

Abstract
The mammalian striatin family consists of three proteins, striatin, S/G2 nuclear autoantigen, and zinedin. Striatin family members have no intrinsic catalytic activity, but rather function as scaffolding proteins. Remarkably, they organize multiple diverse, large signaling complexes that participate in a variety of cellular processes. Moreover, they appear to be regulatory/targeting subunits for the major eukaryotic serine/threonine protein phosphatase 2A. In addition, striatin family members associate with germinal center kinase III kinases as well as other novel components, earning these assemblies the name striatin-interacting phosphatase and kinase (STRIPAK) complexes. Recently, there has been a great increase in functional and mechanistic studies aimed at identifying and understanding the roles of STRIPAK–like complexes in cellular processes of multiple organisms. These studies have identified novel STRIPAK or STRIPAK-like complexes and have explored their roles in specific signaling pathways. Together, the results of these studies have sparked increased interest in striatin family complexes because they have revealed roles in signaling, cell cycle control, apoptosis, vesicular trafficking, Golgi assembly, cell polarity, cell migration, neural and vascular development, and cardiac function. Moreover, STRIPAK complexes have been connected to clinical conditions, including cardiac disease, diabetes, autism, and cerebral cavernous malformation. In this review, we discuss the expression, localization, and protein domain structure of striatin family members. Then we consider the diverse complexes these proteins and their homologs form in various organisms, emphasizing what is known regarding function and regulation. Finally, we will explore possible roles of striatin family complexes in disease, especially cerebral cavernous malformation.
Keywords: STRIPAK, Striatin, CCM, GCKIII, Disease
1. Introduction
The mammalian striatin family consists of three proteins, striatin (HUGO Gene Nomenclature Committee (HGNC) approved symbol, STRN), S/G2 nuclear autoantigen (SG2NA; HGNC approved symbol, STRN3), and zinedin (HGNC approved symbol, STRN4) (Benoist et al., 2006). Of note, there is not a STRN2 member of the striatin family because, confusingly, STRN2 is an alias for an unrelated protein, striamin. While the striatin family of proteins are highly expressed in the central and peripheral nervous systems and are probably important for brain function, they are also expressed in many other tissues. Thus, while they may have specialized functions, they likely also carry out one or more functions common to many different cell types. Striatin family members have no intrinsic catalytic activity, but rather function as scaffolding proteins harboring a variety of protein-protein interaction domains. Strikingly, they are capable of organizing multiple diverse, large signaling complexes, which participate in a variety of cellular processes.
To date, the only striatin family-associated proteins found in nearly all striatin family complexes are the structural A and catalytic C subunits of protein phosphatase 2A (PP2A) (Moreno et al., 2000), a major eukaryotic serine/threonine phosphatase that exists primarily as heterotrimers made of an A subunit, a C subunit, and one of many “B-type” regulatory/targeting subunits. Based on the altered substrate specificity of striatin family-associated PP2A and the lack of other B-type subunits in these complexes, striatin family members were postulated to comprise a novel B‴ family of PP2A B-type regulatory subunits (Moreno et al., 2000).
Recent proteomic analysis of striatin family-associated proteins revealed the additional presence of germinal center kinase III (GCKIII) kinases together with PP2A and other components, earning these striatin family complexes the name striatin-interacting phosphatase and kinase (STRIPAK) complexes (Goudreault et al., 2009). In addition, separate STRIPAK-like complexes have been found that are not yet known to contain both PP2A and a kinase.
In the last few years, members of striatin family complexes have been connected to clinical conditions. These include cerebral cavernous malformation (CCM), a common type of angioma that can cause symptoms ranging from headaches to stroke, cardiac dysfunction, cancer, diabetes, and autism. While striatin family complexes have been linked to these conditions, specific mechanistic roles for STRIPAK complexes in disease remain to be delineated.
Recently, there has been a great increase in the number of functional and mechanistic studies aimed at identifying and understanding the roles of STRIPAK and STRIPAK–like complexes in cellular processes of multiple organisms. Novel complexes have been identified, and their roles in specific signaling pathways have been explored, often by mutating or depleting the striatin family member or associated proteins. Together, the results of these studies have sparked increased interest in striatin family complexes because they have revealed roles in signaling, cell cycle control, apoptosis, vesicular trafficking, Golgi assembly, cell polarity, cell adhesion, cell migration, neural and vascular development, and disease.
In this review, we will first introduce striatin, SG2NA, and zinedin and briefly discuss their expression, localization, and protein domain structure. Next, we will consider the diverse complexes these proteins and their homologs form from yeast to man, including what is known regarding function and regulation. Finally, we will explore possible roles of STRIPAK complexes in disease.
2. The striatin family of proteins
The mammalian striatin family of proteins comprises three highly homologous proteins whose domain structure and intracellular localization are very similar. Functional homologs exist in other species as distant as fungi (Bloemendal et al., 2012, Lisa-Santamaria et al., 2012, Poggeler and Kuck, 2004, Tanabe et al., 2001), implying the conservation of at least one cellular function. The presence of multiple striatin family members in higher eukaryotes and distinct expression patterns for these proteins in different tissues suggests that different striatin family members may have both redundant and specialized functions, which may be cell-type specific.
2.1. Striatin
Striatin, named after the striatum where it is found most abundantly in the brain, was first isolated from a rat brain synaptosomal fraction (Castets et al., 1996). Striatin is a 780 amino acid protein with four protein-protein interaction domains including a caveolin-binding domain, a coiled-coil domain, a Ca2+-calmodulin (CaM)-binding domain, and a Tryptophan-Aspartate (WD)-repeat domain (Fig. 1) (Castets et al., 2000). Consistent with the presence of the caveolin-binding and Ca2+-CaM-binding domains, striatin has been reported to bind caveolin-1 (Cav-1) and binds CaM in the presence of Ca2+ (Castets et al., 1996, Gaillard et al., 2001). Striatin is found throughout the central and peripheral nervous systems, especially in the striatum and motor neurons; however, its expression is also detected in many other tissues including, but not limited to, lung, liver, kidney, skeletal and cardiac muscle, testes, B and T lymphocytes, and fibroblasts (Castets et al., 1996, Castets et al., 2000, Moqrich et al., 1998, Moreno et al., 2000). Because the localization of striatin family members in the central and peripheral nervous systems has been reviewed previously (Benoist et al., 2006), it will only be discussed briefly here. Striatin exhibits a somato-dendritic localization in neurons with a high density in dendritic spines (Castets et al., 1996, Salin et al., 1998). In contrast, striatin is largely absent from axons. The enrichment of striatin within dendritic spines and the presence of a functional Ca2+-CaM-binding domain prompted the early hypothesis that striatin function is likely regulated by Ca2+-dependent signaling in postsynaptic neurons (Castets et al., 1996). Consistent with this possibility, the distribution of striatin in cytosolic, detergent soluble, and detergent-insoluble fractions of brain was found to change depending on the presence or absence of calcium during lysis (Bartoli et al., 1998). Striatin’s distribution in brain structures involved in motor control led to the suggestion that striatin may be involved in control of motor function (Salin et al., 1998). Support for this idea was obtained subsequently through experiments in which striatin was downregulated in rat brains by intracerebro-ventricular infusion of striatin antisense oligonucleotides, resulting in decreased striatin in the striatum and reduced nocturnal locomotor activity (Bartoli et al., 1999). Moreover, downregulation of striatin in motor neurons in vitro impaired the growth of dendrites but not axons, supporting a role for striatin in dendritic growth and remodeling (Bartoli et al., 1999). This latter role is also supported by more recent data indicating that Drosophila Mob4 (dMob4), a functional homolog of the striatin-associated protein, Mob3/phocein (referred to as Mob3 from here on), regulates neurite outgrowth in Drosophila (Schulte et al., 2010). Thus, striatin is implicated broadly in neuronal function.
Fig. 1. Domain structures of striatin family members.
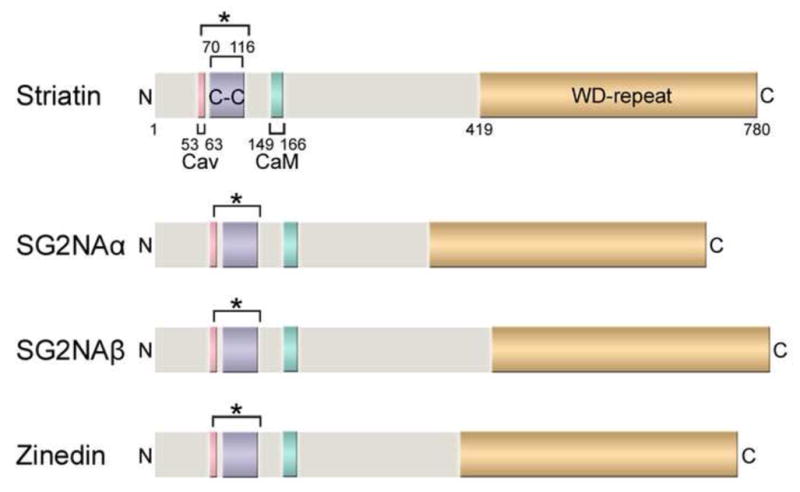
The domain structures of the human striatin family proteins, including striatin (780 amino acids), the two major isoforms of SG2NA (SG2NAα: 713 aa; SG2NAβ: 797 aa), and zinedin (753 aa), are shown drawn to scale. Four protein-protein interaction domains (labeled for striatin and color-coded for comparison in SG2NAα/β and zinedin) are highly conserved among the striatin family members and also throughout different species. Cav: Caveolin-binding domain; C-C: Coiled-coil domain; CaM: Ca2+-CaM-binding domain; WD-repeat: WD-repeat domain. The bracket with an asterisk (*) denotes the putative extended coiled-coil domain regions based on analyses using NCOILS and Paircoil2 algorithms (Gordon et al., 2011). Of note, some of the domains in SG2NA and zinedin are only predicted regions based on sequence comparisons and have not been experimentally verified.
