

Abstract
Membranous nephropathy (MN) is a glomerular disease characterized by a nephrotic syndrome without infiltration of inflammatory cells or proliferation of resident cells. Although the cause of the disease is unknown, the primary pathogenic mechanism responsible for the accumulation of immune deposits on the outer aspect of the glomerular basement membrane involves the generation of autoantibodies against antigen targets expressed at the membrane surface of podocyte. The molecular mechanisms of nephrotic proteinuria, which reflects a profound disorder of podocyte function, remain unclear. We show here that induction of c-mip in passive type Heymann nephritis (PHN) coincides with the occurrence of proteinuria. c-mip expression is not detectable in the glomeruli of PHN rats receiving a single dose of anti-megalin polyclonal antibody, yet the immune complexes are readily present but without triggering of proteinuria. Rats reinjected with anti-megalin develop a few days later heavy proteinuria concomitantly with c-mip overproduction in podocytes. We show that overexpression of c-mip is associated with downregulation of synaptopodin in human MN, PHN rats and c-mip transgenic mice, while the abundance of death-associated protein kinase (DAPK) and integrin linked kinase (ILK) is increased. Finally, cyclosporine treatment reduces significantly proteinuria in PHN rats, concomitantly with downregulation of c-mip abundance in podocytes. These results suggest that c-mip plays an active role in podocyte disorders of MN.
Keywords: Adult; Apoptosis Regulatory Proteins; physiology; Calcium-Calmodulin-Dependent Protein Kinases; physiology; Carrier Proteins; analysis; genetics; physiology; Cyclosporine; therapeutic use; Glomerulonephritis, Membranous; drug therapy; pathology; Humans; Podocytes; pathology; physiology; Protein-Serine-Threonine Kinases; physiology; Up-Regulation
Keywords: glomerular disease, Heymann nephritis, membranous nephropathy, nephrotic syndrome, Pathophysiology of Renal Disease and Progression
Introduction
Idiopathic membranous nephropathy (MN) is a glomerular disease of unknown etiology, commonly associated with nephrotic syndrome. It is histologically characterized by diffuse thickening of the capillary loop, which results from the formation of subepithelial immune deposits consisting of immunoglobulin G (predominantly IgG4), complement components and recently identified antigens. [1–4] MN remains a major cause of nephrotic syndrome in adults, with up to 40% of patients progressing toward end stage renal failure after 10 years. [5] Researches into the molecular mechanisms underlying the pathogenesis of MN were focused many years ago on the identification of target antigens, which initiate the formation of the immune complex. The generation of an experimental model of MN established in rats by Heymann and coworkers fifty years ago, represents the start-point of pathophysiological researches on MN. [6] Active (AHN) and passive type Heymann (PHN) nephritis were induced by direct immunization of Lewis rats with crude preparation of brush border proteins or by injection of rabbit anti-rat brush border antibodies, respectively. Both AHN and PHN closely mimic the human glomerular disease. [7] The identification in this model of the target antigen, megalin, a common component of the tubular and glomerular epithelial cell provided the molecular basis of podocyte disease in MN. [8,9] Nevertheless, since human podocytes do not express megalin, it cannot be considered as the target antigen in human MN. [1] In the last decades, many studies have focused on the identification of the antigen involved in human MN. Several antigens involved in secondary forms of MN caused by infectious disease and cancer have been found in subepithelial immune deposits, without clear evidence for a direct pathogenic role. The first identification of a pathogenic antigen in human MN came from Ronco’s team who identified neutral endopeptidase (NEP) as the podocyte target of nephritogenic antibodies in patients with neonatal MN. [10,11] The anti-NEP antibodies resulted from immunization against the placental NEP of a NEP-deficient mother. In this form of MN, the membrane attack of complement (C5b-9) was also detected within the immune deposits, which indicated that like anti-megalin antibodies, anti-NEP could induce complement activation potentially leading to podocyte injury. [11,12] Subsequently, Beck et al reported that about 70% of patients with idiopathic MN have autoantibodies that react with the M-type phospholipase A2 receptor (PLA2-R), a glycoprotein constitutively expressed in podocytes. [3] Prunotto et al. identified two additional targets of autoimmunity, aldoreductase and SOD2, both induced in glomeruli of patients with MN. [4]
The onset of proteinuria during the course of MN is associated with phenotypic alteration of podocyte and slit diaphragm integrity as demonstrated in a Heymann nephritis model. [13–15] The abundance of nephrin, a major protein of slit diaphragm, is reduced in PHN and in biopsies from patients with MN, while the mRNA and protein expression of cation permeable ion channel TRPC6, which is mutated in the familial form of focal segmental glomerulosclerosis, [16] is increased. [13–15,17,18] However, the mechanisms that lead to these alterations remain partly obscure
The membrane attack complex of complement C5b-9/Mac is thought to play a crucial role in the induction of podocyte injury leading to proteinuria in this model. [12,19] In PHN, proteinuria has been suggested to be entirely dependent on C5b-9/Mac. [20] These data have recently been challenged by the finding that AHN and PHN can be induced in rat deficient in complement component C6, which is unable to form Mac. [21,22] Indeed, in PVG/C6−/− rats, the degree of proteinuria, the extent of cellular infiltrates, the abundance of Ig, C3 and electron-dense deposits were similar to those observed in the PVG/c rats. [22] Anti-podocyte antibodies including anti-megalin, anti-NEP and anti-PLA2-R are directed against functional receptor or enzymatic proteins, so that their binding may induce a cascade of events that may directly alter podocyte biology. [23]
In the wake of studies aimed to understand the molecular pathophysiology of idiopathic nephrotic syndrome, we identified a new gene, c-mip (for c-maf inducing protein), which encodes an 86-kDa protein. [24] The predicted structure of c-mip includes an N-terminal region containing a pleckstrin homology domain (PH), a middle region characterized by the presence of several interacting docking sites including a 14-3-3 module, a PKC domain, an Erk domain, a SH3 domain similar to the p85 regulatory subunit of phosphatidylinositol 3-kinase (PI3K), and a C-terminal region containing a leucin-rich repeat (LRR) domain.
We have recently shown that c-mip abundance is increased in MN during relapse, [25] which led us to study its potential implication in Heymann nephritis. We report here that c-mip protein is not induced at the early stage of PHN, when the immune complex deposits are formed without inducing proteinuria, but increases very quickly after a second injection of anti-megalin polyclonal antibodies, while proteinuria concomitantly rises to reach nephrotic range. We provide evidence that c-mip induces in vivo and in vitro podocyte dysfunctions that are common to MN and PHN.
Results
Renal expression of c-mip in membranous nephropathy and passive Heymann Nephritis
Northern blot analysis showed that basal expression of c-mip in podocyte was scarcely or below the detection limits in control human kidneys (Figure 1a), which suggests that c-mip is transcriptionally repressed in physiological situations. However, quantitative PCR from laser microdissected glomeruli from five control samples and eleven MN biopsy specimens showed that c-mip abundance was significantly increased in MN (Figure 1b). In addition, we confirmed by in situ hybridization (Figure 1c), confocal immunofluorescence (Figure 1d) and immunohistochemistry analysis (supplementary Figure S1) that c-mip was overproduced at the mRNA and protein levels in patients with MN.
Figure 1. c-mip abundance is significantly increased in membranous nephropathy (MN).
