

Abstract
Laminopathies are a heterogeneous group of diseases that are caused by mutations in the nuclear envelope proteins lamins A and C. Laminopathies include dilated cardiomyopathy, Emery-Dreifuss muscular dystrophy, and familial partial lipodystrophy. Despite their near-ubiquitous expression, most laminopathies involve highly tissue-specific phenotypes, often affecting skeletal and cardiac muscle. The underlying mechanism(s) remain incompletely understood. We recently reported that altered actin dynamics in lamin A/C-deficient and mutant cells disturb nuclear shuttling of the transcriptional co-activator MKL1, which is critical for cardiac function. Expression of the inner nuclear membrane protein emerin rescues MKL1 translocation through modulating actin dynamics. Here, we elaborate on these findings, discuss new insights into the role of nuclear actin in MKL1activity, and demonstrate that primary human skin fibroblasts from a patient with dilated cardiomyopathy have impaired MKL1 nuclear translocation. These findings further strengthen the relevance of impaired MKL1 signaling as a potential contributor to the disease mechanism in laminopathies.
Keywords: lamin, MKL1, MAL, MRTF-A, dilated cardiomyopathy, nuclear actin, mechanotransduction
Human Diseases Arising from Lamin Mutations
Inherited or de novo mutations in the LMNA gene encoding the nuclear envelope proteins lamins A and C cause a broad spectrum of human diseases (laminopathies), including the autosomal dominant form of Emery-Dreifuss muscular dystrophy (EDMD), dilated cardiomyopathy (DCM), limb-girdle muscular dystrophy type 1B, familial partial lipodystrophy (FPLD) and Hutchison-Gilford progeria syndrome (HGPS).1 The majority of the more than 460 currently identified LMNA mutations cause diseases affecting skeletal and cardiac muscle (see http://www.umd.be/LMNA/). Interestingly, cardiac and skeletal myopathies can also result from mutations in the inner nuclear membrane protein emerin,2 which is mislocalized from the nuclear envelope in lamin A/C-deficient cells.3 The mechanisms by which the nearly ubiquitously expressed lamins A and C (and also emerin) can cause such tissue-specific phenotypes remain incompletely understood.1 Two non-mutually exclusive hypotheses have been proposed to explain these findings: (1) The structural hypothesis postulates that lamin A/C mutations result in loss of structural function, rendering cells more susceptible to damage in mechanically active tissues such as muscle; (2) the gene regulation hypothesis proposes that lamin A/C mutations alter interactions between lamins and tissue-specific transcriptional regulators. Considering that most of the tissues affected in laminopathies are of mesenchymal origin, a third emerging idea expanding on the gene regulation hypothesis postulates that lamin mutations may impair maintenance or function of adult mesenchymal and muscle stem cells, thereby resulting in loss of tissue function over time.
Unlike mutations responsible for FPLD and HGPS that are mostly restricted to specific nucleotides (C1824T for HGPS) or concentrated around a small region on the surface of the Ig-fold of lamin A/C (as in the case of FPLD), mutations that cause dilated cardiomyopathy and muscular dystrophies are scattered along the full length of the gene. To date, there is no easily identifiable correlation between the position of the mutations on the gene/protein and the diverse diseases they cause. Similarly, morphological analysis of skin fibroblasts from an array of patients with diverse LMNA mutations found no correlation between nuclear shape abnormalities and the specific disease phenotype,4 although the study noticed that LMNA mutations in the lamin head or tail domains result in abnormal nuclear architecture.4 Examining the effect of a broad panel of lamin A mutations on the mechanical stability of cell nuclei, a recent study observed that several mutations associated with muscular and cardiac phenotypes resulted in loss of structural function that caused nuclei to become more deformable and/or exhibit defects in nucleo-cytoskeletal coupling, whereas mutations responsible for FPLD did not diminish the structural function of lamins.5 However, the authors also identified several EDMD- and DCM-causing lamin mutations that retained apparently normal structural function,5 suggesting that additional mechanisms must contribute to the disease development.
