

Natural products have gained renewed interests with the realization that the genetic capacity of microorganisms to produce these molecules is much larger than anticipated. Indeed, genome mining exercises have resulted in the discovery of a variety of new compounds.[1] Whereas the sequenced bacterial and fungal genomes demonstrate the untapped potential of natural products, connecting the biosynthetic genes in genomes to novel compounds is still a challenge. One aspect of this challenge is the difficulty of purifying these molecules from complex spent media. Recently, chemoselective derivatization approaches have been developed to aid in the purification process.[2] Here we illustrate the use of an enzymatic approach to rapidly enrich a phosphonate from the spent medium of Streptomyces sp. WM6372, a strain previously shown to encode an uncharacterized phosphonate biosynthetic gene cluster. Isolation and structure elucidation identified the molecule as methyl 1-hydroxyphosphonoacetic acid (Me-HPnA). Realization that Me-HPnA is present in fosfazinomycin prompted a targeted search after growth of the strain in various media. We also examined Streptomyces XY332, which was previously shown to encode the same phosphonate biosynthetic genes. Both strains produced fosfazinomycin, and heterologous expression of the genes in S. lividans led to production of related phosphonic acids, providing strong support for linking the gene cluster to fosfazinomycin production. Analysis of the genes provides insights into the biosynthetic pathway of hydrazine formation in nature.
Phosphonate natural products form a particular challenge in regards to purification as a consequence of their high polarity and water solubility. These properties may explain why only about 30 phosphonate natural products have been isolated despite the observation that about 5% of randomly isolated actinomycetes contain the genetic capability to produce phosphonates.[3] The phosphonate O-methyltransferase DhpI from the dehydrophos biosynthetic pathway is able to non-specifically methylate other phosphonates such as fosfomycin and fosmidomycin under defined reaction conditions.[4] To determine whether DhpI could methylate these compounds in complex medium, fosfomycin and fosmidomycin were added to international streptomyces project 4 (ISP4) medium. Indeed, when supplied with S-adenosyl methionine (SAM), DhpI methylated both compounds as determined by 31P NMR spectroscopy (Figures 1A and S1, S2 and Tables S1 and S2). The broad substrate specificity and robust activity of DhpI prompted the development of a method that relies on stable isotope labeling of phosphonates in extract (SILPE). The method uses the facilitated identification of pairs of compounds of different stable-isotope composition by mass spectrometry based on their well-defined mass difference, similar to the SILAC method in proteomics.[5] Accordingly, ISP4 media spiked with fosfomycin or fosmidomycin were treated with DhpI and a mixture of SAM and CD3-SAM. The crude material was injected into an LCMS system and the eluent analyzed for compounds giving rise to two ions separated by 3.0188 Da, allowing the facile detection of methylated fosfomycin and fosmidomycin (Figures 1B, S1–S3).
Figure 1.
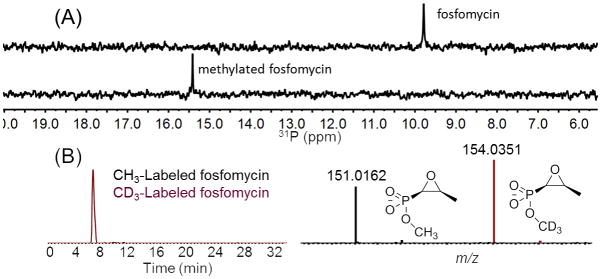
(A) 31P NMR spectrum of fosfomycin (top) and after treatment with DhpI and a mixture of SAM and d3-SAM (1:1) (bottom). (B) LC-MS analysis of the sample in the bottom spectrum of panel A.
