | 13 | ||
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Since the available clinical data support both the safety and clinical activity of third party VSTs, it should be possible to use such banks on a larger scale, particularly given the availability of more rapid VST manufacturing technology (see section ‘Manufacturing limitations for VSTs’ above), thereby making the adoptive transfer of VSTs a standard of care for transplant recipients in which these third party cells may be used as an immediate measure while the need for specific donor‐derived VSTs can be assessed and, if necessary, this additional product made.
Future perspectives
It is likely that VSTs will have an increasing role to play in the prevention and management of post‐transplant viral infections. Our ability to rapidly select VSTs for viruses such as CMV and EBV and to relatively rapidly culture cell lines specific for a broad range of common viral antigens, coupled with the feasibility of developing third‐party off the shelf VSTs will ensure that the advantages of this approach compared with available small molecule therapies will become increasingly evident.
Acknowledgements
This work was supported in parts by NIH grants P50CA126752, PO1CA94237, U54HL081007, N01‐HB‐10‐03, and the Production Assistance for Cellular Therapies (PACT) program (NHLBI contract #HHSN268201000007C). We also appreciate the support of shared resources by the Dan L. Duncan Cancer Center support grant P30CA125123. H. E. H. is supported by a Dan L. Duncan Chair and M. K. B. by a Fayez Sarofim Chair. The authors all have patents in the area of cellular therapy and the authors' Center has a collaborative research agreement with Celgene for genetically modified T cells. The authors declare no conflict of interest.
References
- 1. Navarro WH. CIBMTR Summary slides. 2‐1‐2012. Ref Type: Slide
- 2. Tomblyn M, et al. Guidelines for preventing infectious complications among hematopoietic cell transplantation recipients: a global perspective. Biol Blood Marrow Transplant 2009;15:1143–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Boeckh M, Erard V, Zerr D, Englund J. Emerging viral infections after hematopoietic cell transplantation. Pediatr Transplant 2005;9(Suppl 7):48–54. [DOI] [PubMed] [Google Scholar]
- 4. Kim YJ, Boeckh M, Englund JA. Community respiratory virus infections in immunocompromised patients: hematopoietic stem cell and solid organ transplant recipients, and individuals with human immunodeficiency virus infection. Semin Respir Crit Care Med 2007;28:222–242. [DOI] [PubMed] [Google Scholar]
- 5. Peck AJ, et al. Respiratory virus infection among hematopoietic cell transplant recipients: evidence for asymptomatic parainfluenza virus infection. Blood 2007;110:1681–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kuehnle I, et al. CD20 monoclonal antibody (rituximab) for therapy of Epstein‐Barr virus lymphoma after hemopoietic stem‐cell transplantation. Blood 2000;95:1502–1505. [PubMed] [Google Scholar]
- 7. Heslop HE, How I. treat EBV lymphoproliferation. Blood 2009;114:4002–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boeckh M, Ljungman P. How we treat cytomegalovirus in hematopoietic cell transplant recipients. Blood 2009;113:5711–5719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Porter DL, Roth MS, McGarigle C, Ferrara JL, Antin JH. Induction of graft‐versus‐host disease as immunotherapy for relapsed chronic myeloid leukemia. N Engl J Med 1994;330:100–106. [DOI] [PubMed] [Google Scholar]
- 10. Porter DL, et al. Long‐term follow‐up of patients who achieved complete remission after donor leukocyte infusions. Biol Blood Marrow Transplant 1999;5:253–261. [DOI] [PubMed] [Google Scholar]
- 11. Collins RHJ, et al. Donor leukocyte infusions in 140 patients with relapsed malignancy after allogeneic bone marrow transplantation [see comments]. J Clin Oncol 1997;15:433–444. [DOI] [PubMed] [Google Scholar]
- 12. Kolb HJ, et al. Graft‐versus‐leukemia effect of donor lymphocyte transfusions in marrow grafted patients. Blood 1995;86:2041–2050. [PubMed] [Google Scholar]
- 13. Roush KS, Hillyer CD. Donor lymphocyte infusion therapy. Transfus Med Rev 2002;16:161–176. [DOI] [PubMed] [Google Scholar]
- 14. Hromas R, Cornetta K, Srour E, Blanke C, Broun ER. Donor leukocyte infusion as therapy of life‐threatening adenoviral infections after T‐cell‐depleted bone marrow transplantation. Blood 1994;84:1689–1690. [PubMed] [Google Scholar]