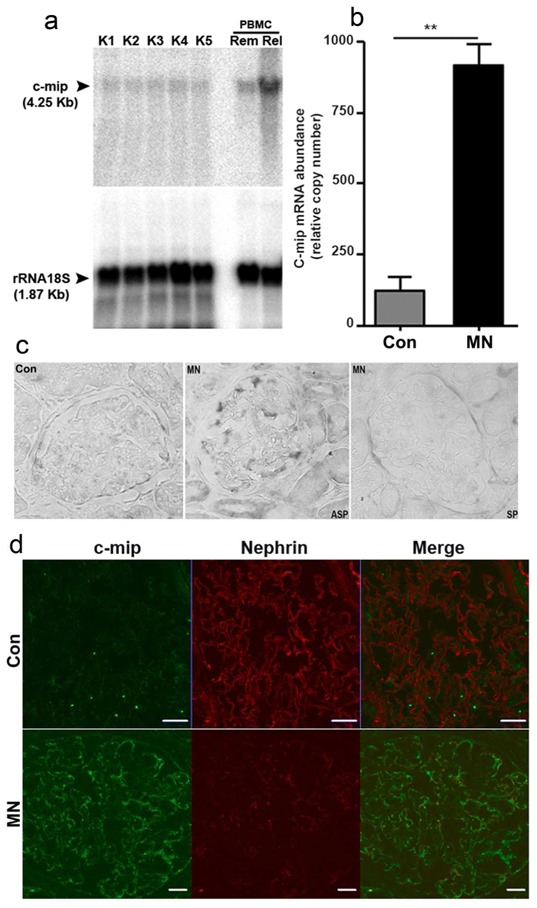
(a) Northern Blot analysis of c-mip expression in control kidneys. Twenty μg of total RNA from kidney were loaded on each lane. The blot was hybridized with a [32P] dCTP-labeled 1200 bp cDNA cmip probe, then with a 18S ribosomal RNA antisens olgonucleotide probe. Positive control consists of total RNA from PBMC of a patient with MCNS relapse (Rel). The remission sample (Rem) from the same patient was included as internal control. (b) Quantitative RT-PCR of laser microdissected glomeruli from MN kidney biopsy specimens (n=11) and control kidneys (n=5). Relative copy numbers were calculated as described in Material and Methods. Mann-Whitney test, ** P <0.01. (c) In situ hybridization with a c-mip probe on control human kidney (NHK: normal human kidney) and kidney biopsy specimens from patients with MN (ASP: antisens probe; SP: sens probe). (d) Confocal double immunofluorescence labeling c-mip-nephrin on kidney biopsy specimens from patients with MN and control human kidney. Scale bars, 20 μm.
The finding that c-mip was highly induced in podocytes of patients with MN led us to study its expression in Heymann nephritis, the experimental model of human MN. We induced PHN by injection of anti-megalin polyclonal antibody. Proteinuria, as tested at day 13 post injection, was very slightly increased (urine protein to creatinine ratio, UPr/UCr, mg/mg± SD: 1.53 ± 0.20) relatively to controls (0.63 ± 0.057) (Figure 2a). At day 12, immunofluorescence analysis of kidney sections showed granular deposits of IgG along the glomerular capillary loops in rats with PHN, while no staining was visualized in control rats (Figure 2b). Following a second injection two weeks after the first one (day 14), rats developed heavy proteinuria that reached a peak at day 19 (UPr/UCr: 9.48 ± 7.64) (Figure 2a). Quantitative RT-PCR from laser microdissected glomeruli (n= 3 rats at each time point) showed that c-mip abundance was markedly increased 24 hours after the second immunization (Figure 2c). C-mip was visualized by immunohistochemistry by day 13 post-injection and increased much at day 15, whereas no signal was detected before (Figure 2d). Overproduction of c-mip persisted until day 42, along the experimental procedure. These results suggest that c-mip induction is paralleled to development of proteinuria.
Figure 2. Induction of passive Heymann’s nephritis (PHN).
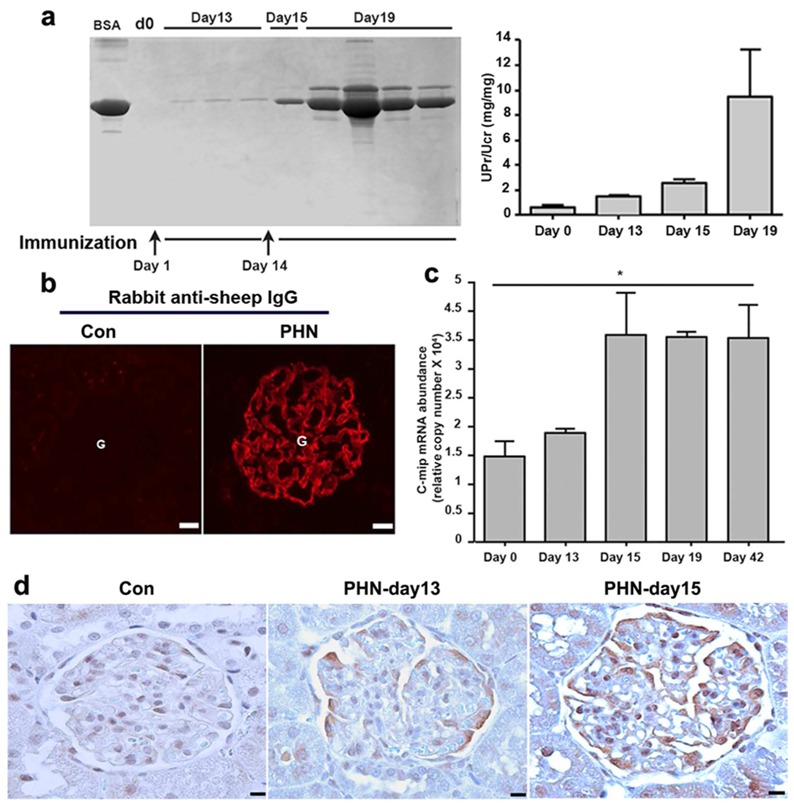
Rat were injected with anti-megalin polyclonal antibody at two week’s interval. (a), left, Urine samples (5 μl) and BSA (10 μg) were resolved by SDS-PAGE and gels stained with Coomassie Blue; right, proteinuria was assessed by calculating the proteinuria/urine creatinine ratio [UPr (mg)/UCr (mg)]. Data are expressed as means ±SD. (b) Immunofluorescence detection of immune complex deposits using anti-sheep IgG in rats injected with anti-megalin antibody (day 12 after the first injection) and in control rats. (c) Quantitative RT-PCR of laser microdissected glomeruli from PHN (n=3 rats in each time point) and control rats (n=5). Relative copy numbers were calculated as described in Material and Methods. Mann-Whitney test, * P <0.05. (d) Immunodetection of c-mip in PHN. Representative immunohistochemical analysis of serial kidney sections from PHN and control rats. C-mip (brown signal) is detected along the external side of the capillary loop in PHN with higher abundance following reimmunization with sheep anti-megalin polyclonal antibody. Scale bars, 20 μm.