We then turned our attention to unknown phosphonates produced by Streptomyces sp. WM6372. This strain was previously identified as a potential phosphonate producer by screening for the presence of the phosphoenolpyruvate mutase gene (pepM), a characteristic marker for phosphonate biosynthesis.[3, 6] Spent medium from this strain grown on ISP2 agar plates displayed two resonances in the phosphonate window of the 31P NMR spectrum (Figure 2A). After several unsuccessful attempts to isolate the compounds using traditional purification methods, we attempted the SILPE method. The solid medium was freeze-thawed and compressed to release metabolites, evaporated to dryness, and taken up in 90% MeOH. Insoluble material was removed by centrifugation. The supernatant was concentrated and partially desalted by Sephadex LH20. The resulting crude mixture was treated with DhpI and a mixture of SAM and CD3-SAM. The two original phosphonate peaks in the 31P NMR spectrum disappeared and two new peaks were generated (Figure 2B and Table S1). Analysis by LCMS using the mass difference between compounds containing a CH3 or a CD3 group as marker allowed the purification of both molecules and determination of their masses. Characterization by 1H and 13C NMR spectroscopy as well as 1H-31P-HMBC and mass spectrometry (Figure S4) allowed assignment of the parent compounds before DhpI-treatment as methyl phosphonoacetate (1) and methyl 1-hydroxyphosphono-acetate (Me-HPnA, 2; Figure 2C), which was confirmed by chemical synthesis (Figure S5). A structure search revealed that these molecules are new phosphonate natural products; however, Me-HPnA is also a substructure in fosfazinomycins (Figure 2C). We therefore cultivated Streptomyces sp. WM6372 in a variety of media and monitored for fosfazinomycin production by LCMS. In most media, production of fosfazinomycins was not observed; however, the strain produces small amounts of both fosfazinomycin A and B after growth in R2AS medium containing phosphonoacetate. Therefore, the organism has all the genes needed to produce fosfazinomycin, although it appears that unknown regulatory or metabolic limitations prevent its de novo synthesis under the conditions examined.
Figure 2.
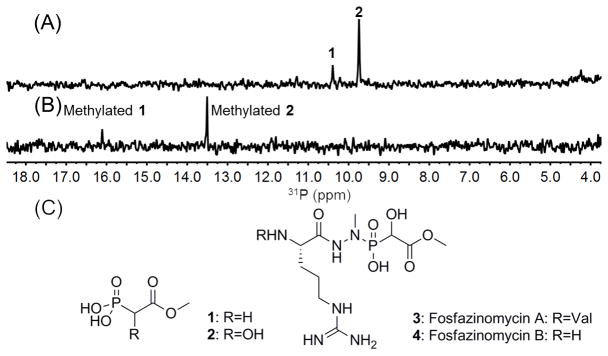
(A) 31P NMR spectrum of spent medium of Streptomyces sp. WM 6372. Only the chemical shift region of interest with respect to phosphonates is depicted. (B) 31P NMR spectrum after treatment with DhpI and SAM. (C) Structures of the phosphonates giving rise to the signals in panel A and the structures of fosfazinomycin A and B.
We previously reported that Streptomyces sp WM6372 and Streptomyces sp. XY332 encode nearly identical phosphonate biosynthetic gene clusters (Figure S6).[6b] Therefore, we tested whether Streptomyces sp. XY332 could produce fosfazinomycins. Evaluation of various growth conditions and analysis by LCMS demonstrated that two compounds were produced with masses consistent with fosfazinomycin A and B. A 31P NMR spectrum of the spent medium also revealed two small new signals with close chemical shifts at 13.7 and 13.5 ppm in addition to the peaks for compounds 1 and 2 (Figure 3A). To provide confirmation that the new peaks represented fosfazinomycin A and B, the compounds were partially purified. A 1H-31P HMBC NMR spectrum displayed correlations consistent with the fosfazinomycin core structure, and MS analysis showed molecular ions consistent with fosfazinomycin A and B (Figure S7). Finally, Streptomyces XY332 was grown on minimal medium containing 15NH4SO4 as sole nitrogen source, and the 31P NMR spectrum was recorded. The two peaks in the 31P spectrum of the spent medium now displayed a doublet splitting pattern with a coupling constant of 11.4 Hz (Figure 3B), and the high resolution mass spectrum indicated the presence of seven 15N atoms, consistent with the assignment to fosfazinomycin A (Figure S8).
Figure 3.
