

Abstract
Inhibition of the nonmevalonate pathway (NMP) of isoprene biosynthesis has been examined as a source of new antibiotics with novel mechanisms of action. Dxr is the best studied of the NMP enzymes and several reports have described potent Dxr inhibitors. Many of these compounds are structurally related to natural products fosmidomycin and FR900098, each bearing retrohydroxamate and phosphonate groups. We synthesized a series of compounds with two to five methylene units separating these groups to examine what linker length was optimal and tested for inhibition against Mtb Dxr. We synthesized ethyl and pivaloyl esters of these compounds to increase lipophilicity and improve inhibition of Mtb growth. Our results show that propyl or propenyl linker chains are optimal. Propenyl analog 22 has an IC50 of 1.07 μM against Mtb Dxr. The pivaloyl ester of 22, compound 26, has an MIC of 9.4 μg/mL, representing a significant improvement in antitubercular potency in this class of compounds.
Keywords: Mycobacterium tuberculosis, Nonmevalonate pathway, Dxr, Antibiotic
Tuberculosis (TB), caused by Mycobacterium tuberculosis (Mtb), remains one of the world's deadliest infectious diseases.1 Emergence of multi-drug (MDR) and extensively-drug (XDR) resistant strains, as well as co-infection with HIV, has made TB both difficult and expensive to treat.2 New TB therapies are needed to shorten treatment, be effective against all strains and metabolic states of the organism, and work well with HIV drugs. Thus, there remains a significant need for new and improved strategies against Mtb. The nonmevalonate pathway (NMP) of isoprene biosynthesis (Figure 1) is essential for Mtb survival and, as it is not present in humans, is an attractive set of targets for novel drug development.3-5 The NMP synthesizes 5-carbon building blocks from pyruvate and glyceraldehyde-3-phosphate. These building blocks are the starting materials for many complex cellular metabolites. 1-Deoxy-D-xylulose-5-phosphate reductoisomerase (Dxr), is the first committed step in the NMP and is responsible for conversion of 1-deoxy-D-xylulose-5-phosphate (DXP) to 2-C-methyl-D-erythritol 4-phosphate (MEP).6 Dxr catalyzes both a reduction and isomerization using NADPH as a cofactor.
Figure 1.
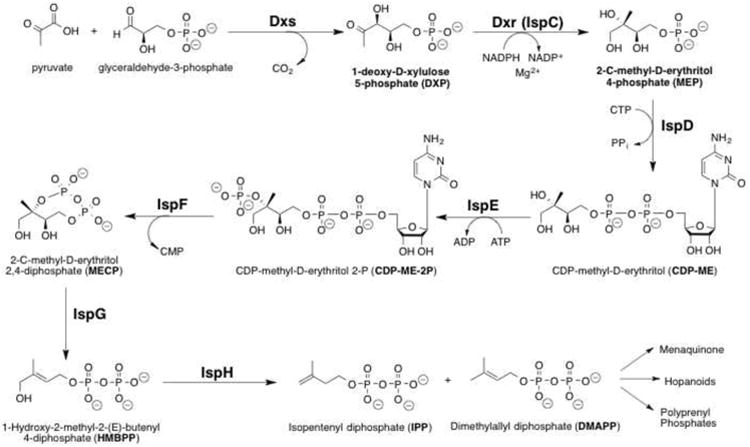
Nonmevalonate Pathway of Isoprenoid Biosynthesis. Dxr (IspC) mediates the conversion of DXP to MEP in the second step.