- 15. Papadopoulos EB, et al. Infusions of donor leukocytes to treat Epstein‐Barr virus‐associated lymphoproliferative disorders after allogeneic bone marrow transplantation. N Engl J Med 1994;330:1185–1191. [DOI] [PubMed] [Google Scholar]
- 16. Heslop HE, Brenner MK, Rooney CM. Donor T cells to treat EBV‐associated lymphoma. N Engl J Med 1994;331:679–680. [DOI] [PubMed] [Google Scholar]
- 17. Mackinnon S, Papadopoulos EB, Carabasi MH, Reich L, Collins NH, O'Reilly RJ. Adoptive immunotherapy using donor leukocytes following bone marrow transplantation for chronic myeloid leukemia: is T cell dose important in determining biological response? Bone Marrow Transplant 1995;15:591–594. [PubMed] [Google Scholar]
- 18. Guinan EC, Gribben JG, Boussiotis VA, Freeman GJ, Nadler LM. Pivotal role of the B7:CD28 pathway in transplantation tolerance and tumor immunity. Blood 1994;84:3261–3282. [PubMed] [Google Scholar]
- 19. Davies JK, Barbon CM, Voskertchian AR, Nadler LM, Guinan EC. Induction of alloantigen‐specific anergy in human peripheral blood mononuclear cells by alloantigen stimulation with co‐stimulatory signal blockade. J Vis Exp 2011;14:pii2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Davies JK, Gribben JG, Brennan LL, Yuk D, Nadler LM, Guinan EC. Outcome of alloanergized haploidentical bone marrow transplantation after ex vivo costimulatory blockade: results of 2 phase 1 studies. Blood 2008;112:2232–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mielke S, et al. A clinical‐scale selective allodepletion approach for the treatment of HLA‐mismatched and matched donor‐recipient pairs using expanded T lymphocytes as antigen‐presenting cells and a TH9402‐based photodepletion technique. Blood 2008;111:4392–4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Heslop HE, et al. Long‐term outcome of EBV‐specific T‐cell infusions to prevent or treat EBV‐related lymphoproliferative disease in transplant recipients. Blood 2010;115:925–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Amrolia PJ, et al. Selective depletion of donor alloreactive T cells without loss of antiviral or antileukemic responses. Blood 2003;102:2292–2299. [DOI] [PubMed] [Google Scholar]
- 24. Albon SJ, et al. Optimization of methodology for production of CD25/CD71 allodepleted donor T cells for clinical use. Cytotherapy 2013;15:109–121. [DOI] [PubMed] [Google Scholar]
- 25. Nonn M, et al. Selective depletion of alloreactive T lymphocytes using patient‐derived nonhematopoietic stimulator cells in allograft engineering. Transplantation 2008;86:1427–1435. [DOI] [PubMed] [Google Scholar]
- 26. Solomon SR, et al. Optimized clinical‐scale culture conditions for ex vivo selective depletion of host‐reactive donor lymphocytes: a strategy for GvHD prophylaxis in allogeneic PBSC transplantation. Cytotherapy 2002;4:395–406. [DOI] [PubMed] [Google Scholar]
- 27. Solomon SR, et al. Selective depletion of alloreactive donor lymphocytes: a novel method to reduce the severity of graft‐versus‐host disease in older patients undergoing matched sibling donor stem cell transplantation. Blood 2005;106:1123–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mavroudis DA, et al. Specific depletion of alloreactivity against haplotype mismatched related individuals by a recombinant immunotoxin: a new approach to graft‐versus‐host disease prophylaxis in haploidentical bone marrow transplantation. Bone Marrow Transplant 1996;17:793–799. [PubMed] [Google Scholar]
- 29. van Dijk AM, Kessler FL, Stadhouders‐Keet SA, Verdonck LF, de Gast GC, Otten HG. Selective depletion of major and minor histocompatibility antigen reactive T cells: towards prevention of acute graft‐versus‐host disease. Br J Haematol 1999;107:169–175. [DOI] [PubMed] [Google Scholar]
- 30. Fehse B, Frerk O, Goldmann M, Bulduk M, Zander AR. Efficient depletion of alloreactive donor T lymphocytes based on expression of two activation‐induced antigens (CD25 and CD69). Br J Haematol 2000;109:644–651. [DOI] [PubMed] [Google Scholar]
- 31. Samarasinghe S, et al. Functional characterization of alloreactive T cells identifies CD25 and CD71 as optimal targets for a clinically applicable allodepletion strategy. Blood 2010;115:396–407. [DOI] [PubMed] [Google Scholar]
- 32. Wehler TC, et al. Targeting the activation‐induced antigen CD137 can selectively deplete alloreactive T cells from antileukemic and antitumor donor T‐cell lines. Blood 2007;109:365–373. [DOI] [PubMed] [Google Scholar]