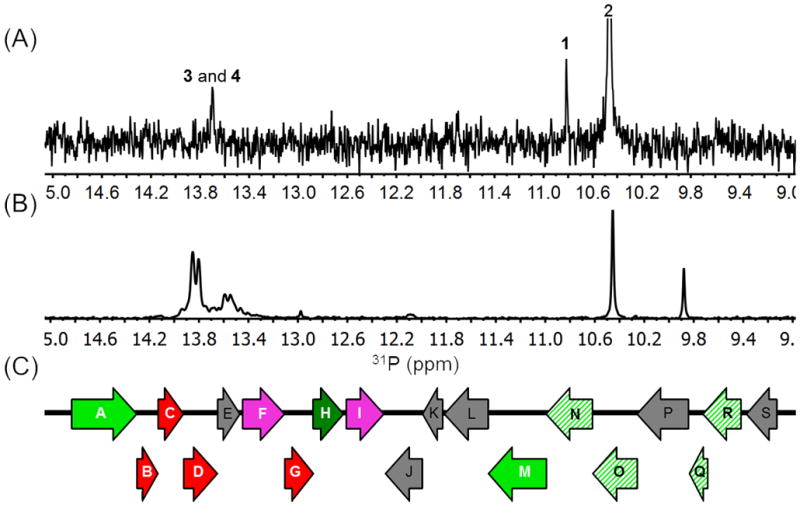
(A) 31P NMR spectrum of spent medium of Streptomyces sp. XY332. (B) 31P NMR spectrum of spent medium of the same strain when grown on 15NH4SO4. The chemical shift variation is the result of small changes in pH as these compounds have pKa values near 7. (C) DNA fragment of the genome from Streptomyces sp. WM 6372 that contains the pepM gene. For color coding, see Figure S6.
Two lines of evidence provide strong support to link the gene cluster shown in Figure 3C with the biosynthesis of fosfazinomycin. First, both organisms contain this gene cluster and both can produce the antibiotic. Moreover, examination of draft genome sequences shows that these clusters are the only PEP mutase-encoding loci in these organisms; PEP mutase is involved in the biosyntheses of all phosphonates that are currently understood. Furthermore, we transferred the cluster to S. lividans, a non-phosphonate producer, and showed that the recombinant strain produced 1 and 2. Thus, the transferred gene cluster clearly directs synthesis of the phosphonate moiety of fosfazinomycin. We suspect that as with the parental strain, regulatory or metabolic limitations prevent de novo synthesis of the final product in S. lividans. Given that all characterized phosphonate biosynthetic pathways require PEP mutase and that the gene cluster in question directs the synthesis of the phosphonate moiety of fosfazinomycin, we believe that it is highly likely, albeit not definitive in the absence of successful heterologous production, that these loci are responsible for fosfazinomycin synthesis.
Fosfazinomycin A and B were first isolated from Streptomyces lavendofoliae 630 as part of a screening program focused on antifungal antibiotics.[7] The two peptide congeners differ at their N-termini (Figure 2C). Fosfazinomycins contain a unique hydrazide linkage between the carboxylic acid of Arg and the phosphonate of Me-HPnA. Nitrogen-nitrogen bonds are relatively rare in nature[8] and the mechanism of their formation is generally still poorly understood.[9] Based on bioinformatics analysis of the putative biosynthetic gene clusters of Streptomyces sp. WM6372 and Streptomyces sp. XY332 (Figures 3C, S6, and Table 1), a putative biosynthetic pathway for fosfazinomycin biosynthesis can be formulated (Figure 4). FzmC is the PEP mutase that installs the C-P bond in phosphonopyruate (PnPy) and FzmD decarboxylates PnPy to generate phosphonoacetaldehyde (PnAA). This intermediate is then proposed to be oxidized to phosphonoacetate (PnA) and methylated to generate 1, one of the observed metabolites. The cluster does not contain a canonical aldehyde dehydrogenase suggesting that a housekeeping enzyme may be used for the conversion of PnAA to PnA, but methylation of the carboxylate is likely catalyzed by the S-adenosylmethionine dependent O- methyltransferase FzmB. FzmG is a likely candidate for the hydroxylation of 1 to produce 2 because its closest homolog is the α-ketoglutarate dependent oxygenase that hydroxylates 2-hydroxyethylphosphonate during dehydrophos biosynthesis.[10] To complete the biosynthesis of fosfazinomycin A, three bonds need to be made between nitrogen nucleophiles and acid electrophiles. Interestingly, the cluster encodes proteins from three different amidoligase families:[11] FzmF, a member of the ATP GRASP enzyme family,[11–12] FzmI, a member of the GCN5-related N-acetyltransferase (GNAT) enzymes that function as aminoacyl tRNA-dependent peptidyl transferases,[11, 13] and FzmN, a member of the glutamine synthase family that is known to catalyze amide bond formation.[11, 14] Further studies are required to determine which of these three enzymes makes which of the bonds colored magenta in Figure 4.