Natural products fosmidomycin (1) and FR900098 (2) inhibit Mtb Dxr by mimicking DXP's polar character and kill many non-mycobacterial organisms reliant on this enzyme (Figure 2).7-9 Our early work in this area showed that lipophilic analogs of 1 and 2 more effectively kill a range of bacterial strains, including Mtb.10-12 Since that time, we and others have reported Dxr inhibitors belonging to several structural families,11, 13-16 but very few of these have displayed potent antitubercular activity. Many of these inhibitors retain key structural features found in the parent compounds 1 and 2: a retrohydroxamic acid, a phosphonate, and an n-propyl carbon chain linking the nitrogen and phosphorus atoms. In the 1980s, a series of Streptomyces-derived and inspired products exchanging the n-propyl chain for ethylene and propenyl chains were described.17, 18 Among these, the propenyl compound was found to be comparable to the propyl analogs 1 and 2 and showed potent antibacterial activity against B. subtilis and E. coli.18 As this work came before the discovery of Dxr as the cellular target of these inhibitors, the inhibitory activity of these carbon chain-modified analogs against the purified enzymer is largely unknown. To fill this gap and expand on the set of analogs examined, we synthesized analogs of 1 and 2, varying the length of the carbon linker from 2-5 methylene groups. We also prepared the propenyl analog to examine the influence of unsaturation within the propyl chain. as our interest is the development of antitubercular agents working through Dxr inhibition, we evaluated these analogs as inhibitors of Mtb Dxr. To study the effects of these structural changes on antitubercular activity, the ethyl and selected pivaloyl esters were prepared. The compounds synthesized and evaluated are shown in Figure 2.
Figure 2.
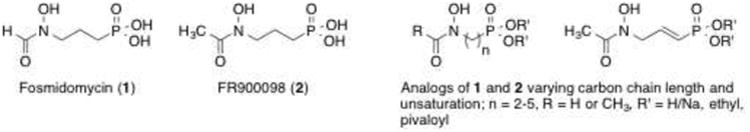
Fosmidomycin (1), FR900098 (2) and the analogs prepared in this work.
Scheme 1 shows the synthetic route used to prepare compounds 7-9, all with a carbon chain of 2 methylene groups. Compound 319 was reacted with N-(diethoxyphosphoryl)-O-benzylhydroxyl-amine20 in the presence of sodium hydride, sodium iodide and tetrabutylammonium bromide to form 4 (25%). Further reaction with concentrated hydrochloric acid gave 5 in quantitative yield.21 Compound 5 was then formylated using acetic anhydride and formic acid to give 6a (71%) or acetylated in the presence of acetyl chloride and triethylamine to give compound 6b (52%). Hydrogenation was used to remove the benzyl group, forming 7 (58%) and 8 (38%). Treatment of 8 with bromotrimethylsilane, water, and sodium hydroxide gave the mono-sodium salt 9 in quantitative yield.
Scheme 1.
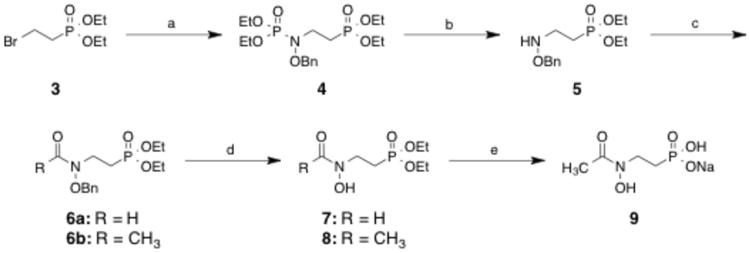
Reagents and conditions: (a) (EtO)2P(O)NHOBn, NaH, Nal, TBABr, THF, reflux, 18 h; (b) HCI, EtOH, reflux, 5 min; (c) AcCI, TEA, CH2CI2, rt, 18 h or Ac2O CH2O2, THF, rt, 2 h; (d) H2, 10% Pd/C, MeOH, 18 h; (e) (i) TMSBr, BSTFA, CH2CI2, 0 °C to rt, 18 h; (ii) H20, rt, 18 h, (iii) NaOH aq., rt, 18 h.