Overexpression of c-mip induces phenotypical and biochemical alterations
To understand the effects of c-mip on podocyte function in PHN, we established stably transfected podocyte cell lines using the inducible T Rex system. Stable c-mip-overexpressing cells exhibited abnormal morphology with retraction of cell body, loss of stress fibers and were more susceptible to cell detachment from collagen matrix, whereas podocytes cultured in the absence of tetracycline proliferate normally and exhibited long intracellular bundles of actin filaments (Figure 3a and supplementary Figure S2). The abundance of synaptopodin, a regulator of podocyte actin cytoskeleton reorganization, was decreased (Figure 3b). Interestingly, the abundance of synaptopodin was reduced in MN glomeruli and in c-mip transgenic mice (Figure 3c).
Figure 3. Stable overexpression of c-mip in podocytes induces phenotypic and biochemical alterations.
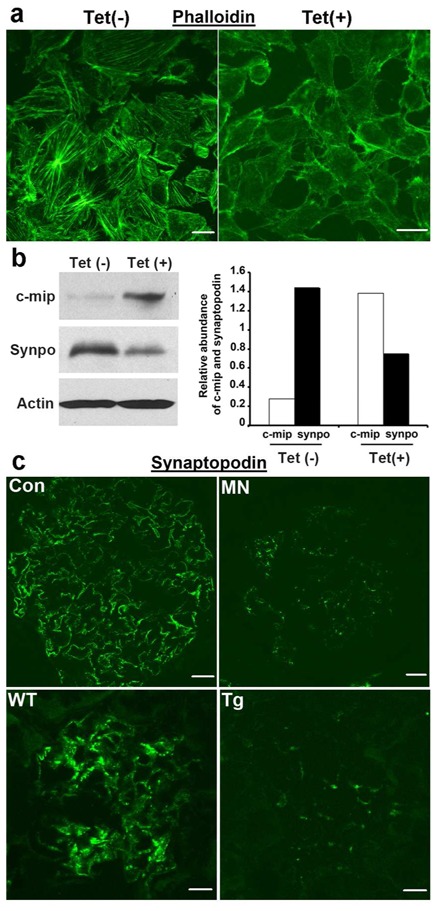
(a) Confocal microscopy analysis of phalloidin staining in stable transfectant murine podocytes without [Tet(-)] and after induction of c-mip by tetracycline [Tet(+)]. c-mip–overexpressing podocytes display a loss of stress fibers, whereas the actin network is well preserved in non-induced stable transfectant cells. Scale bars, 20 μM. (b) Western-blotting of protein lysates from non-induced [Tet(-)] and induced [Tet(+)] c-mip stable transfectants. Overexpression of c-mip induces a downregulation of synaptopodin. (c) Downregulation of synaptopodin in membranous nephropathy (MN) and transgenic mice (Tg). Confocal microscopy analysis of synaptopodin on control human kidney (Con), kidney biopsy specimens from patients with MN, wild type (wt) and c-mip Tg mice. Scale bars, 20 μM.
Because RhoA is required for actin stress fiber formation by interacting notably with synaptopodin, [26] we investigated RhoA activity in stably c-mip-transfected cells. In the absence of tetracycline, RhoA activity was increased in stably transfected podocytes (Figure 4a). However, tetracycline treatment was associated with lower RhoA activity (Figure 4a), but has no effect on Cdc42 activity (Figure 4b), while c-mip was clearly induced (Figure 4c). Tetracycline treatment did not seem to have any influence on RhoA activity in wild-type podocytes (Figure 4d).
Figure 4. Stable overexpression of c-mip in podocytes inhibits RhoA activity but has no effect on Cdc42 activity.
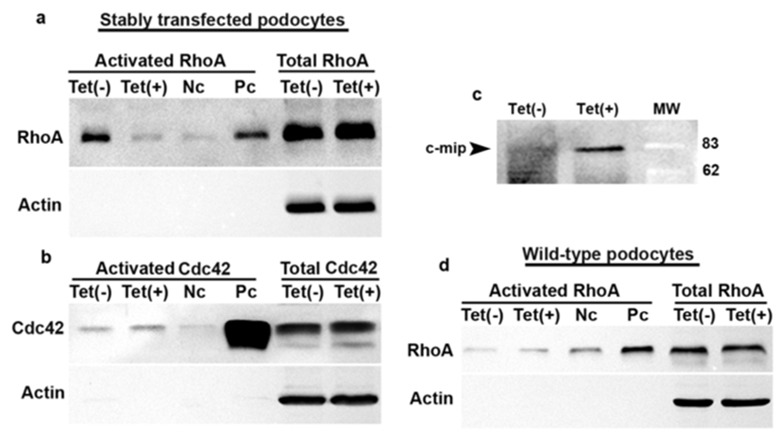
RhoA (a) and Cdc42 (b) activities were measured in stable transfectant podocytes cultured in the absence [Tet(-)] or in the presence of tetracycline [Tet(+)] as indicated in the Material and Method section. Nc: negative control; Pc Positive control. (c) Induction of c-mip by tetracyline in stable transfectant podocytes. (d) Tetracycline has no influence on RhoA activity in wild-type podocytes.
The observation that c-mip alters the adhesion of cultured podocytes to collagen matrix led us to explore whether c-mip affects the abundance of integrin linked kinase (ILK), an adhesion-associated kinase, whose upregulation was shown to cause podocyte detachment. [27,28] Low ILK expression was found in non-induced podocytes, whereas following tetracycline treatment stable c-mip-overexpressing cells exhibited increased ILK levels but podocin abundance did not significantly change (Figure 5a). Because cell detachment from the extracellular matrix alters matrix-transduced survival pathways and cytoskeleton architecture, resulting in release of proapoptotic factors, [29] we tested whether c-mip affects the expression of death-associated protein kinase (DAPK), an actin filament-associated proapoptotic protein. [30] Immunoblotting revealed that podocytes expressing c-mip exhibited higher abundance of DAPK (Figure 5a). The relevance of these findings was tested in vivo both in transgenic mice and PHN rats. Western blot analysis of glomerular lysates showed that c-mip transgenic mice exhibited an increased abundance of DAPK and ILK in c-mip transgenic mice, without significantly change in podocine expression (Figure 5b). Moreover, increased amounts of DAPK and ILK were detected in PHN rats (Figure 5c). We extended this analysis in MN biopsies and control kidneys (n = 5 in each group). Quantitative PCR analysis of laser-microdissected glomeruli showed that DAPK and ILK transcript levels were significantly increased in MN when compared with controls (DAPK: 1.62 ± 0.12 versus 1.18 ± 0.06, p<0.05; ILK 1.52 ± 0.13 versus 1.14 ± 0.14, p<0.05) (Figure 5d). Immunohistochemistry analysis suggests that abundance of both proteins was also increased with a more diffuse pattern for ILK (Figure 5e). Altogether, these results suggest that c-mip overexpression alters both integrin-mediated cell–matrix interaction and the regulation of prosurvival signals.
Figure 5. c-mip induces in vitro and in vivo an upregulation of ILK and DAPK.