Table 1.
Summary of open reading frames (ORF) in the genomic DNA of Streptomyces sp. WM 6372 that includes the fosfazinomycin biosynthetic gene cluster.
| ORF | No. of residues | Highest identity homolog (as of September 1, 2013) | Amino acid identity/ similarity (%) |
|---|---|---|---|
| FzmA | 727 | Actinoplanes sp. SE50/110 asparagine synthase (YP_006269188.1) | 48/64 |
| FzmB | 233 | Haliangium ochraceum DSM 14365 type 11 methyltransferase (YP_003269122.1) | 48/63 |
| FzmC | 284 | Bacillus cereus VD102 PEP phosphomutase (WP_000571606.1) | 52/69 |
| FzmD | 378 | Micromonospora sp. ATCC 39149 PnPy decarboxylase (WP_007071820.1) | 52/64 |
| FzmE | 253 | Streptomyces ambofaciens ATCC 23877 phosphodiesterase (CAJ89340.1) | 55/66 |
| FzmF | 465 | Methanococcoides burtonii DSM 6242 pyruvate carboxylase (YP_567027.1); ATP-GRASP family. | 29/45 |
| FzmG | 317 | Streptomyces luridus dioxygenase (ACZ13452.1) | 45/55 |
| FzmH | 349 | Nonomuraea sp. WU8817 N-methyl transferase (ACS83764.1) | 35/46 |
| FzmI | 426 | Streptomyces rapamycinicus N-acyltransferase (CAA60475.1) | 31/43 |
| FzmJ | 411 | Corynebacterium matruchoti MFS transporter (WP_005523355.1) | 16/30 |
| FzmK | 231 | Desulfovibrio hydrothermalis DSM 14728 thymidylate kinase (YP_007326724.1) | 18/35 |
| FzmL | 492 | Streptomyces bingchenggensis BCW-1 lyase (YP_004961426.1) | 62/70 |
| FzmM | 655 | Streptomyces davawensis JCM 4913 FAD(NAD)-dependent oxidoreductase (YP_007524522.1) | 56/65 |
| FzmN | 519 | uncultured bacterium BAC AB649/1850 glutamine synthetase (AEE65491.1) | 56/67 |
| FzmO | 500 | Salinispora tropica CNB-440 amidase (YP_001159034.1) | 52/61 |
| FzmP | 575 | Staphylococcus epidermidis hypothetical protein (WP_002477807.1) | 24/41 |
| FzmQ | 134 | Streptomyces sp. N-acetyltransferase (WP_008740585.1) | 76/85 |
| FzmR | 435 | Streptomyces ambofaciens ATCC 23877 adenylosuccinate lyase (CAI78075.1) | 64/76 |
| FzmS | 341 | Streptomyces violaceusniger Tu 4113 transcriptional regulator (YP_004814876.1) | 45/57 |
Figure 4.

Proposed biosynthetic pathway of fosfazinomycin A. Putative steps are indicated with dashed arrows. FzmFIN are proposed to be involved in formation of the three magenta bonds, whereas the FzmAMNOQR proteins are proposed to be involved in forming and installing the hydrazine core.
The cluster also provides information on the possible construction of the hydrazide core. The methyl group on the hydrazide is probably installed by the N-methyltransferase FzmH, whereas one or both of the two nitrogen atoms may originate from asparagine/aspartate by the actions of the Asn synthase FzmA and the adenylosuccinate lyase FzmR; the latter delivers a nitrogen atom during de novo purine biosynthesis.[15] Formation of the N-N bond is likely facilitated by initial oxidation of an amine/amido group to the corresponding hydroxylamine/hydroxamate derivative by the flavin dependent oxidoreductase FzmM based on similar activation steps in the N-N bond forming processes during the biosynthesis of valanimycin[9a] and the kutznerides.[9b] Homologs of FzmNOQR (striped green, Figure 3C) are also found in the biosynthetic clusters of kinamycins[16] and lomaiviticin,[17] which contain N-N bonds in diazo groups. The conservation of the Gln synthetase FzmN suggests that the hydrazide group may be assembled on the side-chain of glutamate by these enzymes and that the amidase FzmO may release the hydrazide group. Similar use of glutamate as a molecular scaffold has been reported previously for various processes (Figure S9).[14]
In summary, we used the substrate tolerance of the phosphonate methyltransferase DhpI as a chemoselective tool to purify two phosphonate metabolites that are intermediates in the biosynthesis of fosfazinomycin. Bioinformatics analysis and heterologous production experiments provide support for the involvement of two nearly identical gene clusters in fosfazinomycin biosynthesis, providing insights into how the N-N bond might be fashioned and showing the unusual combination of an ATP-GRASP ligase, a tRNA-dependent peptidyl transferase, and a glutamine synthetase in the biosynthesis of a natural product.