- 33. Hartwig UF, Nonn M, Khan S, Meyer RG, Huber C, Herr W. Depletion of alloreactive T cells via CD69: implications on antiviral, antileukemic and immunoregulatory T lymphocytes. Bone Marrow Transplant 2006;37:297–305. [DOI] [PubMed] [Google Scholar]
- 34. Hartwig UF, Nonn M, Khan S, Link I, Huber C, Herr W. Depletion of alloreactive donor T lymphocytes by CD95‐mediated activation‐induced cell death retains antileukemic, antiviral, and immunoregulatory T cell immunity. Biol Blood Marrow Transplant 2008;14:99–109. [DOI] [PubMed] [Google Scholar]
- 35. Sathe A, Ortega SB, Mundy DI, Collins RH, Karandikar NJ. In vitro methotrexate as a practical approach to selective allodepletion. Biol Blood Marrow Transplant 2007;13:644–654. [DOI] [PubMed] [Google Scholar]
- 36. Guimond M, Balassy A, Barrette M, Brochu S, Perreault C, Roy DC. P‐glycoprotein targeting: a unique strategy to selectively eliminate immunoreactive T cells. Blood 2002;100:375–382. [DOI] [PubMed] [Google Scholar]
- 37. Chen BJ, Cui X, Liu C, Chao NJ. Prevention of graft‐versus‐host disease while preserving graft‐versus‐leukemia effect after selective depletion of host‐reactive T cells by photodynamic cell purging process. Blood 2002;99:3083–3088. [DOI] [PubMed] [Google Scholar]
- 38. Bastien JP, et al. Photodepletion differentially affects CD4+ Tregs versus CD4+ effector T cells from patients with chronic graft‐versus‐host disease. Blood 2010;116:4859–4869. [DOI] [PubMed] [Google Scholar]
- 39. Bastien JP, Roy J, Roy DC. Selective T‐cell depletion for haplotype‐mismatched allogeneic stem cell transplantation. Semin Oncol 2012;39:674–682. [DOI] [PubMed] [Google Scholar]
- 40. Andre‐Schmutz I, et al. Immune reconstitution without graft‐versus‐host disease after haemopoietic stem‐cell transplantation: a phase 1/2 study. Lancet 2002;360:130–137. [DOI] [PubMed] [Google Scholar]
- 41. Amrolia PJ, et al. Add‐back of allodepleted donor T cells to improve immune reconstitution after haplo‐identical stem cell transplantation. Cytotherapy 2005;7:116–125. [DOI] [PubMed] [Google Scholar]
- 42. Amrolia PJ, et al. Adoptive immunotherapy with allodepleted donor T‐cells improves immune reconstitution after haploidentical stem cell transplantation. Blood 2006;108:1797–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mielke S, et al. Selectively T cell‐depleted allografts from HLA‐matched sibling donors followed by low‐dose posttransplantation immunosuppression to improve transplantation outcome in patients with hematologic malignancies. Biol Blood Marrow Transplant 2011;17:1855–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Oliveira G, Greco R, Lupo‐Stanghellini MT, Vago L, Bonini C. Use of TK‐cells in haploidentical hematopoietic stem cell transplantation. Curr Opin Hematol 2012;19:427–433. [DOI] [PubMed] [Google Scholar]
- 45. Bonini C, et al. HSV‐TK gene transfer into donor lymphocytes for control of allogeneic graft versus leukemia. Science 1997;276:1719–1724. [DOI] [PubMed] [Google Scholar]
- 46. Bonini C, et al. The suicide gene therapy challenge: how to improve a successful gene therapy approach. Mol Ther 2007;15:1248–1252. [DOI] [PubMed] [Google Scholar]
- 47. Bonini C, et al. Safety of retroviral gene marking with a truncated NGF receptor. Nat Med 2003;9:367–369. [DOI] [PubMed] [Google Scholar]
- 48. Ciceri F, et al. Antitumor effects of HSV‐TK‐engineered donor lymphocytes after allogeneic stem‐cell transplantation. Blood 2007;109:4698–4707. [DOI] [PubMed] [Google Scholar]
- 49. Tiberghien P, et al. Administration of herpes simplex‐thymidine kinase‐expressing donor T cells with a T‐cell‐depleted allogeneic marrow graft. Blood 2001;97:63–72. [DOI] [PubMed] [Google Scholar]
- 50. Ciceri F, et al. Infusion of suicide‐gene‐engineered donor lymphocytes after family haploidentical haemopoietic stem‐cell transplantation for leukaemia (the TK007 trial): a non‐randomised phase I‐II study. Lancet Oncol 2009;10:489–500. [DOI] [PubMed] [Google Scholar]
- 51. Riddell SR, et al. T‐cell mediated rejection of gene‐modified HIV‐specific cytotoxic T lymphocytes in HIV‐infected patients. Nat Med 1996;2:216–223. [DOI] [PubMed] [Google Scholar]
- 52. Berger C, Flowers ME, Warren EH, Riddell SR. Analysis of transgene‐specific immune responses that limit the in vivo persistence of adoptively transferred HSV‐TK‐modified donor T cells after allogeneic hematopoietic cell transplantation. Blood 2006;107:2294–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mercier‐Letondal P, et al. Early immune response against retrovirally transduced herpes simplex virus thymidine kinase‐expressing gene‐modified T cells coinfused with a T cell‐depleted marrow graft: an altered immune response? Hum Gene Ther 2008;19:937–950. [DOI] [PubMed] [Google Scholar]