Scheme 2 was used to prepare analogs with four or five methylene groups between the nitrogen and phosphorous atoms. Dibromoalkanes 10a and 10b were treated with triethylphosphite in a microwave-assisted Michaelis-Arbuzov reaction to form 11a (61%) and 11b (64%).22 Acetylated O-benzylhydroxylamine23, 24 was treated with sodium hydride and compounds 11a and 11b to form intermediates 12a (79%) and 12b (37%). Compounds 12a and 12b underwent hydrogenation to form compounds 13 (34%) and 14 (49%). Deprotection of the ethyl esters gave compounds 15 and 16 in quantitative yield.
Scheme 2.
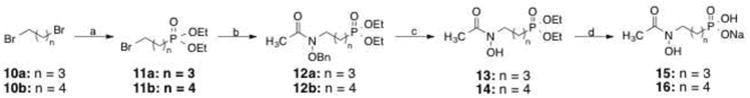
Reagents and conditions: (a) P(OEt)3, microwave 20%, 10-15 min; (b) BnONHAc, NaH, Nal, THF, reflux, 18 h; (c) H2, 10% Pd/C, MeOH, rt, 18 h; (d) (i) TMSBr, CH2CI2, 0 °C to rt, 18 h; (ii) H20, rt, 18 h; (iii) NaOH aq., rt, 18 h.
Synthesis of unsaturated FR900098 analog 22 is shown in Scheme 3. Dibromo compound 1725 was treated with sodium hydride to effect elimination, yielding compound 18 (41%). Boc-protected O-benzylhydroxylamine26 was reacted with sodium hydride and then compound 18 to form substituted product 19 (84%). Alternately, compound 19 was prepared directly from 17 in one step using a single treatment of NaH and the amine in 41% yield. Removal of the BOC protecting group in situ and subsequent acetylation yielded compound 20 (70%).27 To preserve the double bond, BCl3 was used to remove the benzyl group of 20, affording compound 21 (52%).28 Deprotection with bromotrimethylsilane gave α/β-unsaturated phosphonic acid 22 (quantitative).29
Scheme 3.
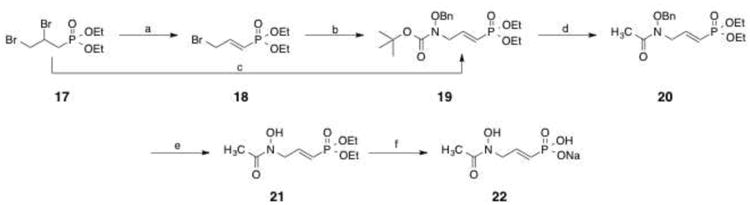
Reagents and conditions: (a) NaH, THF, 60 °C, 18 h; (b) BocNHOBn, NaH, THF, rt, 18 h; (c) BocNHOBn, NaH, Nal, THF, rt, 18 h; (d) (i) AcCI, MeOH, CH2CI2, rt, 30 min; (ii) AcCI, Na2CO3, CH2CI2, rt, 3 h; (e) BCI3, CH2CI2, -50 °C, 2h; (f) (i) TMSBr, BSTFA, CH2CI2, 0 °C to rt, 18 h; (ii) H2O, rt, 18h, (iii) NaOHaq., rt, 18 h.
To assist penetration of compounds across the mycobacterial cell wall10, 30, pivaloyl esters were prepared from two phosphonic acids (Scheme 4). Diethyl protected intermediates 12a and 20 were treated with bromotrimethylsilane yielding compounds 23a (87%) and 23b31 (quantitative). Subsequent reaction with chloromethylpivalate gave esters compounds 24a (6%) and 24b32 (40%). Catalytic hydrogenation removed the benzyl group in saturated analog 24a, yielding compound 25 (85%). Treatment with BCl3 deprotected unsaturated analog 24b to yield compound 26 (13%).33
Scheme 4.