2.2. S/G2 nuclear autoantigen (SG2NA)
Similar to striatin, SG2NA binds to CaM in the presence of Ca2+ and is characterized by the four protein-protein interaction domains common to striatin family members (Fig. 1) (Castets et al., 2000, Moreno et al., 2000). Two major isoforms of SG2NA exist as a result of alternative splicing: a 713 amino acid protein, SG2NAα, which excludes exons 8 and 9, and a full-length 797 amino acid protein, SG2NAβ (Fig. 1) (Benoist et al., 2006). Additional, more minor splice variants also exist (Benoist et al., 2006, Sanghamitra et al., 2008). SG2NA was first cloned using autoantibodies from a cancer patient (Muro et al., 1995). Based on immunofluorescence using both crude and affinity-purified patient sera, SG2NA was first reported to be a nuclear protein whose expression level peaked during the S and G2 phases of the cell cycle (Muro et al., 1995). Seemingly paradoxically, SG2NA was subsequently shown by others to be primarily a cytosolic and membrane-bound protein like striatin (Castets et al., 2000, Moreno et al., 2001). The reason for the almost exclusive nuclear staining using cancer patient antisera is not known, but it is not due to a difference in cell type used since the two different antibody staining patterns were found when the same cell type was used (Baillat et al., 2001, Zhu et al., 2001). The cancer patient serum may recognize an SG2NA epitope only accessible in immunofluorescence staining on a nuclear-localized splice variant of SG2NA. Consistent with this possibility, rSTRN3γ, a novel, nuclear-localized splice variant of rat SG2NA lacking all but one WD-repeat was recently reported to organize an estrogen-inducible complex of PP2A and estrogen receptor α (ERα) (Tan et al., 2008). Also consistent with possible nuclear function, the N-terminal region of SG2NA has been reported to possess transcriptional activation activity, although this activity was largely absent in the context of the full-length protein (Zhu et al., 2001). In brain, SG2NA shows the highest expression in cerebellum and cortex. Like striatin, SG2NA exhibits somato-dendritic localization in neurons with high concentration in dendritic spines and is also found in other tissues (Castets et al., 2000, Moreno et al., 2001).
2.3. Zinedin
Zinedin, a 753 amino acid protein, was identified and cloned through a homology search for proteins highly homologous to striatin and SG2NA (Castets et al., 2000). Like striatin and SG2NA, zinedin binds to CaM in a Ca2+-dependent manner and shares the four protein-protein interaction domains common to striatin family members (Fig. 1) (Castets et al., 2000). In brain, zinedin is expressed most abundantly in the hippocampus (Benoist et al., 2008). Similar to other striatin family members, zinedin shows somato-dendritic localization in neurons with high concentration in dendritic spines and is expressed in a variety of other tissues (Benoist et al., 2008, Castets et al., 2000, Gaillard et al., 2006, Gordon et al., 2011).
3. Domain structure of the striatin family proteins
As mentioned above, four conserved protein-protein interaction domains are found in all three striatin family members (Fig. 1) (Castets et al., 2000). These domains provided some of the first clues as to possible functions of striatin family members as well as to their regulation. Subsequent studies validated the importance of these domains for assembly of striatin family complexes, for proper subcellular localization, and for interaction with important signaling molecules and pathways. Below, we briefly discuss the four main interaction domains and the available data regarding their possible roles in striatin family function.
3.1. The caveolin-binding domain
Caveolins are small integral membrane proteins that are major components of caveolae, specialized, invaginated, cholesterol-rich lipid rafts in the plasma membrane of cells (for reviews, see (Boscher and Nabi, 2012, Chidlow and Sessa, 2010, Parton and Simons, 2007)). The density of caveolae varies with cell type and caveolae are especially abundant in vascular endothelial cells, smooth muscle cells, fibroblasts, adipocytes, and epithelial cells. Caveolin-1 (Cav-1) is synthesized in the endoplasmic reticulum, where it oligomerizes and then is transported to the Golgi apparatus ((Hayer et al., 2010) and references therein). At the Golgi, Cav-1 oligomers interact with cholesterol and the Cav-1-cholesterol complexes are transported to the plasma membrane where additional proteins termed cavins participate in caveolae formation and regulation. In addition, oligomerized Cav-1 scaffolds exist in the plasma membrane apart from caveolae and participate in Cav-1-dependent regulation of signaling not occurring in the caveolae (Lajoie et al., 2009).
Caveolins interact with many signaling proteins via an approximately 20 amino acid stretch of amino acids in their N-termini termed the caveolin scaffolding domain (Li et al., 1996). Striatin family members contain a caveolin scaffolding domain-interaction motif corresponding to the consensus φXXXXφXXφ, where φ is an aromatic amino acid and X is any amino acid (Castets et al., 2000, Couet et al., 1997). This motif is conserved in striatin family homologs from many species, including Drosophila (Chen et al., 2002), which has no recognizable caveolins (Le Lay and Kurzchalia, 2005). Consistent with the presence of this motif, mammalian striatin, SG2NA, and zinedin were reported to interact with Cav-1 (Gaillard et al., 2001).
Little is known about the possible functional role of the striatin-caveolin interaction. This interaction has been hypothesized to be important for localization of striatin to Cav-1-rich dendritic spines of neurons (Benoist et al., 2006) but this possibility needs to be tested. Association of Cav-1 with striatin family members may have relevance to striatin’s role in organizing a membrane signaling complex for rapid, non-genomic activation of endothelial nitric oxide synthase (eNOS) by ERα(Lu et al., 2004) (Fig. 2). Since rapid estrogen activation of eNOS is known to occur in caveolae (Chambliss and Shaul, 2002, Chambliss et al., 2000, Kim et al., 1999) and to be regulated by caveolin (for review, see (Mineo and Shaul, 2012)), one might presume that at least a portion of plasma membrane-bound striatin is localized to caveolae. Thus, like eNOS, it is possible that striatin is targeted to caveolae via its interaction with Cav-1. In turn, striatin likely recruits ERα to caveolae since overexpression of striatin increases the amount of ERα at the plasma membrane (Lu et al., 2004). More research is necessary to investigate these possibilities. In this regard, Cav-1 null mice and derivative cells (Razani et al., 2001) should be very useful for determining whether the loss of Cav-1 alters the localization, assembly, or function of striatin family complexes.
Fig. 2. Model of the STRIPAK complex in signaling pathways.
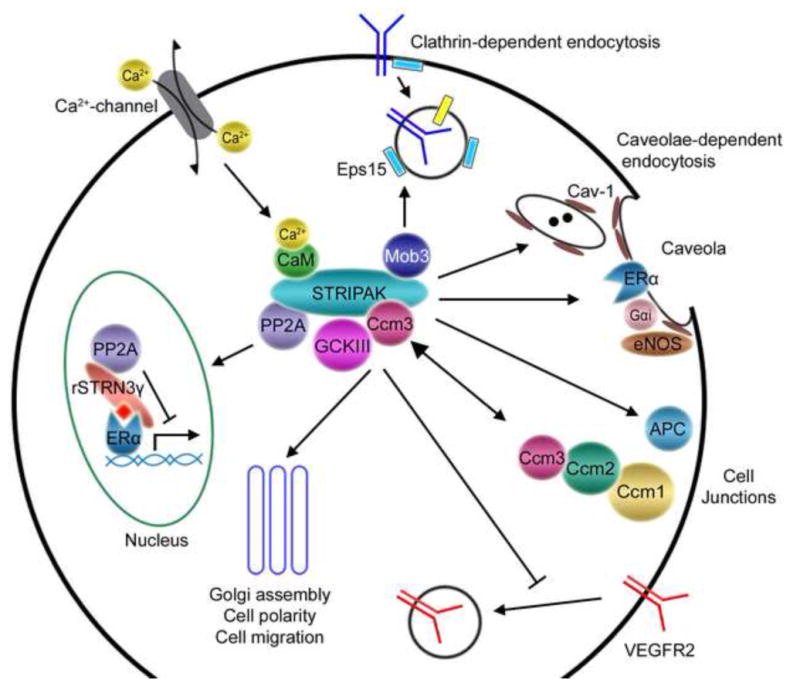
The core STRIPAK complex is depicted in the center of the diagram with some of the STRIPAK components that are thought to mediate several functions of the STRIPAK complex. Other STRIPAK components are omitted for simplicity of the figure. Some of the known or potential connections of STRIPAK components to signaling pathways and cellular events are depicted by arrows and described below starting with clathrin-dependent endocytosis and proceeding clockwise around the depicted cell. First, through Mob3 the STRIPAK complex may associate with components of clathrin-dependent endocytosis (for example, Eps15) and function in this process. Second, the STRIPAK complex might also function in caveolae-dependent endocytosis by interacting with Cav-1. Third, striatin targets ERα to membranes, probably caveolae, and forms a STRIPAK-like complex containing ERα, Gαi, and eNOS to regulate rapid nongenomic ERα signaling. Fourth, striatin binds APC and regulates organization of TJs. Fifth, Ccm3 forms dynamic complexes with either STRIPAK or other Ccm proteins to regulate GCKIII activity and function and cell junction stability. Sixth, Ccm3 stabilizes VEGFR2 on the cell surface by inhibiting its internalization. Seventh, GCKIII kinases, Ccm3, and STRIPAK function in Golgi assembly, cell polarity, and cell migration. Eighth, rat STRN3γ and PP2A modulate ERα-mediated transcriptional activity in nucleus. Ninth, the STRIPAK complex is predicted to be involved in Ca2+ signaling by binding to Ca2+-CaM through the Ca2+-CaM-binding domain, thus responding to changing Ca2+ concentrations in cells. While only changes in Ca2+ concentrations due to influx of extracellular calcium are indicated as an example, Ca2+ concentration changes resulting from release of intracellular calcium such as in response to inositol trisphosphate production might also regulate STRIPAK function.