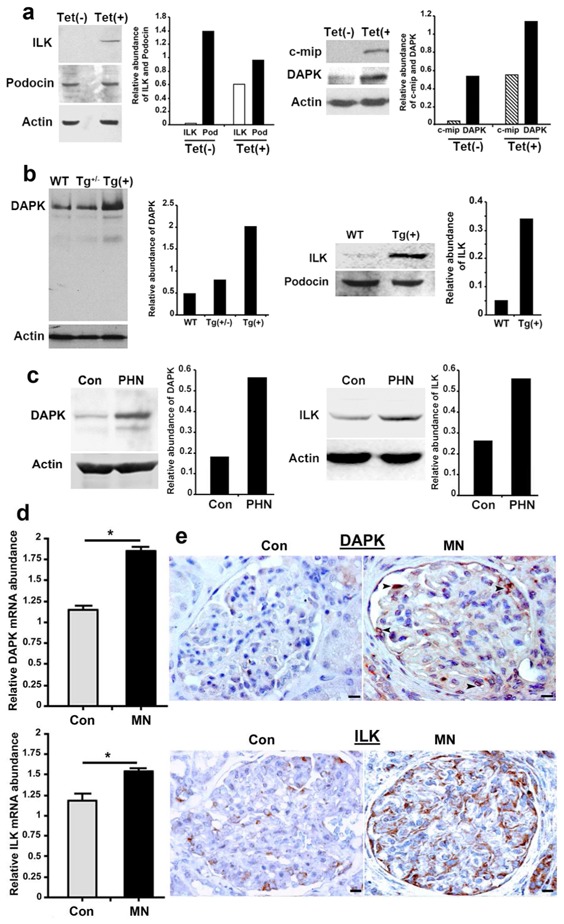
(a) Representative Western blots of ILK, podocin, c-mip and DAPK on protein lysates from non-induced [Tet(-)] and induced [Tet(+)] stable transfectant podocytes. (b) Representative Western blots of DAPK and ILK on glomerular lysates from wild-type and transgenic mice. (c) Representative Western blots of DAPK and ILK on glomerular lysates from PHN and control rat kidneys (Con). (d) Quantitative RT-PCR of laser microdissected glomeruli from MN kidney biopsy specimens (n=5) and control kidneys (n=5); * P <0.05, Mann-Whitney test. (e) Representative immunohistochemical analysis of DAPK and ILK in serial sections of kidney biopsy specimens from patients with MN and control human kidney (Con). Scale bars, 20 μM
Cyclosporine therapy inhibits c-mip expression and reduces proteinuria in Heymann nephritis
To assess the influence of cyclosporine in this model, PHN rats displaying nephrotic proteinuria were separated in two groups: the one was treated intraperitoneally by cyclosporine at 20 mg/kg daily (CsA group) from the day 21 post-first immunization, while the control group received the vehicle (sodium chloride). Proteinuria was analyzed every three days, between day 21 and day 42. UPr/UCr decreased significantly from the third day-post CsA and reached the nadir at day 38 (Figure 6a). In the absence of CsA treatment, proteinuria also decreased over time but remained at nephrotic range in control rats. Quantitative RT-PCR analysis showed that c-mip abundance was reduced by 50% at day 42, as compared with control PHN rats (Figure 6b). Immunochemistry analysis showed that c-mip protein was clearly reduced in CsA-treated rats (Figure 6c). The abundance of active RhoA was increased (Figure 6d), whereas ILK and DAPK abundances were reduced in CsA-treated rats (Figure 6e and 6f). Confocal microscopy analysis showed that nephrin and synaptopodin were mostly downregulated in PHN rats, but restored upon CsA treatment (Figure 7).
Figure 6. Cyclosporine therapy inhibits c-mip expression and reduces proteinuria in PHN rats.
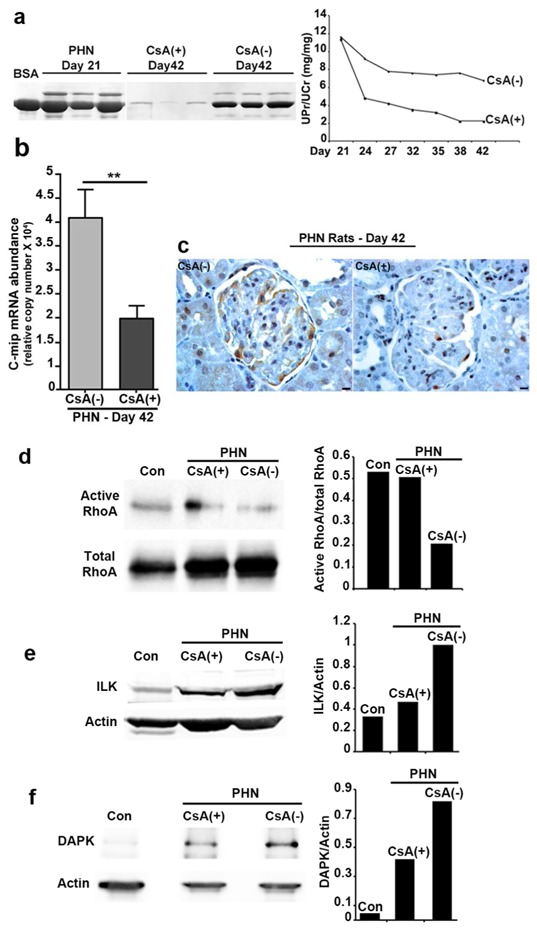
(a) Course of proteinuria during CsA therapy started from the day 21 post-first immunization. left, Urine samples (5 μl) and BSA (10 μg) were resolved by SDS-PAGE and gels stained with Coomassie Blue; right, course of proteinuria as assessed by calculating the proteinuria/urine creatinine ratio (UPr/UCr). (b) Quantitative RT-PCR of laser microdissected glomeruli from untreated [CsA(-)] and CsA-treated [CsA(+)] PHN rats (n=5 rats in each group). Relative copy numbers were calculated as described in Material and Methods; ** P <0.01, Mann-Whitney test. (c) Representative immunohistochemical analysis for c-mip in serial kidney sections from CsA(-) and CsA(+) PHN rats (day 42 post-immunization). Scale bars, 20 μm. (d-f) Western blotting of RhoA, ILK and DAPK on glomerular lysates from CsA(-) and CsA(+) PHN and control rats (Con), respectively.
Figure 7.
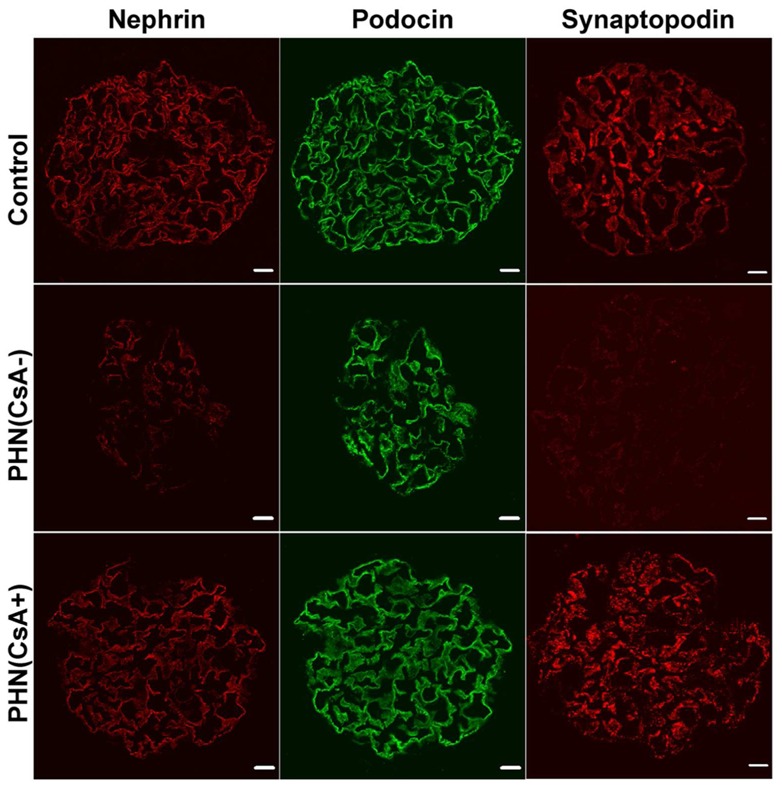
Confocal immunofluorescence labeling of nephrin, podocin and synaptopodin in kidney sections of CsA(-) and CsA(+) PHN rats (both at day 42 post-immunization) and control rats (Con). Scale bars, 20 μm.