- 54. Traversari C, et al. The potential immunogenicity of the TK suicide gene does not prevent full clinical benefit associated with the use of TK‐transduced donor lymphocytes in HSCT for hematologic malignancies. Blood 2007;109:4708–4715. [DOI] [PubMed] [Google Scholar]
- 55. Di SA, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med 2011;365:1673–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Straathof KC, et al. An inducible caspase 9 safety switch for T‐cell therapy. Blood 2005;105:4247–4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tey SK, Dotti G, Rooney CM, Heslop HE, Brenner MK. Inducible caspase 9 suicide gene to improve the safety of allodepleted T cells after haploidentical stem cell transplantation. Biol Blood Marrow Transplant 2007;13:913–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cobbold M, et al. Adoptive transfer of cytomegalovirus‐specific CTL to stem cell transplant patients after selection by HLA‐peptide tetramers. J Exp Med 2005;202:379–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Uhlin M, Okas M, Gertow J, Uzunel M, Brismar TB, Mattsson J. A novel haplo‐identical adoptive CTL therapy as a treatment for EBV‐associated lymphoma after stem cell transplantation. Cancer Immunol Immunother 2010;59:473–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Uhlin M, et al. Rapid salvage treatment with virus‐specific T cells for therapy‐resistant disease. Clin Infect Dis 2012;55:1064–1073. [DOI] [PubMed] [Google Scholar]
- 61. Neudorfer J, et al. Reversible HLA multimers (Streptamers) for the isolation of human cytotoxic T lymphocytes functionally active against tumor‐ and virus‐derived antigens. J Immunol Methods 2007;320:119–131. [DOI] [PubMed] [Google Scholar]
- 62. Schmitt A, et al. Adoptive transfer and selective reconstitution of streptamer‐selected cytomegalovirus‐specific CD8+ T cells leads to virus clearance in patients after allogeneic peripheral blood stem cell transplantation. Transfusion 2011;51:591–599. [DOI] [PubMed] [Google Scholar]
- 63. Feuchtinger T, et al. Safe adoptive transfer of virus‐specific T‐cell immunity for the treatment of systemic adenovirus infection after allogeneic stem cell transplantation. Br J Haematol 2006;134:64–76. [DOI] [PubMed] [Google Scholar]
- 64. Lindemans CA, Leen AM, Boelens JJ. How I treat adenovirus in hematopoietic stem cell transplant recipients. Blood 2010;116:5476–5485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hale GA, et al. Adenovirus infection after pediatric bone marrow transplantation. Bone Marrow Transplant 1999;23:277–282. [DOI] [PubMed] [Google Scholar]
- 66. Myers GD, et al. Reconstitution of adenovirus‐specific cell‐mediated immunity in pediatric patients after hematopoietic stem cell transplantation. Bone Marrow Transplant 2007;39:677–686. [DOI] [PubMed] [Google Scholar]
- 67. Myers GD, et al. Adenovirus infection rates in pediatric recipients of alternate donor allogeneic bone marrow transplants receiving either antithymocyte globulin (ATG) or alemtuzumab (Campath). Bone Marrow Transplant 2005;36:1001–1008. [DOI] [PubMed] [Google Scholar]
- 68. Feuchtinger T, et al. Adoptive transfer of pp65‐specific T cells for the treatment of chemorefractory cytomegalovirus disease or reactivation after haploidentical and matched unrelated stem cell transplantation. Blood 2010;116:4360–4367. [DOI] [PubMed] [Google Scholar]
- 69. Peggs KS, et al. Directly selected cytomegalovirus‐reactive donor T cells confer rapid and safe systemic reconstitution of virus‐specific immunity following stem cell transplantation. Clin Infect Dis 2011;52:49–57. [DOI] [PubMed] [Google Scholar]
- 70. Moosmann A, et al. Effective and long‐term control of EBV PTLD after transfer of peptide‐selected T cells. Blood 2010;115:2960–2970. [DOI] [PubMed] [Google Scholar]
- 71. Icheva V, et al. Adoptive transfer of Epstein‐Barr Virus (EBV) nuclear antigen 1‐specific T cells as treatment for EBV reactivation and lymphoproliferative disorders after allogeneic stem‐cell transplantation. J Clin Oncol 2013;31:39–48. [DOI] [PubMed] [Google Scholar]
- 72. Rickinson AB, Lee SP, Steven NM. Cytotoxic T lymphocyte responses to Epstein‐Barr virus. Curr Opin Immunol 1996;8:492–497. [DOI] [PubMed] [Google Scholar]
- 73. Rickinson AB, Kieff E. Epstein‐Barr virus In: Fields BN, Knipe DM, Howley PM. eds. Fields Virology. Philadelphia, PA: Lipincott‐Raven, 1996:2397–2446. [Google Scholar]
