

Abstract
Ambient particulate matter (PM), including diesel exhaust particles (DEP), promote the development of allergic disorders. Diesel exhaust particles increase oxidative stress and influence human bronchial epithelial cell (HBEC)-dendritic cell (DC) interactions via cytokines including thymic stromal lymphopoietin (TSLP). Upregulation of TSLP results in Th2 responses. Using primary culture human bronchial epithelial cells (pHBEC) and human myeloid DC co-cultures we now show that DEP upregulation of Th2 responses occurred via HBEC-dependent mechanisms that resulted from oxidative stress. Moreover, DEP-treated HBEC and ambient-PM-treated HBEC upregulated OX40L and the Notch ligand Jagged-1 mRNA and expression on mDC. Upregulation of OX40L as well as Jagged-1 on mDC required HBEC and did not occur in the presence of n-acetylcysteine (NAC). Furthermore, OX40L and Jagged-1 upregulation was inhibited when HBEC expression of TSLP was silenced. Thus DEP-treatment of HBEC targeted two distinct pathways in mDC that were downstream of TSLP expression. Upregulation of OX40L and Jagged-1 by mDC resulted in mDC driven Th2 responses. These studies expand our understanding of the mechanism by which ambient pollutants alter mucosal immunity and promote disorders such as asthma.
Keywords: TSLP, diesel exhaust particles, dendritic cells, bronchial epithelial cells, OX40L, Notch ligand, Jagged-1, OX40L, lung
Introduction
Air pollution is associated with an increase in allergic asthma (1). Ambient particulate matter (PM) and diesel exhaust particles (DEP), the largest single source of airborne PM from vehicular traffic, have been documented in both animal and human studies to participate in allergic immune responses (1). Observational human studies, including a recent prospective birth cohort study of over 2,000 children, show an increased risk of atopic diseases and allergic sensitization in relation to exposure to ambient PM (2-6). DEP are adjuvants for allergic inflammation and exposure to DEP in the context of an allergen increases IgE production in human and animal studies (7, 8). Exposure to DEP results in an increase in inflammatory markers in airways and peripheral blood of healthy and asthmatic individuals (9-11). These findings reinforce the need to understand mechanisms by which ambient PM and DEP promote airway immune responses towards an allergic phenotype.
Human bronchial epithelial cells (HBEC) are the first targets for most inhaled pollutants. DEP generate oxidative stress in airway as well as other cells, and this process can promote immune responses (12, 13). Dendritic cells (DC) are abundant in the airway (14) and we have suggested that DEP modifies allergic sensitization by the effect of HBEC on local DC. We have demonstrated that HBEC treated with ambient PM or DEP release chemokines (CCL20) associated with the recruitment of immature DC (15). Furthermore DEP-treated HBEC upregulate cytokines (granulocyte-macrophage colony stimulating factor; GM-CSF, and thymic stromal lymphopoietin; TSLP) associated with the maturation and Th2 polarization of DC (16-19). TSLP is an IL-7-like cytokine that regulates Th2 cell differentiation via its effect on DC (20). We have shown that TSLP is produced by DEP-treated HBEC and that its production results from oxidative stress(19). A recent animal study supports the role of DC in DEP-induced adaptive immunity (21). We now expand upon these studies and suggest that DEP treatment of HBEC results in TSLP and reactive oxygen intermediates that upregulate OX40L and selective Notch pathways, two DC signals that promote a Th2 response. These studies further our understanding of mechanisms by which DEP promote airway immune responses towards those associated with asthma and allergy.
Materials and Methods
Reagents
DMEM, MEM, penicillin-streptomycin, FBS, trypsin-EDTA solution, and PBS were purchased from GIBCO Life Technologies (Grand Island, NY). Bronchial epithelial cell growth medium (BEGM) and bronchial epithelial cell basal medium (BEBM) were purchased from Lonza (Walkersville, MD). Ficoll was obtained from Amersham Bioscience (Piscataway, NJ, USA), and a magnetic cell separator from MACS, Miltenyi Biotech (Auburn, CA). GM-CSF, interleukin (IL) 2, IL-4, IL-1β, IL-6, tumor necrosis factor (TNF)-α and IFNγ were obtained from PeproTech (Princeton, NJ). PMA, ionomycin, mitomycin C and PGE2 were from EMD Chemicals (Gibbstown, NJ). Fluorescent reagents for FACS analyses were obtained from Becton Dickinson Immunocytometry Systems (San Jose, CA), Pharmingen (San Diego, CA), Coulter/Immunotech (Brea, CA), or R&D Systems (Minneapolis, MN). N-acetyl cysteine (NAC) was from MP Biomedicals (Solon, OH).
DEP were derived from a 1.6 l Volkswagen Diesel Engine (40 kW) running under standard city driving cycle conditions according to U.S. test protocol FTP 72 (EPA, 1992) and were a kind gift of D.L. Costa (U.S. EPA). DEP were diluted in cell culture medium, vortexed (5 times, 10 sec), sonicated (1 min) and added to cells in the defined concentrations. Since DEP sediment to the bottom of cell culture dishes, DEP concentrations were based on the available surface area (μg/cm2). Endotoxin activity in DEP (100 μg/ml), a concentration that was 10 times higher than that used in most of the experiments, was below the lower limit of detection (0.01 EU/ml; PyroGene™ Recombinant Factor C Assay; Lonza; Walkersville, MD). Ambient PM samples were collected using a high volume 3-stage impactor (ChemVol model 2400, R&P, Inc.), to simultaneously collect PM10-2.5 (coarse PM), PM2.5-0.15 (fine PM), and PM0.15 (ultrafine) at 900 L/min. The coarse and fine PM fractions were collected on polyurethane foam substrates (McMaster-Carr) whereas the ultrafine PM was collected on polyethylene final filters (G5300, Monandock Non-Wovens LLC). All sampling substrates were pre-cleaned using sterile solutions prior to exposure and extracted after exposure in sterile and pyrogen-free water using sonication. Samples were obtained from midtown Manhattan (Hunter College) and the South Bronx (South Bronx), New York City.
Cells
HBEC included primary human bronchial epithelial cells (pHBEC) or 16HBE14o- cells (16HBEC) as defined for each study. Primary HBEC were purchased from Lonza (Walkersville, MD) and cultured as described (19). 16HBE14o- cells (16HBEC) were generously provided by Dr. D. Gruenert (Univ. of Vermont, Burlington, VT) and cultured as described in MEM supplemented with 10% FBS (22).
Immature myeloid dendritic cells (mDC) were directly isolated from peripheral blood mononuclear cells (PBMC) according to expression of the mDC marker BDCA-1 (CD1c) using the CD1c dendritic cell isolation kit (Miltenyi Biotech, Auburn, CA). Myeloid DC were cultured in DMEM supplemented with FBS (10 %, v/v), β-mercaptoethanol (5 mM), and supplemented with GM-CSF (50 ng/ml) and IL-4 (10 ng/ml). Dendritic cells were routinely checked for immaturity by assessing expression of maturation markers (CD80, 83) by FACS, as well as their inability to induce robust T cell proliferation in an allogeneic mixed lymphocyte reaction (MLR; data not shown). For co-culture experiments immature mDC (1 × 105 cells/well) were cultured with confluent pHBEC or 16HBEC (48 h) and cells subsequently trypsinized (0.5 g/l, 5 min) for analysis. Myeloid DC were discriminated from HBEC by MACS according to their expression of CD11c and CD326. CD4+ CD45RA+ CD4RO- T cells were isolated from PBMC using the CD4 multisort kit in combination with a depletion of CD45RO+ cells using CD45RO-magnetic beads (Miltenyi Biotech, Auburn, CA) according to the manufacturer’s instructions.
Treatment of HBEC with DEP, carbon, fine ambient PM, in the presence or absence of NAC, did not induce cell death compared to untreated HBEC using a cell viability dye (Allamar Blue; Invitrogen, CA) (data not shown). Myeloid DC were recovered from co-culture with pHBEC or 16HBEC after mild trypsination by staining with magnetically labeled anti-EpCAM (CD326) and subsequent magnetic depletion of EpCAM+ pHBEC/16HBEC (Miltenyi Biotech, Auburn, CA) according to the manufacturer’s instructions. In select experiments, purity of mDC rescued from co-culture was assessed by FACS using fluorescent antibodies against CD11c and CD326 and ranged from 85-95% CD11c+ CD326- mDC. From the initial mDC (1 × 105 cells at the start of co-culture) we usually recovered 39-62% mDC after magnetic depletion of pHBEC/16HBEC.
siRNA gene silencing of TSLP
16HBEC were seeded (70% confluence) and after 24h, cells were transfected with a siRNA duplex-pool targeted against human TSLP (Dharmacon siGENOME™ SMARTpool M-011166, Dharmacon, Inc, Lafayette, CO) using lipofectamine (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. Additional cells were transfected with a non-targeting duplex-pool (siCONTROL, Dharmacon). After 48h, cells were again transfected with the same siRNA duplex-pools (48 h). Transfected 16HBEC were subsequently exposed to mDC as indicated.