Discussion
Primary MN defines podocyte disease in which alterations occur commonly in the absence of inflammatory lesions or cell infiltration. Immunofluorescence (IF) findings show typical immunoglobulin and complement deposits along the capillary loop. [31]. Although the pathogenesis remains incompletely elucidated, the underlying mechanism involves the generation of antibodies directed against intrinsic podocyte antigens and subsequent formation of subepithelial dense deposits leading to complement activation and glomerular injuries. [1,3]
In this study, we showed that: i) c-mip abundance is significantly increased in human MN; ii) c-mip protein is slightly and belatedly detectable after the first anti-megalin injection although immune complexes are fully formed; iii) c-mip abundance is strongly increased following a second immunization and precedes the occurrence of nephrotic-proteinuria; iv) stably overexpression of c-mip in podocytes reproduces several podocyte alterations such as those observed in human MN and PHN including downregulation of nephrin and synaptopodin; [17,32,25] v) c-mip induction in MN, PHN and Tg mice is associated with upregulation of DAPK and ILK, and vi) CsA inhibits the expression of c-mip and significantly reduces proteinuria, whereas the abundance of ILK and DAPK is reduced.
The time course of c-mip expression in PHN is closely related to that of proteinuria occurrence. Although the immune complex deposits were clearly formed in glomeruli of PHN rats receiving a single dose of anti-megalin, they did not develop abnormal proteinuria. Conversely, rats receiving a second injection of anti-megalin antibody develop proteinuria in parallel with an overproduction of c-mip. This finding is supported by our mouse model in which targeted expression of c-mip in podocytes induces nephrotic proteinuria. These observations suggest that podocyte damage leading to nephrotic proteinuria in PHN is associated with c-mip induction. It is interesting to note that c-mip is not induced in nephrotic syndromes associated with diabetic nephropathy, HIV-associated nephropathy, or IgA nephropathy, [25] suggesting that c-mip is specifically induced in some primary podocyte diseases of immune origin. Irrespective of the earliest aggression leading to MN, our results suggest that c-mip induction may act as a second, major event resulting in podocyte dysfunction.
We have previously shown that c-mip interacts with Fyn, a member of the Src kinase protein family, and prevents its interaction with nephrin, which results in the inhibition of nephrin phosphorylation. This mechanism may explain the downregulation of nephrin phosphorylation found both in minimal change nephrotic syndrome (MCNS) and in c-mip Tg mice. [33,25] Likewise, it has been reported that nephrin is also hypophosphorylated in MN. [34]
In MN and MCNS, the pattern of nephrin expression shifts from linear epithelial to granular interpodocyte filtration slits with changes in cytoskeleton organization, such as seen in c-mip Tg mice. [25] The downregulation of nephrin expression results at least in part from its shedding from the cell surface and its passage to the urinary space. [35]
According to previous reports, the onset of proteinuria in PHN does not coincide with the formation of immune complex deposits but with the complement activation-mediated podocyte damage. [14] The mechanism by which the membrane attack of complement (C5b-9) alters nephrin signaling and disorganizes the actin cytoskeleton has not been elucidated. Our results showing a clear correlation between c-mip abundance and the occurrence of nephrotic-range proteinuria, suggest that c-mip plays a pivotal role in MN, inasmuch that c-mip reproduces biochemical alterations in Tg mice similar to those observed in MN, in the absence of any complement or immunoglobulin deposits. [25]
Actin cytoskeleton is a highly dynamic structure that plays a central role in the normal morphology of podocytes and establishing a functional link with neighboring foot processes. We have shown that stable overexpression of c-mip in an inducible system (T Rex) alters the morphological characteristics of podocytes and downregulates synaptopodin expression. It has been well established that RhoA regulates contractility and assembly of actin stress fibers and focal adhesions. We show here that c-mip inhibits RhoA activity, in agreement with recent studies showing that synaptopodin regulates the stability of RhoA and modulates actin polymerization activity. [26]
Anti-proteinuric properties of CsA have been demonstrated both in patients with MN and in Heymann nephritis [36,37]. According, we show that CsA reduces proteinuria in PHN, in parallel with downregulation of c-mip and ILK. Although we cannot exclude that antiproteinuric effect could be due to its immunosuppressive action, we found that CsA restores nephrin and synaptopodin abundances in the podocytes. Because calcineurin promotes dephosphorylation of synaptopodin and its subsequent degradation, [38] the antiproteinuric effect involves at least in part the preservation of synaptopodin by CsA, a potent calcineurin inhibitor.
Overexpression of c-mip in vitro and in vivo is associated with an increased expression of ILK, a serine/threonine protein kinase involved in the cellular control of integrin-mediated cell–matrix interaction and cell phenotype regulation. ILK contributes to tightly control the interaction of the podocyte cytoskeleton with the matrix components of the glomerular basement membrane (GBM), which is crucial to maintain the complex capillary architecture against the highly transcapillary pressure gradient. Effacement of foot processes and podocyte retraction suggest that molecules at the interface between cytoskeleton and integrin matrix may play a crucial role in these processes. ILK expression is increased in situations associated with an effacement of foot processes, independently of the causal mechanisms, such as congenital nephrosis of Finnish type and adryamicin/puronomycin models of proteinuria. [39,28] This leads to consider that upregulation of ILK in response to podocyte damage might serve to stabilize the adhesion of unharmed podocytes to GBM, through multiple interactions with cytoskeletal partners. [40] This hypothesis is supported by the fact that ILK is an anti-apoptotic molecule, as opposed to c-mip, which exhibits proapoptotic properties. [41]
In conclusion, this work reveales that c-mip is overproduced in PHN and in human MN. Functional studies suggest that c-mip might play a crucial role in podocyte damage in MN. Overexpression of c-mip in vivo reproduces the biochemical alterations observed in Heymann nephritis as well as in MN, including increased expression ILK, DAPK, downregulation of synaptopodin and inhibition of RhoA activity and alteration of nephrin expression. Taken together, these results suggest that c-mip is a key player in podocyte diseases and may represent a “hot” therapeutic target.
Materials and Methods
Patients
The cohort of adult patients analyzed in this study has been described in a previous report. [25] Normal renal samples were supplied by the department of pathology, from patients undergoing nephrectomy for polar kidney tumor. PBMC were purified through a Ficoll/Hypaque density gradient (Eurobio, France). All experiments were conducted with approval from the INSERM (Institut National de la Santé et de la Recherche Médicale) research ethics committee in accordance to international ethics codes and guidelines. The informed consent was obtained from all participants involved in this study.
