

Abstract
Curcumin is a biologically active component of curry powder. A structurally-related class of mimetics possesses similar anti-inflammatory and anticancer properties. Mechanism has been examined by exploring kinase inhibition trends. In a screen of 50 kinases relevant to many forms of cancer, one member of the series (4, EF31) showed ≥85% inhibition for ten of the enzymes at 5 μM, while twenty-two of the proteins were blocked at ≥40%. IC50’s for an expanded set of curcumin analogs established a rank order of potencies, and analyses of IKKβ and AKT2 enzyme kinetics for 4 revealed a mixed inhibition model, ATP competition dominating. Our curcumin mimetics are generally selective for Ser/Thr kinases. Both selectivity and potency trends are compatible with protein sequence comparisons, while modeled kinase binding site geometries deliver a reasonable correlation with mixed inhibition. Overall, these analogs are shown to be pleiotropic inhibitors that operate at multiple points along cell signaling pathways.
Keywords: curcumin analogs, EF31, kinase screening, kinase interaction network, cancer inhibitors, ATP competition, kinase docking, mixed model inhibition, pleiotropic kinase inhibition
INTRODUCTION
Curcuma longa (turmeric) is a rhizomatous herbaceous perennial plant of the ginger family. The popular spice, curry powder, is isolated from the root of the plant to provide the universally distinctive flavor and yellow color. The primary reactive ingredient refined from turmeric is curcumin (1), a poorly soluble yellow-orange powder. The compound has been studied extensively as a therapeutic agent for its anti-inflammatory, anti-tumor and anti-angiogenesis properties in vitro and in cell culture.1,2,3,4 However, its remarkable pleiotropic properties span a much wider range.5,6 In vivo, curcumin’s applications are unfortunately limited by its low oral bioavailability.7,8 Nonetheless, the substance is or has been the subject of clinical trials for the treatment of colon cancer,9 advanced pancreatic cancer,10 metastatic breast cancer,11 inflammatory bowel disease12 and cognitive impairment, among over 70 others.13,14
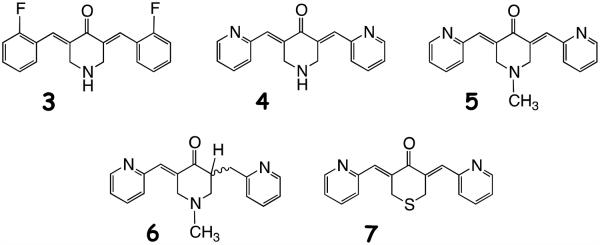
In attempts to improve the solubility, bioavailability and stability characteristics displayed by curcumin, we previously prepared a panel of curcumin analogs in which both the central and terminal sectors of the molecule were modified to give series 2 as illustrated in Figure 1. Namely, the central keto-enol moiety of 1 was replaced by a mono-carbonyl group embedded in a heterocyclic six-membered ring conjugated with the flanking C=C bonds. The terminal oxygenated aromatic rings in 1 were exchanged for fluorophenyl and pyridine moieties among others.15 A representative set of analogs are portrayed by structures 3-7. The conjugated curcumin mimics express Michael acceptor properties by delivering conjugates indefinitely stable at room temperature when treated with glutathione.16 Recently, a convenient NMR assay has provided a simple way to detect Michael adducts and their reversibility,17 curcumin included, although stable conjugates of curcumin are rarely isolated.18,19,20
Figure 1.
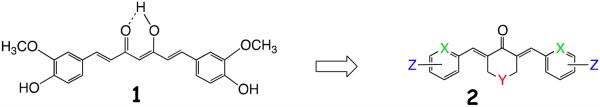
Curcumin 1 modifications to generate a series of curcumin analogs 2.
Initially, a set of novel curcumin analogs embodied by 2 were subjected to the National Cancer Institute in vitro cell line screen. The compounds exhibited anti-cancer and anti-angiogenic activities in cell culture. Some revealed a high degree of cytotoxicity, while most of the agents inhibited tumor cell growth with potency greater than cisplatin, a well-established clinical chemotheraputic agent.15
There are many examples of constitutive or over-activated kinases associated with cancer, and a dozen small molecule kinase blockers have been approved by the FDA for treatment of the disease.21 Full mapping of the kinome landscape is projected to have a major influence on personalized cancer treatment.22 The objective of the present work is to survey the action of 4 on a panel of 50 phosphorylating proteins, including serine/threonine, tyrosine, dual and lipid kinases, in an effort to determine how broadly class 2 penetrates the kinome and to learn if there is a measure of selectivity. We follow-up with IC50 measurements on the most strongly blocked kinases with five diverse analogs (3-7), the selection of which is discussed below. Mechanism of action and SAR are pursued both by kinetic measurements with the most active analog (4 on AKT2) and molecular modeling of the analogs in the AKT2 binding site. Finally, we address the questions of selectivity and pleiotropism by comparing sequences of the ATP binding sites for the kinases screened in the panel of 50. The study does not include curcumin itself, since fluorescence interference from this parent molecule interferes with the readouts of the kinase assays employed (see Methods). However, a number of separate studies have shown that curcumin mimics 3-7 are routinely more potent than curcumin by factors of 5-50 fold under a variety of circumstances: cytotoxicity toward human prostate and breast cancers,23 anticancer and anti-angiogenesis effects,15,24, 25 impairment of NF-kB nuclear translocation in mouse macrophages,26 TNF-α-induced NF-kB activation in osteoblastic MC3T3 precursors,27 Fanconi anemia pathway inhibition28 and stabilization of microtubules in cells.29
RESULTS AND DISCUSSION
To direct the project toward an appropriate selection of cancer kinases, we performed a preliminary screen of 3 and 4 against a small panel of kinases. The compounds had no measurable inhibition of receptor tyrosine kinase IGFR, a weak effect on the cytoplasmic tyrosine kinase Src, moderate inhibition of Ser/Thr kinases (IKKβ, RAF1 and MEK1) and a significantly increased blocking action on AKT1 and AKT2. Results from this screen led to the hypothesis that the current series of curcumin analogs are most likely pleiotropic and, as reported for curcumin, inhibit a range of different kinases.
Kinase Panel Screen
To examine trends and to gain broader knowledge concerning the spread of kinases sensitive to curcumin analogs, we subjected the more potent 4 to screening by a panel of kinases at 5 μM. Since IKKβ and AKT1 were known to be inhibited by the compound prior to conducting the screen, they were inserted as positive controls. Compound 4 indeed proves to be a pleiotropic kinase inhibitor. Of the fifty kinases tested in the profile screen (Figure 2), ten (20% of the kinases tested) were blocked by 4 at >85% and designated as hits. Compound 4 exhibited 40-80% inhibition against twelve kinases, which were categorized as modestly blocked. The other 28 phosphorylating enzymes (56% of the kinases tested) were inhibited by 4 at ≤40% and are likely to be only weakly influenced by the drug, if at all. In sum, 4 appears to be pleiotropic by blocking a range of kinases with varying potency at 5 μM.
Figure 2.

Screening of a 50-kinase panel by curcumin analog 4 (EF31) by the Z’ Lyte in vitro kinase assay. a) 4 was screened against the panel in duplicate at 5 μM with ATP concentrations at Kmapp for each individual kinase. Due to experimental error, values above 100% and below 0% are regarded as maximal or no inhibition respectively. *Adapta and **kinase cascade assays. See Methods. b) The types of kinases among the 50 in both number and % of total. c) Activity ranges: hits (> 80% inhibition), modest activity 40-80% inhibition) and low activity (less than 40% inhibition).
With regard to the selectivity of 4 for specific types of kinases, there is a definite trend. Of the top 10 screening hits, eight were Ser/Thr kinases (25% of Ser/Thr kinases tested), one was a dual function kinase (NEK1, 33% of the dual function kinases tested) and one was a Tyr kinase (KDR, 7% of all Tyr kinases tested). Thus, selectivity for Ser/Thr kinases emerges from this screen. Although the percentage of dual function kinases is higher, it likely results from the small sample size of this kinase class. Examination of the cross section of kinases with “low activity” reveals a somewhat different balance indicating a potential background role for Tyr kinases: five Ser/Thr kinases (10% of all kinases, 15% of Ser/Thr kinases), six Tyr kinases (12% of all kinases, 40% of Tyr kinases), and one dual function kinase (2% of all kinases, 33% of dual function kinases).