Allogeneic MLR
Immature mDC (1 × 105 cells/well) were exposed to confluent pHBEC or 16HBEC (48 h), cells were trypsinized and mDC isolated by magnetic sorting. Myeloid DC were mitomycin C-treated (50 μg/ml; 30min) and used as stimulator cells for allogeneic naive T cells at a stimulator-responder ratio of 1:10 in round bottom 96-well plates (Costar, Cambridge, MA, USA) (5 days). T cells were stimulated with rhIL-2 (10 μg/ml) and expanded (5 days). On day 10, T cells were washed, counted, and viable cells were re-stimulated with PMA (10 ng/ml) and ionomycin (1 μg/ml, 24 h). Cell-free supernatants were analyzed for IL-5 and interferon gamma (IFNγ) using commercially available ELISAs (R&D) with lower detection limits of 3 and 8 pg/ml for each cytokine respectively. To generate polarized mDC for comparison, mDC were exposed to maturation factors (MF) that induce mature DC that support Th1 polarization (MF1) or Th2 polarization (MF2). MF1 includes IL-1β (10 ng/ml), TNF-α (10 ng/ml), IL-6 (100 ng/ml) and IFNγ (50 ng/ml). MF2 includes PGE2 (10 μM) in addition (23).
RNA Quantification
RNA was isolated from mDC using the Micro-to-Midi total RNA purification System (Invitrogen, Carlsbad, CA, USA). Quantification by real-time PCR was performed using the One-Step Quantitech SyberGreen Real Time PCR kit (Qiagen, Valencia, CA) following the manufacturer’s instructions. In vitro transcription was performed using T7/ SP6 RNA polymerase (Invitrogen) followed by complementary DNA (cDNA) digestion (DNAse I, Amplification Grade, Invitrogen) and cDNA purification. Levels of respective transcripts were normalized to glyceraldehyde- 3-phosphate dehydrogenase (GAPDH) transcript level as an internal control ΔCt(target)=Ct(target)-Ct(GAPDH). Data are expressed as relative mRNA expression of target compared to GAPDH (2ΔCt).
Flow Cytometry (FACS)
Myeloid DC isolated by MACS were cultured alone, or co-cultured with HBEC. Myeloid DC were recovered, pelleted and reacted with a cocktail consisting of V450-anti-CD11c, FITC-anti-Jagged-1, PE-anti-OX40L, and PE-Cy5-anti-CD83 (30 min, 0°C), washed (cold PBS) and fixed (PBS containing 1% formaldehyde). Cytometric analyses were performed on a FACSAria (BD Biosciences). Forward and 90° angle scattered 488 nm laser light intensities were used to exclude cellular debris. Fluorescence intensities were detected using 405 nm excitation for V450, 488 nm excitation for FITC and 562 nm excitation for PE and PE-Cy5 through appropriate dichroic and band pass filters (450/50 nm for V450, 530/30 nm for FITC, 585/15 nm for PE and 660/20 nm for PE-Cy5). Spillover detected by inappropriate channels was corrected by electronic compensation. Expression of surface molecules was determined as mean FITC, PE, or PE-Cy5 fluorescence intensity for the V450-positive mDC population. Myeloid DC were identified as CD11c+ cells and Jagged-1, OX40L, and CD83 expression levels were determined as mean FITC, PE, or PE-Cy5 fluorescence intensities on the V450-gated mDC population.
Data Analysis
Data are presented as mean ± standard error (SE). Significance was determined by Student’s t test for comparing two variables or one-way ANOVA for multiple variable comparisons, with a p<0.05 considered significant.
Results
DEP-treated HBEC promote DC dependent Th2 polarization via ROS
We have recently shown that DEP-treated HBEC (pHBEC and 16HBEC) upregulate TSLP and support maturation of immature mDC towards a phenotype that promotes Th2 polarization of naïve T cells (19). We have previously shown that DEP but not carbon, upregulates reactive oxygen species (ROS) in HBEC using carobxy-H2DCF-DA, an oxidation-sensitive fluorescent probe as a measure (19). Since DEP increase oxidative stress in our studies as well as others (19, 24), we examined whether the Th2 polarization induced by mDC exposed to DEP-treated HBEC was dependent upon the generation of reactive oxygen species (ROS). Immature mDC were treated as described, isolated and used as stimulator cells in an MLR and CD4+ T cell polarization measured by the ratio of IL-5/IFNγ in cell supernatants. As shown in Fig. 1A, CD4+ T cells exposed to mDC that had been co-cultured with resting pHBEC released low levels of both IL-5 and IFNγ (IL-5/IFNγ ratio = 0.07 ± 0.02, mean ± SE, n = 3). CD4+ T cell supernatants from an MLR with mDC co-cultured with DEP-treated pHBEC had an increase in the IL-5/IFNγ ratio (0.29 ± 0.13, p < 0.05 DEP-treated vs. resting pHBEC). In contrast, CD4+ T cell supernatants from an MLR with mDC that had been co-cultured with DEP-treated pHBEC in the presence of NAC had a reduced IL-5/IFNγ ratio (0.06 ± 0.02, p < 0.05 vs. DEP-treated pHBEC). No increase in the IL-5/IFNγ ratio was noted for CD4+ T cells exposed to mDC cultured with carbon-treated pHBEC (0.06 ± 0.02, p < 0.05 vs. resting pHBEC). As expected, the IL-5/IFNγ ratio was low in CD4+ T cells exposed to mDC cultured with MF1 and pHBEC (0.01 ± 0.01, p < 0.05 vs. resting pHBEC) and the IL-5/IFNγ ratio was increased in supernatants derived from CD4+ T cells exposed to mDC cultured with MF2 treated HBEC (0.45 ± 0.13, p < 0.05 vs. resting pHBEC). Similar findings were noted for 16HBEC (Fig. 1B). Immature mDC that had not been exposed to HBEC failed to generate detectable levels of IL-5 or IFNγ levels in an MLR (data not shown).
Fig. 1. HBEC upregulate DC dependent Th2 polarization via ROS.
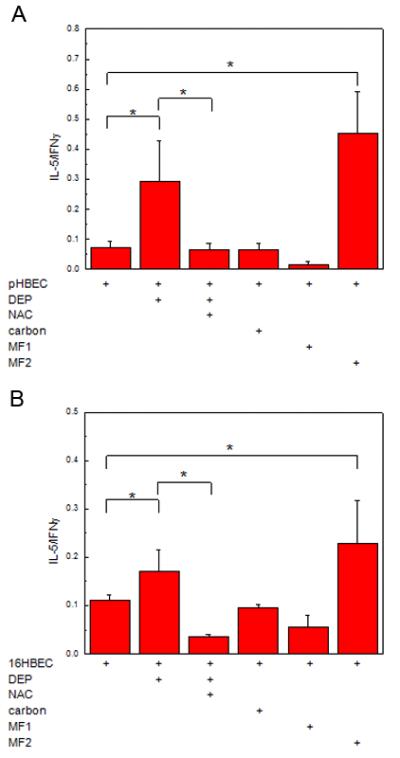
Immature mDC were exposed (48h) to pHBEC treated with DEP (3 μg/cm2) or defined stimuli (A) or 16HBEC and defined stimuli (B). Myeloid DC were subsequently isolated and used in an MLR with allogeneic naïve CD4+ T cells. Supernatants were analyzed for IL-5 and IFNγ and data are expressed as the ratio of IL-5/IFNγ (mean ± SE, * = p < 0.05).
Myeloid DC OX40L mRNA is upregulated by DEP-treated HBEC via TSLP and ROS
We have previously demonstrated that mDC exposed to DEP-treated HBEC induce CD4+ Th2 polarization via TSLP and that TSLP generation is mediated via ROS (19). Since OX40L is associated with mDC-driven Th2 polarization of naïve CD4+ T cells by TSLP (25), we examined whether DEP-treated HBEC upregulated OX40L mRNA in mDC. Immature mDC were cultured alone, or with pHBEC in the absence or presence of DEP (3 μg/cm2). Myeloid DC were isolated and OX40L mRNA assayed (Fig. 2A). OX40L mRNA expression was increased in mDC cultured with resting pHBEC compared to mDC alone (11.3 ± 2.5*103 vs. 0.1 ± 0.1*103 respectively, p < 0.05, n = 3). Exposure of immature mDC to DEP-treated pHBEC further upregulated OX40L mRNA above resting levels (26.9 ± 10.0*103, p < 0.05 vs. resting pHBEC, n = 3). No significant effect was seen with carbon. The increase was similar to that seen after treatment with MF2 (20.7 ± 7.6*103 p < 0.05 vs. resting pHBEC, n = 3) whereas the presence of MF1 during co-culture did not increase OX40L mRNA (0.9 ± 0.5*103, p < 0.05 vs. resting pHBEC, n = 3). Findings were similar for mDC exposed to 16HBEC (Fig. 2B).
Fig. 2. Myeloid OX40L mRNA is upregulated by DEP-treated HBEC.