- 74. Hislop AD, Taylor GS, Sauce D, Rickinson AB. Cellular responses to viral infection in humans: lessons from Epstein‐Barr virus. Annu Rev Immunol 2007;25:587–617. [DOI] [PubMed] [Google Scholar]
- 75. Tan LC, et al. A re‐evaluation of the frequency of CD8+ T cells specific for EBV in healthy virus carriers. J Immunol 1999;162:1827–1835. [PubMed] [Google Scholar]
- 76. Callan MF, et al. Direct visualization of antigen‐specific CD8+ T cells during the primary immune response to Epstein‐Barr virus In vivo. J Exp Med 1998;187:1395–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hongeng S, et al. Outcomes of transplantation with matched‐sibling and unrelated‐donor bone marrow in children with leukemia. Lancet 1997;350:767–770. [DOI] [PubMed] [Google Scholar]
- 78. Smith CA, et al. Production of genetically modified Epstein‐Barr virus‐specific cytotoxic T cells for adoptive transfer to patients at high risk of EBV‐associated lymphoproliferative disease. J Hematother 1995;4:73–79. [DOI] [PubMed] [Google Scholar]
- 79. Heslop HE, et al. Long‐term restoration of immunity against Epstein‐Barr virus infection by adoptive transfer of gene‐modified virus‐specific T lymphocytes. Nat Med 1996;2:551–555. [DOI] [PubMed] [Google Scholar]
- 80. Rooney CM, et al. Use of gene‐modified virus‐specific T lymphocytes to control Epstein‐Barr virus‐related lymphoproliferation. Lancet 1995;345:9–13. [DOI] [PubMed] [Google Scholar]
- 81. Rooney CM, et al. Infusion of cytotoxic T cells for the prevention and treatment of Epstein‐Barr virus‐induced lymphoma in allogeneic transplant recipients. Blood 1998;92:1549–1555. [PubMed] [Google Scholar]
- 82. Melenhorst JJ, et al. Allogeneic virus‐specific T cells with HLA alloreactivity do not produce GVHD in human subjects. Blood 2010;116:4700–4702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Doubrovina E, et al. Adoptive immunotherapy with unselected or EBV‐specific T cells for biopsy‐proven EBV+ lymphomas after allogeneic hematopoietic cell transplantation. Blood 2012;119:2644–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Gottschalk S, et al. An Epstein‐Barr virus deletion mutant associated with fatal lymphoproliferative disease unresponsive to therapy with virus‐specific CTLs. Blood 2001;97:835–843. [DOI] [PubMed] [Google Scholar]
- 85. Comoli P, et al. Preemptive therapy of EBV‐related lymphoproliferative disease after pediatric haploidentical stem cell transplantation. Am J Transplant 2007;7:1648–1655. [DOI] [PubMed] [Google Scholar]
- 86. Comoli P, et al. Infusion of autologous Epstein‐Barr virus (EBV)‐specific cytotoxic T cells for prevention of EBV‐related lymphoproliferative disorder in solid organ transplant recipients with evidence of active virus replication. Blood 2002;99:2592–2598. [DOI] [PubMed] [Google Scholar]
- 87. Gustafsson A, et al. Epstein‐Barr virus (EBV) load in bone marrow transplant recipients at risk to develop posttransplant lymphoproliferative disease: prophylactic infusion of EBV‐specific cytotoxic T cells. Blood 2000;95:807–814. [PubMed] [Google Scholar]
- 88. Lucas KG, et al. Semiquantitative Epstein‐Barr virus (EBV) polymerase chain reaction for the determination of patients at risk for EBV‐induced lymphoproliferative disease after stem cell transplantation. Blood 1998;91:3654–3661. [PubMed] [Google Scholar]
- 89. Boeckh M, Nichols WG, Papanicolaou G, Rubin R, Wingard JR, Zaia J. Cytomegalovirus in hematopoietic stem cell transplant recipients: current status, known challenges, and future strategies. Biol Blood Marrow Transplant 2003;9:543–558. [DOI] [PubMed] [Google Scholar]
- 90. Walter EA, et al. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T‐cell clones from the donor. N Engl J Med 1995;333:1038–1044. [DOI] [PubMed] [Google Scholar]
- 91. Perruccio K, et al. Transferring functional immune responses to pathogens after haploidentical hematopoietic transplantation. Blood 2005;106:4397–4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Einsele H, et al. Infusion of cytomegalovirus (CMV)‐specific T cells for the treatment of CMV infection not responding to antiviral chemotherapy. Blood 2002;99:3916–3922. [DOI] [PubMed] [Google Scholar]
- 93. Peggs KS, et al. Adoptive cellular therapy for early cytomegalovirus infection after allogeneic stem‐cell transplantation with virus‐specific T‐cell lines. Lancet 2003;362:1375–1377. [DOI] [PubMed] [Google Scholar]