In the present study, any kinase eliciting ≥85% inhibition at 5 μM, was evaluated further by determining IC50 values for 3 (EF24), 4 (EF31), 5 (UBS109), 6 (mono-C=C reduction product of 5) and the sulfur analog 7 (SEF31). Justification for this choice has its origin in 3, a compound that performed particularly well in the NCI in vitro cell screen.15 It was subsequently shown to induce apoptosis in two different cancer cell lines21 and inhibit activation of the NF-κB pathway by blocking IKKβ kinase.22 This behavior has been posited to contribute to the compound’s anti-inflammatory properties. Analog 4 has also been shown to inhibit inflammation pathways by inhibiting NF-κB binding in mouse RAW264.7 macrophages with a 10-fold greater potency than 324 and to inhibit the growth of head and neck squamous cell carcinoma xenografts i.p.30 The inclusion of 5 was based on the outcome of a successful oral efficacy study in mice bearing a human tumor xenograft,31 while addition of metabolite 6 resulted from its noteworthy but reduced cellular cytotoxicity relative to 5. Both compounds show a modest suppressive effect on osteoclastogenesis.25 Compound 7, a phosphorylation inhibitor of proteins in the Fanconi anemia pathway,26 is an analog that introduces a large heteroatom into the central ring that occasions a significant change in molecular polarity and simultaneously serves as a poor proton acceptor and weak electron donor by comparison with 3-6 (i.e. 2, Y = NH and NMe). The selection permits evaluation of the importance of proton donor and acceptor interactions at the binding sites of the target kinases as well as the influence of protonation and molecular size. These points are taken up in the molecular modeling section below.
Kinase IC50 values and an emerging SAR
Screened enzymes not previously reported as targets for 3-7 are listed in Figure 3 along with IC50 values for each agent (Table 1). As mentioned above, AKT1 and IKKβ were added as positive controls. The kinases represent a wide variety of cellular activities: regulation of protein synthesis and cell proliferation (RPS6KB1), energy sensing and sensitization of cancer cells to cisplatin treatment (AMPK), DNA damage repair and maintenance of mitochondrial membrane potential (NEK1), mediation of cellular responses to VEGF (KDR/VEGFR2), environmental stress relief and assistance of production of certain cytokines (MAPK14/p38α), cooperation with the latter kinase to expedite nuclear export, gene expression and cell proliferation (MAPKAPK2), development, cell proliferation and apoptosis regulation (RAF1, MEK1 and ERK2). Two kinases blocked by 4 (5 μM, ≥85%), but not examined by dose-response, are CHEK1 and PRKCβ1. The former mediates cell cycle arrest in response to DNA irregularities, while the latter, activated by diacylglycerol, phosphorylates a range of cellular proteins and serves as the receptor for phorbol esters, a class of tumor promoters. Figure 4 captures the network of relationships among the twelve most readily blocked kinases portrayed in Figures 2 and 3. A somewhat more elaborate variation provided by Ingenuity Pathway Analysis32 is provided in the Supporting Information. Given the tightly connected associations, simultaneous inhibition of these enzymes will exert pairwise-protein influence as well as a major network perturbation. As suggested by the multiple actions of the weaker parent curcumin, 4 is clearly a pleiotropic kinase blocker in addition to exhibiting a ≥10 fold greater kinase suppression by comparison with curumin.24 At the same time, on average, the molecule exhibits only modest potency across the range of enzymes (sub-μM to 7 μM, Table 1), the exceptions being 4 against AKT and p53α (MAPK14; see kinase cascade below). Other analogs furnish a different profile (Figure 3, Table 1) suggesting an opportunity for reduced toxicity coupled to variable pan-kinase outcomes.
Figure 3.

IC50 values for inhibited kinases. a) The analogs were tested at nine different concentrations using serial dilutions:  3 (EF24),
3 (EF24),  4 (EF31),
4 (EF31),  5 (UBS109),
5 (UBS109),  6,
6,  7. Assays for RAF1 and MEK used the kinase cascade method diagramed in Figure 4 and discussed in the Materials and Methods section.
7. Assays for RAF1 and MEK used the kinase cascade method diagramed in Figure 4 and discussed in the Materials and Methods section.
Table 1.
| Kinase |
Compound |
||||
|---|---|---|---|---|---|
| 3 | 4 | 5 | 6 | 7 | |
| AKT1 (PKB alpha) | 0.78 | 0.02 | 2.6 | >100 | 3.6 |
| AKT-2 | 0.72 | 0.02 | 1.9 | >100 | 3.3 |
| IKBKB (IKKβ) | 72 | 1.1 | 4 | 15 | 34 |
| RPS6K1 | 20 | 1.4 | 6 | >100 | 12 |
| AMPK (A1/B1/G1) | 46 | 1.5 | 2.5 | 10 | 26 |
| RAF1a | 24 | 1.6 | 6.5 | >100 | 8.5 |
| MEK1a | 12.8 | 1.1 | 9 | >100 | 30 |
| ERK2b | 13 | 4 | 27 | >100 | 20 |
| NEK1 | 77 | 0.5 | 3.5 | 83 | 14 |
| KDR (VEGFR2) | 0.77 | 0.66 | 1.3 | >100 | 6.5 |
| MAPK14 (p38α) | 92 | 35 | >100 | >100 | >100 |
| MAPKAPK2 | >100 | 6.7 | 9.8 | >100 | 16 |
Assays for RAF1 and MEK used the kinase cascade method diagramed in Figure 5 and discussed in the Materials and Methods section.
The IC50 values for ERK2 resulted from large experimental variability and should be regarded as only semiquantitative.
Figure 4.

Phosphorylation and other interactions of transcription factors and enzymes leading to cell proliferation, differentiation, and survival. Kinases highlighted in blue have been studied in this work (Figs. 2 and 3). Alternative kinase names: ERK2 = MAPK1 = MAPK2; p38 = MAPK14; MEK1 = MAP2K; COT = MAP3KB; RSK1 = RPS6KA1; AMPK = PRKAA1; IKKb = IKBKB; KDR = VEGFR-2; RPS6KB1 = S6K1; Chek1 = Chk1.
Not surprisingly, most of these cellular functions are involved in the promotion of cancer and inflammation. Without yet understanding the details linking the wide diversity of biological responses, modest pleiotropic blockade of this set of kinases appears at the heart of the ability of the curcumin analogs to arrest cancer, angiogenesis and inflammation both in vitro15 and in vivo.33 A pertinent example is the behavior of NEK1, the only dual function kinase that is significantly inhibited by 4 in the kinase screen (Figure 2). Among other functions, it contributes to maintenance of the mitochondrial membrane potential. Inhibition of NEK1 causes a decrease in phosphorylation of voltage-dependent anion-selective channel protein 1 (VDAC1), which leads to an uncoupling of the mitochondrial membrane potential.34 Later stages in this event are identical to the manner in which 3 induces apoptosis in two cancer cell lines.21
Blockade of Signal Transduction Pathways
We have commented on both individual kinase functions and the potential coupling of kinases in subsections of signaling pathways. The dose-response assays performed for the strongly inhibited kinases involve activation of a given kinase, phosphorylation of a peptide substrate and disruption of a FRET signal upon cleavage of the peptide to provide the assay readout (Z’ Lyte in vitro kinase assay; see Methods and Figure 3). For example, when measuring inhibition of RAF1, the procedure takes advantage of the cellular mitogen activated protein (MAP) kinase cascade involving sequential phosphorylation and activation of inactive MEK1 and ERK2 followed by ERK2 phosphorylation of the substrate (Figure 5). By examining individual kinases, it is clear that curcumin analogs 3, 4, 5 and 7 block all three MAP kinases to within a factor of three (Table 1 and Figure 3). This leads to a damping of the entire pathway and eliminates dependence on any one specific kinase.