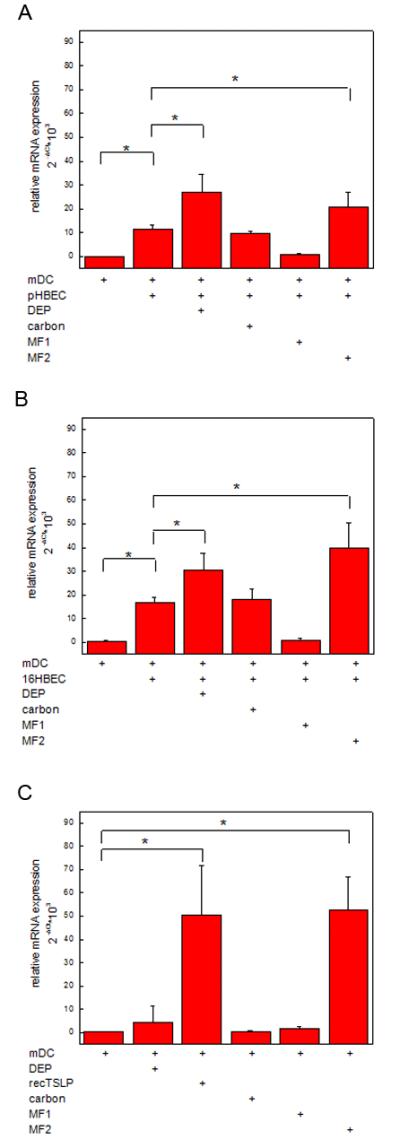
Immature mDC were cultured (48h) with pHBEC (2A), 16HBEC (2B) or alone (2C) in the absence or presence of DEP (3 μg/cm2), carbon (3 μg/cm2), MF1 or MF2 and for mDC alone, recombinant TSLP. Myeloid DC were isolated and OX40L mRNA measured by RT-PCR. Data are expressed as relative mRNA expression of OX40L compared to GAPDH (2ΔCt) (mean ± SE, n = 3, * = p < 0.05).
To confirm that HBEC were required for the DEP effect, immature mDC were exposed to DEP (6, 48h) in the absence of HBEC and OX40L mRNA assayed (Fig. 2C). OX40L mRNA was minimal in immature mDC and was not significantly increased after DEP (3 μg/cm2) or carbon exposure (data not shown for 6h; data yielded similar results). OX40L mRNA was significantly increased after exposure to recombinant TSLP (15ng/ml) and MF2 but not after MF1.
We next examined whether OX40L up-regulation by DEP-treated HBEC was a result of HBEC-derived TSLP and was ROS-dependent. As shown in Fig. 3, anti-TSLP, but not control IgG reduced OX40L mRNA expression in mDC exposed to DEP-treated 16HBEC. To confirm that the increase in mDC OX40L mRNA expression was due to epithelial cell-derived TSLP, 16HBEC were transfected with an siTSLP duplex pool to silence TSLP expression as described (19). OX40L mRNA expression was significantly reduced in mDC exposed to DEP-treated TSLP-silenced 16HBE compared to those that were exposed to DEP-treated 16HBE or DEP-treated 16HBE with siCONTROL (Fig. 3). OX40L mRNA expression was also significantly reduced in mDC that were exposed to DEP-treated 16HBE in the presence of NAC. These data suggested that OX40L mRNA expression in mDC was due to TSLP derived from DEP-treated HBEC and that this process was associated with the generation of ROS.
Fig. 3. Myeloid DC OX40L mRNA is upregulated by DEP-treated 16HBCE via TSLP and ROS.
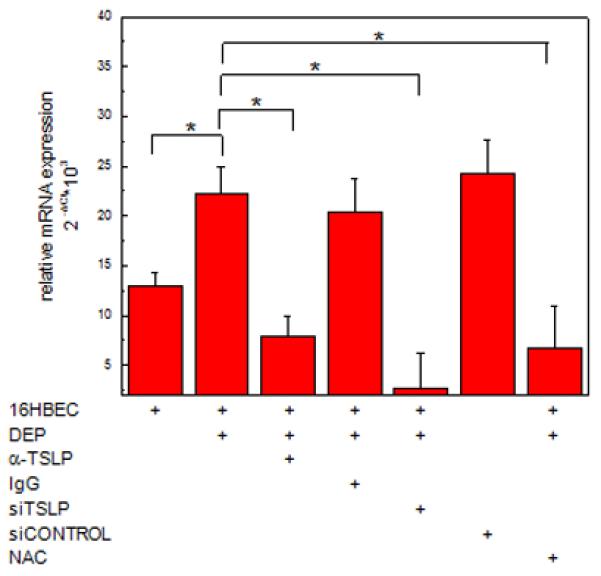
Myeloid DC were co-cultured (48h) with 16HBEC treated with DEP (3 μg/cm2) in the absence or presence of anti-TSLP, siTSLP or NAC and the appropriate controls. Myeloid DC were isolated and OX40L mRNA measured by RT-PCR. Data are expressed as relative mRNA expression of OX40L compared to GAPDH (2ΔCt) (mean ± SE, n = 3, * = p < 0.05).
Selective mDC Notch ligand mRNA is upregulated by DEP-treated HBEC via TSLP and ROS
Th2 polarization of T cells requires multiple IL-4-dependent and independent signals (26). IL-4 independent signals include those derived via distinct Notch ligands expressed by DC with Notch receptors on T cells (27). We therefore examined whether exposure of mDC to DEP-treated pHBEC upregulated Jagged-1, Jagged-2 and Dll4 mRNA. Jagged-1 was significantly upregulated in mDC that had been co-cultured with DEP-treated pHBEC compared to mDC co-cultured with resting pHBEC (3.3 ± 0.4*103 vs.1.5 ± 0.4*103, p < 0.05, n = 3) (Fig. 4A). MF2, but neither carbon nor MF1, upregulated Jagged-1 mRNA. Dll4 mRNA was upregulated by MF1, but no increase was seen with either DEP-treated 16HBE or MF2 (Fig. 4A). This finding was similar for mDC exposed to 16HBEC (Fig. 4B) in which DEP increased Jagged-1 mRNA. Jagged-2 mRNA in mDC was not increased by DEP-treated HBEC (data not shown).
Fig. 4. Jagged-1 but not Dll4 is upregulated by DEP-treated HBEC.

Immature mDC were cultured (48h) with DEP (3 μg/cm2), MF1, MF2, carbon (3 μg/cm2) or recombinant TSLP (15 ng/ml) in the presence of untreated or treated pHBEC (4A) or 16HBEC (4B) or alone (4C). Myeloid DC were isolated and Jagged-1 or Dll4 mRNA measured by RT-PCR. Data are expressed as relative mRNA expression of Jagged-1 or Dll4 compared to GAPDH (2ΔCt) (mean ± SE, n = 3, * = p < 0.05).
To confirm that DEP upregulation of Jagged-1 was associated with mDC exposure to HBEC, immature mDC were treated with DEP (3 μg/cm2), recombinant TSLP, or appropriate stimuli in the absence of HBEC and Notch ligand mRNA measured by RT-PCR. Resting mDC expressed low level Jagged-1, which was not increased by DEP at 6h (data not shown) or 48h (Fig. 4C). TSLP and MF2, but not MF1, significantly increased Jagged-1 mRNA in mDC. Dll4 mRNA was increased in mDC treated with MF1, but not DEP or TSLP (Fig. 4C). Jagged-2 expression was not significantly upregulated by any of these stimuli (data not shown).
To determine whether the increase in Jagged-1 induced by DEP-treated 16HBEC resulted from epithelial cell-derived TSLP and was ROS-dependent, immature mDC were exposed to DEP-treated 16HBEC in the presence of anti-TSLP or TSLP-silenced 16HBEC (Fig. 5). Anti-TSLP reduced Jagged-1 mRNA compared to mDC exposed to DEP-treated 16HBEC (1.3 ± 0.5*103 vs. 5.2 ± 1.7*103 respectively, p < 0.05, n = 3). Myeloid DC exposed to TSLP-silenced 16HBEC also had a significant reduction in Jagged-1 mRNA compared to mDC exposed to DEP-treated 16HBEC (1.0 ± 1.0*103 vs. 4.9 ± 2.3*103 respectively, p < 0.05, n = 3). Myeloid DC co-cultured with DEP-treated 16HBEC and NAC also had a reduction in Jagged-1 mRNA (1.3 ± 0.2*103).
Fig. 5. Myeloid DC Jagged-1mRNA is upregulated by DEP-treated 16HBEC via TSLP and ROS.

Myeloid DC were co-cultured (48h) with 16HBEC treated with DEP (3 μg/cm2) in the absence or presence of anti-TSLP, siTSLP or NAC and the appropriate controls. Myeloid DC were isolated and Jagged-1 mRNA measured by RT-PCR. Data are expressed as relative mRNA expression of Jagged-1 compared to GAPDH (2ΔCt) (mean ± SE, n = 3, * = p < 0.05).