Lamins, Mechanosensing, and Cardiomyopathies
The fact that many laminopathies display primarily muscular and/or cardiac phenotypes suggests a potential mechanosensitive component at work, which is further supported by previous findings that lamin A/C- and emerin-deficient cells have defects in the activation of mechanosensitive genes6,7 and mice with reduced levels of lamins A/C (Lmna+/–) have a markedly weaker hypertrophic response to ventricular pressure overload than wild-type littermates.8 Moreover, Lmna–/– mice develop severe muscular dystrophy and cardiomyopathy and die at 4 to 8 weeks of age.9 The LmnaN195K/N195K mouse model also develops severe DCM but lacks skeletal muscle involvement.10 Motivated by these earlier reports, we recently conducted an in-depth analysis into mechanotransduction pathways relevant to cardiac function, using cells and tissues from lamin A/C-deficient (Lmna–/–) and Lmna N195K mutant mice. We reported that cells and cardiac tissue from Lmna–/– and Lmna N195K mice had impaired nuclear translocation and downstream signaling of the transcription factor megakaryoblastic leukemia 1 protein (MKL1, also known as MRTF-A and MAL), a myocardin family member that is pivotal in cardiac development and function.11
MKL1 is a mechanosensitive transcription factor with important functions in the cardiovascular system.12 MKL1 mediates the TCF-independent activation of serum response factor (SRF), which controls vascular smooth muscle cell and cardiomyocyte differentiation.12,13 MKL1 shares partially overlapping functions with myocardin and MRTF-B during cardiac development and regulates cardiovascular patterning;14 MKL1 also plays an important role in the pathogenesis of cardiomyopathies, as deletion of MKL1 results in lethality due to dilated cardiomyopathy and heart failure in approximately a third of MKL1-null embryos15 and MKL1 mediates activation of a hypertrophic gene program in response to mechanical stress in cardiomyocytes.16 Intracellular localization and activity of MKL1 is regulated via changes in the polymerization of cellular actin.13 At baseline (e.g., in resting or serum-starved cells), MKL1 is mainly sequestered in the cytoplasm through binding to cytoplasmic G-actin. At the same time, MKL1 is constitutively exported from the nucleus via chromosome region maintenance-1 (CRM1)-dependent transport, further contributing to the predominantly cytoplasmic localization of MKL1.13 Upon mitogenic or mechanical stimulation, RhoA activation promotes actin polymerization, which liberates MKL1 from G-actin and exposes a nuclear localization sequence (NLS) within the actin-binding domain of MKL1, inducing MKL1 to translocate to the nucleus.17,18 The increased nuclear import, coupled with decreased nuclear export, results in accumulation of MKL1 in the nucleus, where it acts as a co-activator of SRF to turn on genes regulating cellular motility and contractility, including talin, vinculin, actin, and SRF itself.13 The subsequent increase in actin levels feeds back to turn off the MKL1-SRF circuitry.13 Importantly, SRF is highly expressed in skeletal and cardiac muscle of adult mice19 and controls a multitude of genes relevant to cardiac muscle, including cardiac α-actin, α-myosin heavy chain (α-MHC) and β-MHC.20-22 Underscoring the critical role of SRF in the regulation of cardiac structure and function, targeted disruption of SRF is embryonically lethal due to defective mesoderm formation,23 and cardiac-specific deletion of SRF in adult mice results in dilated cardiomyopathy with abnormal expression of cardiac structural genes.24
Given this prominent role of MKL1/SRF signaling in striated muscle function, our findings that Lmna–/– and Lmna N195K cells, as well as skeletal and cardiac muscle derived from the corresponding mice, have impaired MKL1/SRF signaling11 are particularly relevant to the dilated cardiomyopathy present in several laminopathies. Considering the striking similarity of these phenotypes with those observed in human EDMD and DCM patients, it is tempting to hypothesize that impaired MKL1-SRF signaling and the resulting deficiencies in cytoskeletal organization could contribute to the manifestation of cardiac and muscular phenotypes in various laminopathies. This idea is further supported by previous reports that cells and tissues from Lmna–/– mice have defects in cytoskeletal organization9,25,26 consistent with a disturbed role of MKL1-SRF signaling.27
MKL1 Signaling in the Context of Human Laminopathies
Since the results in our original study11 were obtained from cells and tissue from mouse models of laminopathies, we decided to test whether similar defects in MKL1 translocation could also be observed in cells from laminopathy patients, particularly those with skeletal and cardiac muscle phenotypes. Unlike the mouse models, which often require homozygous expression of the mutant lamin A/C,28 most human laminopathies patients carry only one mutated lamin allele and also express wild-type lamin, in addition to mutant lamin.1 We compared MKL1 signaling in cells from a patient with DCM (LMNA K219T mutation),29 cells from a patient with congenital muscular dystrophy (CMD) with both skeletal and cardiac symptoms (LMNA ΔK32),4,30 and a patient with FPLD (LMNA R482Q),4,30 as well as healthy controls.
MKL1 signaling was assessed based on nuclear localization of MKL1 upon serum stimulation and in serum-starved controls (baseline). Under baseline conditions, MKL1 was cytoplasmic in all cells, regardless of genotype (data not shown). In response to serum stimulation, MKL1 rapidly translocated to the nucleus in cells from healthy controls (Fig. 1A, top two rows). In contrast, fibroblasts carrying the DCM-causing LMNA K219T mutation had decreased nuclear localization of MKL1 (Fig. 1A, bottom row), indicating impaired MKL1 signaling, similar to our previous findings obtained in the Lmna–/– and Lmna N195K mouse models. On the other hand, cells from patients with FPLD and CMD had normal MKL1 translocation (Fig. 1A, third and fourth rows). The fact that the LMNA ΔK32 cells had normal MKL1 translocation was somewhat surprising, as laminopathy patients with muscular dystrophy also develop cardiovascular phenotypes,31 including in the case of the LMNA ΔK32 mutation studied here.30 Furthermore, LmnaΔK32/+ heterozygous mice develop muscular dystrophy accompanied by cardiac phenotypes, in part due degradation of the toxic Lmna ΔK32 protein that results in haploinsufficiency of lamin A/C.32 Given the limited number of currently available primary patient cells, along with the heterogeneity of patient samples with regards to genetic background, handling, passage number and other compounding factors, further experiments with a wider panel of patient samples, as well as animal models and genetically modified cells expressing various lamin mutations, will be necessary to establish whether deregulation of MKL1/SRF signaling is specific to a subset of laminopathies.