Supplementary Material
Footnotes
This work was supported by the National Institutes of Health (GM PO1 GM077596 to W.W.M. and W.A.V.) NMR spectra were recorded on a 600-MHz instrument purchased with support from NIH Grant S10 RR028833.
Supporting information for this article is available on the WWW under http://www.angewandte.org.
Contributor Information
Dr. Jiangtao Gao, Institute for Genomic Biology, University of Illinois at Urbana-Champaign, 1206 West, Gregory Drive, Urbana, Illinois 61801, USA
Dr. Kou-San Ju, Institute for Genomic Biology, University of Illinois at Urbana-Champaign, 1206 West, Gregory Drive, Urbana, Illinois 61801, USA
Xiaomin Yu, Department of Microbiology, 601 South Goodwin Avenue, Urbana, Illinois 61801, USA.
Dr. Juan E. Velásquez, Howard Hughes Medical Institute and Department of Chemistry, University of Illinois at Urbana-Champaign, 600, South Mathews Avenue, Urbana, Illinois 61801, USA
Subha Mukherjee, Howard Hughes Medical Institute and Department of Chemistry, University of Illinois at Urbana-Champaign, 600, South Mathews Avenue, Urbana, Illinois 61801, USA.
Dr. Jaeheon Lee, Institute for Genomic Biology, University of Illinois at Urbana-Champaign, 1206 West, Gregory Drive, Urbana, Illinois 61801, USA
Dr. Changming Zhao, Institute for Genomic Biology, University of Illinois at Urbana-Champaign, 1206 West, Gregory Drive, Urbana, Illinois 61801, USA
Dr. Bradley S. Evans, Institute for Genomic Biology, University of Illinois at Urbana-Champaign, 1206 West, Gregory Drive, Urbana, Illinois 61801, USA
Dr. James R. Doroghazi, Institute for Genomic Biology, University of Illinois at Urbana-Champaign, 1206 West, Gregory Drive, Urbana, Illinois 61801, USA
Prof. William W. Metcalf, Email: metcalf@illinois.edu, Department of Microbiology, 601 South Goodwin Avenue, Urbana, Illinois 61801, USA
Prof. Wilfred A. van der Donk, Email: vddonk@illinois.edu, Howard Hughes Medical Institute and Department of Chemistry, University of Illinois at Urbana-Champaign, 600, South Mathews Avenue, Urbana, Illinois 61801, USA
References
- 1.a) Bode HB, Müller R. Angew Chem, Int Ed Engl. 2005;44:6828–6846. doi: 10.1002/anie.200501080. [DOI] [PubMed] [Google Scholar]; b) Challis GL. J Med Chem. 2008;51:2618–2628. doi: 10.1021/jm700948z. [DOI] [PubMed] [Google Scholar]; c) Velásquez JE, van der Donk WA. Curr Opin Chem Biol. 2011;15:11–21. doi: 10.1016/j.cbpa.2010.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Trader DJ, Carlson EE. Mol Biosyst. 2012;8:2484–2493. doi: 10.1039/c2mb25122a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Metcalf WW, van der Donk WA. Annu Rev Biochem. 2009;78:65–94. doi: 10.1146/annurev.biochem.78.091707.100215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee JH, Bae B, Kuemin M, Circello BT, Metcalf WW, Nair SK, van der Donk WA. Proc Natl Acad Sci U S A. 2010;107:17557–17562. doi: 10.1073/pnas.1006848107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Mol Cell Proteom. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 6.a) Peck SC, Gao J, van der Donk WA. Methods Enzymol. 2012;516:101–123. doi: 10.1016/B978-0-12-394291-3.00029-0. [DOI] [PubMed] [Google Scholar]; b) Yu X, Doroghazi JR, Janga SC, Circello BT, Griffin BM, Zhang JK, Labeda DP, Metcalf WW. Proc Natl Acad Sci U S A. 2013 doi: 10.1073/pnas.1315107110. accepted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Kuroda Y, Okuhara M, Goto T, Okamoto M, Terano H, Kohsaka M, Aoki H, Imanaka H. J Antibiot (Tokyo) 1980;33:29–35. doi: 10.7164/antibiotics.33.29. [DOI] [PubMed] [Google Scholar]; b) Ogita T, Gunji S, Fukuzawa Y, Terahara A, Kinoshita T, Nagaki H, Beppu T. Tetrahedron Lett. 1983;24:2283–2286. [Google Scholar]