OX40L and Jagged-1 mRNA in response to HBEC treated with urban ambient PM
DEP are derived from a single source engine. To examine whether ambient PM, which is comprised of DEP as well as particles with other chemical components had a similar effect, we used fine ambient PM from two urban sources: midtown Manhattan (Hunter College) and the South Bronx. Myeloid DC were exposed to resting or ambient PM-treated HBEC and mRNA for OX40L, Jagged-1, and Dll4 was measured as described for cells exposed to DEP. Hunter College and South Bronx PM were used at 3 μg/cm2. As shown in Fig. 6, mDC mRNA for OX40L was upregulated after exposure to resting HBEC, and further increased in the presence of DEP, Hunter College, and South Bronx-treated HBEC, but not carbon. Myeloid DC mRNA for Jagged-1 but not DLL4 was upregulated after exposure of mDC to ambient PM-treated HBEC. There was no significant difference in the effect of DEP, Hunter College PM and South Bronx PM when used at the same concentration.
Fig. 6. Ambient PM-treated 16HBEC upregulated the expression of mDC OX40L and Jagged-1, but not and Dll4 in mDC.
Immature mDC were cultured (48h) with DEP, Hunter College, or South Bronx fine ambient PM (3 μg/cm2), in the presence of untreated or treated 16HBEC and mRNA for OX40L (6A), Jagged-1 (6B), or Dll4 (6C) measured by RT-PCR. Data are presented as a representative experiment performed in triplicate and expressed as relative mRNA expression of OX40L, Jagged-1 or Dll4 compared to GAPDH (2ΔCt) (mean ± SE, in triplicate, * = p < 0.05).
DEP-treated HBEC upregulated Jagged-1 and OX40L expression on the surface of mDC
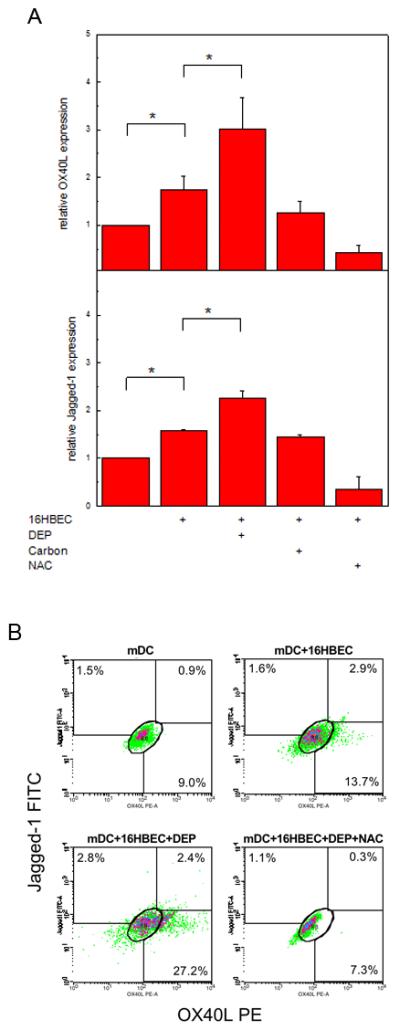
To confirm that mRNA upregulation was association with surface expression of OX40L and Jagged-1, we performed FACS analyses as described and monitored mean fluorescence intensity. As shown in Fig. 6A, OX40L expression was increased in mDC exposed to 16HBEC (1.74 ± 0.29 fold increase compared to resting mDC alone, n = 3, p < 0.05). Consistent with mRNA data, exposure of mDC to DEP-treated 16HBEC resulted in a further increase in OX40L expression (3.02 ± 0.67 fold increase compared to mDC exposed to resting 16HBEC, n = 3, p < 0.05). An increase in Jagged-1 expression was seen when mDC were co-cultured with 16HBEC (1.59 ± 0.03 fold increase compared to mDC alone) with a further increase in Jagged-1 in mDC exposed to DEP-treated 16HBEC (2.26 ± 0.16 fold compared to mDC and resting 16HBEC, n = 3, p < 0.05). Up-regulation of OX40L and Jagged-1 was inhibited in the presence of NAC. Neither OX40L nor Jagged-1were upregulated in mDC exposed to carbon-treated 16HBEC.
Figure 6B is one of three experiments illustrating the heterogeneity in the mDC responses revealed by FACS. A small increase in the frequency of cells expressing OX40L (1.9 ± 1.4%) and Jagged-1 (3.3 ± 2.2%) alone, or co-expressing OX40L and Jagged-1 (4.1 ± 1.1%) was seen in mDC exposed to untreated 16HBEC (detected as the difference between mDC cultured with untreated 16HBEC and mDC cultured alone, mean ± SE, n = 3). Similar heterogeneity in the mDC response to DEP-treated 16HBEC (detected as the difference between mDC cultured with DEP-treated 16HBEC and mDC cultured alone) was also seen in the increase of OX40L+ cells (7.5 ± 5.4%), Jagged-1+ cells (6.0 ± 3.6%), and OX40L+Jagged-1+ cells (5.0 ± 2.0%). It is interesting to note that only a minority of mDC upregulated their expression of OX40L or Jagged-1 in response to untreated 16HBEC (9.3 ± 2.4%) or DEP-treated 16HBEC (18.5 ± 3.0%), an increase that was associated with a similar rise in the number of CD11+ cells that upregulated their expression of the activation marker CD83 (data not shown).
DEP-treated HBEC upregulated Th2 polarization by mDC Jagged-1 and OX40L
Having demonstrated the up-regulation of two pathways associated with CD4+ Th2 polarization by DC, we next examined whether these pathways were required for CD4+ Th2 polarization by mDC exposed to DEP-treated 16HBEC. Immature mDC were exposed to DEP-treated 16HBEC in the absence or presence of anti-OX40L or anti-Jagged-1. Myeloid DC were isolated, cultured with allogeneic CD4+T cells and CD4+T cell polarization determined by the IL-5/IFNγ ratio (Fig. 7). Up-regulation of the IL-5/IFNγ ratio induced by mDC exposed to DEP-treated 16HBEC was significantly reduced in mDC in the presence of anti-Jagged-1 or anti-OX40L. These data suggested that the DEP-treated HBEC induced expression of OX40L and Jagged-1 in mDC was associated with a functional effect resulting in Th2 cell skewing of naïve CD4+ T cells.
Fig. 7. DEP-treated 16HBEC upregulated the expression of Jagged-1 and OX40L on the surface of mDC.

Myeloid DC were cultured (48h) alone, or in the presence of untreated 16HBEC, or 16HBEC treated with DEP (3 μg/cm2), carbon (3 μg/cm2), or DEP and NAC. Cells were recovered and analyzed by flow cytometry. Jagged-1 and OX40L expression were detected as FITC and PE mean fluorescence intensity (MFI) on CD11c+ cells. (A) Relative expression (mean ± SE, n = 3, * = p < 0.05) was obtained by normalization against the MFI detected on mDC cultured alone. (B) % mDC expressing Jagged-1 alone, OX40L alone, or both Jagged-1 and OX40L, are shown in linear density plots for CD11c+ cells from one of three experiments.
Discussion
Increasing epidemiologic data suggest that ambient PM, and in particular, DEP, promote the development of allergic disorders (1). To understand the impact of these pollutants on human health and to gain insights into mechanisms leading to potential interventions, it is important to understand how these pollutants affect adaptive immune responses at mucosal sites. DC direct T cells to respond as Treg, Th1, Th2 or Th17 cells and the instruction depends on the signals DC receive as immature cells (28, 29). We have suggested that DEP perturb the interaction between airway epithelial cells and DC resulting in DC that promote Th2 polarization (19, 30). We have previously shown that DEP promote a Th2 response by stimulating HBEC to produce GM-CSF and TSLP and up-regulate mature mDC that support T cell proliferation and Th2 skewing (30). Our current data expand our previous observations and we now demonstrate that DEP-treated HBEC induce mDC that promote Th2 polarization via TSLP and ROS regulation of two distinct pathways involving OX40L and specific Notch receptors.
The role of oxidative stress in DEP-stimulated airway inflammation has been well described (1, 24, 31, 32). We have demonstrated the production of ROS in HBEC in response to DEP and the addition of NAC inhibits many DEP effects in HBEC (19). We now show that NAC inhibited DEP-treated HBEC induced mDC Th2 skewing. Furthermore, we demonstrated that upregulation of both mDC OX40L and Jagged-1 mRNA by DEP-treated HBEC was inhibited in the presence of NAC. We demonstrated this effect using both primary and transformed epithelial cells to confirm the transformed cell model for subsequent studies. These findings suggest that ROS generation is associated with epithelial cell-derived TSLP resulting in OX40L and Jagged-1 signaling in mDC and subsequent Th2 skewing.