3.2. The Ca2+-Calmodulin-binding domain
All striatin family members bind CaM in a Ca2+-dependent manner (Castets et al., 1996, Castets et al., 2000, Moreno et al., 2000). Bartoli and colleagues used deletion mapping and site-directed mutagenesis to narrow down the domain on striatin responsible for Ca2+-CaM-binding and to identify critical residues (Bartoli et al., 1998). They designated striatin amino acids 149-166 as the Ca2+-CaM-binding domain because of the basic amphiphilic helical nature of this amino acid stretch, a feature common to many Ca2+-CaM-binding domains in proteins, and because of the ability of a W155G/Q158P double mutation in striatin to completely abolish Ca2+-CaM binding (Bartoli et al., 1998). However, the exact limits of the Ca2+-CaM-binding domain need to be established experimentally.
Currently, it is not clear how the striatin family of proteins functions in Ca2+ signaling. It has been hypothesized that they might function as Ca2+ sensors that respond to changes in intracellular Ca2+ concentration and convey the signals to other proteins (Benoist et al., 2006) (Fig. 2). Interestingly, the presence of calcium reduced the interaction of striatin with GST-Cav-1 (Gaillard et al., 2001) while, in separate experiments, the presence of physiologically relevant calcium levels during cell lysis increased the amount of striatin found in the cytosol (Bartoli et al., 1998). Together, these results suggest that calcium signaling may modulate striatin family interaction with Cav-1 and thus regulate striatin family subcellular localization. Of note, this may be analogous to what occurs with eNOS regulation by Cav-1 and Ca2+-CaM. Cav-1 inhibits eNOS both in vitro and in vivo, and Ca2+/CaM plays a positive role in dissociation of eNOS from Cav-1 (Garcia-Cardena et al., 1997, Ju et al., 1997, Michel et al., 1997, Razani et al., 2001). Moreover, estradiol has been reported to cause Ca2+-dependent translocation of eNOS from the plasma membrane to intracellular sites proximal to the nucleus (Goetz et al., 1999). Thus, based on these data one might hypothesize that the estrogen-activated striatin-ERα-eNOS rapid signaling complex (Lu et al., 2004) might be regulated at the level of both striatin and eNOS by opposing effects of Cav-1 and Ca2+-CaM binding.
Recently, it was found that deletion of the Ca2+-CaM-binding domain of striatin greatly enhances the binding of the GCKIII subfamily sterile 20-like kinases, Mst3 and Mst4, to striatin (Gordon et al., 2011). Binding of another member of the complex, Mob3, was unaffected. Similar deletions on either side of the Ca2+-CaM-binding domain had little to no effect on Mst3 binding (Mst4 binding was not assayed). Thus, the Ca2+-CaM-binding domain may negatively regulate the binding of Mst3 and Mst4 to wild-type striatin. Although direct testing of an effect of Ca2+-CaM binding on Mst3 and Mst4 association with striatin has yet to be done, it is tempting to speculate based on calcium effects on striatin localization that Ca2+-CaM binding may regulate Mst3 and Mst4 binding to striatin by altering striatin subcellular localization.
3.3. The coiled-coil domain
Oligomerization of a wide variety of proteins is enabled by α-helical coiled-coils in which two or more α-helices intertwine like strands of a rope (for a review, see (Burkhard et al., 2001)). Hetero-oligomerization of striatin family members was first discovered for striatin and SG2NA by coimmunoprecipitation of the endogenous proteins (Moreno et al., 2001). Subsequently, this finding was confirmed and extended to demonstrate homo-oligomerization of SG2NA and hetero-oligomerization of zinedin with SG2NA and striatin (Gaillard et al., 2006). Based on these data, all striatin family members likely form homo-and hetero-oligomers. By deletion analysis of SG2NA, the coiled-coil domain was shown to be critical for these higher order associations to occur (Gaillard et al., 2006).
Initially, amino acids 70-116 of striatin were identified using the computer algorithms Coil and Paircoil as the likely coiled-coil domain (Castets et al., 2000). However, when the predicted coiled-coil domain of SG2NA (corresponding to amino acids 65-115 of striatin) was fused to GFP, oligomerization did not occur (Castets et al., 2000). More recently, it was found that deletion of striatin amino acids 53-66, which include the caveolin-binding domain (53-63), abolished striatin oligomerization (Gordon et al., 2011). These findings prompted a re-examination of the N- and C-terminal limits of striatin family oligomerization domains using NCOILS and Paircoil2 algorithms, resulting in the conclusion that the coiled-coil domain of striatin family members has a very high probability of including striatin amino acids 64-120 (Gordon et al., 2011). Moreover, there was a high probability that the N-terminal end of the domain might extend even further to include part or the entire caveolin-binding domain (Fig. 1, brackets with asterisk). Finally, it was suggested that the complete loss of striatin oligomerization caused by loss of amino acids 53-66 was consistent with the possibility that a trigger sequence, a short sequence absolutely necessary for coiled-coil formation, might exist at the N-terminal end of the striatin family coiled-coil (Gordon et al., 2011). These possibilities will need to be investigated further to better understand the mechanism of, and residues involved in, striatin family oligomerization.
Determining the higher order oligomeric state of striatin family members could provide important insights into the organization of striatin family complexes and into the molecular mechanisms of their regulation and function. Being composed of heptad repeats, the striatin family coiled-coil domain likely forms a left-handed coiled-coil. However, whether striatin family members oligomerize in a parallel or antiparallel manner and form dimers, trimers, or even higher order oligomers is not known. An antiparallel association of striatin family members has been proposed based on sequence analysis (Gaillard et al., 2006). Such an arrangement might help explain how Ca2+-CaM binding on one side of the coiled-coil domain might reduce caveolin binding on the other side because these domains would be in proximity to one another (Gaillard et al., 2006). Analyses with Multicoil, PrOCoil, and SCORER 2.0 suggest that striatin family coiled-coil domains are more likely to form a parallel dimer than a parallel trimer (Armstrong et al., 2011, Mahrenholz et al., 2011, Wolf et al., 1997); however, these algorithms do not address antiparallel versus parallel topology. Analysis with LOGICOIL (Vincent et al., 2013), a more recent algorithm designed for the first time to predict multiple coiled-coil oligomeric states (antiparallel dimer, parallel dimer, trimer, or tetramer) solely from protein sequence, yielded scores indicating that either a parallel dimer or a trimer was most likely, but an anti-parallel dimer was also possible. Striatin members contain a conserved ‘trimerization motif’ (Gordon et al., 2011, Kammerer et al., 2005), but it was shown recently that this type of oligomerization state determinant needs to be located within the trigger sequence of a coiled-coil to influence the topology of the coiled-coil (Ciani et al., 2010). As neither a trigger sequence nor precise ends of the striatin family coiled-coil domain are known, additional experimentation will be necessary to determine the higher order oligomerization state of these proteins.
The coiled-coil domain of striatin family members has been proposed to be a ‘routing motif’, targeting striatin family members to dendritic spines (Gaillard et al., 2006). However, while deletion analysis showed that the coiled-coil domain is necessary for targeting of SG2NA to dendritic spines, it was not shown to be sufficient (Gaillard et al., 2006). Furthermore, the coiled-coil is required for oligomerization, which is essential for PP2A binding (Gordon et al., 2011), and may be necessary for binding of other striatin family-associated proteins such as CTTNBP2, which interacts within the first 200 amino acids of striatin (Chen et al., 2012). Therefore, there are multiple reasons that loss of the coiled-coil domain might alter the subcellular localization of SG2NA in neurons, and more research will be required to elucidate the molecular basis for this observation.
3.4. The Tryptophan-Aspartate (WD)-repeat domain
WD-repeat domains consist of four or more copies of a conserved, approximately 40 amino acid sequence motif usually having a glycine-histidine (GH) dipeptide at the beginning and a tryptophan-aspartate (WD) dipeptide at the C-terminus (for reviews, see (Li and Roberts, 2001, Smith, 2008)). All WD-repeat domains are thought to fold into βpropeller structures that create a stable platform for interaction with other proteins. Because of its basic and versatile function, this domain is found in a wide variety of proteins involved in multiple cellular processes. Among WD-repeat proteins, the striatin family proteins are unique in the fact that they are the only members that associate with CaM (Castets et al., 1996).
The WD-repeat domain of striatin family members is likely to be important for association of several different striatin family member-associated proteins. First, the armadillo repeat domain (ARD) of the adenomatous polyposis coli (APC) protein has been reported to bind to striatin via the striatin WD-repeat domain (Breitman et al., 2008). Second, Mob3 may interact with this domain because deletion mutants of striatin that remove most or all of the WD-repeats significantly reduced Mob3 binding in cells (Gordon et al., 2011). Third, a number of CCT/TRiC chaperonin proteins (also called tailless complex polypeptide-1 or TCP-1 proteins) have been identified as striatin-associated proteins (Goudreault et al., 2009). The presence of TCP-1 proteins in striatin complexes suggests that striatin associates with the TCP-1 ring complex, TRiC, which is composed of multiple TCP-1 proteins. The TRiC complex is involved in the folding of a number of proteins, including WD-repeat-containing proteins (Valpuesta et al., 2002). Therefore, TCP-1 proteins may be involved in the folding of striatin WD-repeats and may bind to those WD-repeats. Indeed, using deletion mutagenesis and co-immunoprecipitation assays we have found that striatin’s WD-repeat domain is required for association with TCP-1 proteins (K. Bockbrader and D. Pallas, unpublished), suggesting that CCT/TRiC chaperonin proteins bind to striatin family WD-repeat domains and assist in their folding. Finally, a fourth protein that may interact with the WD-repeat domain of striatin family members is Gαi. Striatin forms an estrogen-inducible complex containing PP2A, ERα, Gαi, and eNOS in endothelial cells (Lu et al., 2004). While ERα binds within the first 203 amino acids of striatin, Gαi does not, indicating that Gαi binds to more C-terminal amino acids of striatin. Based on this data and the role of WD-repeat domains in mediating association of heterotrimeric G proteins, it was hypothesized that Gαi likely binds to the WD-repeat domains of striatin (Lu et al., 2004). In summary, the striatin family WD-repeat domains are likely critical for striatin family regulation of estrogen receptor signaling and APC and Mob3 function, which will be discussed below in section 4.