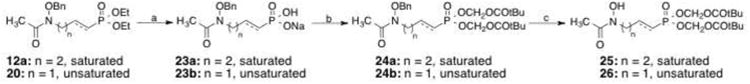
Reagents and conditions: (a) (i) TMSBr, CH2CI2, 0 °C to rt, 3-18 h; (ii) H2O, rt, 18 h for 23a or H2O, NaOH, rt, 18 h for 23b; (b) chloromethylpivalate, 60 °C, TEA/DMF/6-16 h; (c) H2, 10% Pd/C, THF, rt, 18 h for 25 or BCI3, CH2CI2, -70 °C, 10 h for 26.
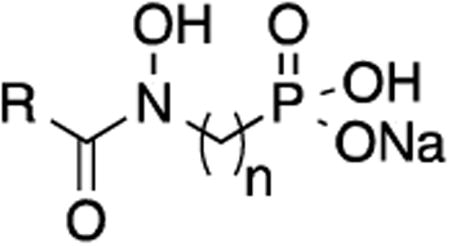
The analogs were evaluated for inhibition of Mtb Dxr and growth of Mtb (Tables 1-3). All of the saturated compounds, with chain lengths between two and five methylene groups, inhibited Mtb Dxr to some extent (Table 1). Among these acids, compounds with three methylene groups separating the nitrogen and phosphorus atoms (that is, compounds 1 and 2) were the most active. Not surprisingly, these compounds did not inhibit mycobacterial growth in nutrient-rich media (>200 μg/mL in 7H9), although 9 had a very slight effect when minimal media was used (150 μg/mL in GAST). The polarity of these compounds diminishes penetration of the lipophilic mycobacterial cell wall.10, 30
Table 1. Effect of chain length on Mtb Dxr inhibition and Mtb MIC.
| ||||
|---|---|---|---|---|
| Compound | R | n | Mtb Dxr IC50, μM (% inh at 100 μM) | MIC, μg/mL 7H9 (GAST) |
| Fosmidomycin (1) | H | 3 | 0.44 | >500 |
| FR900098 (2) | CH3 | 3 | 2.39 | >500 |
| 9 | CH3 | 2 | (74%) | >200 (t50) |
| 15 | CH3 | 4 | (80%) | >200 (>200) |
| 16 | CH3 | 5 | (86%) | >200 (>200) |
Mtb = Mycobacterium tuberculosis; IC50 = inhibitory concentration at 50%; inh = inhibition; MIC = minimum inhibitory concentration; 7H9 = rich media; GAST = minimal media
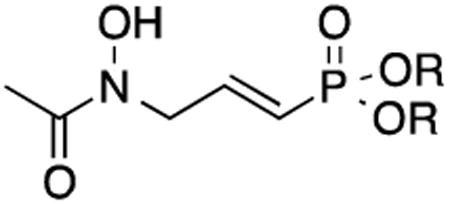
Table 3. Effect of unsaturation on Mtb Dxr inhibition and Mtb MIC.
| |||
|---|---|---|---|
| Compound | R | Mtb Dxr IC50, μM | MIC, μg/mL 7H9 (GAST) |
| 22 | H/Na | 1.07 | >200 (150) |
| 21 | CH2CH3 | ND* | >200 (150) |
| 26 | CH2OCOtBu | ND | 9.4 (12.5) |
ND = not determined
Diethyl and dipivaloyl esterification of these compounds improved antimycobacterial activity (Table 2). As previously shown, diethyl esters of 1 and 2 (27 and 28, respectively) are weakly potent inhibitors of Mtb growth with MIC values of 200-400 μg/mL.10 Pivaloyl ester 29 showed improved potency with an MIC of 50-100 μg/mL, and this compound was the most potent in the saturated series. Taken together, these data show that linker chains of two, four or five methylene units are not advantageous for Mtb Dxr inhibition or inhibition of Mtb cell growth.
Table 2. Effect of esterification on Mtb MIC.