Our previous studies suggest the importance of soluble mediators derived from HBEC after DEP treatment in DC maturation and Th2 polarization and a recent murine study supports the role of DC-epithelial cell interactions in DEP-induced Th2 skewing (21, 30). We have previously shown that DEP up-regulate TSLP in HBEC and that this expression results in Th2 polarization of mDC by DEP-treated HBEC (19). TSLP, predominantly derived from epithelial cells, has been considered a “master switch” of the mucosal immune response (33, 34). Human and murine mDC express high levels of the TSLP receptors (TSLPR/CRLF2 and IL-7Ra) and via signal transducer and activator of transcription (STAT)3 and STAT5 signaling, their engagement results in mDC proliferation and the production of inflammatory Th2 promoting cytokines (25, 34-36). TSLP stimulated DC activate naïve T cells and up-regulate Th2 memory cells (37, 38). Interestingly, TSLP only conditions the lung for a Th2 response and lung-specific TSLP transgene expression requires antigenic stimulation for the full inflammatory response (39). In humans, bronchial epithelial cell expression of TSLP is associated with the severity of asthma (33, 40, 41). Our new data expand upon our previous observations and suggest that DEP promote a Th2 response via epithelial cell-derived TSLP that results in mDC expression of Th2 promoting signals.
We now show OX40L upregulation in immature mDC exposed to DEP-treated HBEC and the importance of TSLP and ROS in this process. OX40L (CD252, TNFSF4, glycoprotein 34kDa; GP34), a member of the TNF superfamily, is selectively expressed by TSLP-activated DC (42) and binds OX40 expressed on activated naïve CD4+ and CD8+ T cells (43). Engagement of OX40 results in activation of signaling pathways that preferentially lead to generation of inflammatory Th2 CD4+ T cells (42-44)(40). OX40L regulates memory as well as naïve T cells (43, 45-47) and blocks the generation of peripheral Tregs and regulatory activity of naturally occurring CD4+ CD25+ Foxp3+ Treg (43, 48). Thus the OX40 pathway promotes Th2 diseases by multiple mechanisms. Animal models support the importance of the OX40 pathway in asthma since OX40L−/− mice have reduced Th2 responses and allergen sensitization (49, 50) and anti-OX40L decreases inflammation, CD4+ effector and memory T cells in animal models of TSLP-driven asthma (51). Allergen-specific Foxp3+ T regulatory cells and airway tolerance are also reduced by OX40L expression (52). Our studies now suggest that OX40L, via TSLP, may also be involved in the DEP promotion of allergic diseases.
We also showed that DEP-treatment of HBEC resulted in TSLP and ROS-mediated upregulation of Jagged-1 on mDC. Notch1 signaling is essential in T cell fate specification and instructs lymphoid progenitors to adopt T versus B cell fates (53). Recent studies also demonstrate that Notch signaling is required for both effector T cell and regulatory T cell differentiation (26, 53, 54). In DC, Dll promote DC-mediated induction of Th1-cell differentiation of naïve T cells (53, 55, 56), whereas Jagged-1 Notch receptor expression is associated with Th2 cell differentiation (26, 53, 57, 58) and is up-regulated by Th2-cell promoting stimuli (26, 27, 59, 60). Engagement of Jagged-1 promotes Th2 polarization in both naïve and memory T cells via an increase in the GATA3/T-bet ratio and subsequent STAT-6 phosphorylation (27). IL-4 expression is downstream of Notch signaling (26, 61, 62) however Th2 cell differentiation is induced via GATA3 even in situations in which IL-4 signaling is inhibited (58, 63). Thus Notch signaling via Jagged supports Th2-cell differentiation in an IL-4-independent as well as IL-4-dependent manner (53). In humans, mDC constitutively express Jagged-1 in levels higher than any of the other Notch ligands (27). Inhibition of Notch signaling by a gamma-secretase inhibitor results in the inhibition of the asthma phenotype and downregulation of GATA-3 (64). Our data suggest that HBEC treated with DEP selectively up-regulated Jagged-1 in immature mDC and that Jagged-1 expression was associated with functional DC that supported Th2 differentiation of naïve T cells. Moreover, Jagged-1 up-regulation was dependent on TSLP and the generation of ROS. Our finding that DEP-treated HBEC promoted DC that up-regulated Jagged-1, but not Dll4 provide an additional mechanism by which DEP can promote Th2 responses and suggest the potential for an IL-4-independent process.
FACS analyses confirmed increased surface expression of both OX40L and Jagged-1 in mDC exposed to DEP-treated HBEC. FACS analyses also demonstrated that the mDC response to untreated and DEP-treated HBEC was heterogeneous with mDC subsets expressing OX40L and Jagged-1 alone or expressing both ligands. These data also showed that only a minor percentage of myeloid DC isolated from peripheral blood had the potential to respond to HBEC signals. These data suggest the possibility that a maturation threshold may be required for mDC-HBEC interaction and that there are subsets of mDC with the ability to respond.
Dendritic cells have potential to directly access and phagocytose particles such as DEP and they have been well shown to sample through epithelial cell tight junctions (65-68). DEP have also been reported to directly stimulate some cytokines (IL-1, TNF-α) in human monocyte-derived DC (69) and ambient PM obtained from Baltimore air has been shown to directly induce maturation of human monocyte-derived DC (70-72). Studies of isolated bone marrow-derived DC from murine models suggest a response of DC to carbon black particles or DEP and suggest that this is a complex process (73, 74). We, and others did not identify direct activation of mDC by DEP or ambient PM in the absence of HBEC (19, 21, 30, 75, 76). In contrast, we demonstrated the importance of DEP upregulated HBEC-derived signals, specifically TSLP, in the mDC expression of OX40L and Jagged-1. One explanation for the discrepancy in findings may be due to differences in the source of the particles and their chemical composition. The level of endotoxin in PM may also play a role in the discrepant findings. Endotoxin was below the limit of detection in the DEP used for our studies (19, 30) but was detected in Baltimore ambient PM. The possibility exists that endotoxin serves as an adjuvant in the presence of Baltimore ambient PM. We therefore suggest that the microenvironment provided by epithelial-cell derived cytokines is important for the Th2 polarization regulated by DC in response to DEP.
Our previous work, and the initial experiments for this study were performed in both human primary HBEC and confirmed in 16HBEC, suggesting that for this area of study, the 16HBE transformed cell line functions in a manner that is similar to primary cells. DEP was used at a physiologically relevant concentration (3 μg/cm2), previously shown to induce cell activation without cell toxicity (30). Recent studies identify different origins of DC and responses between monocyte-derived DC, plasmacytoid DC, and blood derived mDC may differ (77, 78). We used human blood-derived mDC for these studies and the possibility exists that different responses may be identified for other DC. We identified Th2 skewing in the absence of allergen stimulation, and thus suggest that DEP up-regulation of TSLP and its downstream pathways may condition the lung for an enhanced allergen-regulated response.
In summary, our data, in combination with our previous studies, suggest that DEP-treated HBEC upregulate TSLP in a ROS-dependent manner, and that this upregulation results in functional mDC that support Th2 polarization. Th2-cell differentiation by mDC exposed to DEP-treated HBEC occurs via multiple pathways, including expression of OX40L and selective Notch ligands. These findings have implications for furthering our understanding of how pollutants may generate a permissive environment at the mucosal interface that supports the generation of a Th2 airway response.
Fig. 8. Th2 polarization by mDC exposed to DEP-treated HBEC is mediated via OX40L and Jagged-1.
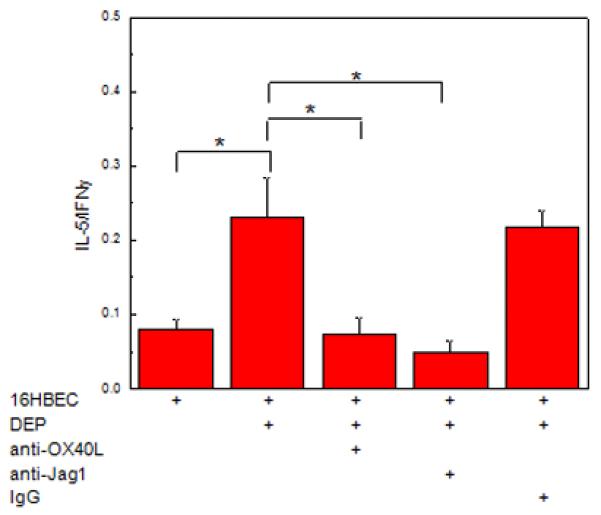
Myeloid DC were exposed to DEP (3 μg/cm2)-treated pHBEC (48 h) in the absence or presence of anti-OX40L or anti-Jagged-1 or appropriate control. DC were subsequently isolated and used in an MLR with allogeneic naïve CD4+ T cells. Supernatants were analyzed for IL-5 and IFNγ and data were expressed as the ratio of IL-5/IFNγ (mean ± SE, * = p < 0.05).
Acknowledgements
We would like to thank Robert Devlin PhD (US EPA) for his assistance in the sampling and extraction of the PM samples.
Footnotes
Sources of Support: This work was supported by National Institutes of Health Grants ES010187 6 (JR), T32 ES007267 (RS), AI27742-129007 (DT), RR020950 (DT), ES00260 (TG), and Environmental Protection Agency grants R827351 (TG) and RD-83374201 (TG).