Light microscopy studies and immunohistochemical analyses
In situ hybridization (ISH) experiments using c-mip riboprobes have been previously described. [25] For immunohistochemistry analysis, kidney samples were fixed for 16 hours in Dubosq Brazil, and subsequently dehydrated and embedded in paraffin. Antigen retrieval was performed by immersing the slides in boiling 0.01 M citrate buffer in a 500 W microwave oven for 15 min. The endogenous peroxidase activity was blocked with 0,3% H2O2 in methanol for 30 min. Slides were incubated with the blocking reagents consisting of the Avidin–biotin solution for 30 min and the normal blocking serum for 20 min. For detection of c-mip, the slides were incubated overnight with a polyclonal antibody at a final concentration of 15 mg/ml, then with a biotinylated secondary antibody. An avidin-biotinylated horseradish peroxidase complex (Vectastain ABC Reagent, Vector Laboratories; Burlingame, CA) and 3,3′-diaminobenzidine (Sigma Biochemicals; St Louis, MO) as a chromogen were applied for visualization of the immunoreaction. Slides were counterstained with hematoxylin. Omission of the primary antibody was considered as a negative control.
Immunofluorescence labeling was carried out on 4-mm thick cryostat sections of kidney tissue fixed in acetone for 10 min, air-dried for 30 min at room temperature, then incubated in PBS for 3 min and blocked in 1% BSA in PBS. The sections were incubated with the indicated antibodies for 1 hour at room temperature, washed in PBS and incubated with Red Texas-conjugated secondary antibodies. Sections were examined by fluorescence microscopy (Zeiss).
Northern blots and stable transfections
Total RNA was prepared from glomerular fractions isolated by graded sieving, using Rnaeasy kit (Quiagen, France). Northern blots were performed as previously described (Sahali, 2002 #3714}, using human kidney samples isolated from normal tissues of total nephrectomy for polar tumors. Briefly, twenty μg of total RNA from kidney were loaded on each lane. The blot was hybridized with a [32P] dCTP-labeled 1200 bp cDNA c-mip probe, then with a 18S ribosomal RNA antisens olgonucleotide probe. Conditionally immortalized mouse podocyte cell line is a gift of Peter Mundel. [42] Generation of stably transfected podocytes was performed as previously reported. [25]
Immunoprecipitation and Western blotting
The primary antibodies used in this study include anti-ILK, anti-RhoA and anti-DAPK (Upstate Cell signaling, USA), anti-synaptopodin and anti-nephrin (Progen Biotechnik GmbH, Germany), anti-actin (Sigma, Aldrich, France), anti-sheep-IgG (Santa Cruz, Biotechnology, Inc, USA). The anti-podocin antibody was a gift from Dr Corinne Antignac (Inserm U983, Necker Hospital, France). The anti-c-mip polyclonal antibody was produced in rabbits immunized with acrylamide gel sections containing the c-mip protein. Immunoprecipitation and Western blotting were performed as previously reported. [25]
RhoA and Cdc42 activation assays
Activated RhoA and Cdc42 pull-down kits were purchased from Cytoskeleton (Cytoskeleton, Inc., Denver, USA) and the assays performed following supplied protocols. Briefly, stably transfected podocytes were incubated without or with tetracycline (1 μg/ml) for 48 hours as previously described, then cells were lysed for protein extraction. The protein concentration of lysates was determined by Bradford reaction assay. Equal amount of protein lysates were incubated with Rhotekin-RBD (RhoA assay) or PAK-PBD (Cdc42 assay) affinity beads for 1 hour at 4°C, followed by two washes in lysis buffer and three washes in the supplied wash buffer. Bound proteins were eluted in 1x Laemmli buffer and examined by 15% SDS-PAGE and western blot analysis.
Induction of passive Heymann Nephritis
PHN was induced in male Lewis rats weighting 100 g by intravenous injection of sheep anti-rat megalin antiserum (kindly provided by Pr Pierre Verroust, INSERM U538, St Antoine Hospital). Two injections at fifteen day’s-interval were performed. Twenty rats were injected with anti-megalin antiserum and five control rats received equal volume of normal sheep serum. Individual rats were housed in metabolic cages (Techniplast, France). Urines were collected after 24 hours and this was repeated five times for each individual. Proteinuria was performed using appropriate kits from Advia Chemistry 1650 (Bayer Healthcare AG, Leverkusen, Germany). Rats were sacrificed between 15 and 28 days following the second injection.
Cyclosporine (CsA) treatment
At day 21 post-first immunization, PHN rats with well established nephrotic proteinuria were separated into two groups (n=5 each). The first one (CsA group) was treated with CsA (SandimmumR, Novartis) by daily intraperitoneal injection of CsA (20 mg/kg body weight) until proteinuria decreased significantly (Day 42). The second group (control) received intraperitoneal injection of an equal volume of sodium chloride solution (NaCl 0,9%). At day 42, rats were sacrificed and kidneys were processed for analysis.
Laser capture microdissection, reverse transcription and real time quantitative PCR (RT-qPCR)
Serial sections (25 μm-thick rat frozen tissue specimens) were placed on membrane slides (Carl Zeiss MicroImaging). Cresyl violet staining was performed by using a protocol from Zeiss Labs, Munich, Germany. [43] Glomerular structures were selectively dissected using the PALM MicroBeam system (Carl Zeiss). Each rat kidney sample was entirely microdissected, giving around 500 glomeruli from which RNAs were extracted in triplicate (about 170 glomeruli/tube) with PicoPure™ RNA Isolation Kit (Arcturus, Applied Biosystem, USA)). For human kidney biopsy specimens from patients with membranous nephropathy, glomeruli were isolated as previously described. [44] A representative figure of human and rat glomeruli before and after laser microdissection is shown in supplementary material (Figure S2). Reverse transcription (RT) was performed with Maxima First Strand cDNA synthesis Kit (Fermentas). Real time quantitative polymerase chain reaction (qPCR) was performed using the oligonucleotides listed in Table 1. Samples (2 μl of the RT reaction mixture, corresponding to 10 ng of equivalent total RNA) were amplified in a 20 μl reaction mixture containing 0.5 mM levels of each primer and 1 X Quantitect Sybr Green PCR mix (QIAGEN GmbH, Hilden, Germany). PCR was performed in a Light Cycler 480 apparatus (Roche Diagnostic). The c-mip primers amplified a 176-bp sequence (Table 1). A standard curve was generated for qPCR by using serial dilutions of linearized pdonr221-c-mip plasmid. The copy number was calculated as described (http://www.uri.edu/research/gsc/resources/cndna.html). PCR was initiated by denaturation at 95°C for 15 min, followed by 32 three-step cycles (95°C for 10 s, 63°C for 30 s, and 72°C for 30 s). A dissociation run (95°C for 5 sec followed by 65°C for one min) was performed at the end of PCR program allowing the generation of melting curve. The relative value for each sample was calculated using LightCycler analysis software. All PCR data were normalized to 18S rRNA expression, to control for variations in RT reactions.
Table 1.