Figure 5.
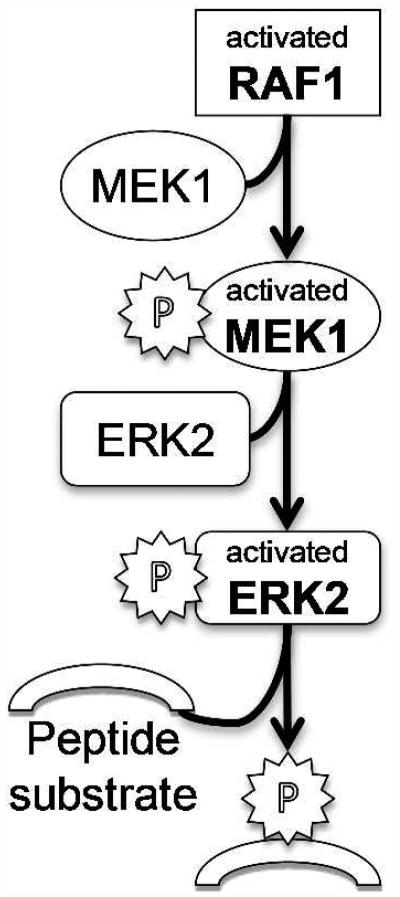
Diagram of kinase cascade used in the Z’Lyte kinase assay. The RAF1 assay is a three tiered cascade initiated with RAF1, which subsequently phosphorylates and activates MEK1 and ERK2 leading to peptide substrate phosphorylation employed to measure reaction progress (see Z’Lyte in vitro kinase assay in Materials and Methods.
A second example from the MAP kinase cascade concerns p38α (MAPK14) and its dependence on MAPKAPK2. Labeling p38α (MAPK14) as a hit in the kinase screen is somewhat misleading, since both kinases are pathway-linked in that screen (Figures 2 and 5b). Further examination of p38α (MAPK14) inhibition, however, using a direct kinase assay, shows its blockade to be significantly reduced (Figure 3 and Table 1). Thus, identification of p38α (MAPK14) as a hit in the kinase screen is primarily due to the inhibition of MAPKAPK2 downstream of p38α (MAPK14) in the kinase cascade. It is presently unknown whether assay constitution precisely mirrors these actions in a living cell.
These examples highlight drug action against multiple steps in a localized kinase interaction network by a family of readily accessible curcumin analogs. They simultaneously demonstrate the pleiotropic nature of the analogs both within a select segment of a pathway and among kinases in a more diffuse network (Figures 4 and S1). In a similar manner, the parent compound curcumin, inhibits cell signaling cascades at multiple nodes. Since these cascades are involved in cell proliferation and apoptosis, curcumin affects the overall process of carcinogenesis35 as do substances 3-5 and 7.
Mechanism of Kinase Inhibition
The curcumin mimics can exert their effects as kinase inhibitors by several possible mechanisms: reversible or irreversible, competition with ATP or the peptide substrate, or allosteric modulation. Since a great many such inhibitors function by competing with ATP and occupying the ATP binding pocket, we sought to explore this possibility by analyzing reaction rates at different ATP and inhibitor concentrations. AKT2 and analog 4 were selected to probe this question as the most potent combination among kinases and blockers (Table 1 and Figure 3). The corresponding Lineweaver-Burk plot (Figure S3, SI) reveals a clustering of lines on the Y axis indicative of a competitive inhibition model.
To achieve a more quantitative evaluation, we plotted the data on a Michaelis-Menten graph. By selecting specific models that force shared parameters in the global analysis (Km, Vmax, Ki) with GraphPad Prism,36 a compromise solution applied to each model spreads the data across all the curves providing a framework that allows mechanistic distinctions to be made; namely, competitive or mixed models.37
When performing a global analysis that fits the reaction rates to a Michaelis-Menten graph, the optimized value for Ki is shared by all inhibitor concentration curves. In a competitive model, in addition to the shared Ki constraint, all curves must also maintain a common optimized Vmax. Furthermore, Ki and Vmax must also be greater than zero in both models. Execution of the global analysis under the constraints of a competitive model as illustrated by the corresponding Michaelis-Menten graph significantly worsens the fit of the curves to the data (Figure 6a). By contrast, casting the data in the context of a mixed mechanism provides a superior fit for the data (Figure 6b). Comparison of these two models favors the mixed inhibition model with a p-value <0.01. This suggests that competitive reversible inhibition is not the only mechanistic path traveled by 4 in its inhibition of AKT2. The observation is consistent with a recent study reporting a mixed mechanism for the blockade of IKKβ by analog 3.22
Figure 6.
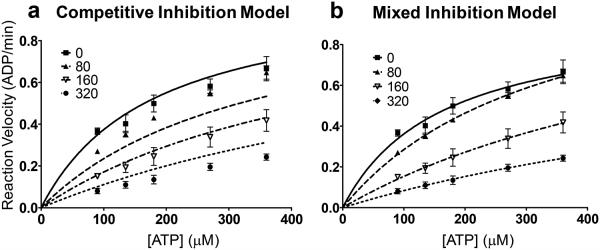
Analysis of enzyme kinetics data for 4 and AKT2. a) Michaelis-Menten graph of kinetic data using a competitive model of inhibition. b) Michaelis-Menten plot using a mixed model of inhibition.
To measure the quantitative extent of parallel mechanisms and the impact of substrate concentration (ATP) on kinase inhibition, the parameter α must be examined in the global analysis.38 Its value determines the degree to which the binding of inhibitor alters the affinity of the enzyme for its substrate and is always greater than zero. When α is very small (but greater than zero), binding of the inhibitor enhances substrate binding to the enzyme, and the mixed model becomes nearly identical to an uncompetitive model. When α = 1, the inhibitor does not alter binding of substrate to the enzyme, and the mixed-model is identical to noncompetitive inhibition. With increasing α, the inhibitor increasingly prevents binding of the substrate, and ultimately the mixed-model becomes identical to competitive inhibition.
Using global analysis of the Michaelis-Menten graph, we calculate an α of 31 for the inhibitory action of 4 on AKT2. This value >1 demonstrates that, while compound 4 has multiple mechanisms of inhibition, competition with ATP dominates action against this kinase under these experimental conditions. As mentioned, the mixed inhibition model has been reported for 3 and IKKβ by Kasinski et al.,22 although α was not calculated.
It is tempting to hypothesize that the other mode of inhibition may corresponds to covalent binding of inhibitor to enzyme as a result of Michael addition of a free cysteine to the electrophilic enone moiety of 4. This is consistent with our previous demonstration that two glutathione (GSH) molecules are able to combine with a single molecule of 3 and 4 rapidly and quantitatively in aqueous media followed by isolation of the bis-GSH adducts.16 The same study demonstrated reversibility39 in water by exchange of GSH between the two compounds and by the equal effectiveness of the parents and their conjugates (3-(GSH)2 and 4-(GSH)2) to serve as cytotoxins against MDA-MB-435 human breast cancer cells. This mechanistic option is discussed below from the viewpoint of the explicit interaction between 4 and AKT2 kinase.
Analysis of IC50 data by molecular modeling
We have sought to understand the variation of the ligand IC50 values (Table 1) by assuming they block the kinases primarily by occupying the ATP binding pocket. Both the kinetic measurements described above for 4 and AKT2, as well as the previously described action of 3 on IKKβ22 support competitive displacement of ATP as the dominant inhibitory mechanism accompanied by a second action we ascribe to covalent coupling between cysteine and the inhibitor enones. Among the kinases sampled, AKT2 exhibits the lowest IC50 values (Table 1). Accordingly, we selected the crystal structure of the corresponding kinase domain (PDB code 3E88) for pose prediction, once adjusted by the Maestro Protein Preparation Wizard. Ligands 3-7 were docked by Glide into the ATP binding site and optimized by MacroModel.