- 8.a) Blair LM, Sperry J. J Nat Prod. 2013;76:794–812. doi: 10.1021/np400124n. [DOI] [PubMed] [Google Scholar]; b) Fujimori DG, Hrvatin S, Neumann CS, Strieker M, Marahiel MA, Walsh CT. Proc Natl Acad Sci U S A. 2007;104:16498–16503. doi: 10.1073/pnas.0708242104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Garg RP, Alemany LB, Moran S, Parry RJ. J Am Chem Soc. 2009;131:9608–9609. doi: 10.1021/ja901243p. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Neumann CS, Jiang W, Heemstra JR, Jr, Gontang EA, Kolter R, Walsh CT. Chembiochem. 2012;13:972–976. doi: 10.1002/cbic.201200054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Circello BT, Eliot AC, Lee JH, van der Donk WA, Metcalf WW. Chem Biol. 2010;17:402–411. doi: 10.1016/j.chembiol.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iyer LM, Abhiman S, Maxwell Burroughs A, Aravind L. Mol Biosyst. 2009;5:1636–1660. doi: 10.1039/b917682a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a) Fawaz MV, Topper ME, Firestine SM. Bioorg Chem. 2011;39:185–191. doi: 10.1016/j.bioorg.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Galperin MY, Koonin EV. Protein Sci. 1997;6:2639–2643. doi: 10.1002/pro.5560061218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Garg RP, Gonzalez JM, Parry RJ. J Biol Chem. 2006;281:26785–26791. doi: 10.1074/jbc.M603675200. [DOI] [PubMed] [Google Scholar]; b) Garg RP, Qian XL, Alemany LB, Moran S, Parry RJ. Proc Natl Acad Sci U S A. 2008;105:6543–6547. doi: 10.1073/pnas.0708957105. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhang W, Ntai I, Kelleher NL, Walsh CT. Proc Natl Acad Sci U S A. 2011;108:12249–12253. doi: 10.1073/pnas.1109539108. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Bougioukou DJ, Mukherjee S, van der Donk WA. Proc Natl Acad Sci U S A. 2013;110:10952–10957. doi: 10.1073/pnas.1303568110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a) de Azevedo Wäsch SI, van der Ploeg JR, Maire T, Lebreton A, Kiener A, Leisinger T. Appl Environ Microbiol. 2002;68:2368–2375. doi: 10.1128/AEM.68.5.2368-2375.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kurihara S, Oda S, Kato K, Kim HG, Koyanagi T, Kumagai H, Suzuki H. J Biol Chem. 2005;280:4602–4608. doi: 10.1074/jbc.M411114200. [DOI] [PubMed] [Google Scholar]; c) Takeo M, Ohara A, Sakae S, Okamoto Y, Kitamura C, Kato DI, Negoro S. J Bacteriol. 2013 doi: 10.1128/jb.00397-00313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ratner S. In: The Enzymes. Boyer P, editor. Vol. 7. Academic Press; New York: 1972. pp. 167–197. [Google Scholar]
- 16.a) Gould SJ, Hong ST, Carney JR. J Antibiot. 1998;51:50–57. doi: 10.7164/antibiotics.51.50. [DOI] [PubMed] [Google Scholar]; b) Bunet R, Song L, Mendes MV, Corre C, Hotel L, Rouhier N, Framboisier X, Leblond P, Challis GL, Aigle B. J Bacteriol. 2011;193:1142–1153. doi: 10.1128/JB.01269-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kersten RD, Lane AL, Nett M, Richter TK, Duggan BM, Dorrestein PC, Moore BS. ChemBioChem. 2013;14:955–962. doi: 10.1002/cbic.201300147. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.