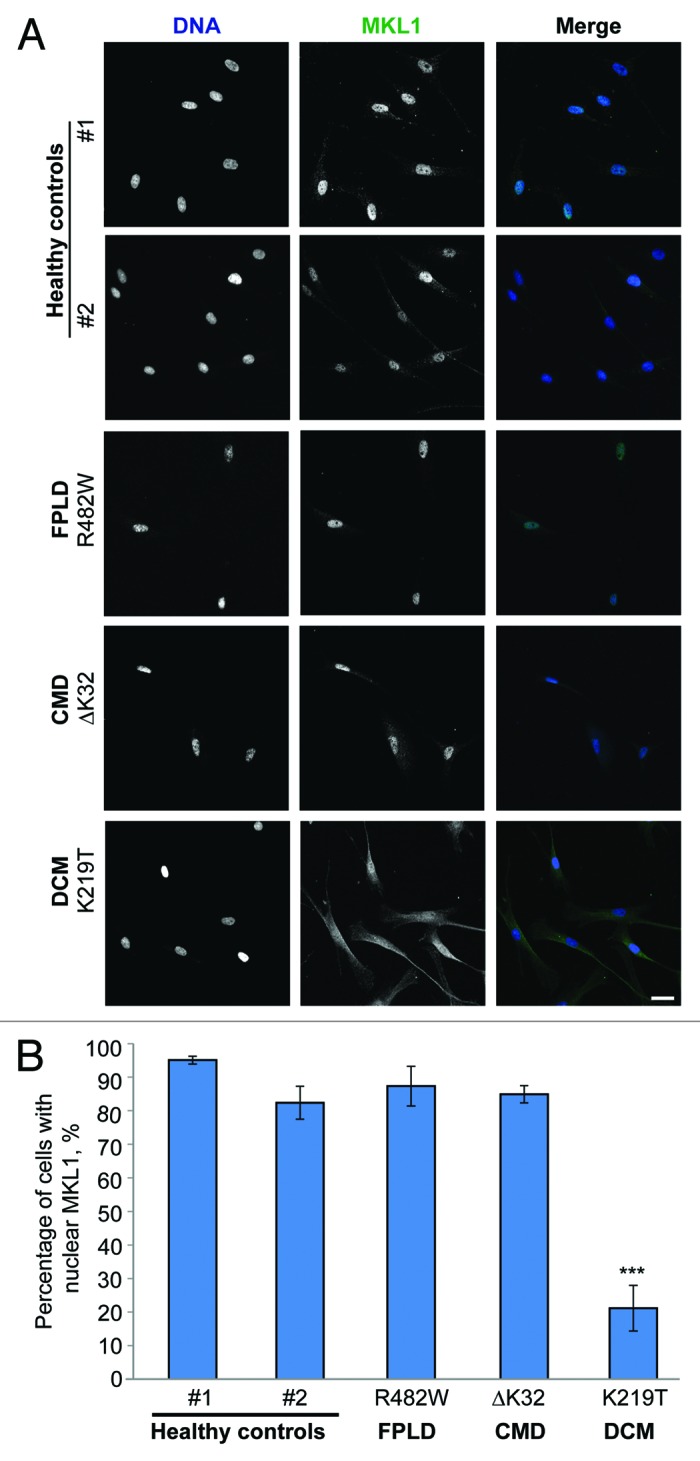
Figure 1. MKL1 translocation in primary human skin fibroblasts from diverse laminopathy patients and healthy controls. (A) Representative images of MKL1 staining in healthy control fibroblasts and human laminopathy patient fibroblasts stimulated for 15 min with 15% fetal bovine serum and subsequently fixed with 4% paraformaldehyde and immunofluorescently labeled for MKL1. DNA is shown in blue and MKL1 is shown in green. Scale bar, 20 µm. (B) Quantitative analysis of cells with MKL1 nuclear localization after serum stimulation (n = 3, *** P < 0.005).
Further complicating any attempt to identify tissue-specific defects associated with particular lamin mutations in human samples is the fact that laboratory studies are often confined to the use of primary skin fibroblasts, which may not adequately reflect cardiac-specific defects. The growing use of induced pluripotent stem cells generated from patient fibroblasts that can be differentiated into specific lineages, including cardiomyocytes, is expected to yield more detailed insights into tissue-specific defects arising from LMNA mutations in human cells.33 At the same time, at least in the homozygous Lmna–/– and LmnaN195K/N195K mouse models, embryonic fibroblasts and bone-marrow derived mesenchymal stem cells showed similar defects in MKL1 nuclear localization as cells in skeletal and cardiac muscle sections.11 Furthermore, apart from their known roles in cardiomyocytes, MKL1-SRF have been shown to regulate myofibroblast activation and fibrosis in response to the renin-angiotensin system and cardiac remodeling following myocardial infarction by turning on genes involved in extracellular matrix synthesis and smooth muscle cell differentiation.34 The ability of MKL1-SRF to regulate smooth muscle cell phenotypes is especially significant in the context of cardiac-related pathologies. In lung fibroblasts, MKL1-SRF play a role in collagen synthesis, and deregulation of MKL1-SRF signaling can lead to pulmonary fibrosis characterized by excessive collagen deposition.35 These findings highlight the broad relevance of MKL1-SRF signaling across diverse cell types and suggest that impaired MKL1 signaling could also contribute to other defects in various laminopathies.