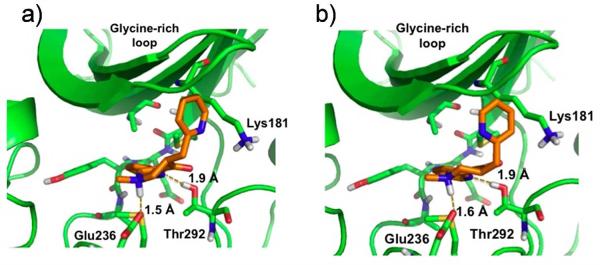
Figure 7a illustrates the best-scored docking pose for 4 in the ATP binding pocket of AKT2 in which several non-covalent interactions anchor the ligand to the protein. Hydrogen-bonds are established between one of the pyridine nitrogen atoms and Thr292 on the receptor and between the ligand carbonyl group and Lys181. A salt bridge is formed between Glu236 and the protonated nitrogen within the central ring of the ligand. One of the pyridine rings of 4 resides in a relatively hydrophobic pocket cirumscribed by Ala179, Met229, Glu230, Tyr231, Ala232 and Met282. For fluorinated 3, the top Glide pose is similar to that of 4 (Figure 7b). H-bonds with Glu236 and Lys181 are maintained and one of the phenyl rings resides in the same hydrophobic pocket. However, consistent with the higher IC50 values of 3, the favorable hydrogen bond with Thr292 is lost. To some extent the latter is compensated by electrostatic association between the aromatic fluorines and the proton of the axial NH+ bond in the central ring, although the association are weak (r(aro) F(δ−)---H(δ+)(N) = 3.1 Å).
Figure 7.

Top predicted curcumin analog poses from Glide docking of ligands on AKT2. a) 4 (EF31), b) 3 (EF24), c) 5 (UBS109).
Figure 7c shows that the N-methyl analog of protonated 5, like 4, enjoys a hydrogen bond between one of the pyridine nitrogen atoms and Thr292. The N-Me group on the central ring assumes an equatorial conformation, causing the NH+ to form a salt bridge with Glu236 from the axial position. The latter obligates the ligand to move towards Glu236 and away from Lys181 by comparison with 3 and 4 (Figures 7a,b), essentially deleting the hydrogen bond interaction with this residue. In addition, as shown in Figure 7c, two hydrogen atoms in 5 are separated by only 2.0 Å (black stipled line), somewhat below the sum of Van der Waals radii (2.4 Å) and, thereby, introducing internal ligand strain energy. The same H---H distance for 4 (2.3 Å) is at the minimum acceptable van der Waals contact. The diminished hydrogen bonding and ligand strain energy can explain the relatively higher IC50 values of 5 relative to 4.
Partially saturated 6 exists as two epimers, 6R and 6S. As shown in Figures 8a and 8b, respectively, the left half of each ligand and the equatorial N-methyl orientations are identical to that of 5. While the stereoisomer poses retain the non-covalent interactions with Thr292 and Glu236, the hydrogen bond with Lys181 is lost as observed for 5. Critical new contacts arise, however, because the C=C saturation in 6, requires the CH2-pyridine moiety to be relocated, placing the relatively hydrophobic edge of the pyridine ring into a polar sector of the protein’s glycine-rich loop. Furthermore, in 6R the pendant CH2-pyridine group fits into the pocket only by adopting a near eclipsed conformation with the adjacent C-H bond of the central 6-membered ring. Comparison of the Glide SP docking scores for 5 and 6S/6R predicts the former to be more tightly bound by 2.1 and 2.4 kcal/mol, respectively. Thus, the loss of a key H-bond; the predicted placement of the benzylic pyridine rings into unfavorable pockets, in one case in an equally unfavorable conformation; and the incorporation of only one electrophilic enone as compared to the two present in 3-5 appear to contribute to the significantly reduced activity of the epimers of 6 on AKT2 as has been reported for prostaglandin enones and dienones.40
Figure 8.
Top predicted curcumin analog poses for enantiomers of 6. a) 6R, b) 6S.
Analog 7 possesses a sulfur atom in the central ring instead of nitrogen. In the most favorable pose depicted by Figure 9, the hydrogen bond shared between ligand and Thr292 remains, while association between C=O and Lys181 NH increases to 2.7 Å, weakening the H-bond significantly. The NH to S replacement naturally not only eliminates the hydrogen bond with Glu236 but also causes the ligand to retreat as a consequence of the unfavorable electrostatic contact between the bulky electron-pair bearing sulfur atom and Glu236. The lack of two anchoring H-bonds and the electrostatic disconnect are believed to contribute to the increased IC50 values of 7 compared to 4. In sum, a semi-quantitative explanation of the various IC50 values for AKT2 inhibition can be developed by analysis of the docking poses.
Figure 9.

Top predicted pose for 7 (SEF31) on AKT2.
Since all the curcumin analogs carry an α,β-unsaturated ketone Michael acceptor, covalent bonds might be formed between kinase cysteines and these compounds. Previously, we reported the modification of cysteine-rich thioredoxin-1 by 3 in the presence of GSH.21 Similarly, Yamakoshi and coworkers have labeled the nuclear fusion protein KSRP/FUBP2 with a biotinylated dienone related to 2 and identified the covalently bound cysteine.41 More recently, Taunton and colleagues have outlined a strategy for targeting noncatalytic cysteines in kinases with reversible covalent inhibitors, suggesting it can be applied generally to this class of enzymes, assuming there is an exposed cysteine near the active site.42,43 Given the thiol reversibility of our analogs,16 this is a credible, albeit tentative, means to understand the mixed-inhibition observed herein. For example, Figure 10 shows that a cysteine (Cys311 AKT2) is located near the substrate binding site. Consistent with the design of the bioassay, the formation of a covalent bond between ligand and cysteine would most likely interrupt substrate binding, making the target peptide reagent susceptible to cleavage as monitored by subsequent separation of the FRET pairs. Under these circumstances, an apparent high IC50 value will be observed. Unfortunately, such an event can hinder quantification of the non-covalent binding affinity of the ATP competitive inhibitor unless this mechanism is a small contribution to the overall blockade.
Figure 10.

Cys311 in the cleft where the substrate binds on AKT2
Sequence comparison of kinase binding sites
We have attempted to understand the pleiotropic aspects of 4 by sequence alignment and identity/similarity comparison of the residues around the ATP binding sites for various kinases. The 3D structures of the proteins were aligned using the protein structure alignment tool in the Schrodinger Maestro Software package. Employing the docking pose of 4 in AKT2, key residues within the ATP binding sites were selected for identity comparison with the corresponding residues in other kinases: Ala179, Met229, Glu230, Tyr231, Ala232, Met282, Glu236, Lys181 and Thr292 (Figure 11). The comparison reveals that most of the active kinases (inhibition >80% in Figure 2) share >55% identity and >75% similarity with these residues in the AKT2 binding environment, while AKT1 and RPS6KB1 share >75% identity and >85% similarity, respectively. KDR is an exception to the >55% and 75% rule; i.e. 33 and 67%, respectively. Further examination of the KDR binding site reveals that two cysteines at the binding site (Cys919 and Cys1045 equivalent to the spatial positions for Ala232 and Thr292 in AKT2) adopt an orientation that favors covalent Michael addition to the α,β-unsaturated ketone moiety. It is conceivable that covalent binding dominates the binding affinities in this case and, thereby, rationalizes the similar KDR IC50 values for the four curcumin analogs 3, 4, 5 and 7 (Table 1). However, examination of other kinases with low activity in Figure 2 also reveals similar cysteines at the ATP binding sites (example: KIT), suggesting that the corresponding cysteines alone do not contribute the majority of the observed activity of KDR. Further work is still required to understand why KDR and NEK1 are exceptions.
Figure 11.