Sequence of primers and PCR conditions.
| Primers | Sequence | Accession number | Expected size | Ann Temp (°C) | PCR cycles |
|---|---|---|---|---|---|
| Rat c-mip | |||||
| Forward: TGTGTGCCTGGCTGCCATATATTCCTGCTATG | NM_001163273.1 | 176 | 64 | 32 | |
| Reverse: GACAATGTGGCTTCCTGAGACACCAGGTC | |||||
| 18S | |||||
| Forward: GTAACCCGTTGAACCCCATT | NR_003278 | 151 | 60 | 16 | |
| Reverse: CCATCCAATCGGTAGTAGCG | |||||
| Human c-mip | Forward: CGTGTGCCTGGCTGCCATCTACTCCTGCTATG | NM_198390 | 176 | 68 | 32 |
| Reverse: GACAGCGTGGCTTCCTGAGACACCAGGTC | |||||
| Human DAPK | Forward: CTCAGGCGCATTGCTCAGCAGCTC | 248 | |||
| Reverse: GATGTCCATGGCATCGAGGATCTGC | NM_004938 | ||||
| Human ILK | Forward: GCAACGCAGTCGCCGTTCGCCT | ||||
| Forward: CACGGTGTCCATGACTGGCTGCCA | NM_001014795 | 210 | 58 | 35 |
The PCR conditions for ILK and DAPK are shown in Table 1.
Generation of c-mip transgenic mice
The generation of c-mip transgenic mice has been previously described. [25] All experiments involving animals were conducted in accordance with French laws.
Statistics
The data presented are means ± SD and were prepared with GraphPad Prism software, version 4.0. Mann-Whitney test was used to evaluate the significance of differences. Values of P < 0.05 were considered significant.
Supplementary Material
Acknowledgments
This work was supported in part by an Avenir Program from INSERM, a grant from the French Kidney Foundation and Association pour l’Utilisation du Rein Artificiel (AURA).
We are grateful to Dr Peter Mundel for providing us with the mouse podocyte cell line and to Dr Corinne Antignac (INSERM U574, Hôpital Necker Enfant-Malades, Paris, France) for the gift of the anti-podocin antibody. We thank Dr Esmilaire Liliane (Biochemistry department, Henri Mondor Hospital) for biochemical dosages. We thank Dr Yves Allory (Pathology department) for providing us with renal samples. This work was supported in part by an Avenir Program from INSERM, a grant from the French Kidney Foundation and Association pour l’Utilisation du Rein Artificiel (AURA). V Audard is a recipient of a poste d’accueil INSERM. K Sendeyo was supported by grants of Ministère de la recherche et de la Technologie (MRT).
Footnotes
The authors have declared that no conflict of interest exists
References
- 1.Ronco P, Debiec H. Molecular pathomechanisms of membranous nephropathy: from Heymann nephritis to alloimmunization. J Am Soc Nephrol. 2005;16:1205–1213. doi: 10.1681/ASN.2004121080. [DOI] [PubMed] [Google Scholar]
- 2.Imai H, Hamai K, Komatsuda A, et al. IgG subclasses in patients with membranoproliferative glomerulonephritis, membranous nephropathy, and lupus nephritis. Kidney Int. 1997;51:270–276. doi: 10.1038/ki.1997.32. [DOI] [PubMed] [Google Scholar]
- 3.Beck LH, Jr, Bonegio RG, Lambeau G, et al. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med. 2009;361:11–21. doi: 10.1056/NEJMoa0810457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prunotto M, Carnevali ML, Candiano G, et al. Autoimmunity in membranous nephropathy targets aldose reductase and SOD2. J Am Soc Nephrol. 21:507–519. doi: 10.1681/ASN.2008121259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Glassock RJ. Diagnosis and natural course of membranous nephropathy. Semin Nephrol. 2003;23:324–332. doi: 10.1016/s0270-9295(03)00049-4. [DOI] [PubMed] [Google Scholar]
- 6.Heymann W, Hackel DB, Harwood S, et al. Production of nephrotic syndrome in rats by Freund’s adjuvants and rat kidney suspensions. Proc Soc Exp Biol Med. 1959;100:660–664. doi: 10.3181/00379727-100-24736. [DOI] [PubMed] [Google Scholar]
- 7.Ronco P, Debiec H. Target antigens and nephritogenic antibodies in membranous nephropathy: of rats and men. Semin Immunopathol. 2007 doi: 10.1007/s00281-007-0091-2. [DOI] [PubMed] [Google Scholar]
- 8.Kerjaschki D, Farquhar MG. The pathogenic antigen of Heymann nephritis is a membrane glycoprotein of the renal proximal tubule brush border. Proc Natl Acad Sci U S A. 1982;79:5557–5561. doi: 10.1073/pnas.79.18.5557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kerjaschki D, Farquhar MG. Immunocytochemical localization of the Heymann nephritis antigen (GP330) in glomerular epithelial cells of normal Lewis rats. J Exp Med. 1983;157:667–686. doi: 10.1084/jem.157.2.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Debiec H, Guigonis V, Mougenot B, et al. Antenatal membranous glomerulonephritis due to anti-neutral endopeptidase antibodies. N Engl J Med. 2002;346:2053–2060. doi: 10.1056/NEJMoa012895. [DOI] [PubMed] [Google Scholar]
- 11.Debiec H, Nauta J, Coulet F, et al. Role of truncating mutations in MME gene in fetomaternal alloimmunisation and antenatal glomerulopathies. Lancet. 2004;364:1252–1259. doi: 10.1016/S0140-6736(04)17142-0. [DOI] [PubMed] [Google Scholar]
- 12.Salant DJ, Belok S, Madaio MP, et al. A new role for complement in experimental membranous nephropathy in rats. J Clin Invest. 1980;66:1339–1350. doi: 10.1172/JCI109987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yuan H, Takeuchi E, Taylor GA, et al. Nephrin dissociates from actin, and its expression is reduced in early experimental membranous nephropathy. J Am Soc Nephrol. 2002;13:946–956. doi: 10.1681/ASN.V134946. [DOI] [PubMed] [Google Scholar]
- 14.Saran AM, Yuan H, Takeuchi E, et al. Complement mediates nephrin redistribution and actin dissociation in experimental membranous nephropathy. Kidney Int. 2003;64:2072–2078. doi: 10.1046/j.1523-1755.2003.00305.x. [DOI] [PubMed] [Google Scholar]
- 15.Nakatsue T, Koike H, Han GD, et al. Nephrin and podocin dissociate at the onset of proteinuria in experimental membranous nephropathy. Kidney Int. 2005;67:2239–2253. doi: 10.1111/j.1523-1755.2005.00328.x. [DOI] [PubMed] [Google Scholar]
- 16.Reiser J, Polu KR, Moller CC, et al. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat Genet. 2005;37:739–744. doi: 10.1038/ng1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doublier S, Ruotsalainen V, Salvidio G, et al. Nephrin redistribution on podocytes is a potential mechanism for proteinuria in patients with primary acquired nephrotic syndrome. Am J Pathol. 2001;158:1723–1731. doi: 10.1016/S0002-9440(10)64128-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moller CC, Wei C, Altintas MM, et al. Induction of TRPC6 channel in acquired forms of proteinuric kidney disease. J Am Soc Nephrol. 2007;18:29–36. doi: 10.1681/ASN.2006091010. [DOI] [PubMed] [Google Scholar]