- 94. Micklethwaite K, et al. Ex vivo expansion and prophylactic infusion of CMV‐pp65 peptide‐specific cytotoxic T‐lymphocytes following allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2007;13:707–714. [DOI] [PubMed] [Google Scholar]
- 95. Micklethwaite KP, et al. Prophylactic infusion of cytomegalovirus‐specific cytotoxic T lymphocytes stimulated with Ad5f35 pp65 gene‐modified dendritic cells after allogeneic hemopoietic stem cell transplantation. Blood 2008;112:3974–3981. [DOI] [PubMed] [Google Scholar]
- 96. Blyth E, et al. Donor‐derived CMV‐specific T cells reduce the requirement for CMV‐directed pharmacotherapy after allogeneic stem cell transplantation. Blood 2013;121:3745–3758. [DOI] [PubMed] [Google Scholar]
- 97. Sili U, et al. Production of good manufacturing practice‐grade cytotoxic T lymphocytes specific for Epstein‐Barr virus, cytomegalovirus and adenovirus to prevent or treat viral infections post‐allogeneic hematopoietic stem cell transplant. Cytotherapy 2012;14:7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Leen A, et al. Contact‐activated monocytes: efficient antigen presenting cells for the stimulation of antigen‐specific T cells. J Immunother 1997;2007:96–107. [DOI] [PubMed] [Google Scholar]
- 99. Leen AM, et al. Fiber‐modified adenoviruses generate subgroup cross‐reactive, adenovirus‐specific cytotoxic T lymphocytes for therapeutic applications. Blood 2004;103:1011–1019. [DOI] [PubMed] [Google Scholar]
- 100. Leen AM, et al. Identification of hexon‐specific CD4 and CD8 T‐cell epitopes for vaccine and immunotherapy. J Virol 2008;82:546–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Leen AM, et al. Cytotoxic T lymphocyte therapy with donor T cells prevents and treats adenovirus and Epstein‐Barr virus infections after haploidentical and matched unrelated stem cell transplant. Blood 2009;114:4283–4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Leen AM, et al. Monoculture‐derived T lymphocytes specific for multiple viruses expand and produce clinically relevant effects in immunocompromised individuals. Nat Med 2006;12:1160–1166. [DOI] [PubMed] [Google Scholar]
- 103. Leen AM, et al. Conserved CTL epitopes on the adenovirus hexon protein expand subgroup cross‐reactive and subgroup‐specific CD8+ T cells. Blood 2004;104:2432–2440. [DOI] [PubMed] [Google Scholar]
- 104. Gerdemann U, et al. Nucleofection of DCs to generate multivirus‐specific T cells for prevention or treatment of viral infections in the immunocompromised host. Mol Ther 2009;17:1616–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Gerdemann U, et al. Rapidly generated multivirus‐specific cytotoxic T lymphocytes for the prophylaxis and treatment of viral infections. Mol Ther 2012;20:1622–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Vera JF, et al. Accelerated production of antigen‐specific T cells for preclinical and clinical applications using gas‐permeable rapid expansion cultureware (G‐Rex). J Immunother 2010;33:305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Lapteva N, Vera JF. Optimization manufacture of virus‐ and tumor‐specific T cells. Stem Cells Int 2011;2011:434392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Lapteva N, et al. Large‐scale ex vivo expansion and characterization of natural killer cells for clinical applications. Cytotherapy 2012;14:1131–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Jin J, et al. Simplified method of the growth of human tumor infiltrating lymphocytes in gas‐permeable flasks to numbers needed for patient treatment. J Immunother 2012;35:283–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Vella AT, Dow S, Potter TA, Kappler J, Marrack P. Cytokine‐induced survival of activated T cells in vitro and in vivo. Proc Natl Acad Sci USA 1998;95:3810–3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Vella A, Teague TK, Ihle J, Kappler J, Marrack P. Interleukin 4 (IL‐4) or IL‐7 prevents the death of resting T cells: stat6 is probably not required for the effect of IL‐4. J Exp Med 1997;186:325–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Gerdemann U, et al. Safety and clinical efficacy of rapidly‐generated trivirus‐directed T cells as treatment for adenovirus, EBV, and CMV infections after allogeneic hematopoietic stem cell transplant. Mol Ther 2013. doi: 10.1038/mt.2013.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Gerdemann U, et al. Immunotherapeutic strategies to prevent and treat human herpesvirus 6 reactivation after allogeneic stem cell transplantation. Blood 2013;121:207–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Hanley PJ, et al. Functionally active virus‐specific T cells that target CMV, adenovirus, and EBV can be expanded from naive T‐cell populations in cord blood and will target a range of viral epitopes. Blood 2009;114:1958–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Haque T, et al. Allogeneic cytotoxic T‐cell therapy for EBV‐positive posttransplantation lymphoproliferative disease: results of a phase 2 multicenter clinical trial. Blood 2007;110:1123–1131. [DOI] [PubMed] [Google Scholar]