Key residues around the ATP binding sites of various kinases. Residue type and number according to AKT2: 1: Ala179 2: Met229, Glu230, Tyr231, Ala232 3: Met282 4: Glu236 5:Lys181 6:Thr292. Upper panel, >85% inhibition; lower panel, ≤10% inhibition.
The inactive kinases (≤10% inhibition, Figure 2), by contrast, exhibit < 35% identity and <70% similarity on-average for the nine residues listed above compared to AKT2. Since these residues are critically important for the binding of 4, we surmise that while the ATP binding site side-chain constitution is not the full story, it appears to capture a major structural feature behind much of the pleiotropism of 4, although exceptions such as KDR and NEK1 exist. Other important features of the kinases not captured by Glide docking include the plasticity of the DLG motif and the glycine-rich loop.44 These aspects are under active investigation for the kinases considered here.45
The sequence comparisons of Figure 11 likewise address the predominant selectivity for Ser/Thr kinases by the monocarbonyl inhibitors. Of the ≥85% inhibition hits from 4’s interrogation of the kinase panel, the sequence identities for the Ser/Thr kinases (Figure 2, orange color) range from 56-100% relative to AKT2, while the KDR (Tyr kinase) and NEK1 (dual function kinase) exceptions fall at 33%. In a similar fashion, the corresponding sequence identities for Ser/Thr kinases that were poorly blocked (≤10% inhibition, PIM1, IRAK4, CDK7, ACVR1B, CDK2, EGFR), also fall in the 22-33% range. Accordingly, kinase binding site structure trends with both the Ser/Thr selectivity and the observed pleiotropic behavior.
CONCLUSIONS
By examination of 50 kinases, we have identified a family of enzymes that are inhibited by a small panel of curcumin mimics, nearly a dozen of which are blocked at micromolar or submicromolar concentrations. Two MAP kinase cascade pathways identified from the present kinase inhibition data are at the heart of tumor development (p38α-MAPKAPK2 and RAF1-MEK1-ERK2). Various oncogenic factors activate these pathways and, by this means, relay proliferative, survival, chemoresistance and angiogenic signals essential to tumor maintenance.46 The present observations suggest that the pleiotropic nature of the curcumin analogs leads to damping of pathways without dependence on a single kinase for inhibition (Figures 4 and S1).
Enzyme kinetics studies reveal that kinase inhibition by compounds within series 2 operates by multiple mechanisms, with ATP competition appearing to be a major factor. Complementary molecular modeling at the ATP binding site of AKT2 provides a qualitative explanation for ligand activity. Reversible Michael addition to curcumin mimetics by cysteine residues in the kinases is compatible with the observed mixed mechanism kinetics and the flat IC50 values for the KDR kinase. Sequence comparisons for residues at the kinase active sites suggest that site geometry is an important feature both for the pleiotropic character of the inhibitors and the Ser/Thr selectivity.
In a cancer context, one might expect drug resistance to be significantly diminished in cells following treatment with such inhibitors. With the rising importance of the “omes” (genome, proteome, kinome, transcriptome, etc.) and our ability to understand and map the complexities of intracellular signaling, curcumin analogs may be an example of a molecular class that is able to target multiple enzymes with a “magic shotgun”.47 This would be especially valuable for diseases like cancer that are complex, inflammation associated and often evolve mutations in multiple genes. Not surprisingly, the class parent, curcumin, shares these properties. In spite of its minimal bioavailability and poor solubility, the compound perturbs numerous cellular proteins5 and has achieved clinical status in more than 30 different cancer trials.14 The ability of a single molecule to target multiple cellular proteins and pathways represents an emerging philosophy in cancer drug discovery, one in which “promiscuous” drugs that target multiple enzymes or pathways may be superior to mono therapies selective for only one target.48,49,50 Gleevec (imatinib), a highly successful anticancer agent attenuating, among others, the Ras/MapK pathway, has been approved by the FDA to treat ten different cancers.51,52 It serves as a prime example of the phenomenon.53,54
EXPERIMENTAL SECTION
Chemicals, Reagents and Assay Kits
Compounds 3,15 4,24 5,25 6,25 and 726 were prepared at Emory University as previously described. All compounds were recrystallized and judged to be > 95% pure as determined by HPLC (various concentrations of MeOH in 0.1% aqueous formic acid solution; Agilent Zorbax 50 mm C18 column; 1 mL/min; monitoring at 254 and 340 nm). For kinase profiling, ADP Quest™ (Cat # 90-0071) and ATP Gold (Cat #90-0099) were obtained from DiscoveRx,55 while the Crosstide peptide substrate (95% pure) was available from Millipore (Cat # 12-331).56
Z’-LYTE™ Kinase Assay Kits were provided by Invitrogen:57 Ser/Thr 6 (Cat # PV3179), Ser/Thr 05 (Cat # PV3178), Tyr 02 (Cat # PV3191), Ser/Thr 03 (Cat # PV3176), Tyr 01 (Cat # PV3190), Ser/Thr 07 (Cat # PV3180), Ser/Thr 23 (Cat # PV4644), Ser/Thr 04 (Cat # PV 3177) and Ser/Thr 15 (Cat # PV3799).
Recombinant proteins came from Invitrogen:58 AKT1 (PKB alpha) (Cat #P2999), AKT2 (PKB beta) (Cat #PV3184), IKBKB (IKK Beta) (Cat# PV3836), SRC (Cat# PV3044), RAF1 (cRAF) Y340D Y341D (Cat #PV3805), inactive MAP2K1 (MEK1) (Cat #3093), MAP2K1 (MEK1) (Cat# P3093), inactive MAPK1 (ERK2) (Cat #3314), MAPK1 (ERK2) (Cat #PV3313), IGF1R (Cat# PV3250), RPS6KB1 (p70S6K) (Cat# PV3815), AMPK A1/B1/G1 (Cat# PV4672), NEK1 (Cat# PV4202), KDR (VEGFR2) (Cat# PV3660), p38α (MAPK14) (Cat# PV3304), MAPKAPK2, Inactive (Cat #PV3316), MAPKAPK2 (Cat #PV3317),
Z’Lyte In vitro kinase assay (Invitrogen)
In a 10 μL kinase reaction, the IKKβ transfers the gamma-phosphate of ATP to a single serine/threonine residue in a specific synthetic peptide substrate (2 μM). The peptide is labeled with the two fluorophores coumarin and fluorescein, one at each end of the peptide to make up a FRET pair. In the development reaction, 5 μL of a site-specific protease recognizes and cleaves any non-phosphorylated peptides. Cleavage disrupts FRET between the coumarin and the fluorescein on the peptide. Phosphorylation of the peptide suppresses cleavage by the protease. Uncleaved, phosphorylated peptides maintain the FRET pair. Five μL of stop reagent is added to halt the development reaction before the plate is read by an Envision 2102 plate reader from Perkin Elmer. During detection, a ratiometric read-out of the donor emission over the acceptor emission quantitates reaction progress. The ratio is low if the peptide is phosphorylated, and high if the peptide is non-phosphorylated. Percent phosphorylation was calculated using controls, and each compound concentration was run in triplicate. Results were graphed using GraphPad Prism, and IC50 values were calculated using non-linear regression.
Fluorescence Interference
Curcumin could not be compared in the Z’ Lyte assay due to fluorescence interference from curcumin in the readouts of the assays used. For instance, the Z’ lyte assay uses excitation at 400 nm, and measures fluorescence of the fluorophores coumarin and fluorescein at 445 nm and 520 nm respectively. Curcumin, depending on solvent, absorbs light from 350-475 nm with maximal absorption around 425 nm. It then fluoresces in the 450-700 nm range59 using the 355 nm excitation to measure fluorescence. This would be sufficient to disrupt all aspects of the readout from absorbing the excitation light to absorbing the fluoresced light from coumarin, as well as curcumin fluorescing at wave lengths that are similar to fluorescein. These effects were observed when the “test compound fluorescence interference” control was run according the Z’ Lyte assay protocol. The curcumin analogs, however did not show any fluorescence interference at concentrations up to 50 uM.