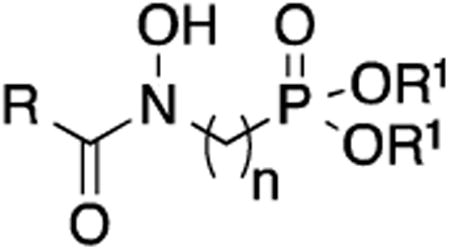
| ||||
|---|---|---|---|---|
| Compound | R | R1 | n | MIC, μg/mL 7H9 (GAST) |
| 27 | H | CH2CH3 | 3 | 400 |
| 7 | H | CH2CH3 | 2 | >500 |
| 8 | CH3 | CH2CH3 | 2 | >500 |
| 28 | CH3 | CH2CH3 | 3 | 200-400 |
| 29 | CH3 | CH2OCOtBu | 3 | 50-100 |
| 13 | CH3 | CH2CH3 | 4 | >200 (75) |
| 25 | CH3 | CH2OCOtBu | 4 | ≥200 (150) |
| 14 | CH3 | CH2CH3 | 5 | >200 (200) |
The compounds listed in Table 3 were synthesized to examine the effect of unsaturation on Mtb Dxr inhibition and cell growth. Interestingly, α/β-unsaturated compound 22 is a potent inhibitor of Mtb Dxr with an IC50 of 1.07 μM. Indeed, 22 is more active than parent compound 2. While 21 and 22 do not inhibit Mtb, the more lipophilic pivaloyl ester of 22 (compound 26) is a potent inhibitor of mycobacterial growth with an MIC of 9.4 μg/mL in rich media and 12.5 μg/mL in minimal media. To our knowledge, compound 26 displays the most potent antitubercular activity of all compounds that work through a Dxr-mediated mechanism.
Overall, the results collectively indicate that a carbon propyl or propenyl chain between the nitrogen and phosphorus atoms of fosmidomycin/FR900098 analogs yields the highest potency. Lipophilic esters of these compounds improve their antitubercular activity. α/β-Unsaturated compound 22 and its lipophilic pivaloyl ester 26 show higher potency than the parent compound FR900098 (2) on Mtb Dxr inhibition and antitubercular activity. These data improve our understanding of the Mtb Dxr active site and its tolerance to length variation between the phosphonate and retrohydroxamate groups. These results are significant for aiding the rational design of Mtb Dxr inhibitors using the phosphonate/retrohydroxamate scaffold and guide the development of Dxr inhibitors as antitubercular agents.
Acknowledgments
This work was supported by the Intramural Research Program of NIAID/NIH, the George Washington University Department of Chemistry, the GWU University Facilitating Fund, and NIH (AI086453 to CSD). RDC was supported by George Mason University's Department of Chemistry and Biochemistry and the U.S. Army Medical Research and Materiel Command W23RYX1291N601.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Dye C, Glaziou P, Floyd K, Raviglione M. Annu Rev Public Health. 2013;34:271. doi: 10.1146/annurev-publhealth-031912-114431. [DOI] [PubMed] [Google Scholar]
- 2.WHO. Global Tuberculosis Report. 2012 [Google Scholar]
- 3.Rohmer M. Lipids. 2008;43:1095. doi: 10.1007/s11745-008-3261-7. [DOI] [PubMed] [Google Scholar]
- 4.Perez-Gil J, Rodriguez-Concepcion M. Biochem J. 2013;452:19. doi: 10.1042/BJ20121899. [DOI] [PubMed] [Google Scholar]
- 5.Grawert T, Groll M, Rohdich F, Bacher A, Eisenreich W. Cell Mol Life Sci. 2011;68:3797. doi: 10.1007/s00018-011-0753-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jackson ER, Dowd CS. Curr Top Med Chem. 2012;12:706. doi: 10.2174/156802612799984599. [DOI] [PubMed] [Google Scholar]