- 116. Haque T, et al. Treatment of Epstein‐Barr‐virus‐positive post‐transplantation lymphoproliferative disease with partly HLA‐matched allogeneic cytotoxic T cells. Lancet 2002;360:436–442. [DOI] [PubMed] [Google Scholar]
- 117. Gandhi MK, et al. Immunity, homing and efficacy of allogeneic adoptive immunotherapy for posttransplant lymphoproliferative disorders. Am J Transplant 2007;7:1293–1299. [DOI] [PubMed] [Google Scholar]
- 118. Barker JN, et al. Successful treatment of EBV‐associated posttransplantation lymphoma after cord blood transplantation using third‐party EBV‐specific cytotoxic T lymphocytes. Blood 2010;116:5045–5049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Leen AM, et al. Multicenter study of banked third party virus‐specific T‐cells to treat severe viral infections after hematopoietic stem cell transplantation. Blood 2013;121:5113–5523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Betts BC, Young JA, Ustun C, Cao Q, Weisdorf DJ. Human herpesvirus 6 infection after hematopoietic cell transplantation: is routine surveillance necessary? Biol Blood Marrow Transplant 2011;17:1562–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. de Pagter PJ, Schuurman R, Meijer E, van Baarle D, Sanders EA, Boelens JJ. Human herpesvirus type 6 reactivation after haematopoietic stem cell transplantation. J Clin Virol 2008;43:361–366. [DOI] [PubMed] [Google Scholar]
- 122. de Pagter PJ, et al. Human herpes virus 6 plasma DNA positivity after hematopoietic stem cell transplantation in children: an important risk factor for clinical outcome. Biol Blood Marrow Transplant 2008;14:831–839. [DOI] [PubMed] [Google Scholar]
- 123. Dulery R, et al. Early human herpesvirus type 6 reactivation after allogeneic stem cell transplantation: a large‐scale clinical study. Biol Blood Marrow Transplant 2011;18:1080–1089. [DOI] [PubMed] [Google Scholar]
- 124. Kadakia MP, et al. Human herpesvirus 6: infection and disease following autologous and allogeneic bone marrow transplantation. Blood 1996;87:5341–5354. [PubMed] [Google Scholar]
- 125. Zerr DM, Corey L, Kim HW, Huang ML, Nguy L, Boeckh M. Clinical outcomes of human herpesvirus 6 reactivation after hematopoietic stem cell transplantation. Clin Infect Dis 2005;40:932–940. [DOI] [PubMed] [Google Scholar]
- 126. Hubacek P, et al. Incidence of HHV7 in donors and recipients of allogeneic hematopoietic stem cell transplantation. Pediatr Blood Cancer 2008;50:935. [DOI] [PubMed] [Google Scholar]
- 127. Smith C, Khanna R. Immune regulation of human herpesviruses and its implications for human transplantation. Am J Transplant 2013;13(Suppl 3):9–23. [DOI] [PubMed] [Google Scholar]
- 128. Rahiala J, et al. Human parvoviruses B19, PARV4 and bocavirus in pediatric patients with allogeneic hematopoietic SCT. Bone Marrow Transplant 2013;48:1308–1312. [DOI] [PubMed] [Google Scholar]
- 129. Plentz A, Hahn J, Holler E, Jilg W, Modrow S. Long‐term parvovirus B19 viraemia associated with pure red cell aplasia after allogeneic bone marrow transplantation. J Clin Virol 2004;31: 16–19. [DOI] [PubMed] [Google Scholar]
- 130. Eid AJ, Brown RA, Patel R, Razonable RR. Parvovirus B19 infection after transplantation: a review of 98 cases. Clin Infect Dis 2006;43: 40–48. [DOI] [PubMed] [Google Scholar]
- 131. Klumpen HJ, Petersen EJ, Verdonck LF. Severe multiorgan failure after parvovirus B19 infection in an allogeneic stem cell transplant recipient. Bone Marrow Transplant 2004;34:469–470. [DOI] [PubMed] [Google Scholar]
- 132. Comoli P, Hirsch HH, Ginevri F. Cellular immune responses to BK virus. Curr Opin Organ Transplant 2008;13:569–574. [DOI] [PubMed] [Google Scholar]
- 133. Comoli P, Binggeli S, Ginevri F, Hirsch HH. Polyomavirus‐associated nephropathy: update on BK virus‐specific immunity. Transpl Infect Dis 2006;8:86–94. [DOI] [PubMed] [Google Scholar]
- 134. Laskin BL, et al. BK viremia precedes hemorrhagic cystitis in children undergoing allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2013;19:1175–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Egli A, et al. Cytomegalovirus and polyomavirus BK posttransplant. Nephrol Dial Transplant 2007;22(Suppl 8):viii72–viii82. [DOI] [PubMed] [Google Scholar]