4. Composition and function of striatin family complexes
Numerous clues about the function of striatin family proteins have emerged since the discovery of striatin nearly two decades ago. These clues have come from the identification and study of striatin family proteins and their associated proteins (Table 1), and the elucidation and study of different striatin family complexes. Early studies of striatin complexes demonstrated that mammalian striatin family members associate with Ca2+-calmodulin (Castets et al., 1996, Castets et al., 2000, Moreno et al., 2000), Cav-1 (Gaillard et al., 2001), PP2A (Moreno et al., 2000), and Mob3 (Baillat et al., 2001, Moreno et al., 2001). In addition, two-dimensional gel analyses revealed the presence of a number of additional striatin family-associated proteins (Moreno et al., 2001, Moreno et al., 2000). Subsequently, the discovery that mammalian striatin family complexes contained not only PP2A but also GCKIII kinases led to these complexes being termed STRIPAK (striatin interacting phosphatase and kinase) complexes. In the last decade, the number of new proteins identified in striatin family complexes has exploded, revealing multiple distinct complexes with diverse functions in multiple organisms.
Table 1.
STRIPAK components and other relevant striatin family member-associated proteins.
| Protein Name | Description | Reference(s) |
|---|---|---|
|
STRIPAK components
| ||
| PP2AC | Protein phosphatase 2A catalytic subunit | (Moreno et al., 2000) |
| PP2AA | Protein phosphatase 2A structural subunit | (Moreno et al., 2000) |
| Striatin | Striatin family member; putative protein phosphatase 2A B‴ regulatory subunit; STRN | (Moreno et al., 2000) |
| SG2NA | Striatin family member; putative protein phosphatase 2A B‴ regulatory subunit; STRN3 | (Moreno et al., 2000) |
| Zinedin | Striatin family member; putative protein phosphatase 2A B‴ regulatory subunit; STRN4 | (Castets et al., 2000) |
| Mob3 | Monopolar spindle-one-binder family 3; Phocein | (Baillat et al., 2001, Moreno et al., 2001) |
| Ccm3 | Cerebral cavernous malformation 3; Programmed cell death 10 (PDCD10) | (Goudreault et al., 2009) |
| Mst3 | Member of germinal center kinase III (GCKIII) subfamily of sterile 20-like kinases; Mammalian sterile-20-like kinase 3; Sterile 20-like kinase 24 (STK24) | (Goudreault et al., 2009) |
| Mst4 | Member of germinal center kinase III (GCKIII) subfamily of sterile 20-like kinases; Mammalian sterile-20-like kinase 4; Mst3 and Sok1-related kinase (MASK) | (Goudreault et al., 2009) |
| Ysk1 | Member of germinal center kinase III (GCKIII) subfamily of sterile 20-like kinases; yeast Sps1/Sterile-20-related kinase 1; Sterile 20-like kinase 25 (Stk25); Sterile 20/oxidant stress- response kinase 1 (Sok1) | (Goudreault et al., 2009) |
| STRIP1/2 | Striatin-interacting protein 1/2; previously called Fam40a/Fam40b | (Goudreault et al., 2009) |
| SLMAP | Sarcolemmal membrane-associated protein | (Goudreault et al., 2009) |
| CTTNBP2/NL | Cortactin-binding protein 2/Cortactin-binding protein 2, N-terminal-like | (Goudreault et al., 2009) |
| SIKE | Suppressor of IKKε | (Goudreault et al., 2009) |
| FGFR1OP2 | FGFR1 (fibroblast growth factor receptor 1) Oncogene Partner 2 | (Goudreault et al., 2009) |
| Mink1 | Misshapen-like kinase 1 | (Hyodo et al., 2012) |
|
| ||
|
Other proteins that associate with STRIPAK or one of its components
| ||
| Map4k4 | Mitogen-activated protein kinase kinase kinase kinase 4; Nck-interacting kinase (NIK); Hepatocyte progenitor kinase-like/germinal center kinase-like kinase (HGK) | (Frost et al., 2012, Herzog et al., 2012, Hyodo et al., 2012) |
| Tnik | TRAF2- and NCK-interacting kinase | (Hyodo et al., 2012) |
| Dynein | A motor protein | (Goudreault et al., 2009) |
| APC | Adenomatous polyposis coli protein | (Breitman et al., 2008) |
| ERα | Estrogen receptor alpha | (Lu et al., 2004) |
| Gαi | Heterotrimeric guanine nucleotide binding (G) protein subunit | (Lu et al., 2004) |
| eNOS | Endothelial Nitric Oxide synthase | (Lu et al., 2004) |
| Cav-1 | Caveolin-1 | (Gaillard et al., 2001) |
| CaM | Calmodulin | (Castets et al., 1996) |
| GIPC | GAIP-interacting protein, C terminus | (Varsano et al., 2006) |
| Eps15 | Epidermal growth factor receptor substrate 15 | (Baillat et al., 2002) |
| NDPK | Nucleoside-diphosphate kinase | (Baillat et al., 2002) |
| Dynamin I | GTPase involved in endocytosis | (Baillat et al., 2002) |
| TCP-1 proteins | Members of the chaperonin-containing TCP1 complex (CCT) | (Goudreault et al., 2009) |
While our knowledge of the composition of the striatin family complexes has increased greatly, much remains to be determined regarding the function of these complexes and the roles of the various striatin family-associated proteins. The fact that striatin family members associate with numerous proteins (Table 1) involved in multiple signaling pathways suggests that striatin family complexes have a range of functions in cellular regulation, some of which are illustrated in Fig. 2 and discussed further below. Moreover, how striatin complexes are targeted to different subcellular locations and functions is only beginning to be understood, and much of what we know is inferred from what we know about the localization of proteins that associate with STRIPAK.
Insights have also been obtained from what is known about the function of homologues in other organisms. Model organisms such as budding yeast, fission yeast, filamentous fungi, and fruit flies serve as useful tools for gaining insights that have application to cellular and developmental processes in higher eukaryotes. Homologs of striatin family members and several other components of STRIPAK complexes have been identified in these organisms and have been shown to be important in cellular and developmental processes (see Table 2 for homologs of STRIPAK components in various species). In a couple instances, mammalian STRIPAK components have been shown to functionally complement these homologs (Lisa-Santamaria et al., 2012, Poggeler and Kuck, 2004), increasing the confidence that many of the insights gained from these model systems will have applicability to mammalian systems.
Table 2.
Homologs of STRIPAK components in different organisms.
| Homo sapiens | Striatin, SG2NA, Zinedin | Mob3 | STRIP1, STRIP2 | SLMAP | PP2A A subunit | PP2A C subunit |
|---|---|---|---|---|---|---|
| Drosophila melanogaster | Cka | Mob4 | CG11526 | CG17494 | Pp2A-29B | Mts Pph21, Pph22, Pph3, Ppg1 |
| Saccharomyces cerevisiae | Far8 | -a,b | Far11 | Far9, Far10 | Tpd3 | |
| Schizosaccharomyces pombe | Csc3 | - | Csc2 | Csc1 | Paa1 | Ppa3 |
| Sordaria macrospora | Pro11 | SmMob3 | Pro22 | Pro45c | SmPP2AA | SmPP2AC |
| Neurospora crassa | Ham-3 | Mob-3 | Ham-2 | Ham-4 | PP2AA | PP2AC/Ppg1 |
| Aspergillus nidulans | StrA | - | - | - | - | - |
| Fusarium verticillioides | Fsr1 | - | - | - | - | - |
| Fusarium graminearum | FgFsr1 | - | - | - | - | - |
A dash (−) indicates that a corresponding homolog for that mammalian STRIPAK component has not yet been published.
In the case of S. cerevisiae, a homolog for Mob3 may not exist.
In this section, we will discuss the major complexes that striatin family members form in different organisms, the cellular signaling pathways in which they have been implicated, and what is known about targeting and regulation of these complexes. While most of this discussion will be grouped by organism in the subsections 4.1.1–4.1.4 below, in some cases related results from other organisms will be integrated to emphasize the generality of the results.