- 19.Cunningham PN, Quigg RJ. Contrasting roles of complement activation and its regulation in membranous nephropathy. J Am Soc Nephrol. 2005;16:1214–1222. doi: 10.1681/ASN.2005010096. [DOI] [PubMed] [Google Scholar]
- 20.Couser WG. Pathogenesis of glomerular damage in glomerulonephritis. Nephrol Dial Transplant. 1998;13:10–15. doi: 10.1093/ndt/13.suppl_1.10. [DOI] [PubMed] [Google Scholar]
- 21.Leenaerts PL, Hall BM, Van Damme BJ, et al. Active Heymann nephritis in complement component C6 deficient rats. Kidney Int. 1995;47:1604–1614. doi: 10.1038/ki.1995.224. [DOI] [PubMed] [Google Scholar]
- 22.Spicer ST, Tran GT, Killingsworth MC, et al. Induction of passive Heymann nephritis in complement component 6-deficient PVG rats. J Immunol. 2007;179:172–178. doi: 10.4049/jimmunol.179.1.172. [DOI] [PubMed] [Google Scholar]
- 23.Ronco P, Debiec H. Antigen identification in membranous nephropathy moves toward targeted monitoring and new therapy. J Am Soc Nephrol. 2010;21:564–569. doi: 10.1681/ASN.2009121220. [DOI] [PubMed] [Google Scholar]
- 24.Sahali D, Pawlak A, Valanciute A, et al. A novel approach to investigation of the pathogenesis of active minimal- change nephrotic syndrome using subtracted cDNA library screening. J Am Soc Nephrol. 2002;13:1238–1247. doi: 10.1681/ASN.V1351238. [DOI] [PubMed] [Google Scholar]
- 25.Zhang SY, Kamal M, Dahan K, et al. c-mip impairs podocyte proximal signaling and induces heavy proteinuria. Sci Signal. 2010;3:ra39. doi: 10.1126/scisignal.2000678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Asanuma K, Yanagida-Asanuma E, Faul C, et al. Synaptopodin orchestrates actin organization and cell motility via regulation of RhoA signalling. Nat Cell Biol. 2006;8:485–491. doi: 10.1038/ncb1400. [DOI] [PubMed] [Google Scholar]
- 27.Hannigan GE, Leung-Hagesteijn C, Fitz-Gibbon L, et al. Regulation of cell adhesion and anchorage-dependent growth by a new beta 1-integrin-linked protein kinase. Nature. 1996;379:91–96. doi: 10.1038/379091a0. [DOI] [PubMed] [Google Scholar]
- 28.de Teixeira VP, Blattner SM, Li M, et al. Functional consequences of integrin-linked kinase activation in podocyte damage. Kidney Int. 2005;67:514–523. doi: 10.1111/j.1523-1755.2005.67108.x. [DOI] [PubMed] [Google Scholar]
- 29.Puthalakath H, Huang DC, O’Reilly LA, et al. The proapoptotic activity of the Bcl-2 family member Bim is regulated by interaction with the dynein motor complex. Mol Cell. 1999;3:287–296. doi: 10.1016/s1097-2765(00)80456-6. [DOI] [PubMed] [Google Scholar]
- 30.Wang WJ, Kuo JC, Yao CC, et al. DAP-kinase induces apoptosis by suppressing integrin activity and disrupting matrix survival signals. J Cell Biol. 2002;159:169–179. doi: 10.1083/jcb.200204050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wiggins RC. The spectrum of podocytopathies: a unifying view of glomerular diseases. Kidney Int. 2007;71:1205–1214. doi: 10.1038/sj.ki.5002222. [DOI] [PubMed] [Google Scholar]
- 32.Honkanen E, von Willebrand E, Koskinen P, et al. Decreased expression of vascular endothelial growth factor in idiopathic membranous glomerulonephritis: relationships to clinical course. Am J Kidney Dis. 2003;42:1139–1148. doi: 10.1053/j.ajkd.2003.08.014. [DOI] [PubMed] [Google Scholar]
- 33.Uchida K, Suzuki K, Iwamoto M, et al. Decreased tyrosine phosphorylation of nephrin in rat and human nephrosis. Kidney Int. 2008 doi: 10.1038/ki.2008.19. [DOI] [PubMed] [Google Scholar]
- 34.Ohashi T, Uchida K, Asamiya Y, et al. Phosphorylation status of nephrin in human membranous nephropathy. Clin Exp Nephrol. 14:51–55. doi: 10.1007/s10157-009-0241-z. [DOI] [PubMed] [Google Scholar]
- 35.Yu D, Petermann A, Kunter U, et al. Urinary podocyte loss is a more specific marker of ongoing glomerular damage than proteinuria. J Am Soc Nephrol. 2005;16:1733–1741. doi: 10.1681/ASN.2005020159. [DOI] [PubMed] [Google Scholar]
- 36.Ambalavanan S, Fauvel JP, Sibley RK, et al. Mechanism of the antiproteinuric effect of cyclosporine in membranous nephropathy. J Am Soc Nephrol. 1996;7:290–298. doi: 10.1681/ASN.V72290. [DOI] [PubMed] [Google Scholar]
- 37.Blume C, Heise G, Hess A, et al. Different effect of cyclosporine A and mycophenolate mofetil on passive Heymann nephritis in the rat. Nephron Exp Nephrol. 2005;100:e104–112. doi: 10.1159/000085029. [DOI] [PubMed] [Google Scholar]
- 38.Faul C, Donnelly M, Merscher-Gomez S, et al. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat Med. 2008;14:931–938. doi: 10.1038/nm.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kretzler M, Teixeira VP, Unschuld PG, et al. Integrin-linked kinase as a candidate downstream effector in proteinuria. Faseb J. 2001;15:1843–1845. doi: 10.1096/fj.00-0832fje. [DOI] [PubMed] [Google Scholar]
- 40.Wu C, Dedhar S. Integrin-linked kinase (ILK) and its interactors: a new paradigm for the coupling of extracellular matrix to actin cytoskeleton and signaling complexes. J Cell Biol. 2001;155:505–510. doi: 10.1083/jcb.200108077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ory V, Fan Q, Hamdaoui N, Zhang SY, Desvaux D, Audard V, Candelier M, Noel LH, Lang P, Guellaen G, Pawlak A, Sahali D. c-mip Down-Regulates NF-kappaB Activity and Promotes Apoptosis in Podocytes. Am J Pathol. 2012;180:2284–2292. doi: 10.1016/j.ajpath.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 42.Mundel P, Reiser J, Zuniga Mejia Borja A, et al. Rearrangements of the cytoskeleton and cell contacts induce process formation during differentiation of conditionally immortalized mouse podocyte cell lines. Exp Cell Res. 1997;236:248–258. doi: 10.1006/excr.1997.3739. [DOI] [PubMed] [Google Scholar]
- 43.Aaltonen KE, Ebbesson A, Wigerup C, et al. Laser capture microdissection (LCM) and whole genome amplification (WGA) of DNA from normal breast tissue --- optimization for genome wide array analyses. BMC Res Notes. 2011;4:69. doi: 10.1186/1756-0500-4-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Audard V, Zhang SY, Copie-Bergman C, et al. Occurrence of minimal change nephrotic syndrome in classical Hodgkin lymphoma is closely related to the induction of c-mip in Hodgkin-Reed Sternberg cells and podocytes. Blood. 2010;115:3756–3762. doi: 10.1182/blood-2009-11-251132. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.