Mechanisms for Altered Actin Dynamics and MKL1 Signaling in Laminopathy Cells
How can mutations in nuclear envelope proteins affect the nuclear localization of MKL1? In our original report,11 we described that the altered nuclear import and export of MKL1 could be attributed to changes in nuclear and cytoskeletal actin dynamics in the lamin A/C-deficient and mutant cells, and that these defects were mediated by the inner nuclear membrane emerin and its role in promoting actin polymerization.36 Ectopic expression of wild-type emerin, but not of emerin mutations that cannot bind to actin and do not promote actin polymerization, was sufficient to restore MKL1 nuclear translocation and actin dynamics in Lmna–/–, Lmna N195K, and emerin-deficient cells.11 These data suggest a novel mechanism for nuclear envelope proteins to regulate MKL1-SRF signaling by modulating actin polymerization and organization, which controls nuclear import and export of MKL1. Since the levels of emerin on the outer nuclear membrane are generally low and based on our findings that Lmna–/– and Lmna N195K cells show increased segregation of emerin into the endoplasmic reticulum, we hypothesize that emerin primarily affects nuclear actin polymerization, which controls nuclear export and transcriptional activity of MKL1,13 and that changes in cytoskeletal actin dynamics arise as a consequence of altered MKL1/SRF signaling (Fig. 2).

Figure 2. Schematic model of the interplay between nuclear envelope proteins and MKL1 signaling in wild-type and lamin A/C-deficient or mutant cells. Top: Sequence of events in a serum-stimulated wild-type cell. (1) Upon mechanical or serum stimulation, Rho/ROCK activity promotes cytoplasmic actin polymerization. (2) After being released from cytoplasmic G-actin, MKL1 translocates into the nucleus, and, together with SRF, binds to the CArG box to induce expression or MKL1/SRF-target genes, including actin, vinculin and SRF. (3) Emerin facilitates polymerization of (nuclear) actin. This reduces nuclear export of MKL1 which requires binding to nuclear monomeric actin. (4) Transcription of SRF downstream target genes occurs. Bottom: Sequence of events in a serum-stimulated lamin A/C-deficient cell. (1) Emerin is lost from the nuclear envelope and is unable to modulate nuclear actin polymerization. (2) G-actin binds to MKL1. (3) Export of MKL1 from the nucleus occurs. (4) SRF binding to its target element on the DNA and transcription of downstream target genes are impaired.
Importantly, mutations in emerin itself can also cause (X-linked) EDMD that includes cardiac phenotypes,2 and proper targeting of emerin to the nuclear envelope requires lamin A/C.3 This latter fact is evident as emerin is redistributed from the nuclear envelope to the endoplasmic reticulum in the Lmna–/– and Lmna N195K models.11 To evaluate the effect of specific lamin A mutations on emerin localization, Raharjo and colleagues37 ectopically expressed a panel of lamin A mutations in a Lmna–/– mouse embryo fibroblast background. They found that the DCM-associated (L85R and N195K) and EDMD-associated (L530P) lamin A mutants failed to rescue abnormal localization of emerin, whereas the FPLD-causing mutation R482W successfully restored emerin localization to the nuclear envelope.37 It is noteworthy that both DCM-associated mutations displayed mislocalization of emerin, consistent with our model that the role of emerin in modulating (nuclear) actin dynamics and thereby MKL1/SRF signaling can result in skeletal and cardiac muscle defects.
To assess whether mislocalization or loss of emerin expression could also explain the impaired nuclear localization of MKL1 in the LMNA K219T fibroblasts derived from a DCM patient, we analyzed emerin localization in these and other laminopathy patient cells by immunofluorescence staining. Surprisingly, even in the LMNA K219T fibroblasts, which displayed distinct defects in MKL1 nuclear localization upon serum stimulation (Fig. 1), the majority of cells showed the typical nuclear rim staining of emerin (data not shown). While we did observe a few LMNA K219T fibroblasts with reduced emerin expression, western blot analysis did not reveal any significant differences in emerin levels between the LMNA K219T fibroblasts compared with healthy control fibroblasts.
A potential explanation for these results could be the fact that—unlike the mouse models studied previously—most human laminopathy patients, including those studied here, are heterozygous for the LMNA mutations and also produce wild-type lamin A/C, which may result in less severe defects on emerin distribution. A similar effect can be observed in cells from heterozygous Lmna+/– mice, in which mislocalization of emerin is substantially milder compared with cells from homozygous Lmna–/– mice, but Lmna+/– cells nonetheless show defects in MKL1 nuclear translocation. At the same time, we cannot exclude the possibility that species-related differences or other factors, e.g., involvement of additional signaling pathways that provide further regulation for MKL1 signaling, are responsible for the observed discrepancy between apparently normal emerin localization and yet impaired MKL1 nuclear translocation. These findings further demonstrate the complexity and heterogeneity of the underlying mechanisms of laminopathies in humans and in model organisms created to recapitulate these phenotypes.