- 7.Mine Y, Kamimura T, Nonoyama S, Nishida M, Goto S, Kuwahara S. J Antibiot. 1980;33:36. doi: 10.7164/antibiotics.33.36. [DOI] [PubMed] [Google Scholar]
- 8.Kuroda Y, Okuhara M, Goto T, Okamoto M, Terano H, Kohsaka M, Aoki H, Imanaka H. J Antibiot. 1980;33:29. doi: 10.7164/antibiotics.33.29. [DOI] [PubMed] [Google Scholar]
- 9.Iguchi E, Okuhara M, Kohsaka M, Aoki H, Imanaka H. J Antibiot. 1980;33:19. [PubMed] [Google Scholar]
- 10.Uh E, Jackson ER, San Jose G, Maddox M, Lee RE, Boshoff HI, Dowd CS. Bioorg Med Chem Lett. 2011;21:6973. doi: 10.1016/j.bmcl.2011.09.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.San Jose G, Jackson ER, Uh E, Johny C, Haymond A, Lundberg L, Pinkham C, Kehn-Hall K, Boshoff HI, Couch RD, Dowd CS. MedChemComm. 2013;4:1099. doi: 10.1039/C3MD00085K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McKenney ES, Sargent M, Khan H, Uh E, Jackson ER, San Jose G, Couch RD, Dowd CS, van Hoek ML. PLoS One. 2012;7:e38167. doi: 10.1371/journal.pone.0038167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Andaloussi M, Henriksson LM, Wieckowska A, Lindh M, Bjorkelid C, Larsson AM, Suresh S, Iyer H, Srinivasa BR, Bergfors T, Unge T, Mowbray SL, Larhed M, Jones TA, Karlen A. J Med Chem. 2011;54:4964. doi: 10.1021/jm2000085. [DOI] [PubMed] [Google Scholar]
- 14.Deng L, Diao J, Chen P, Pujari V, Yao Y, Cheng G, Crick DC, Prasad BV, Song Y. J Med Chem. 2011;54:4721. doi: 10.1021/jm200363d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jansson AM, Wieckowska A, Bjorkelid C, Yahiaoui S, Sooriyaarachchi S, Lindh M, Bergfors T, Dharavath S, Desroses M, Suresh S, Andaloussi M, Nikhil R, Sreevalli S, Srinivasa BR, Larhed M, Jones TA, Karlen A, Mowbray SL. J Med Chem. 2013;56:6190. doi: 10.1021/jm4006498. [DOI] [PubMed] [Google Scholar]
- 16.Verbrugghen T, Vandurm P, Pouyez J, Maes L, Wouters J, Van Calenbergh S. J Med Chem. 2013;56:376. doi: 10.1021/jm301577q. [DOI] [PubMed] [Google Scholar]
- 17.Hashimoto M, Hemmi K, Takeno H, Kamiya T. Tetrahedron Lett. 1980;21:99. [Google Scholar]
- 18.Hemmi K, Takeno H, Hashimoto M, Kamiya T. Chem Pharm Bull. 1982;30:111. doi: 10.1248/cpb.30.111. [DOI] [PubMed] [Google Scholar]
- 19.Balczewski P, Pietrzykowski WM. Tetrahedron. 1996;52:13681. [Google Scholar]
- 20.Zwierzak A, Osowska K. Synthesis. 1984;3:223. [Google Scholar]
- 21.Blazewska K, Gajda T. Tetrahedron. 2003;59:10249. [Google Scholar]
- 22.Villemin D, Simeon F, Decreus H, Jaffres PA. Phosphorus, Sulfur Silicon Relat Elem. 1998;133:209. [Google Scholar]
- 23.Ortmann R, Wiesner J, Reichenberg A, Henschker D, Beck E, Jomaa H, Schlitzer M. Arch Pharm. 2005;338:305. doi: 10.1002/ardp.200500976. [DOI] [PubMed] [Google Scholar]
- 24.Reichenberg A, Wiesner J, Weidemeyer C, Dreiseidler E, Sanderbrand S, Altincicek B, Beck E, Schlitzer M, Jomaa H. Bioorg Med Chem Lett. 2001;11:833. doi: 10.1016/s0960-894x(01)00075-0. [DOI] [PubMed] [Google Scholar]