4.1. STRIPAK and STRIPAK-like complexes
4.1.1. STRIPAK and STRIPAK-like complexes in mammals
The core mammalian STRIPAK complex contains the A and C subunits of PP2A, the mammalian Mps one binder homolog, Mob3, the GCKIII subfamily of the mammalian sterile 20-like (Mst) kinases including Mst3 (Stk24), Mst4 (MASK), and Ysk1 (Sok1; Stk25), cerebral cavernous malformation 3 (Ccm3; also called programmed cell death 10 or PDCD10) protein, and Fam40a/Fam40b, which were renamed striatin interacting proteins 1 and 2 (STRIP1/STRIP2) (Goudreault et al., 2009, Kean et al., 2011) (Fig. 3 and Table 1). This core complex binds additional proteins in a mutually exclusive manner to form distinct STRIPAK complexes containing either a cortactin-binding protein 2 family member (CTTNBP2 or CTTNBP2NL) or sarcolemmal membrane-associated protein (SLMAP) and a suppressor of IKKε (SIKE) family member (Goudreault et al., 2009) (Fig. 3 and Table 1). In mammalian cells, striatin family members have also been reported to complex with other proteins, such as dynein (Goudreault et al., 2009), GIPC (GAIP-interacting protein, C terminus) (Varsano et al., 2006), APC protein (Breitman et al., 2008), the deubiquitinase (DUB), Trabid (Tran et al., 2013, Tran et al., 2008), and CCT/TCP-1 chaperonin proteins (Goudreault et al., 2009) (Table 1). The compositions of the striatin family complexes containing these latter proteins remain to be determined, but they may be part of classic STRIPAK complexes described above (Goudreault et al., 2009). Alternatively, some of them may be members of what we term STRIPAK-like complexes, which share some basic elements with STRIPAK complexes, but have not been shown to contain both PP2A and a kinase. In our discussions below, we will make this distinction in terminology, realizing that in some cases the association of PP2A or a kinase may occur and may simply not have been demonstrated yet. Examples of STRIPAK-like complexes include both nuclear (Tan et al., 2008) and plasma membrane (Lu et al., 2004) striatin family complexes that regulate genomic and non-genomic estrogen signaling, respectively. These will be discussed below in section 4.2.
Fig. 3. Mammalian STRIPAK complexes.
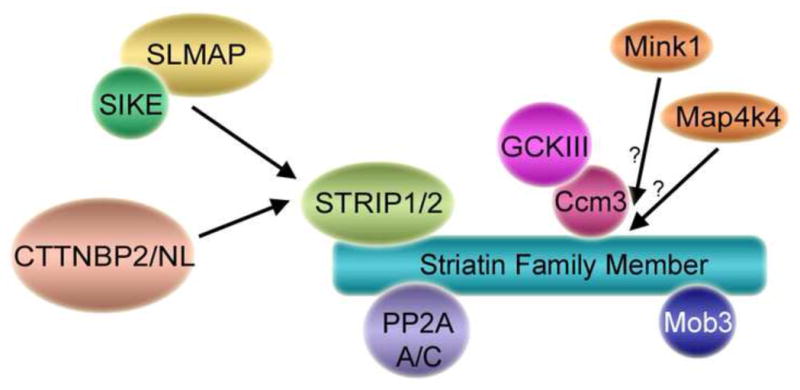
Shown is a schematic of the core mammalian STRIPAK complex (Goudreault et al., 2009) and some of its various accessory proteins. Core STRIPAK components include a striatin family member, the PP2A A/C heterodimer, Mob3, STRIP1 or STRIP2, and a GCKIII kinase bound via Ccm3. Binding of different, mutually exclusive accessory proteins to the STRIPAK core results in the formation of different STRIPAK complexes. For example, SLMAP and SIKE are not detected in STRIPAK complexes containing CTTNBP2/NL and vice versa. In addition, some STRIPAK complexes contain alternative kinases (see text) such as Mink1 and Map4k4 and Tnik (latter not shown in figure). Association of GCKIII, Mink1, and Map4k4 kinases to the STRIPAK core is likely mutually exclusive. For this reason their binding is shown as competitive (arrows). However, whether Mink1 and Map4k4 bind via Ccm3 or directly to striatin family members is not known. Additional proteins that bind to striatin family members including Ca2+-CaM, Cav-1, ERα, APC, MR, CCT, dynein and others (see text and Table 1) are not shown for simplicity and because the composition of the complexes in which some of these proteins participate are not known. Together, however, it is clear that well over a hundred variations of STRIPAK and STRIPAK-like complexes exist in mammalian cells.
4.1.1.1. Negative regulation of kinases by STRIPAK-associated protein phosphatase 2A
Given that a phosphatase and several kinases have been identified in STRIPAK complexes, it seems likely that the functions of these complexes are regulated by reversible phosphorylation carried out by these enzymes. Many of the STRIPAK constituents are phosphoproteins (Goudreault et al., 2009, Moreno et al., 2001), and the phosphorylation of Striatin, SG2NA, Mob3, and a number of other striatin-family-associated proteins is enhanced by treatment of cells with the okadaic acid at concentrations that inhibit PP2A (Moreno et al., 2001). These results indicate that STRIPAK components are indeed being reversibly phosphorylated and are consistent with the idea that striatin-associated PP2A may be the relevant phosphatase. This hypothesis has been validated for the GCKIII kinase, Mst3, by demonstrating that point mutants in striatin that reduce PP2A binding cause hyperphosphorylation and activation of Mst3 (Gordon et al., 2011). The other GCKIII kinases also appear to be regulated by PP2A. Mst4 undergoes a gel shift consistent with hyperphosphorylation in okadaic acid-treated cells (Gordon et al., 2011) and Ysk1 is dephosphorylated and partially inactivated by purified PP2A in vitro (Pombo et al., 1996). Thus, STRIPAK complexes negatively regulate Mst3 and likely the other related GCKIII kinases as well by recruiting them together with PP2A (Gordon et al., 2011, Goudreault et al., 2009). Since the GCKIII kinases play important roles in multiple cellular processes including cell cycle, cell growth and transformation, cell survival, apoptosis, Golgi assembly, cell polarity, and cell migration (for review, see (Ling et al., 2008)), STRIPAK likely regulates these events at least in part through PP2A-dependent regulation of these kinases (see also section 5.5 below).
STRIPAK may also regulate members of the GCKIV subfamily of kinases. For example, the GCKIV subfamily kinase Misshapen (Msn)-like kinase 1 (Mink1) was identified as a new STRIPAK component (Fig. 3 and Table 1) on the basis that it associates with zinedin and a number of the other established components of STRIPAK complexes (Hyodo et al., 2012). Mink1 is important for cytokinesis, particularly for the abscission process, and knockdown of either Mink1 or zinedin in HeLa cells produces multinucleated cells as a result of abnormal abscission (Hyodo et al., 2012). Zinedin enhances the dephosphorylation of Mink1 by PP2A in vitro (Hyodo et al., 2012), suggesting that, similar to the negative regulation of GCKIII kinases by striatin (Gordon et al., 2011), zinedin may coordinate the negative regulation of Mink1 by PP2A. Mink1 phosphorylation increases during mitosis (Hyodo et al., 2012), a time when PP2A is known to be inhibited (Wurzenberger and Gerlich, 2011). Based on gel shift experiments, Mink1 begins to be dephosphorylated within one hour after release of cells from nocodazole-induced cell cycle arrest (Hyodo et al., 2012), consistent with the reactivation of PP2A at mitotic exit (Wurzenberger and Gerlich, 2011). These results suggest that negative regulation of Mink1 by zinedin-associated PP2A may be required for Mink1 function in abscission.
Other STRIPAK complexes likewise appear to function in regulation of cytokinesis based on several additional studies. A genome-wide phenotypic profiling study found that loss of striatin in HeLa cells results in an increase in binuclear cells (Neumann et al., 2010). GFP-SG2NA stably expressed in HeLa cells associates with Mink, and knockdown of SG2NA or of STRIP1 in HeLa cells increases DNA content, generating binuclear cells as well as dysmorphic and fragmented nuclei (Frost et al., 2012). Mob3 knockdown also causes a striking increase in cellular DNA content, and induces abnormal spindles, mitotic failure, and cell death (Frost et al., 2012). Consistent with these results, in Drosophila S2 cells, dMob4 localizes to mitotic centrosomes and kinetochores and depletion of dMob4 results in defective centrosome separation and abnormal spindles with disorganized poles and splayed kinetochore (K) fibers (Trammell et al., 2008). Knockdown of SLMAP in HeLa cells causes only a minor increase in cellular DNA content, but increases the quantity of pericentrin foci in interphase cells (Frost et al., 2012). Given that striatin family association with PP2A increases its activity towards Cdk-mediated phosphorylations (Moreno et al., 2000), STRIPAK complexes may regulate multiple mitotic substrates to help coordinate mitotic progression. Together, these findings clearly indicate that STRIPAK function is important for mitotic progression and cytokinesis.
Other findings suggest that STRIPAK complexes are involved in additional functions in mammals. For example, because zinedin knockdown cells are defective in attachment and spreading after mitosis, zinedin-scaffolded STRIPAK complexes may have a role in focal adhesion formation following mitosis (Hyodo et al., 2012). As this phenotype is not seen upon Mink1 knockdown (Hyodo et al., 2012), another STRIPAK-associated kinase may be involved. SG2NA and zinedin have been reported to bind the GCKIV subfamily kinase Map4k4 (Frost et al., 2012, Hyodo et al., 2012) (Fig. 3), which is a mediator of tumor necrosis factor alpha (Tnf-α) and interleukin-1β production in response to an inflammatory stimulus (Aouadi et al., 2009). Map4k4 has also been identified as a modulator of cancer cell motility and invasion (Collins et al., 2006). Therefore, it will be important to investigate whether STRIPAK complexes may play a role in the regulation of inflammation, cancer cell migration, and other functions in which Map4k4 has been implicated.