Nuclear Actin and Transcriptional Regulation
Our original work directly implicated altered actin dynamics—modulated by emerin at the nuclear envelope—in regulating MKL1 nuclear translocation. Further support for the idea that polymerization of nuclear actin plays a pivotal role in regulating MKL1 activity comes from a recent report by the Grosse group that found that serum-dependent MKL1-SRF activity is driven by actin polymerization inside the nucleus.38 In particular, they showed that nuclear expression of mDia formin protein promotes MKL1-SRF transcriptional activity. Activation of mDia induces polymerization of nuclear actin filaments and nuclear accumulation of MKL1 as part of the serum response.38 These findings point to an emerging role of nuclear actin as an important player in modulating transcriptional activity and in regulation of gene expression.39 The study of nuclear actin in live cells, however, has been traditionally hampered by the difficulty to visualize nuclear actin in living cells without disturbing its normal dynamics and structural organization, for example, when ectopically expressing fluorescently labeled actin. Consequently, the exact structure, organization, and function of nuclear actin remain a matter of intense debate. Novel fluorescent probes for nuclear actin recently developed in the Mullins laboratory may help overcome this limitation. These probes, which are based on fusing fluorescent proteins with carefully selected (and as necessary, modified) actin-binding domains, are capable of distinguishing polymeric and monomeric forms of nuclear actin and enable monitoring nuclear actin organization in living cells.40 Application of these probes to a variety of somatic cells revealed distinct nuclear populations of monomeric and filamentous actin: monomeric nuclear actin primarily colocalized with nuclear speckles, which are involved in RNA processing; in contrast, filamentous nuclear actin was found in punctate structures distributed across the nucleoplasm that could be perturbed by latrunculin treatment,40 a known actin disruptor. Time-lapse microscopy with probes for filamentous actin revealed two distinct populations with different diffusive behavior, including one with a sub-diffusive and non-directed mobility, suggesting that filamentous actin may be part of a viscoelastic network within the inter-chromatin domain.40 In the context of MKL1 signaling, future application of these probes could potentially be useful in visualizing dynamic changes in nuclear actin and its role in orchestrating MKL1 nuclear translocation in response to stimulation in normal and lamin A/C-deficient cells.
Prospects of Pharmacological Interventions
A better understanding of the disease etiology of laminopathies has important implications in the development of therapeutic approaches for affected patients. While structural deficiencies caused by lamin mutations might prove to be difficult to correct pharmacologically, disturbed cell signaling is more amendable to small molecule treatment approaches. Extensive research on altered signaling pathways in laminopathies, driven by the Worman group, identified upregulation of mitogen-activated protein kinase (MAPK) signaling in lamin A/C-41,42 as well as in emerin-mutant models.43 Intervention with MAPK inhibitors has already proven beneficial in alleviating cardiac phenotypes in these mouse models.44,45 Particularly, selumetinib, a highly specific MEK1/2 inhibitor that is currently undergoing Phase II clinical trials for anti-cancer therapy, has already produced encouraging results in LmnaH222P/H222P mice, improving cardiac function and prolonging survival.46,47 Our recent findings suggest that the positive outcomes may also (at least in part) be due to the drugs’ effect on MKL1-SRF signaling: activity of the downstream kinase of MEK1, ERK1/2, promotes nuclear export of MKL1;48 hence inhibition of this cascade is expected to increase the nuclear fraction of MKL1. While it is a logical next step to test the efficacy and clinical benefits of MAPK inhibitors on MKL1-SRF signaling, another worthwhile approach might be to target MKL1/SRF directly to restore its downstream signaling.
Conclusion
The recent identification of impaired MKL1/SRF signaling in lamin A/C- and emerin-deficient cells due to disturbed actin dynamics connects the structural and gene regulation hypotheses that have been proposed to explain tissue-specific phenotypes in laminopathies. In particular, the ability of emerin to promote (nuclear) actin polymerization can directly modulate the intracellular localization and activity of the transcriptional coactivator MKL1, which, together with SRF, regulates a number of cytoskeletal and adhesion molecules, thereby further affecting cellular structure and function, particularly in the cardiovascular system. Importantly, these findings shed new light on the disease etiology of many laminopathies, specifically those that present striated muscle phenotypes, but impaired MKL1 signaling could also affect diverse other cell types. At the same time, given the growing number of signaling pathways affected by lamin mutations,1 studies investigating only select pathways in vitro may not fully reflect the complex interactions found in vivo, where cells and tissues are exposed to a broad range of mechanical and biochemical stimuli that can activate a variety of overlapping and interacting signaling pathways. Future studies are expected to provide further insights into the complex role of lamins in mechanotransduction and muscle-specific function. Nonetheless, the increasing identification of specific signaling pathways impaired in laminopathies provides a glimmer of hope to affected patients, as normalizing MKL1-SRF and/or MAPK signaling through small molecules could present an attractive therapeutic target to treat the devastating cardiac disease associated with many of the laminopathies.49
Acknowledgments
We thank Dr Colin Stewart for providing the mouse models and mouse cell lines. The LMNA K219T/+ patient fibroblasts were kindly provided by Dr Gianluigi Condorelli (University of Milan, Italy). The LMNA R482Q and LMNA ΔK32 mutant cells were generated in the laboratory of Dr Gisele Bonne (Institut de Myologie, France) and graciously provided by Drs Antoine Muchir and Howard Worman (Columbia University).