References
- 1.Riedl MA. The effect of air pollution on asthma and allergy. Curr Allergy Asthma Rep. 2008;8:139–146. doi: 10.1007/s11882-008-0024-8. [DOI] [PubMed] [Google Scholar]
- 2.Morgenstern V, Zutavern A, Cyrys J, Brockow I, Koletzko S, Kramer U, Behrendt H, Herbarth O, von Berg A, Bauer CP, Wichmann HE, Heinrich J. Atopic diseases, allergic sensitization, and exposure to traffic-related air pollution in children. Am J Respir Crit Care Med. 2008;177:1331–1337. doi: 10.1164/rccm.200701-036OC. [DOI] [PubMed] [Google Scholar]
- 3.McConnell R, Berhane K, Yao L, Jerrett M, Lurmann F, Gilliland F, Kunzli N, Gauderman J, Avol E, Thomas D, Peters J. Traffic, susceptibility, and childhood asthma. Environ Health Perspect. 2006;114:766–772. doi: 10.1289/ehp.8594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Janssen NA, Brunekreef B, van Vliet P, Aarts F, Meliefste K, Harssema H, Fischer P. The relationship between air pollution from heavy traffic and allergic sensitization, bronchial hyperresponsiveness, and respiratory symptoms in Dutch schoolchildren. Environ Health Perspect. 2003;111:1512–1518. doi: 10.1289/ehp.6243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gergen PJ, Turkeltaub PC, Kovar MG. The prevalence of allergic skin test reactivity to eight common aeroallergens in the U.S. population: results from the second National Health and Nutrition Examination Survey. J Allergy Clin Immunol. 1987;80:669–679. doi: 10.1016/0091-6749(87)90286-7. [DOI] [PubMed] [Google Scholar]
- 6.Penard-Morand C, Raherison C, Kopferschmitt C, Caillaud D, Lavaud F, Charpin D, Bousquet J, Annesi-Maesano I. Prevalence of food allergy and its relationship to asthma and allergic rhinitis in schoolchildren. Allergy. 2005;60:1165–1171. doi: 10.1111/j.1398-9995.2005.00860.x. [DOI] [PubMed] [Google Scholar]
- 7.Takenaka H, Zhang K, Diaz-Sanchez D, Tsien A, Saxon A. Enhanced human IgE production results from exposure to the aromatic hydrocarbons from diesel exhaust: direct effects on B-cell IgE production. J Allergy Clin Immunol. 1995;95:103–115. doi: 10.1016/s0091-6749(95)70158-3. [DOI] [PubMed] [Google Scholar]
- 8.Diaz-Sanchez D, Tsien A, Fleming J, Saxon A. Combined diesel exhaust particulate and ragweed allergen challenge markedly enhances human in vivo nasal ragweed-specific IgE and skews cytokine production to a T helper cell 2-type pattern. J Immunol. 1997;158:2406–2413. [PubMed] [Google Scholar]
- 9.Devouassoux G, Saxon A, Metcalfe DD, Prussin C, Colomb MG, Brambilla C, Diaz-Sanchez D. Chemical constituents of diesel exhaust particles induce IL-4 production and histamine release by human basophils. J Allergy Clin Immunol. 2002;109:847–853. doi: 10.1067/mai.2002.122843. [DOI] [PubMed] [Google Scholar]
- 10.Salvi S, Blomberg A, Rudell B, Kelly F, Sandstrom T, Holgate ST, Frew A. Acute inflammatory responses in the airways and peripheral blood after short-term exposure to diesel exhaust in healthy human volunteers. Am J Respir Crit Care Med. 1999;159:702–709. doi: 10.1164/ajrccm.159.3.9709083. [DOI] [PubMed] [Google Scholar]
- 11.Nordenhall C, Pourazar J, Ledin MC, Levin JO, Sandstrom T, Adelroth E. Diesel exhaust enhances airway responsiveness in asthmatic subjects. Eur Respir J. 2001;17:909–915. doi: 10.1183/09031936.01.17509090. [DOI] [PubMed] [Google Scholar]
- 12.Gilmour MI, Jaakkola MS, London SJ, Nel AE, Rogers CA. How exposure to environmental tobacco smoke, outdoor air pollutants, and increased pollen burdens influences the incidence of asthma. Environ Health Perspect. 2006;114:627–633. doi: 10.1289/ehp.8380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diaz-Sanchez D, Riedl M. Diesel effects on human health: a question of stress? Am J Physiol Lung Cell Mol Physiol. 2005;289:L722–723. doi: 10.1152/ajplung.00217.2005. [DOI] [PubMed] [Google Scholar]
- 14.Holt PG, Schon-Hegrad MA, Oliver J, Holt BJ, McMenamin PG. A contiguous network of dendritic antigen-presenting cells within the respiratory epithelium. Int Arch Allergy Appl Immunol. 1990;91:155–159. doi: 10.1159/000235107. [DOI] [PubMed] [Google Scholar]
- 15.Reibman J, Hsu Y, Chen LC, Bleck B, Gordon T. Airway epithelial cells release MIP-3alpha/CCL20 in response to cytokines and ambient particulate matter. Am J Respir Cell Mol Biol. 2003;28:648–654. doi: 10.1165/rcmb.2002-0095OC. [DOI] [PubMed] [Google Scholar]
- 16.Ohtoshi T, Takizawa H, Okazaki H, Kawasaki S, Takeuchi N, Ohta K, Ito K. Diesel exhaust particles stimulate human airway epithelial cells to produce cytokines relevant to airway inflammation in vitro. J Allergy Clin Immunol. 1998;101:778–785. doi: 10.1016/S0091-6749(98)70307-0. [DOI] [PubMed] [Google Scholar]
- 17.Boland S, Baeza-Squiban A, Fournier T, Houcine O, Gendron MC, Chevrier M, Jouvenot G, Coste A, Aubier M, Marano F. Diesel exhaust particles are taken up by human airway epithelial cells in vitro and alter cytokine production. Am J Physiol. 1999;276:L604–613. doi: 10.1152/ajplung.1999.276.4.L604. [DOI] [PubMed] [Google Scholar]
- 18.Reibman J, Hsu Y, Chen LC, Kumar A, Su WC, Choy W, Talbot A, Gordon T. Size fractions of ambient particulate matter induce granulocyte macrophage colony-stimulating factor in human bronchial epithelial cells by mitogen-activated protein kinase pathways. Am J Respir Cell Mol Biol. 2002;27:455–462. doi: 10.1165/rcmb.2001-0005OC. [DOI] [PubMed] [Google Scholar]
- 19.Bleck B, Tse DB, Curotto de Lafaille MA, Zhang F, Reibman J. Diesel Exhaust Particle-Exposed Human Bronchial Epithelial Cells Induce Dendritic Cell Maturation and Polarization via Thymic Stromal Lymphopoietin. J Clin Immunol. 2008;28:147–156. doi: 10.1007/s10875-007-9149-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu YJ, Soumelis V, Watanabe N, Ito T, Wang YH, Malefyt Rde W, Omori M, Zhou B, Ziegler SF. TSLP: an epithelial cell cytokine that regulates T cell differentiation by conditioning dendritic cell maturation. Annu Rev Immunol. 2007;25:193–219. doi: 10.1146/annurev.immunol.25.022106.141718. [DOI] [PubMed] [Google Scholar]
- 21.Provoost S, Maes T, Willart MA, Joos GF, Lambrecht BN, Tournoy KG. Diesel exhaust particles stimulate adaptive immunity by acting on pulmonary dendritic cells. J Immunol. 2010;184:426–432. doi: 10.4049/jimmunol.0902564. [DOI] [PubMed] [Google Scholar]
- 22.Cozens AL, Yezzi MJ, Kunzelmann K, Ohrui T, Chin L, Eng K, Finkbeiner WE, Widdicombe JH, Gruenert DC. CFTR expression and chloride secretion in polarized immortal human bronchial epithelial cells. Am J Respir Cell Mol Biol. 1994;10:38–47. doi: 10.1165/ajrcmb.10.1.7507342. [DOI] [PubMed] [Google Scholar]
- 23.Bender A, Sapp M, Schuler G, Steinman RM, Bhardwaj N. Improved methods for the generation of dendritic cells from nonproliferating progenitors in human blood. J Immunol Methods. 1996;196:121–135. doi: 10.1016/0022-1759(96)00079-8. [DOI] [PubMed] [Google Scholar]
- 24.Baulig A, Garlatti M, Bonvallot V, Marchand A, Barouki R, Marano F, Baeza-Squiban A. Involvement of reactive oxygen species in the metabolic pathways triggered by diesel exhaust particles in human airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2003;285:L671–679. doi: 10.1152/ajplung.00419.2002. [DOI] [PubMed] [Google Scholar]
- 25.Liu YJ. Thymic stromal lymphopoietin and OX40 ligand pathway in the initiation of dendritic cell-mediated allergic inflammation. J Allergy Clin Immunol. 2007;120:238–244. doi: 10.1016/j.jaci.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 26.Amsen D, Blander JM, Lee GR, Tanigaki K, Honjo T, Flavell RA. Instruction of distinct CD4 T helper cell fates by different notch ligands on antigen-presenting cells. Cell. 2004;117:515–526. doi: 10.1016/s0092-8674(04)00451-9. [DOI] [PubMed] [Google Scholar]