- 25.Laureyn I, Stevens CV, Soroka M, Malysa P. ARKIVOC. 2003;iv:102. [Google Scholar]
- 26.Kadi N, Oves-Costales D, Barona-Gomez F, Challis GL. Nat Chem Biol. 2007;3:652. doi: 10.1038/nchembio.2007.23. [DOI] [PubMed] [Google Scholar]
- 27.Compound 20. Acetyl chloride (1.8mL, 25.0mmol) and dry MeOH (0.5mL) were added dropwise to 19 (0.19g, 2.5mmol) in dry CH2Cl2 (6mL) under N2. The reaction mixture was allowed to stir at rt for 30 min. Dry Na2CO3 (0.5g, 5.0mmol) and additional acetyl chloride (0.7mL, 9.8mmol) were added, and the reaction mixture was allowed to stir at rt for 3h. The reaction mixture was filtered over celite, and the solvent was removed under reduced pressure. The crude oil was purified by column chromatography using silica gel (CH2Cl2:MeOH, 49:1) to yield 20 (0.13g, 0.36mmol, 77%) as a clear, colorless oil. 1H NMR (CDCl3, 200MHz), δ (ppm): 7.37 (s, 5H), 6.82-6.57 (m, 1H), 5.90-5.72 (m, 1H), 4.83 (s, 2H), 4.34 (bs, 2H), 4.13-3.99 (m, 4H), 2.13 (s, 3H), 1.34-1.27 (m, 6H). LCMS (ESI) m/z: 705.1 (2M+Na).
- 28.Compound 21. A solution of 20 (0.117g, 0.34mmol) in dry CH2Cl2 (6.0 mL) was cooled to -50°C and BCl3 (1.4 mL, 1M in CH2Cl2) was added dropwise, and the reaction mixture was allowed to stir for 2h. The reaction was quenched with saturated NaHCO3 (aq, 9.0 mL) and allowed to warm to rt. The aqueous solution was extracted with CH2Cl2. The organic fractions were combined, dried over MgSO4, filtered and the solvent was removed under reduced pressure. The resulting crude residue was purified using an Isolera Flash Chromatography system and a silica column (EtOAcMeOH, 49:1) to yield 21 (45mg, 0.18mmol, 52%) as a light yellow oil. 1H NMR (200 MHz, CDCl3) δ (ppm): 1.28 (t, 6H), 2.15 (s, 3H), 4.02 (q, 4H),4.43 – 4.25 (m, 2H), 5.84 (t, J = 18.8 Hz, 1H), 6.83 - 6.52 (m, 1H), 9.88 (bs, 1H). 13C NMR (50 MHz, CDCl3) δ (ppm): 16.30, 20.36, 50.47 (d, J = 27.2 Hz), 62.37 (d, J = 7.0 Hz), 120.02, 147.62, 172.66. LCMS (ESI) m/z 252.1 (M+H).
- 29.Compound 22. N,O-bis(trimethylsilyl)trifluoroacetamide (0.18mL, 0.67mmol) was added to 21 (0.03g, 0.12mmol) in CH2Cl2 (0.60mL) under N2. The reaction mixture was allowed to stir at rt for 20 min. The reaction mixture was cooled to 0 °C, and bromotrimethylsilane (0.18mL, 1.34mmol) was added dropwise. The reaction was allowed to warm to rt and was stirred overnight under N2. Ethyl bromide and excess silylating agent were removed under reduced pressure, and the residue was dissolved in aqueous NaOH (0.68mL, 7.8mg/mL) and stirred overnight. The mixture was extracted between H2O and CH2Cl2. The aqueous portions were combined, and the solvent was removed by lyophilization to give 22 (0.03g, 0.12mmol, quant.) as a yellow solid. 1H NMR (400 MHz, D2O) δ (ppm): 2.36 (s, 3H), 4.61 – 4.46 (m, 2H), 6.19 – 6.06 (m, 1H), 6.56 – 6.43 (m, 1H). 13C NMR (101 MHz, D2O) δ (ppm): 19.34, 50.81 (d, J = 23.7 Hz), 126.91, 137.79, 174.18. HRMS (ESI) m/z calcd. for C10H18N2NaO10P2 (2M + Na]): 411.0328, found: 411.0334.