Finally, zinedin also appears to interact with the Mink-related GCKIV subfamily kinase, Tnik (Hyodo et al., 2012). Although the significance of this interaction has yet to be examined, a recent study showed that Tnik functions as an effector of the small G protein, Rap2A, to induce brush border formation upon polarization of intestinal epithelial cells (Gloerich et al., 2012). Rap2A activation of Tnik causes relocalization of Mst4 from Golgi to the apical membrane of polarized intestinal epithelial cells, promoting Mst4-mediated phosphorylation of the actin binding protein, ezrin, and subsequent microvilli formation. Tnik phosphorylates Mst4 in vitro, suggesting that phosphorylation of Mst4 by Tnik in polarized epithelial cells may mediate the effects of Tnik on Mst4. Interestingly, the STRIPAK component, Ccm3, is known to enhance the amount of Mst4 outside the Golgi (Kean et al., 2011) and increases Mst4 phosphorylation of ezrin/radixin/moesin (ERM) proteins (Fidalgo et al., 2012). Thus, we speculate that Ccm3 will be important for Mst4 phosphorylation of ezrin and thus for microvilli formation in intestinal epithelial cells. Since striatin family members promote localization of Mst4 to Golgi (Kean et al., 2011) and likely negatively regulate Mst4 (Gordon et al., 2011), striatin family members may negatively regulate Mst4 function in brush border formation. Therefore, It will be of great interest to investigate a potential role of Tnik association with zinedin in Tnik-induced relocalization of Mst4 and induction of brush border formation. In summary, one common function of mammalian STRIPAK complexes is to regulate (and perhaps be regulated by) a variety of kinases that play roles in diverse cellular functions.
4.1.1.2. STRIPAK-like adenomatous polyposis coli complexes
Striatin, SG2NA, and Zinedin have been reported to complex with the APC tumor suppressor protein, and striatin has been proposed to function together with APC in regulation of tight junctions (TJs) (Breitman et al., 2008) (Fig. 2). APC associates with diverse proteins and thus regulates multiple cellular processes including Wnt signaling, cell-cell adhesion, migration, and polarity (Buda and Pignatelli, 2011). The association between APC and striatin appears to be mediated by interaction of the APC ARD domain and the striatin WD-repeat domain (Breitman et al., 2008). In epithelial cells, striatin co-localizes at sites of cell-cell contact with both APC and the TJ protein, ZO-1, but not with the adherens junction (AJ) protein, β-catenin (Breitman et al., 2008). Consistent with this result, initial evidence suggests that striatin complexes with ZO-1. Intriguingly, the localization of striatin and APC is interdependent. Moreover, actin filament integrity is important for APC and striatin localization to cell junctions, and conversely, striatin and APC are important for the normal organization of filamentous actin and the localization of ZO-1, suggesting a role for striatin and APC in TJ function (Breitman et al., 2008). Whether striatin mediates its effects through APC’s known function in regulation of actin is not known. Initial experiments suggest that striatin is not necessary for formation of TJs (Breitman et al., 2008). However, more thorough testing for a role for striatin function in TJ formation needs to be performed using not only striatin depletion, but also striatin overexpression, depletion and overexpression of other striatin family members (since they also bound APC and could be redundant), time-courses of TJ reformation, and measurements of transepithelial resistance to monitor TJ function.
While striatin family members have been demonstrated to bind APC, it remains to be determined whether other STRIPAK components are also present. Very recently, STRIPAK components, including all three striatin family proteins, STRIP1 (Fam40a), SLMAP, CTTNBP2, and CTTNBP2NL, were found associated with the APC-deubiquitinating enzyme, Trabid (Tran et al., 2013, Tran et al., 2008). Trabid also associates with the E3 ligase, HectD1, which is thought to transfer K63 polyubiquitin to APC, increasing APC binding to Axin (Tran et al., 2013). Like striatin, Trabid and HectD1 both bind to the ARD domain of APC (Tran et al., 2013, Tran et al., 2008). Striatin proteins appear to be required for HectD1 binding to the APC ARD domain, and it has been proposed that Trabid, striatin, and HectD1 might be in a complex together (Tran et al., 2013). In addition, striatin family proteins are important for proper subcellular localization of APC (Breitman et al., 2008, Tran et al., 2013). Thus, STRIPAK may function together with Trabid and HectD1 to regulate APC localization and function. Of note, knockdown of striatin family members had little effect on β-catenin protein levels and signaling, suggesting that they may not function in APC regulation of β-catenin stability (Tran et al., 2013). However, the ARD domain of APC also interacts with a B′-directed PP2A complex (Seeling, 1999), and it is possible that B′-directed PP2A may be able to compensate for striatin loss. Interestingly, Trabid knockdown in PC3 prostate cancer cells increases stress fiber formation and cell spreading and decreases cell migration (Bai et al., 2011), demonstrating that Trabid has an important role in regulation of the actin cytoskeleton. Whether this function is connected to the effects of striatin knockdown on actin cytoskeleton organization is not known. In addition, whether the Trabid-STRIPAK-like complex is equivalent to the striatin-APC complex studied in epithelial cells by Breitman and colleagues (Breitman et al., 2008) remains to be seen. In any case, many questions remain to be addressed regarding STRIPAK function in this system.
4.1.1.3. Possible functions of sarcolemmal membrane-associated protein (SLMAP), and suppressor of IKKε (SIKE), and cortactin-binding protein 2 (CTTNBP2) family members in STRIPAK complexes
Other potential functions of STRIPAK complexes are suggested by studies of individual STRIPAK components. For example, SLMAP is actually a family of integral membrane proteins containing C-terminal regions of coiled-coil structure, which mediate SLMAP homo-oligomerization (Guzzo et al., 2005, Guzzo et al., 2004b, Wigle et al., 1997). Multiple SLMAP isoforms are expressed by alternative splicing from a single gene in a tissue-specific and developmentally regulated manner, including a larger, apparently ubiquitous isoform (SLMAP3), and smaller isoforms (SLMAP1 and SLMAP2) predominantly found in cardiac, slow-twitch, and smooth muscle (Guzzo et al., 2005, Wielowieyski et al., 2000, Wigle et al., 1997). SLMAP3 consists of two isoforms (M1 and M2), generated by use of two alternative initiator codons (Guzzo et al., 2004a). Moreover, SLMAP isoforms can contain either of two possible transmembrane domains, TM1 or TM2, which are critical for subcellular targeting, generating even more diversity (Byers et al., 2009, Guzzo et al., 2005, Wielowieyski et al., 2000). Because of the diversity of SLMAP isoforms, it will be important to determine which of these isoforms can associate with the core STRIPAK complex. The answer to this question will provide insight as to which SLMAP functions might be mediated by SLMAP-containing STRIPAK complexes and which SLMAP isoforms might target STRIPAK complexes to different subcellular locations.
Several studies have addressed the subcellular localization of SLMAPs. SLMAPs appear to reside in the sarcolemma, transverse (T)-tubules, and sarcoplasmic reticulum (SR) of muscle cells and in the outer nuclear envelope, endoplasmic reticulum, mitochondria, and centrosomes of non-muscle cells, but not in Golgi (Byers et al., 2009, Frost et al., 2012, Guzzo et al., 2005, Guzzo et al., 2004b, Wigle et al., 1997). In addition, SLMAP binds to, and co-localizes with, myosin in cardiomyocytes (Guzzo et al., 2005). Interestingly, SLMAP has been implicated in myoblast fusion (Guzzo et al., 2004b). Myoblast differentiation induces expression of a new SLMAP isoform while exogenous expression of SLMAP3 or SLMAP1 inhibits myoblast fusion (Guzzo et al., 2004b). Given the role of STRIPAK in cell fusion in other organisms, it will be key to determine whether other STRIPAK components also play a role in myoblast fusion. Very recently, Map4k4 has been identified as a negative regulator of myoblast differentiation, including myoblast fusion (Wang et al., 2013). Since SG2NA and zinedin bind Map4k4 (Frost et al., 2012, Hyodo et al., 2012), it is tempting to speculate that a STRIPAK complex that includes SLMAP and Map4k4 might be involved. However, a STRIPAK complex containing CTTNBP2NL may also be involved, since CTTNBP2NL was recently found complexed with Map4k4 (Herzog et al., 2012).
SIKE, another STRIPAK component, is a small coiled-coil-containing protein that was originally identified as a novel suppressor of toll-like receptor 3 (TLR3)- and virus-initiated activation of interferon regulatory factor 3 (IRF-3) (Huang et al., 2005). Thus, a SIKE-directed STRIPAK complex might function to regulate activation of IRF-3. SIKE binds to the IKK-related kinases, IKKε and TBK1, and inhibits their ability to interact with the double stranded RNA sensor protein, RIG-1, the adaptor protein, TRIF, and IRF-3. Virus infection or double stranded RNA activation of TLR3 greatly reduces the interaction of SIKE with TBK1 and induces a portion of SIKE to move to a monomeric/homodimeric pool (Huang et al., 2005). Therefore, it will be important to determine whether these same effectors modulate the association of SIKE with the STRIPAK core proteins or whether SIKE functions in this pathway independent of STRIPAK.
FGFR1 (fibroblast growth factor receptor 1) Oncogene Partner 2 (FGFR1OP2) is a SIKE-related protein that is also found in STRIPAK complexes, probably mutually exclusively with SIKE (Goudreault et al., 2009). FGFR1OP2 was identified originally as a protein whose mRNA transcript (wit3.0) was induced in rat gingiva undergoing wound healing after extraction of a tooth (Sukotjo et al., 2002). Consistent with this, existing data indicate that FGFR1OP2 helps to facilitate wound healing. FGFR1OP2 is specifically induced in wound-associated oral fibroblasts, concomitant with an increase in the ability of these fibroblasts to contract collagen gel in vitro (Sukotjo et al., 2002). Increased FGFR1OP2 expression upon oral wounding is necessary and perhaps sufficient for the increased ability of wound fibroblasts to contract collagen gel in vitro (Lin et al., 2010, Sukotjo et al., 2002, Sukotjo et al., 2003). Moreover, FGFR1OP2 has also been reported to be important for fibroblast cell migration and to be induced to associate with the cytoskeleton upon oral wounding (Lin et al., 2010). In skin, where FGFR1OP2 is not induced upon wounding, wound closure was accelerated by exogenous expression of FGFR1OP2 via lentivirus infection (Lin et al., 2010). Together, these results support the hypothesis that FGFR1OP2 plays a role in wound healing in vivo, perhaps in facilitating the closure of the wound (Lin et al., 2010). Results of additional studies suggest that particular FGFR1OP2 single nucleotide polymorphisms may correlate with excessive jawbone atrophy (Kim et al., 2012, Suwanwela et al., 2011). Therefore, efforts have begun to identify small molecules that regulate FGF1OP2 expression (Cheng and Nishimura, 2012). Given the fact that FGFR1OP2 is a STRIPAK component, future experiments should address the possible involvement of FGFR1OP2-directed STRIPAK function in oral wound healing.