- 27.Liotta F, Frosali F, Querci V, Mantei A, Fili L, Maggi L, Mazzinghi B, Angeli R, Ronconi E, Santarlasci V, Biagioli T, Lasagni L, Ballerini C, Parronchi P, Scheffold A, Cosmi L, Maggi E, Romagnani S, Annunziato F. Human immature myeloid dendritic cells trigger a TH2-polarizing program via Jagged-1/Notch interaction. J Allergy Clin Immunol. 2008;121:1000–1005. e1008. doi: 10.1016/j.jaci.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 28.Kalinski P, Hilkens CM, Wierenga EA, Kapsenberg ML. T-cell priming by type-1 and type-2 polarized dendritic cells: the concept of a third signal. Immunol Today. 1999;20:561–567. doi: 10.1016/s0167-5699(99)01547-9. [DOI] [PubMed] [Google Scholar]
- 29.Kabelitz D, Wesch D, Oberg HH. Regulation of regulatory T cells: role of dendritic cells and toll-like receptors. Crit Rev Immunol. 2006;26:291–306. doi: 10.1615/critrevimmunol.v26.i4.10. [DOI] [PubMed] [Google Scholar]
- 30.Bleck B, Tse DB, Jaspers I, Curotto de Lafaille MA, Reibman J. Diesel exhaust particle-exposed human bronchial epithelial cells induce dendritic cell maturation. J Immunol. 2006;176:7431–7437. doi: 10.4049/jimmunol.176.12.7431. [DOI] [PubMed] [Google Scholar]
- 31.Li N, Kim S, Wang M, Froines J, Sioutas C, Nel A. Use of a stratified oxidative stress model to study the biological effects of ambient concentrated and diesel exhaust particulate matter. Inhal Toxicol. 2002;14:459–486. doi: 10.1080/089583701753678571. [DOI] [PubMed] [Google Scholar]
- 32.Kumagai Y, Arimoto T, Shinyashiki M, Shimojo N, Nakai Y, Yoshikawa T, Sagai M. Generation of reactive oxygen species during interaction of diesel exhaust particle components with NADPH-cytochrome P450 reductase and involvement of the bioactivation in the DNA damage. Free Radic Biol Med. 1997;22:479–487. doi: 10.1016/s0891-5849(96)00341-3. [DOI] [PubMed] [Google Scholar]
- 33.Liu YJ. Thymic stromal lymphopoietin: master switch for allergic inflammation. J Exp Med. 2006;203:269–273. doi: 10.1084/jem.20051745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang YH, Liu YJ. Thymic stromal lymphopoietin, OX40-ligand, and interleukin-25 in allergic responses. Clin Exp Allergy. 2009;39:798–806. doi: 10.1111/j.1365-2222.2009.03241.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Quentmeier H, Drexler HG, Fleckenstein D, Zaborski M, Armstrong A, Sims JE, Lyman SD. Cloning of human thymic stromal lymphopoietin (TSLP) and signaling mechanisms leading to proliferation. Leukemia. 2001;15:1286–1292. doi: 10.1038/sj.leu.2402175. [DOI] [PubMed] [Google Scholar]
- 36.Soumelis V, Reche PA, Kanzler H, Yuan W, Edward G, Homey B, Gilliet M, Ho S, Antonenko S, Lauerma A, Smith K, Gorman D, Zurawski S, Abrams J, Menon S, McClanahan T, de Waal-Malefyt Rd R, Bazan F, Kastelein RA, Liu YJ. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat Immunol. 2002;3:673–680. doi: 10.1038/ni805. [DOI] [PubMed] [Google Scholar]
- 37.Wang YH, Angkasekwinai P, Lu N, Voo KS, Arima K, Hanabuchi S, Hippe A, Corrigan CJ, Dong C, Homey B, Yao Z, Ying S, Huston DP, Liu YJ. IL-25 augments type 2 immune responses by enhancing the expansion and functions of TSLP-DC-activated Th2 memory cells. J Exp Med. 2007;204:1837–1847. doi: 10.1084/jem.20070406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang YH, Ito T, Wang YH, Homey B, Watanabe N, Martin R, Barnes CJ, McIntyre BW, Gilliet M, Kumar R, Yao Z, Liu YJ. Maintenance and polarization of human TH2 central memory T cells by thymic stromal lymphopoietin-activated dendritic cells. Immunity. 2006;24:827–838. doi: 10.1016/j.immuni.2006.03.019. [DOI] [PubMed] [Google Scholar]
- 39.Headley MB, Zhou B, Shih WX, Aye T, Comeau MR, Ziegler SF. TSLP conditions the lung immune environment for the generation of pathogenic innate and antigen-specific adaptive immune responses. J Immunol. 2009;182:1641–1647. doi: 10.4049/jimmunol.182.3.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rochman Y, Leonard WJ. Thymic stromal lymphopoietin: a new cytokine in asthma. Curr Opin Pharmacol. 2008;8:249–254. doi: 10.1016/j.coph.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ying S, O’Connor B, Ratoff J, Meng Q, Mallett K, Cousins D, Robinson D, Zhang G, Zhao J, Lee TH, Corrigan C. Thymic Stromal Lymphopoietin Expression Is Increased in Asthmatic Airways and Correlates with Expression of Th2-Attracting Chemokines and Disease Severity. J Immunol. 2005;174:8183–8190. doi: 10.4049/jimmunol.174.12.8183. [DOI] [PubMed] [Google Scholar]
- 42.Ito T, Wang YH, Duramad O, Hori T, Delespesse GJ, Watanabe N, Qin FX, Yao Z, Cao W, Liu YJ. TSLP-activated dendritic cells induce an inflammatory T helper type 2 cell response through OX40 ligand. J Exp Med. 2005;202:1213–1223. doi: 10.1084/jem.20051135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Croft M, So T, Duan W, Soroosh P. The significance of OX40 and OX40L to T-cell biology and immune disease. Immunol Rev. 2009;229:173–191. doi: 10.1111/j.1600-065X.2009.00766.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rogers PR, Song J, Gramaglia I, Killeen N, Croft M. OX40 promotes Bcl-xL and Bcl-2 expression and is essential for long-term survival of CD4 T cells. Immunity. 2001;15:445–455. doi: 10.1016/s1074-7613(01)00191-1. [DOI] [PubMed] [Google Scholar]
- 45.Gramaglia I, Jember A, Pippig SD, Weinberg AD, Killeen N, Croft M. The OX40 costimulatory receptor determines the development of CD4 memory by regulating primary clonal expansion. J Immunol. 2000;165:3043–3050. doi: 10.4049/jimmunol.165.6.3043. [DOI] [PubMed] [Google Scholar]
- 46.Salek-Ardakani S, Song J, Halteman BS, Jember AG, Akiba H, Yagita H, Croft M. OX40 (CD134) controls memory T helper 2 cells that drive lung inflammation. J Exp Med. 2003;198:315–324. doi: 10.1084/jem.20021937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sugamura K, Ishii N, Weinberg AD. Therapeutic targeting of the effector T-cell co-stimulatory molecule OX40. Nat Rev Immunol. 2004;4:420–431. doi: 10.1038/nri1371. [DOI] [PubMed] [Google Scholar]
- 48.So T, Croft M. Cutting edge: OX40 inhibits TGF-beta- and antigen-driven conversion of naive CD4 T cells into CD25+Foxp3+ T cells. J Immunol. 2007;179:1427–1430. doi: 10.4049/jimmunol.179.3.1427. [DOI] [PubMed] [Google Scholar]
- 49.Arestides RS, He H, Westlake RM, Chen AI, Sharpe AH, Perkins DL, Finn PW. Costimulatory molecule OX40L is critical for both Th1 and Th2 responses in allergic inflammation. Eur J Immunol. 2002;32:2874–2880. doi: 10.1002/1521-4141(2002010)32:10<2874::AID-IMMU2874>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 50.Hoshino A, Tanaka Y, Akiba H, Asakura Y, Mita Y, Sakurai T, Takaoka A, Nakaike S, Ishii N, Sugamura K, Yagita H, Okumura K. Critical role for OX40 ligand in the development of pathogenic Th2 cells in a murine model of asthma. Eur J Immunol. 2003;33:861–869. doi: 10.1002/eji.200323455. [DOI] [PubMed] [Google Scholar]
- 51.Seshasayee D, Lee WP, Zhou M, Shu J, Suto E, Zhang J, Diehl L, Austin CD, Meng YG, Tan M, Bullens SL, Seeber S, Fuentes ME, Labrijn AF, Graus YM, Miller LA, Schelegle ES, Hyde DM, Wu LC, Hymowitz SG, Martin F. In vivo blockade of OX40 ligand inhibits thymic stromal lymphopoietin driven atopic inflammation. J Clin Invest. 2007;117:3868–3878. doi: 10.1172/JCI33559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Duan W, So T, Croft M. Antagonism of airway tolerance by endotoxin/lipopolysaccharide through promoting OX40L and suppressing antigen-specific Foxp3+ T regulatory cells. J Immunol. 2008;181:8650–8659. doi: 10.4049/jimmunol.181.12.8650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Amsen D, Antov A, Flavell RA. The different faces of Notch in T-helper-cell differentiation. Nat Rev Immunol. 2009;9:116–124. doi: 10.1038/nri2488. [DOI] [PubMed] [Google Scholar]