Cascade Reaction assay
Some of the assays require a cascade reaction in which inactivated kinase is activated by an upstream kinase. Two cascades were important for our studies: the RAF1-MEK1-ERK2 cascade and the p38α (MAPK14)-MAPKAPK2 cascade. A direct assay was used for p38α (MAPK14) when generating its nine point IC50 curve.
RAF1 (cRAF) Kinase Cascade
The final 10 μL Kinase Reaction consists of 0.001 – 0.005 ng RAF1 (cRAF), 10 ng inactive MAP2K1 (MEK1), and 100 ng inactive MAPK1 (ERK2).
MAP2K1 (MEK1) Kinase Cascade
The final 10 μL Kinase Reaction consists of 1.0 - 4.0 ng MAP2K1 (MEK1) and 105 ng inactive MAPK1 (ERK2).
p38α (MAPK14)-Kinase Cascade
The final 10 μL Kinase Reaction consists of 0.01 - 0.02 ng p38α (MAPK14) and 5 ng inactive MAPKAPK2.
Kinase profile screen
Compound 4 was tested against a panel of kinases at a 5 μM concentration. Each data point was measured in duplicate using Z’ Lyte substrates with two control wells. One control well contains a protease control to determine if the compound interferes with the development reaction. The second well encloses a fluorescence control to ascertain if the compound interferes with the fluorescense reading of the FRET pair.
Adapta Assay
After addition of inhibitor and all other kinase reaction components, the reaction was incubated for one hour. During this period, the kinase reaction produces phosphorylated peptide and ADP. A detection solution of Europium-labeled anti-ADP antibodies, a labeled ADP tracer and an EDTA to stop-kinase reaction were added. ADP formed by the kinase reaction will displace the ADP tracer from the antibody, resulting in a TR-FRET signal decrease. When an inhibitor is used, the amount of ADP formed by the kinase reaction is reduced, and the resulting intact antibody-tracer interaction results in a high TR-FRET signal. By comparing this to a control, the amount of inhibition can be calculated.
ADP Quest
Kinase assays for the enzyme kinetics were accomplished with ADP Quest assays. Using purified recombinant AKT2 from Invitrogen (PV3184) and the Crosstide substrate from Millipore (Cat # 12-331), kinase reactions were run from 10-90 minutes using fresh samples of varying ATP concentrations. When the Crosstide substrate molecule is phosphorylated, an ADP molecule is generated. The ADP Quest procedure measures total ADP by an enzyme coupled reaction that causes a fluorescent signal. The latter is compared with a known control.
Michaelis-Menten analysis of enzyme kinetics
Analysis of reaction velocities was performed by first determining reaction rates using the ADP quest assay. The reaction rates were measured by following ADP produced over time. The reaction velocities were graphed on a Lineweaver-Burk plot and two Michaelis-Menten plots with appropriate constraints for the type of model represented. Calculations were performed with GraphicPad Prism 5.30
Molecular Modeling
Docking
The structures of compounds 3, 4, 5, 6 and 7 were drawn in 2D with Chemdraw, and then submitted to Ligprep in Maestro 9.1 to obtain 3D structures. Protein complexes were processed by the protein preparation wizard in Maestro followed by removal of the ligands. The receptor grid was generated at the ATP binding sites. Ligand-docking was accomplished with: SP precision (Maestro 9.0) for flexible docking of the ligands.60,61 The side chain of Lys181 was adjusted to accommodate an H-bond interaction with C=O of the ligand after docking. The pdb protein codes used in the analysis follow: AKT2: 3E88, AKT1: 3MVH, IKKB: 3QAD, NEK1:4B9D, RPS6KB1: 3A60, AMPK: 3AQV, PRKCB1: 2I0E, CHEK1: 2YEX, KDR: 2XIR, MAPKAPK2: 3W2M, PIM1: 3QF9, IRAK4: 2OIC, CDK7: 1UA2, KIT: 3G0E, ACVR1B: 1RW8, CDK2: 2XMY, EGFR: 3BEL, PLK1: 3THB, ERBB2: 3PP0.
Generation of a kinase interaction network
A network of the ten most inhibited kinases by 4 (≥85% inhibition at 5 uM, Figure 2a) was generated using Ingenuity Pathway Analysis with direct connections (Ingenuity Systems, Inc.).26
Supplementary Material
Acknowledgments
We are grateful to Dr. Dale Edmundson and Erika Milczek (Biochemistry Department, Emory University) for their invaluable assistance and patience concerning analysis of data from the enzyme kinetics studies. We also thank Ms. Jessica Paulishen for editing the manuscript. This study was partially supported by NCI R01 CA 165306 (Shim, H.).
Abbreviations
- IKKβIκB
kinase
- IκB
inhibitor of NF-κB
- AKT
protein kinase B
- ATP
adenosine triphosphate
- HT-29 cells
human colonic adenocarcinoma cells
- mTOR
mammalian target of rapamycin
- NF-kB
nuclear factor κ B
- RAW264.7
mouse macrophage-like cell line
- PI3K
phosphoinositide 3 kinase
- PKA
protein kinase A
- BCR
breakpoint cluster region protein
- ABL
Ableson leukemia oncogene cellular homolog
- Src
Rous sarcoma oncogene cellular homolog
- RAF
rapidly accelerated fibrosarcoma
- MEK
MAPK/Erk kinase
- IGFR
insulin- like growth factor receptor
- Ser/Thr
serine/threonine
- Tyr
tyrosine
- Cys
cysteine
- KDR
kinase domain receptor
- RPS6K1
ribosomal protein S6 kinase 1
- AMPK
adenosine monophosphate-activated protein kinase
- NEK1
never in mitosis gene A-related kinase 1
- VEGF
vascular endothelial growth factor
- VEGFR2
vascular endothelial growth factor receptor-2
- MAPK
mitogen-activated protein kinase
- MAPKAP
MAP kinase activated protein kinase
- ERK
extracellular signal-regulated kinase
- SAR
structure-activity relationship
- PRKC
protein kinase C
- p38
stress-activated protein kinase
- COT
cancer osaka thyroid
- RSK1
ribosomal S6 kinase 1
- S6K1
S6 kinase 1
- IKBKB
I kappa B kinase beta
- Chek1, Chk1
checkpoint kinase
- VDAC1
voltage-dependent anion-selective channel protein 1
- FRET
fluorescence resonance energy transfer
- PDB
protein database.