- 30.Brown AC, Parish T. BMC Microbiol. 2008;8:78. doi: 10.1186/1471-2180-8-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Compound 23b. Trimethylsilylbromide (1.75mL, 11.7mmol) was added dropwise to a stirring solution of 20 (0.5g, 1.5mmol) in dry CH2Cl2 (20mL) under N2 at 0°C. The reaction mixture was allowed to warm to room temperature. After 3.5 h, the mixture was evaporated to dryness, dissolved in dry CH2Cl2, and evaporated to dryness again (×3). The resulting residue was stirred overnight in water (3mL) and NaOH (5.5mL, 3mmol, aq.). After 20 hours, the aqueous mixture was washed with CH2Cl2. The organic portion was separated, and the water was removed by lyophilization to give 23b (0.53g, quant.) as white crystals. 1H NMR (CDCl3, 400MHz), δ (ppm): 2.28 (s, 3H), 4.53 (s, 2H), 5.17 (s, 2H), 6.07-6.15 (m, 1H), 6.30-6.40 (m, 1H), 7.65-7.68 (m, 5H). LCMS (ESI) m/z: 286 (Macid+H), 571 (2Macid+H), 856 (3Macid+H).
- 32.Compound 24b. Chloromethylpivalate (2.15mL, 15mmol) was added to a stirred solution of 23b (0.49g, 1.5mmol) and triethylamine (0.45mL, 3mmol) in DMF (40mL). The reaction mixture was heated to 60°C for 16 hours. Water (50mL) was added, and the aqueous layer was extracted with CH2Cl2. The organic layer was washed with brine, dried over MgSO4, and the solvent was removed under reduced pressure. The crude oil was purified by column chromatography using silica gel and CH2Cl2/EtOAc to yield 24b (0.22g, 28%). 1H NMR (CDCl3, 400MHz), δ (ppm): 1.20 (s, 18H), 2.12 (s, 3H), 4.33 (bs, 2H), 4.82 (s, 2H), 5.65 (d,/= 12.8 Hz, 4H), 5.80-5.89 (m, 1H), 6.70-6.81 (m, 1H), 7.35-7.38 (m, 5H). LCMS (ESI) m/z: 536 (M+Na).
- 33.Compound 26. BCl3 (1M in CH2Cl2, 0.88mL) was added dropwise to a stirred solution of 24b (190mg, 0.37mmol) in dry CH2Cl2 (5mL) under N2 at -78°C. After 10 hours, the reaction mixture was poured into satd. NaHCO3 (aq.) and was extracted with CH2Cl2. The organic layers were combined and washed with brine, dried over MgSO4, and the solvent was removed under reduced pressure. The crude oil was purified by column chromatography using EtOAc, CH2Cl2, and MeOH. The oil was further purified over a silica plug, washed with hexanes and CH2Cl2, and then eluted with EtOAc to give 26 as a pale yellow oil (21mg, 13.4%). 1H NMR (CDCl3, 400MHz), δ (ppm): 1.22 (s, 18H), 2.19 (s, 3H), 4.41 (s, 2H), 5.61-5.69 (m, 4H), 5.87-5.97 (m, 1H), 6.71-6.84 (m, 1H), 8.61 (s, 1H). 13C NMR (CDCl3, 100MHz), δ (ppm): 20.47, 26.93, 38.88, 50.30 (d,/= 26.5 Hz), 81.70 (d,/= 5.3 Hz), 117.95 (d,/= 188.2 Hz), 148.50, 172.87, 177.30. LCMS (ESI) m/z: 446 (M+Na), 847 (2M+H), 869 (2M+Na).