- 54.Hoyne GF, Dallman MJ, Lamb JR. T-cell regulation of peripheral tolerance and immunity: the potential role for Notch signalling. Immunology. 2000;100:281–288. doi: 10.1046/j.1365-2567.2000.00073.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Napolitani G, Rinaldi A, Bertoni F, Sallusto F, Lanzavecchia A. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat Immunol. 2005;6:769–776. doi: 10.1038/ni1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Skokos D, Nussenzweig MC. CD8- DCs induce IL-12-independent Th1 differentiation through Delta 4 Notch-like ligand in response to bacterial LPS. J Exp Med. 2007;204:1525–1531. doi: 10.1084/jem.20062305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kubo M. Notch: filling a hole in T helper 2 cell differentiation. Immunity. 2007;27:3–5. doi: 10.1016/j.immuni.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 58.Fang TC, Yashiro-Ohtani Y, Del Bianco C, Knoblock DM, Blacklow SC, Pear WS. Notch directly regulates Gata3 expression during T helper 2 cell differentiation. Immunity. 2007;27:100–110. doi: 10.1016/j.immuni.2007.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Krawczyk CM, Sun J, Pearce EJ. Th2 differentiation is unaffected by Jagged2 expression on dendritic cells. J Immunol. 2008;180 doi: 10.4049/jimmunol.180.12.7931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Worsley AG, LeibundGut-Landmann S, Slack E, Phng LK, Gerhardt H, Reis e Sousa C, MacDonald AS. Dendritic cell expression of the Notch ligand jagged2 is not essential for Th2 response induction in vivo. Eur J Immunol. 2008;38:1043–1049. doi: 10.1002/eji.200737335. [DOI] [PubMed] [Google Scholar]
- 61.Minter LM, Turley DM, Das P, Shin HM, Joshi I, Lawlor RG, Cho OH, Palaga T, Gottipati S, Telfer JC, Kostura L, Fauq AH, Simpson K, Such KA, Miele L, Golde TE, Miller SD, Osborne BA. Inhibitors of gamma-secretase block in vivo and in vitro T helper type 1 polarization by preventing Notch upregulation of Tbx21. Nat Immunol. 2005;6:680–688. [PubMed] [Google Scholar]
- 62.Tanaka S, Tsukada J, Suzuki W, Hayashi K, Tanigaki K, Tsuji M, Inoue H, Honjo T, Kubo M. The interleukin-4 enhancer CNS-2 is regulated by Notch signals and controls initial expression in NKT cells and memory-type CD4 T cells. Immunity. 2006;24:689–701. doi: 10.1016/j.immuni.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 63.Amsen D, Antov A, Jankovic D, Sher A, Radtke F, Souabni A, Busslinger M, McCright B, Gridley T, Flavell RA. Direct regulation of Gata3 expression determines the T helper differentiation potential of Notch. Immunity. 2007;27:89–99. doi: 10.1016/j.immuni.2007.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kang JH, Kim BS, Uhm TG, Lee SH, Lee GR, Park CS, Chung IY. Gamma-secretase inhibitor reduces allergic pulmonary inflammation by modulating Th1 and Th2 responses. Am J Respir Crit Care Med. 2009;179 doi: 10.1164/rccm.200806-893OC. [DOI] [PubMed] [Google Scholar]
- 65.Jahnsen FL, Strickland DH, Thomas JA, Tobagus IT, Napoli S, Zosky GR, Turner DJ, Sly PD, Stumbles PA, Holt PG. Accelerated antigen sampling and transport by airway mucosal dendritic cells following inhalation of a bacterial stimulus. J Immunol. 2006;177:5861–5867. doi: 10.4049/jimmunol.177.9.5861. [DOI] [PubMed] [Google Scholar]
- 66.Sung SS, Fu SM, Rose CE, Jr., Gaskin F, Ju ST, Beaty SR. A major lung CD103 (alphaE)-beta7 integrin-positive epithelial dendritic cell population expressing Langerin and tight junction proteins. J Immunol. 2006;176:2161–2172. doi: 10.4049/jimmunol.176.4.2161. [DOI] [PubMed] [Google Scholar]
- 67.Rothen-Rutishauser BM, Kiama SG, Gehr P. A three-dimensional cellular model of the human respiratory tract to study the interaction with particles. Am J Respir Cell Mol Biol. 2005;32:281–289. doi: 10.1165/rcmb.2004-0187OC. [DOI] [PubMed] [Google Scholar]
- 68.Blank F, Wehrli M, Lehmann A, Baum O, Gehr P, von Garnier C, Rothen-Rutishauser BM. Macrophages and dendritic cells express tight junction proteins and exchange particles in an in vitro model of the human airway wall. Immunobiology. 2010 doi: 10.1016/j.imbio.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 69.Ohtani T, Nakagawa S, Kurosawa M, Mizuashi M, Ozawa M, Aiba S. Cellular basis of the role of diesel exhaust particles in inducing Th2-dominant response. J Immunol. 2005;174:2412–2419. doi: 10.4049/jimmunol.174.4.2412. [DOI] [PubMed] [Google Scholar]
- 70.Williams MA, Porter M, Horton M, Guo J, Roman J, Williams D, Breysse P, Georas SN. Ambient particulate matter directs nonclassic dendritic cell activation and a mixed TH1/TH2-like cytokine response by naive CD4+ T cells. J Allergy Clin Immunol. 2007;119:488–497. doi: 10.1016/j.jaci.2006.10.022. [DOI] [PubMed] [Google Scholar]
- 71.Porter M, Karp M, Killedar S, Bauer SM, Guo J, Williams D, Breysse P, Georas SN, Williams MA. Diesel-enriched particulate matter functionally activates human dendritic cells. Am J Respir Cell Mol Biol. 2007;37:706–719. doi: 10.1165/rcmb.2007-0199OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Williams MA, Rangasamy T, Bauer SM, Killedar S, Karp M, Kensler TW, Yamamoto M, Breysse P, Biswal S, Georas SN. Disruption of the transcription factor Nrf2 promotes pro-oxidative dendritic cells that stimulate Th2-like immunoresponsiveness upon activation by ambient particulate matter. J Immunol. 2008;181:4545–4559. doi: 10.4049/jimmunol.181.7.4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.de Haar C, Kool M, Hassing I, Bol M, Lambrecht BN, Pieters R. Lung dendritic cells are stimulated by ultrafine particles and play a key role in particle adjuvant activity. J Allergy Clin Immunol. 2008;121:1246–1254. doi: 10.1016/j.jaci.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 74.Inoue K, Koike E, Takano H, Yanagisawa R, Ichinose T, Yoshikawa T. Effects of diesel exhaust particles on antigen-presenting cells and antigen-specific Th immunity in mice. Exp Biol Med (Maywood) 2009;234:200–209. doi: 10.3181/0809-RM-285. [DOI] [PubMed] [Google Scholar]
- 75.Verstraelen S, Van Den Heuvel R, Nelissen I, Witters H, Verheyen G, Schoeters G. Flow cytometric characterisation of antigen presenting dendritic cells after in vitro exposure to diesel exhaust particles. Toxicol In Vitro. 2005;19:903–907. doi: 10.1016/j.tiv.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 76.Braun A, Bewersdorff M, Lintelmann J, Matuschek G, Jakob T, Gottlicher M, Schober W, Buters JT, Behrendt H, Mempel M. Differential impact of diesel particle composition on pro-allergic dendritic cell function. Toxicol Sci. 2010;113:85–94. doi: 10.1093/toxsci/kfp239. [DOI] [PubMed] [Google Scholar]
- 77.Liu K, Victora GD, Schwickert TA, Guermonprez P, Meredith MM, Yao K, Chu FF, Randolph GJ, Rudensky AY, Nussenzweig M. In vivo analysis of dendritic cell development and homeostasis. Science. 2009;324:392–397. doi: 10.1126/science.1170540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bogunovic M, Ginhoux F, Helft J, Shang L, Hashimoto D, Greter M, Liu K, Jakubzick C, Ingersoll MA, Leboeuf M, Stanley ER, Nussenzweig M, Lira SA, Randolph GJ, Merad M. Origin of the lamina propria dendritic cell network. Immunity. 2009;31:513–525. doi: 10.1016/j.immuni.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]