Footnotes
Supporting Information Available: Ingenuity Pathway Analysis interaction network for the 12 most inhibited kinases. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Jurenka JS. Anti-inflammatory properties of curcumin, a major constituent of Curcuma longa: a review of preclinical and clinical research. Altern. Med. Rev. 2009;14:141. [PubMed] [Google Scholar]
- 2.Perry MC, Demeule M, Regina A, Moumdjian R, Beliveau R. Curcumin inhibits tumor growth and angiogenesis in glioblastoma xenografts. Mol. Nutr. Food Res. 2010;54:1192. doi: 10.1002/mnfr.200900277. [DOI] [PubMed] [Google Scholar]
- 3.Lee YK, Park SY, Kim YM, Park OJ. Regulatory effect of the AMPK-COX-2 signaling pathway in curcumin-induced apoptosis in HT-29 colon cancer cells. Ann. N Y Acad. Sci. 2009;1171:489. doi: 10.1111/j.1749-6632.2009.04699.x. [DOI] [PubMed] [Google Scholar]
- 4.Yang CL, Liu YY, Ma YG, Xue YX, Liu DG, Ren Y, Liu XB, Li Y, Li Z. Curcumin Blocks Small Cell Lung Cancer Cells Migration, Invasion, Angiogenesis, Cell Cycle and Neoplasia through Janus Kinase-STAT3 Signalling Pathway. PLoS One. 2012;7:e37960. doi: 10.1371/journal.pone.0037960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aggarwal BB, Surh Y-J, Shishodia S. Springer Science+Business Media, LLC, NY, 10013; USA: 2007. The Molecular Targets and Therapeutic Uses of Curcumin in Health and Disease. [Google Scholar]
- 6.Zhou H, Beevers CS, Huang S. The targets of curcumin. Curr. Drug. Targets. 2010;12:332. doi: 10.2174/138945011794815356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharma RA, Steward WP, Gescher AJ. Pharmacokinetics and pharmacodynamics of curcumin. Adv. Exp. Med. Biol. 2007;595:453. doi: 10.1007/978-0-387-46401-5_20. [DOI] [PubMed] [Google Scholar]
- 8.Ji JL, Huang XF, Zhu HL. Curcumin and its formulations: potential anti-cancer agents. Anticancer Agents Med. Chem. 2011;12:210. doi: 10.2174/187152012800228733. [DOI] [PubMed] [Google Scholar]
- 9.Sun SH, Huang HC, Huang C, Lin JK. Cycle arrest and apoptosis in MDA-MB-231/Her2 cells induced by curcumin. Eur. J. Pharmacol. 2010;690:22–30. doi: 10.1016/j.ejphar.2012.05.036. [DOI] [PubMed] [Google Scholar]
- 10.Epelbaum R, Schaffer M, Vizel B, Badmaev V, Bar-Sela G. Curcumin and gemcitabine in patients with advanced pancreatic cancer. Nutr. Cancer. 2010;62:1137. doi: 10.1080/01635581.2010.513802. [DOI] [PubMed] [Google Scholar]
- 11.Bayet-Robert M, Kwiatkowski F, Leheurteur M, Gachon F, Planchat E, Abrial C, Mouret-Reynier MA, Durando X, Barthomeuf C, Chollet P. Phase I dose escalation trial of docetaxel plus curcumin in patients with advanced and metastatic breast cancer. Cancer Biol. Ther. 2010;9:8. doi: 10.4161/cbt.9.1.10392. [DOI] [PubMed] [Google Scholar]
- 12.Taylor RA, Leonard MC. Curcumin for inflammatory bowel disease: a review of human studies. Altern. Med. Rev. 2011;16:152. [PubMed] [Google Scholar]
- 13.Gupta SC, Patchva S, Aggarwal BB. Therapeutic roles of curcumin: lessons learned from clinical trials. Amer. Asso. Pharm. Sci. J. 2013;15:195–218. doi: 10.1208/s12248-012-9432-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.National Institutes of Health http://www.clinicaltrials.gov/ct2/results?term=curcumin&Search=Search; last accessed 02/10/2013.
- 15.Adams BK, Ferstl EM, Davis MC, Herold M, Kurtkaya S, Camalier RF, Hollingshead MG, Kaur G, Sausville EA, Rickles FR, Snyder JP, Liotta DC, Shoji M. Synthesis and biological evaluation of novel curcumin analogs as anti-cancer and anti-angiogenesis agents. Bioorg. Med. Chem. 2004;12:3871. doi: 10.1016/j.bmc.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 16.Sun A, Lu YJ, Hu H, Shoji M, Liotta DC, Snyder JP. Curcumin analog cytotoxicity against breast cancer cells: exploitation of a redox-dependent mechanism. Bioorg. Med. Chem. Lett. 2009;19:6627. doi: 10.1016/j.bmcl.2009.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Avonto C, Taglialatela-Scafati O, Pollastro F, Minassi A, Di Marzo V, De Petrocellis L, Appendino G. An NMR Spectroscopic Method to Identify and Classify Thiol-Trapping Agents: Revival of Michael Acceptors for Drug Discovery? Angew. Chem. Int. Edn. 2011;50:467–471. doi: 10.1002/anie.201005959. [DOI] [PubMed] [Google Scholar]
- 18.Usta M, Wortelboer HM, Vervoort J, Boersma MG, Rietjens IM, van Bladeren PJ, Cnubben NH. Human glutathione S-transferase-mediated glutathione conjugation of curcumin and efflux of these conjugates in Caco-2 cells. Chem. Res. Toxicol. 2007;20:1895–1902. doi: 10.1021/tx7002245. [DOI] [PubMed] [Google Scholar]
- 19.Wortelboer HM, Usta M, van der Velde AE, Boersma MG, Spenkelink B, van Zanden JJ, Rietjens IM, van Bladeren PJ, Cnubben NH. Interplay between MRP inhibition and metabolism of MRP inhibitors: the case of curcumin. Chem. Res. Toxicol. 2003;16:1642–1651. doi: 10.1021/tx034101x. [DOI] [PubMed] [Google Scholar]
- 20.Awasthi S, Pandya U, Singhal SS, Lin JT, Thiviyanathan V, Seifert WE, Jr., Awasthi YC, Ansari GA. Curcumin-glutathione interactions and the role of human glutathione S-transferase P1-1. Chem. Biol. Interact. 2000;128:19. doi: 10.1016/s0009-2797(00)00185-x. [DOI] [PubMed] [Google Scholar]
- 21.Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nature Rev. Can. 2009;9:28–39. doi: 10.1038/nrc2559. [DOI] [PubMed] [Google Scholar]
- 22.Kothari V, Wei I, Shanker S, Kalyana-Sundaram S, Wang L, Ma LW, Vats P, Grasso CS, Robinson DR, Wu YM, Cao X, Simeone DM, Chinnaiyan AM, Kumar-Sinha C. Outlier Kinase Expression by RNA Sequencing as Targets for Precision Therapy. Cancer Discov. 2013;3:1–14. doi: 10.1158/2159-8290.CD-12-0336. (AACR) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adams BK, Cai J, Armstrong J, Herold M, Lu YJ, Sun A, Snyder JP, Liotta DC, Jones DP, Shoji M. EF24, a novel synthetic curcumin analog, induces apoptosis in cancer cells via a redox-dependent mechanism. Anticancer Drugs. 2005;16:263. doi: 10.1097/00001813-200503000-00005. [DOI] [PubMed] [Google Scholar]
- 24.Kasinski AL, Du Y, Thomas SL, Zhao J, Sun SY, Khuri FR, Wang CY, Shoji M, Sun A, Snyder JP, Liotta D, Fu H. Inhibition of IkappaB kinase-nuclear factor-kappaB signaling pathway by 3,5-bis(2-flurobenzylidene)piperidin-4-one (EF24), a novel monoketone analog of curcumin. Mol. Pharmacol. 2008;74:654. doi: 10.1124/mol.108.046201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thomas SL, Zhao J, Li Z, Lou B, Du Y, Purcell J, Snyder JP, Khuri FR, Liotta D, Fu H. Activation of the p38 pathway by a novel monoketone curcumin analog, EF24, suggests a potential combination strategy. Biochem. Pharmacol. 2010;80:1309–1316. doi: 10.1016/j.bcp.2010.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Olivera A, Moore TW, Hu F, Brown AP, Sun A, Liotta DC, Snyder JP, Yoon Y, Shim H, Marcus AI, Miller AH, Pace TW. Inhibition of the NF-kappaB signaling pathway by the curcumin analog, 3,5-Bis(2-pyridinylmethylidene)-4-piperidone (EF31): anti-inflammatory and anti-cancer properties. Int. Immunopharmacol. 2012;12:368. doi: 10.1016/j.intimp.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamaguchi M, Moore TW, Sun A, Snyder JP, Shoji M. Novel curcumin analogue UBS 109 potently stimulates osteoblastogenesis and suppresses osteoclastogenesis: involvement in Smad activation and NF-κB inhibition. Integr. Biol. 2012;4:905–913. doi: 10.1039/c2ib20045g. (Camb) [DOI] [PubMed] [Google Scholar]
- 28.Landis I, McCarroll M, Yang C, Sun A, Hiddingh S, Turker MS, Snyder JP, Hoatlin ME. Monoketone analogs of curcumin, a new class of potent Fanconi anemia pathway inhibitors. Molec. Cancer. 2009;8(133):1–13. doi: 10.1186/1476-4598-8-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thomas SL, Zhong D, Zhou W, Malik S, Liotta D, Snyder JP, Hamel E, Giannakakou P. EF24, a Novel Curcumin Analog, Disrupts the Microtubule Cytoskeleton and Inhibits HIF-1. Cell Cycle. 2008;7:2409–2417. doi: 10.4161/cc.6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu S, Moore TW, Lin X, Morii N, Mancini A, Howard RB, Culver D, Arrendale RF, Reddy P, Evers TJ, Zhang H, Sica G, Chen ZG, Sun A, Fu H, Khuri FR, Shin DM, Snyder JP, Shoji M. Synthetic curcumin analog EF31 inhibits the growth of head and neck squamous cell carcinoma xenografts. Integr. Biol. (Camb.) 2012;4:633. doi: 10.1039/c2ib20007d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu S, Moore TW, Snyder JP, Shoji M. in preparation.