STRIP1 and STRIP2 were recently implicated as having roles in cytoskeletal organization, cell morphology and migration (Bai et al., 2011). Intriguingly, depletion of each of these proteins revealed distinct phenotypes that varied with cell type. For example, STRIP1 knockdown in PC3 prostate cancer cells increased cortical actin, lamellipodia formation, and reduced cell spreading, while STRIP2 knockdown in the same cells altered microtubule organization and induced cell elongation. Thus, STRIP1 and STRIP2 may target distinct STRIPAK complexes to regulate cytoskeletal organization and function.
Different STRIPAK complexes function in different subcellular compartments, but little is known about how they are directed to distinct locations. Likely candidates for targeting STRIPAK include the additional proteins bound by the core STRIPAK complex such as SLMAP, SIKE, FGFR1OP2, CTTNBP2, and CTTNBP2NL. While striatin, SG2NA, Mob3, and STRIP1 localize primarily to the Golgi, SLMAP localizes to the outer nuclear envelope, some endoplasmic reticulum structures, and to centrosomes and associated membranous material, but not to Golgi (Frost et al., 2012, Guzzo et al., 2005, Guzzo et al., 2004a). Localization of SLMAP can be isoform-dependent (Frost et al., 2012, Guzzo et al., 2005, Guzzo et al., 2004a). Therefore, different SLMAPs have the potential to target STRIPAK complexes to distinct subcellular locations, although this remains to be directly demonstrated. Based on the localizations of the different STRIPAK components and the effects of their knockdown, it has been speculated that STRIPAK complexes may have roles such as linking centrosomes to the Golgi, targeting Golgi fragments to the centrosome at mitosis, regulating Golgi fragmentation at the G2/M transition, and regulating centrosome duplication (Frost et al., 2012).
CTTNBP2 and CTTNBP2NL may also target STRIPAK complexes to distinct locations. Although both CTTNBP2 and CTTNBP2NL are cortactin-binding proteins that bind the STRIPAK core complex, the subcellular distribution of these related proteins is different because they target cortactin to different populations of actin fibers (Chen et al., 2012). CTTNBP2 colocalizes with cortactin in the cell cortex of COS cells more than CTTNBP2NL does, while CTTNBP2NL colocalizes with cortactin at the stress fibers more than CTTNBP2 does (Chen et al., 2012, Goudreault et al., 2009). In neurons, CTTNBP2, but not CTTNBP2NL, stably localizes to spines of neuronal dendrites, regulates dendritic spine density, and is important for targeting striatin and zinedin to dendritic spines (Chen et al., 2012). Consistent with this distribution of function, CTTNBP2 is highly expressed in brain while CTTNBP2NL is only expressed at a low level. Overexpression of CTTNBP2NL is unable to rescue the spine density phenotype in CTTNBP2 knockdown neurons, suggesting that it may not target STRIPAK to dendritic spines (Chen et al., 2012). Thus, it is possible that CTTNBP2 and CTTNBP2NL target STRIPAK complexes to different subcellular compartments. However, more experiments are needed to address this important topic.
While striatin family members are concentrated at dendritic spines, no definitive data exist that demonstrate a role for striatin in dendritic spinogenesis or maintenance. Since CTTNBP2 is important for the proper size and density of dendritic spines and targets STRIPAK to dendritic spines, STRIPAK may contribute to CTTNBP2 function in dendritic spines. However, CTTNBP2 also targets cortactin to dendritic spines, and cortactin overexpression completely rescues spine density defects in CTTNBP2 knockdown neurons, while CTTNBP2 defective in binding cortactin does not (Chen et al., 2012, Chen and Hsueh, 2012). Thus, while cortactin is clearly important for CTTNBP2 function in dendritic spines, the role of the core STRIPAK complex in CTTNBP2 function remains to be determined.
Interestingly, recent studies reveal that synaptic signaling modulates the localization of striatin and zinedin to dendritic spines. Three minutes after treatment of neurons with N-methyl-D-aspartate (NMDA), the immunoreactivity of striatin family members in dendritic spines decreases as does the colocalization of striatin and zinedin with CTTNBP2, which remains in dendritic spines (Chen et al., 2012). Moreover, fifteen minutes after NMDA treatment, the amount of CTTNBP2 and striatin in complex together was reduced. We speculate that NMDA treatment may alter striatin and zinedin localization by increasing the concentration of calcium in dendritic spines, thus promoting Ca2+-CaM binding to STRIPAK complexes and, consequently, reducing the association of striatin and zinedin with proteins that bind proximal to their Ca2+-CaM-binding domain. NMDA receptors (NMDARs) are glutamate-gated ion channels that are very permeable to calcium and their activation causes rapid increases in calcium from a resting concentration of approximately 50 nM to well over 1 μM in dendritic spines (Connor et al., 1994, Muller and Connor, 1991). As mentioned previously, calcium reduces the interaction of striatin with GST-Cav-1 (Gaillard et al., 2001) and increases the amount of striatin found in the brain cytosol (Bartoli et al., 1998). Because CTTNBP2 interacts within the N-terminal 200 amino acids of striatin (Chen et al., 2012) that contain the striatin family Ca2+-CaM-binding domain, calcium might also reduce CTTNBP2 binding to striatin and zinedin. Given that the affinity of brain striatin for Ca2+-CaM is very low in 0.1 μM calcium and greatly increased in 1 μM calcium (Bartoli et al., 1998), the previous results are consistent with the idea that increased calcium levels from NMDAR activation reduce striatin association with Cav-1 and CTTNBP2 and help promote redistribution of striatin family members to the cytosol of neurons. In addition, other changes that occur upon NMDA treatment such as redistribution of cortactin and actin to the dendritic shaft may contribute to striatin relocalization as well (Chen et al., 2012, Chen and Hsueh, 2012, Hering and Sheng, 2003).
4.1.2. STRIPAK and STRIPAK-like complexes in Drosophila
4.1.2.1. STRIPAK-like complex regulation of the Drosophila Jnk pathway
The sole Drosophila striatin family homolog, Cka (Connector of Kinase to AP-1) (Table 2), was originally discovered as a novel scaffolding protein that positively regulates Drosophila Jun N-terminal kinase (dJnk) signaling in two developmental events: epithelial sheet movement required for embryonic dorsal closure and apoptosis of cells in wing imaginal disks (Chen et al., 2002). Cka forms complexes containing the dJnk pathway components Hep (Hemipterous; dJnk kinase), Bsk (Basket; dJnk), and the Jra (Jun-related antigen; dJun) and Kay (kayak; dFos) proteins that dimerize to make up the AP-1 transcription factor (Chen et al., 2002). Cka promotes dJnk signaling by enhancing both phosphorylation of dJnk by Hep and phosphorylation of dJun and dFos by dJnk, resulting in activation of AP-1-regulated transcription, which functions to promote both proper dorsal closure and apoptosis in wing imaginal disks. Expression of Hep or dJun and dFos increases the nuclear localization of Cka in 293T cells, suggesting that the Cka complex may translocate to the nucleus to promote AP-1-dependent transcription (Chen et al., 2002). However, further experimentation will be necessary to dissect how Cka localization may play a role in dJnk signaling. Also, because Drosophila PP2A binds to Cka (Horn et al., 2011, Ribeiro et al., 2010) and negatively regulates the Jnk pathway, it will be important to determine whether PP2A functions in Cka-dJnk pathway signaling complexes.
4.1.2.2. STRIPAK complex regulation of the Drosophila Erk pathway
In a recent study mapping signaling networks in Drosophila by identifying synthetic interactions, Cka was implicated as a positive regulator of Ras-Raf-Erk (extracellular signal-regulated kinase) signaling (Horn et al., 2011). Cka was shown to function downstream of the epidermal growth factor receptor (EGFR), to maintain basal phosphorylation of Drosophila Erk (dErk), and to enhance expression of Ras-Raf-Erk pathway target genes (Horn et al., 2011). Consistent with results from others (Ribeiro et al., 2010), proteomic analysis of Cka complexes revealed that Cka forms a complex with Drosophila GckIII (dGckIII) kinase and the Drosophila PP2A C subunit, Mts (Horn et al., 2011). Like Cka, dGckIII functions positively downstream of EGFR in the Ras-Raf-Erk signaling pathway and interacts with Mts (Friedman and Perrimon, 2006, Horn et al., 2011). Together, these results support the idea of a Drosophila STRIPAK (dSTRIPAK) complex containing dGckIII and PP2A that functions as a positive regulator of Ras-Raf-Erk signaling.
A similar STRIPAK complex may also positively regulate Ras-Raf-Erk signaling in mammalian cells. Mammalian striatin family members bind PP2A (Moreno et al., 2000) and the mammalian GCKIII kinases, Mst3, Mst4, and Ysk1 (Goudreault et al., 2009). Moreover, SG2NA and striatin, as well as Mst3 and Mst4, are important for maintaining basal Erk phosphorylation in mammalian cells (Friedman and Perrimon, 2006, Horn et al., 2011). Because dGckIII also interacts with dRaf, dGckIII and Cka have been hypothesized to be constituents of the Raf activation complex known to contain PP2A (Friedman and Perrimon, 2006, Horn et al., 2011). However, despite the common binding partners shared between dRaf and Cka, neither Cka and dRaf nor mammalian striatin family members and Raf have been demonstrated to exist in the same complex. Thus, the role of striatin family members in the Ras-Raf-Erk pathway in these organisms needs to be clarified further.