This work was supported by National Institutes of Health awards [R01 NS059348 and R01 HL082792]; the Department of Defense Breast Cancer Idea Award [BC102152]; a National Science Foundation CAREER award to Lammerding J [CBET-1254846]; an award from the Progeria Research Foundation [PRF2011–0035]; postdoctoral fellowships from the American Heart Association to Jaalouk DE [AHA 09POST2320042] and to Ho CY. [AHA 13POST16740004]; a Faculty Development Grant from AUB to Jaalouk DE; and a Pilot Project Award by the Cornell Center on the Microenvironment & Metastasis through Award Number U54CA143876 from the National Cancer Institute. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
Glossary
Abbreviations:
- CMD
congenital muscular dystrophy
- DCM
dilated cardiomyopathy
- EDMD
Emery-Dreifuss muscular dystrophy
- FPLD
familial partial lipodystrophy
- HGPS
Hutchison-Gilford progeria syndrome
- MKL1
megakaryoblastic leukemia 1
- MRTF-A
myocardin-related transcription factor-1
- MAL
megakaryocytic acute leukemia protein
- NLS
nuclear localization signal
- SRF
serum response factor
Citation: Ho CY, Jaalouk DE, Lammerding J. Novel insights into the disease etiology of laminopathies. Rare Diseases 2013; 1:e27002;
Disclosure of potential conflicts of interest
The authors declare that no conflicts of interest exist.
References
- 1.Schreiber KH, Kennedy BK. When lamins go bad: nuclear structure and disease. Cell. 2013;152:1365–75. doi: 10.1016/j.cell.2013.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bione S, Maestrini E, Rivella S, Mancini M, Regis S, Romeo G, Toniolo D. Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat Genet. 1994;8:323–7. doi: 10.1038/ng1294-323. [DOI] [PubMed] [Google Scholar]
- 3.Vaughan A, Alvarez-Reyes M, Bridger JM, Broers JL, Ramaekers FC, Wehnert M, Morris GE, Hutchison CJ, Hutchison CJ, Whitfield WGF Both emerin and lamin C depend on lamin A for localization at the nuclear envelope. J Cell Sci. 2001;114:2577–90. doi: 10.1242/jcs.114.14.2577. [DOI] [PubMed] [Google Scholar]
- 4.Muchir A, Medioni J, Laluc M, Massart C, Arimura T, van der Kooi AJ, Desguerre I, Mayer M, Ferrer X, Briault S, et al. Nuclear envelope alterations in fibroblasts from patients with muscular dystrophy, cardiomyopathy, and partial lipodystrophy carrying lamin A/C gene mutations. Muscle Nerve. 2004;30:444–50. doi: 10.1002/mus.20122. [DOI] [PubMed] [Google Scholar]
- 5.Zwerger M, Jaalouk DE, Lombardi ML, Isermann P, Mauermann M, Dialynas G, Herrmann H, Wallrath LL, Lammerding J. Myopathic lamin mutations impair nuclear stability in cells and tissue and disrupt nucleo-cytoskeletal coupling. Hum Mol Genet. 2013;22:2335–49. doi: 10.1093/hmg/ddt079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lammerding J, Schulze PC, Takahashi T, Kozlov S, Sullivan T, Kamm RD, Stewart CL, Lee RT. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J Clin Invest. 2004;113:370–8. doi: 10.1172/JCI19670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lammerding J, Hsiao J, Schulze PC, Kozlov S, Stewart CL, Lee RT. Abnormal nuclear shape and impaired mechanotransduction in emerin-deficient cells. J Cell Biol. 2005;170:781–91. doi: 10.1083/jcb.200502148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cupesi M, Yoshioka J, Gannon J, Kudinova A, Stewart CL, Lammerding J. Attenuated hypertrophic response to pressure overload in a lamin A/C haploinsufficiency mouse. J Mol Cell Cardiol. 2010;48:1290–7. doi: 10.1016/j.yjmcc.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sullivan T, Escalante-Alcalde D, Bhatt H, Anver M, Bhat N, Nagashima K, Stewart CL, Burke B. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J Cell Biol. 1999;147:913–20. doi: 10.1083/jcb.147.5.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mounkes LC, Kozlov SV, Rottman JN, Stewart CL. Expression of an LMNA-N195K variant of A-type lamins results in cardiac conduction defects and death in mice. Hum Mol Genet. 2005;14:2167–80. doi: 10.1093/hmg/ddi221. [DOI] [PubMed] [Google Scholar]
- 11.Ho CY, Jaalouk DE, Vartiainen MK, Lammerding J. Lamin A/C and emerin regulate MKL1-SRF activity by modulating actin dynamics. Nature. 2013;497:507–11. doi: 10.1038/nature12105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parmacek MS. Myocardin-related transcription factors: critical coactivators regulating cardiovascular development and adaptation. Circ Res. 2007;100:633–44. doi: 10.1161/01.RES.0000259563.61091.e8. [DOI] [PubMed] [Google Scholar]
- 13.Olson EN, Nordheim A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat Rev Mol Cell Biol. 2010;11:353–65. doi: 10.1038/nrm2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Small EM. The actin-MRTF-SRF gene regulatory axis and myofibroblast differentiation. J Cardiovasc Transl Res. 2012;5:794–804. doi: 10.1007/s12265-012-9397-0. [DOI] [PubMed] [Google Scholar]