- 32.Systems I. Ingenuity Systems, Inc., 1700 Seaport Blvd, 3rd Floor; Redwood City, CA 94063: 2012. http://www.ingenuity.com/, accessed 9/20/2012. [Google Scholar]
- 33.Zhu S, Moore TW, Lin X, Morii N, Mancini A, Howard RB, Culver D, Arrendale RF, Reddy P, Evers TJ, Zhang H, Sica G, Chen ZG, Sun A, Fu H, Khuri FR, Shin DM, Snyder JP, Shoji M. Synthetic curcumin analog EF31 inhibits the growth of head and neck squamous cell carcinoma xenografts. Integr. Biol. (Camb.) 2012;4:633. doi: 10.1039/c2ib20007d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen Y, Craigen WJ, Riley DJ. Nek1 regulates cell death and mitochondrial membrane permeability through phosphorylation of VDAC1. Cell Cycle. 2009;8:257. doi: 10.4161/cc.8.2.7551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sa G, Das T. Anti cancer effects of curcumin: cycle of life and death. Cell. Div. 2008;3:14. doi: 10.1186/1747-1028-3-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.GraphPad Software, Inc. 2236 Avenida de la Playa, La Jolla, CA 92037 USA: http://www.graphpad.com/, accessed 9/20/2012. [Google Scholar]
- 37. Global analysis is a non-linear regression treatment for multiple curves corresponding to different inhibitor concentrations. The Km, Vmax and Ki parameters for each curve are adjusted for the best global fit of the data using GraphicPad,36 but constrained to the same value for each curve. Thus, for example, the resulting Vmax in a competitive model is forced to be shared across all curves.
- 38. The parameter α derived from the global analysis of the data in GraphPad Prism36 differs from the similarly-named variables α and α’ in the classic Michaelis-Menten equations.
- 39.Moore T, Snyder JP. Compound 3 has also been shown to exhibit reversibility by the NMR method of Appendino et al.17. unpublished.
- 40.Suzuki M, Mori M, Niwa T, Hirata R, Furuta K, Ishikawa T, Noyori R. Chemical Implications for Antitumor and Antiviral Prostaglandins: Reaction of Δ7-Prostaglandin A1 and Prostaglandin A1 Methyl Esters with Thiols. J. Am. Chem. Soc. 1997;119:2376–2385. [Google Scholar]
- 41.Yamakoshi H, Kanoh N, Kudo C, Sato A, Ueda K, Muroi M, Kon S, Satake M, Ohori H, Ishioka C, Oshima Y, Osada H, Chiba N, Shibata H, Iwabuchi Y. KSRP/FUBP2 Is a Binding Protein of GO-Y086, a Cytotoxic Curcumin Analogue. ACS Med. Chem. Lett. 2010;1:273–276. doi: 10.1021/ml1000454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Serafimova IM, Pufall MA, Krishnan S, Duda K, Cohen MS, Maglathlin RL, McFarland JM, Miller RM, Frodin M. Taunton. Reversible targeting of noncatalytic cysteines with chemically tuned electrophiles. J. Nat. Chem. Biol. 2012;8:471–476. doi: 10.1038/nchembio.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee C-U, Grossmann TN. Reversible Covalent Inhibition of a Protein Target. Angew. Chem. Int. Ed. 2012;51:8699–8700. doi: 10.1002/anie.201203341. [DOI] [PubMed] [Google Scholar]
- 44.Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer. 2009;9:28–39. doi: 10.1038/nrc2559. [DOI] [PubMed] [Google Scholar]
- 45.Hu H, Snyder JP. Models for Predicting IKKA and IKKB Blockade. J. Chem. Inf. Model. 2012;52:310–3199. doi: 10.1021/ci300287t. [DOI] [PubMed] [Google Scholar]
- 46.Yang Q, Lee J-D. Targeting the BMK1 MAP Kinase Pathway in Cancer Therapy. Clin. Cancer Res. 2011;17:3527–3532. doi: 10.1158/1078-0432.CCR-10-2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roth BL, Sheffler DJ, Kroeze WK. Magic shotguns versus magic bullets: selectively non-selective drugs for mood disorders and schizophrenia. Nat Rev Drug Discov. 2004;3:353. doi: 10.1038/nrd1346. [DOI] [PubMed] [Google Scholar]
- 48.Fojo T. Commentary: Novel therapies for cancer: why dirty might be better. Oncologist. 2008;13:277. doi: 10.1634/theoncologist.2007-0090. [DOI] [PubMed] [Google Scholar]
- 49.Hopkins AL. Drug discovery: Predicting promiscuity. Nature. 2009;462:167. doi: 10.1038/462167a. [DOI] [PubMed] [Google Scholar]
- 50.von Eichborn J, Murgueitio MS, Dunkel M, Koerner S, Bourne PE, Preissner R. PROMISCUOUS: a database for network-based drug-repositioning. Nucleic Acids Res. 2011;39:D1060. doi: 10.1093/nar/gkq1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee SJ, Wang JYJ. Exploiting the promiscuity of imatinib. J. Biol. 2009;8:30.1–30.4. doi: 10.1186/jbiol134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. http://en.wikipedia.org/wiki/Imatinib; accessed 9/21/2012.
- 53.Hopkins AL, Mason JS, Overington JP. Can we rationally design promiscuous drugs? Curr. Opin. Struc. Biol. 2006;16:127–136. doi: 10.1016/j.sbi.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 54.Besnard J, Ruda GF, Setola V, Abecassis K, Rodriguiz RM, Huang X-P, Norvall S, Sassano MF, Shin AI, Webster LA, Simeons FRC, Stojanovski L, Prat A, Seidah NG, Constam DB, Bickerton GR, Read KD, Wetsel WC, Gilbert IH, Roth BL, Hopkins AL. Automated design of ligands to polypharmacological profiles. Nature. 2012;492:215–222. doi: 10.1038/nature11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. http://www.discoverx.com/kinases/adp-quest_adp-quest-hs.php; accessed 9/21/2012.
- 56. http://www.millipore.com/catalogue/item/12-331; accessed 9/21/2012.
- 57. http://www.invitrogen.com/site/us/en/home/Products-and-Services/Applications/Drug-Discovery/Target-and-Lead-Identification-and-Validation/KinaseBiology/Kinase-Activity-Assays/Z-LYTE.html?CID=fl-zlyte; accessed 9/21/2012.
- 58. http://www.invitrogen.com/site/us/en/home/Products-and-Services/Services/custom-services/Screening-and-Profiling-Services/SelectScreen-Profiling-Service/SelectScreen-Kinase-Profiling-Service.html; accessed 9/21/2012.
- 59.Khopde SM, Priyadarsini KI, Palit DK, Mukherjee T. Effect of Solvent on the Excited-state Photophysical Properties of Curcumin. Photochem. Photobiol. 2000;72:625–631. doi: 10.1562/0031-8655(2000)072<0625:eosote>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 60.Schrödinger, LLC; New York, NY: 2009. Maestro modeling package. http://www.schrodinger.com/products/14/12/; accessed 9/20/2012. [Google Scholar]
- 61.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shaw DE, Shelley M, Perry JK, Francis P, Shenkin PS. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]; Schrödinger, LLC; New York, NY: 2009. http://www.schrodinger.com/products/14/5/; accessed 9/20/2012. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.