- 136. Koskenvuo M, et al. BK polyomavirus‐associated hemorrhagic cystitis among pediatric allogeneic bone marrow transplant recipients: treatment response and evidence for nosocomial transmission. J Clin Virol 2013;56:77–81. [DOI] [PubMed] [Google Scholar]
- 137. Choi JH, et al. Respiratory viral infections after hematopoietic stem cell transplantation in children. J Korean Med Sci 2013;28:36–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Hirsch HH, Martino R, Ward KN, Boeckh M, Einsele H, Ljungman P. Fourth European Conference on Infections in Leukaemia (ECIL‐4): guidelines for diagnosis and treatment of human respiratory syncytial virus, parainfluenza virus, metapneumovirus, rhinovirus, and coronavirus. Clin Infect Dis 2013;56:258–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Srinivasan A, et al. Detection of respiratory viruses in asymptomatic children undergoing allogeneic hematopoietic cell transplantation. Pediatr Blood Cancer 2013;60:149–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Casey J, Morris K, Narayana M, Nakagaki M, Kennedy GA. Oral ribavirin for treatment of respiratory syncitial virus and parainfluenza 3 virus infections post allogeneic haematopoietic stem cell transplantation. Bone Marrow Transplant 2013;48:1558–1561. [DOI] [PubMed] [Google Scholar]
- 141. Elizaga J, Olavarria E, Apperley J, Goldman J, Ward K. Parainfluenza virus 3 infection after stem cell transplant: relevance to outcome of rapid diagnosis and ribavirin treatment. Clin Infect Dis 2001;32:413–418. [DOI] [PubMed] [Google Scholar]
- 142. Seo S, et al. Outcome of respiratory syncytial virus lower respiratory tract disease in hematopoietic cell transplant recipients receiving aerosolized ribavirin: significance of stem cell source and oxygen requirement. Biol Blood Marrow Transplant 2013;19:589–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Lo MS, Lee GM, Gunawardane N, Burchett SK, Lachenauer CS, Lehmann LE. The impact of RSV, adenovirus, influenza, and parainfluenza infection in pediatric patients receiving stem cell transplant, solid organ transplant, or cancer chemotherapy. Pediatr Transplant 2013;17:133–143. [DOI] [PubMed] [Google Scholar]
- 144. Khanna N, et al. Outcome of influenza infections in outpatients after allogeneic hematopoietic stem cell transplantation. Transpl Infect Dis 2009;11:100–105. [DOI] [PubMed] [Google Scholar]
- 145. Lehners N, et al. Risk factors and containment of respiratory syncytial virus outbreak in a hematology and transplant unit. Bone Marrow Transplant 2013;48:1548–1553. [DOI] [PubMed] [Google Scholar]
- 146. Renaud C, et al. Mortality rates of human metapneumovirus and respiratory syncytial virus lower respiratory tract infections in hematopoietic cell transplantation recipients. Biol Blood Marrow Transplant 2013;19:1220–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Souza JS, Watanabe A, Carraro E, Granato C, Bellei N. Severe metapneumovirus infections among immunocompetent and immunocompromised patients admitted to hospital with respiratory infection. J Med Virol 2013;85:530–536. [DOI] [PubMed] [Google Scholar]
- 148. Waggoner JJ, Soda EA, Deresinski S. Rare and emerging viral infections in transplant recipients. Clin Infect Dis 2013;57:1182–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Chatzidimitriou D, Gavriilaki E, Sakellari I, Diza E. Hematopoietic cell transplantation and emerging viral infections. J Med Virol 2010;82:528–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Norja P, Ubillos I, Templeton K, Simmonds P. No evidence for an association between infections with WU and KI polyomaviruses and respiratory disease. J Clin Virol 2007;40:307–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Kuypers J, et al. WU and KI polyomaviruses in respiratory samples from allogeneic hematopoietic cell transplant recipients. Emerg Infect Dis 2012;18:1580–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Hirsch HH, Kardas P, Kranz D, Leboeuf C. The human JC polyomavirus (JCPyV): virological background and clinical implications. APMIS 2013;121:685–727. [DOI] [PubMed] [Google Scholar]
- 153. Balduzzi A, et al. Polyomavirus JC‐targeted T‐cell therapy for progressive multiple leukoencephalopathy in a hematopoietic cell transplantation recipient. Bone Marrow Transplant 2011;46:987–992. [DOI] [PubMed] [Google Scholar]