- 15.Sun Y, Boyd K, Xu W, Ma J, Jackson CW, Fu A, Shillingford JM, Robinson GW, Hennighausen L, Hitzler JK, et al. Acute myeloid leukemia-associated Mkl1 (Mrtf-a) is a key regulator of mammary gland function. Mol Cell Biol. 2006;26:5809–26. doi: 10.1128/MCB.00024-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuwahara K, Kinoshita H, Kuwabara Y, Nakagawa Y, Usami S, Minami T, Yamada Y, Fujiwara M, Nakao K. Myocardin-related transcription factor A is a common mediator of mechanical stress- and neurohumoral stimulation-induced cardiac hypertrophic signaling leading to activation of brain natriuretic peptide gene expression. Mol Cell Biol. 2010;30:4134–48. doi: 10.1128/MCB.00154-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirano H, Matsuura Y. Sensing actin dynamics: structural basis for G-actin-sensitive nuclear import of MAL. Biochem Biophys Res Commun. 2011;414:373–8. doi: 10.1016/j.bbrc.2011.09.079. [DOI] [PubMed] [Google Scholar]
- 18.Pawłowski R, Rajakylä EK, Vartiainen MK, Treisman R. An actin-regulated importin α/β-dependent extended bipartite NLS directs nuclear import of MRTF-A. EMBO J. 2010;29:3448–58. doi: 10.1038/emboj.2010.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Belaguli NS, Schildmeyer LA, Schwartz RJ. Organization and myogenic restricted expression of the murine serum response factor gene. A role for autoregulation. J Biol Chem. 1997;272:18222–31. doi: 10.1074/jbc.272.29.18222. [DOI] [PubMed] [Google Scholar]
- 20.Chien R. Signaling mechanisms for the activation of an embryonic gene program during the hypertrophy of cardiac ventricular muscle. Basic Res Cardiol. 1992;87(Suppl 2):49–58. doi: 10.1007/978-3-642-72477-0_5. [DOI] [PubMed] [Google Scholar]
- 21.Colucci WS. Molecular and cellular mechanisms of myocardial failure. Am J Cardiol. 1997;80(11A):15L–25L. doi: 10.1016/S0002-9149(97)00845-X. [DOI] [PubMed] [Google Scholar]
- 22.Durand JB. Genetic basis of cardiomyopathy. Curr Opin Cardiol. 1999;14:225–9. doi: 10.1097/00001573-199905000-00006. [DOI] [PubMed] [Google Scholar]
- 23.Arsenian S, Weinhold B, Oelgeschläger M, Rüther U, Nordheim A. Serum response factor is essential for mesoderm formation during mouse embryogenesis. EMBO J. 1998;17:6289–99. doi: 10.1093/emboj/17.21.6289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parlakian A, Charvet C, Escoubet B, Mericskay M, Molkentin JD, Gary-Bobo G, De Windt LJ, Ludosky MA, Paulin D, Daegelen D, et al. Temporally controlled onset of dilated cardiomyopathy through disruption of the SRF gene in adult heart. Circulation. 2005;112:2930–9. doi: 10.1161/CIRCULATIONAHA.105.533778. [DOI] [PubMed] [Google Scholar]
- 25.Nikolova V, Leimena C, McMahon AC, Tan JC, Chandar S, Jogia D, Kesteven SH, Michalicek J, Otway R, Verheyen F, et al. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. J Clin Invest. 2004;113:357–69. doi: 10.1172/JCI19448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hale CM, Shrestha AL, Khatau SB, Stewart-Hutchinson PJ, Hernandez L, Stewart CL, Hodzic D, Wirtz D. Dysfunctional connections between the nucleus and the actin and microtubule networks in laminopathic models. Biophys J. 2008;95:5462–75. doi: 10.1529/biophysj.108.139428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morita T, Mayanagi T, Sobue K. Reorganization of the actin cytoskeleton via transcriptional regulation of cytoskeletal/focal adhesion genes by myocardin-related transcription factors (MRTFs/MAL/MKLs) Exp Cell Res. 2007;313:3432–45. doi: 10.1016/j.yexcr.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 28.Stewart CL, Kozlov S, Fong LG, Young SG. Mouse models of the laminopathies. Exp Cell Res. 2007;313:2144–56. doi: 10.1016/j.yexcr.2007.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roncarati R, Viviani Anselmi C, Krawitz P, Lattanzi G, von Kodolitsch Y, Perrot A, di Pasquale E, Papa L, Portararo P, Columbaro M, et al. Doubly heterozygous LMNA and TTN mutations revealed by exome sequencing in a severe form of dilated cardiomyopathy. Eur J Hum Genet. 2013;21:1105–11. doi: 10.1038/ejhg.2013.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Quijano-Roy S, Mbieleu B, Bönnemann CG, Jeannet PY, Colomer J, Clarke NF, Cuisset JM, Roper H, De Meirleir L, D’Amico A, et al. De novo LMNA mutations cause a new form of congenital muscular dystrophy. Ann Neurol. 2008;64:177–86. doi: 10.1002/ana.21417. [DOI] [PubMed] [Google Scholar]
- 31.Vytopil M, Ricci E, Dello Russo A, Hanisch F, Neudecker S, Zierz S, Ricotti R, Demay L, Richard P, Wehnert M, et al. Frequent low penetrance mutations in the Lamin A/C gene, causing Emery Dreifuss muscular dystrophy. Neuromuscul Disord. 2002;12:958–63. doi: 10.1016/S0960-8966(02)00178-5. [DOI] [PubMed] [Google Scholar]
- 32.Cattin ME, Bertrand AT, Schlossarek S, Le Bihan MC, Skov Jensen S, Neuber C, Crocini C, Maron S, Lainé J, Mougenot N, et al. Heterozygous LmnadelK32 mice develop dilated cardiomyopathy through a combined pathomechanism of haploinsufficiency and peptide toxicity. Hum Mol Genet. 2013;22:3152–64. doi: 10.1093/hmg/ddt172. [DOI] [PubMed] [Google Scholar]
- 33.Zhang J, Lian Q, Zhu G, Zhou F, Sui L, Tan C, Mutalif RA, Navasankari R, Zhang Y, Tse HF, et al. A human iPSC model of Hutchinson Gilford Progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell. 2011;8:31–45. doi: 10.1016/j.stem.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 34.Small EM, Thatcher JE, Sutherland LB, Kinoshita H, Gerard RD, Richardson JA, Dimaio JM, Sadek H, Kuwahara K, Olson EN. Myocardin-related transcription factor-a controls myofibroblast activation and fibrosis in response to myocardial infarction. Circ Res. 2010;107:294–304. doi: 10.1161/CIRCRESAHA.110.223172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luchsinger LL, Patenaude CA, Smith BD, Layne MD. Myocardin-related transcription factor-A complexes activate type I collagen expression in lung fibroblasts. J Biol Chem. 2011;286:44116–25. doi: 10.1074/jbc.M111.276931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holaska JM, Kowalski AK, Wilson KL. Emerin caps the pointed end of actin filaments: evidence for an actin cortical network at the nuclear inner membrane. PLoS Biol. 2004;2:E231. doi: 10.1371/journal.pbio.0020231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Raharjo WH, Enarson P, Sullivan T, Stewart CL, Burke B. Nuclear envelope defects associated with LMNA mutations cause dilated cardiomyopathy and Emery-Dreifuss muscular dystrophy. J Cell Sci. 2001;114:4447–57. doi: 10.1242/jcs.114.24.4447. [DOI] [PubMed] [Google Scholar]
- 38.Baarlink C, Wang H, Grosse R. Nuclear actin network assembly by formins regulates the SRF coactivator MAL. Science. 2013;340:864–7. doi: 10.1126/science.1235038. [DOI] [PubMed] [Google Scholar]
- 39.de Lanerolle P. Nuclear actin and myosins at a glance. J Cell Sci. 2012;125:4945–9. doi: 10.1242/jcs.099754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Belin BJ, Cimini BA, Blackburn EH, Mullins RD. Visualization of actin filaments and monomers in somatic cell nuclei. Mol Biol Cell. 2013;24:982–94. doi: 10.1091/mbc.E12-09-0685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muchir A, Pavlidis P, Decostre V, Herron AJ, Arimura T, Bonne G, Worman HJ. Activation of MAPK pathways links LMNA mutations to cardiomyopathy in Emery-Dreifuss muscular dystrophy. J Clin Invest. 2007;117:1282–93. doi: 10.1172/JCI29042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Muchir A, Wu W, Worman HJ. Reduced expression of A-type lamins and emerin activates extracellular signal-regulated kinase in cultured cells. Biochim Biophys Acta. 2009;1792:75–81. doi: 10.1016/j.bbadis.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Muchir A, Pavlidis P, Bonne G, Hayashi YK, Worman HJ. Activation of MAPK in hearts of EMD null mice: similarities between mouse models of X-linked and autosomal dominant Emery Dreifuss muscular dystrophy. Hum Mol Genet. 2007;16:1884–95. doi: 10.1093/hmg/ddm137. [DOI] [PubMed] [Google Scholar]
- 44.Muchir A, Shan J, Bonne G, Lehnart SE, Worman HJ. Inhibition of extracellular signal-regulated kinase signaling to prevent cardiomyopathy caused by mutation in the gene encoding A-type lamins. Hum Mol Genet. 2009;18:241–7. doi: 10.1093/hmg/ddn343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu W, Muchir A, Shan J, Bonne G, Worman HJ. Mitogen-activated protein kinase inhibitors improve heart function and prevent fibrosis in cardiomyopathy caused by mutation in lamin A/C gene. Circulation. 2011;123:53–61. doi: 10.1161/CIRCULATIONAHA.110.970673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kirkwood JM, Bastholt L, Robert C, Sosman J, Larkin J, Hersey P, Middleton M, Cantarini M, Zazulina V, Kemsley K, et al. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin Cancer Res. 2012;18:555–67. doi: 10.1158/1078-0432.CCR-11-1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Muchir A, Reilly SA, Wu W, Iwata S, Homma S, Bonne G, Worman HJ. Treatment with selumetinib preserves cardiac function and improves survival in cardiomyopathy caused by mutation in the lamin A/C gene. Cardiovasc Res. 2012;93:311–9. doi: 10.1093/cvr/cvr301. [DOI] [PubMed] [Google Scholar]
- 48.Muehlich S, Wang R, Lee SM, Lewis TC, Dai C, Prywes R. Serum-induced phosphorylation of the serum response factor coactivator MKL1 by the extracellular signal-regulated kinase 1/2 pathway inhibits its nuclear localization. Mol Cell Biol. 2008;28:6302–13. doi: 10.1128/MCB.00427-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cattin ME, Muchir A, Bonne G. ‘State-of-the-heart’ of cardiac laminopathies. Curr Opin Cardiol. 2013;28:297–304. doi: 10.1097/HCO.0b013e32835f0c79. [DOI] [PubMed] [Google Scholar]