In the synthetic interaction study described above (Horn et al., 2011), genetic interactions found for Cka in regard to effects on cell proliferation included Msn, the Drosophila homolog of the mammalian Mink1 kinase, as well as PP2A C subunit (Mts) and Rho1, the Drosophila homolog of the mammalian GTPase, RhoA (ras homolog gene family member A). For each of these proteins, the combined effect of their knockdown with Cka depletion was less than predicted if no genetic interaction was manifested. A genetic interaction between Rho1 and Cka is very interesting, given the possible role of mammalian RhoA in CCM discussed below in section 5. Although Msn knockdown alone had no effect on cell proliferation, it substantially rescued the negative effect of Cka knockdown on cell proliferation (Horn et al., 2011). Msn is a positive upstream regulator of dJnk signaling in dorsal closure (Liu et al., 1999, Su et al., 1998) and has been reported to complex with Cka (Ribeiro et al., 2010). We speculate that under limiting Cka concentrations, Msn knockdown might increase the available Cka to function positively in the Ras-Raf-Erk pathway. In support of this possibility, evidence suggesting the coexistence of independent Cka complexes in the same cell has been reported (Ribeiro et al., 2010) (see section 4.1.2.3 below). The existence of multiple STRIPAK and STRIPAK-like complexes in the same cell type suggests that experiments knocking down common components should be interpreted with caution.
4.1.2.3. Negative regulation of kinases by Drosophila STRIPAK-associated PP2A
Further evidence in support of the idea that STRIPAK complexes target PP2A to negatively regulate a variety of kinases comes from the study of the Hippo (Hpo) pathway in Drosophila (Ribeiro et al., 2010). Hpo is the Drosophila homolog of the mammalian GCKII kinases, Mst1 and Mst2, and is critically involved in the control of tissue size (Wu et al., 2003). Although Mst1 and Mst2 were not found in proteomic analyses of mammalian STRIPAK complexes (Glatter et al., 2009, Goudreault et al., 2009), Hpo associates with, and is negatively regulated by, PP2A in the dSTRIPAK complex (Ribeiro et al., 2010). The dSTRIPAK complex containing Hpo is organized by Cka and appears to contain at a minimum the Drosophila PP2A A (PP2A-29B) and C (Mts) subunits, the Mob3 functional homolog, dMob4, and homologs of mammalian STRIP, FGR10P2/SIKE, and Ccm3 (Ribeiro et al., 2010). Consistent with its role as a negative regulator of Hpo, the Hpo-associated protein dRassf (Drosophila ras association domain family protein) complexes with Hpo and Cka and promotes the association of Hpo with Cka. Similar dSTRIPAK components were found associated with dRassf by proteomic analysis, with the major exception that dRassf associates with both a CTTNBP2 homolog and a FGR10P2/SIKE homolog (Ribeiro et al., 2010), which are thought to be in mutually exclusive complexes in mammalian cells (Goudreault et al., 2009). Interestingly, while affinity purifications of proteins associated with either Cka or dMob4 from Drosophila Kc167 cells contained, in addition to Hpo, the STE20-like kinases, dGckIII/Stlk3 and Msn, complexes recovered by affinity purification of Hpo or dRassf did not include these kinases. These results suggest that, as for mammalian striatin family members, dSTRIPAK forms mutually exclusive complexes with different kinases to regulate diverse cellular processes.
4.1.3. STRIPAK-like complexes in yeasts
4.1.3.1. STRIPAK-like complexes in Saccharomyces cerevisiae
Homologs of components of the STRIPAK complex are also found in the budding yeast, Saccharomyces cerevisiae (S. cerevisiae) (Table 2). The yeast FAR (Factor arrest) complex is composed of six FAR family proteins (Far3 and Far7-11), some of which are homologs of mammalian STRIPAK components, including Far8 (striatin), Far9/Far10 (SLMAP), and Far11 (STRIP1/2) (Goudreault et al., 2009, Kemp and Sprague, 2003). Far11 interacts with the PP2A A (Tpd3) and C subunits (Pph21, Pph22, Pph3) (Lisa-Santamaria et al., 2012, Pracheil et al., 2012, Uetz et al., 2000), suggesting that, like mammalian STRIPAK, the yeast FAR complex targets PP2A to regulate different cellular processes. Although the FAR complex was originally discovered as being necessary for pheromone-induced cell cycle arrest in budding yeast (Kemp and Sprague, 2003), additional functions have been ascribed to it. Recently, all of the FAR complex proteins and yeast PP2A catalytic subunits were identified as being important for caspase-10-induced death in yeast (Lisa-Santamaria et al., 2012). In this study, loss of Far11 was partially complemented by mammalian STRIP1 and STRIP2, supporting the idea that the FAR complex is functionally similar to the mammalian STRIPAK complex. Also analogous to mammalian cells, FAR complex components were found to be concentrated in the endoplasmic reticulum-Golgi. The phenotype of caspase-10-induced death includes some attributes of autophagy and apoptosis as well as impairment of the intra-S checkpoint (Lisa-Santamaria et al., 2012). Caspase-10 induces the dephosphorylation of the autophagy inducer protein, Atg13, and of Rad53 (yeast CHK2), which participates in the intra-S checkpoint. Far11 is necessary for caspase-10-induced dephosphorylation of Atg13, and the induction of autophagy by caspase-10 expression, nitrogen starvation, or rapamycin treatment (Lisa-Santamaria et al., 2012). Far11 is also required for caspase-10-induced dephosphorylation of Rad53 and prevention of cell cycle arrest initiated by the intra-S checkpoint (Lisa-Santamaria et al., 2012). Thus, the FAR complex likely targets PP2A to regulate Atg13 and Rad53 phosphorylation, and thus autophagy and DNA damage-induced arrest.
The fact that loss of Far11 rescues rapamycin-induced autophagy suggests that the FAR complex is involved in the rapamycin-sensitive TORC1 (target of rapamycin complex 1) kinase signaling pathway, which is regulated by nutrient availability and is known to regulate autophagy (Lisa-Santamaria et al., 2012, Wullschleger et al., 2005). However, results from other recent studies indicate that the FAR complex also negatively regulates the less rapamycin-sensitive TORC2 signaling pathway, because loss of any member of the FAR complex at least partially suppresses the lethality of a TORC2-deficient mutant (Baryshnikova et al., 2010, Pracheil et al., 2012). Far11 does not regulate TORC2 but rather functions downstream of this complex, at a minimum to negatively regulate the phosphorylation of Slm1, a TORC2 substrate required together with the functionally redundant protein, Slm2, for polarization of the actin cytoskeleton (Audhya et al., 2004, Fadri et al., 2005, Pracheil et al., 2012). Loss of actin polarization due to Slm1/Slm2 mutation can be partially rescued by overexpression of Rho1, indicating that Slm1/Slm2 may regulate actin polarization at least in part through regulation of Rho GTPases (Fadri et al., 2005). Thus, Far11 may also function upstream of Rho GTPases. Deletion of the gene encoding Far11 (Baryshnikova et al., 2010, Pracheil et al., 2012) or Sac7 (Pracheil et al., 2012), a Rho GTPase activating protein (GAP) that negatively regulates actin cytoskeleton dynamics, restores polarization of the actin cytoskeleton lost in a TORC2-deficient mutant. Moreover, deletion of the gene encoding the Rho activator, Rom2, greatly impaired the ability of loss of Far11 to rescue the lethality of a mutant TORC2, suggesting that Far11 may function upstream of Rho GTPases (Pracheil et al., 2012). Thus, Far11 clearly functions downstream of TORC2 to negatively regulate actin polarization, perhaps in part by modulating Rho GTPase signaling.
An important question is whether Far11 functions in TORC2 regulation of actin polarization as part of the FAR complex or whether Far11 has FAR complex-independent roles in TORC2 signaling. While loss of any FAR complex member at least partially suppressed the lethality of a TORC2-deficient mutant, loss of Far11 was more effective than loss of any other FAR complex member (Pracheil et al., 2012). However, this cannot be equated with differences in rescue of actin polarization. Unfortunately, the effects of deletion of the other FAR complex members on Slm1 phosphorylation and actin polarization have not been assayed. Comparable to loss of Far11, loss of the PP2A family catalytic subunit, Ppg1, partially rescued actin polarization in a TORC2-deficient mutant (Baryshnikova et al., 2010). Thus, Far11 may target Ppg1 to regulate actin polarization. Deletion of Rts1, the yeast PP2A B′ regulatory/targeting subunit, was also found to negatively regulate Slm1 phosphorylation and to suppress the lethality of a TORC2 mutant, leading the authors to propose that Rts1 might interact with PP2A A and C subunits in the FAR complex to regulate Slm1 phosphorylation (Pracheil et al., 2012). However, no data evidencing a physical interaction between Far11 and Rts1 exists to support this conclusion. Moreover, the phosphatase calcineurin also counteracts TORC2 phosphorylation of Slm1 (Bultynck et al., 2006, Daquinag et al., 2007). Thus, multiple phosphatase complexes oppose TORC2-mediated phosphorylation of Slm1 and further experimentation will be necessary to determine whether Rts1 interacts physically with the FAR complex to function in TORC2 regulation of actin polarity, or perhaps functions redundantly with the FAR complex.
Finally, the results from the study by Pracheil et al. are consistent with the possibility that the yeast FAR complex may function upstream of Rho GTPases (Pracheil et al., 2012). Interestingly, a high throughput two-hybrid screen identified an interaction between Far11 and Rho4 (Uetz et al., 2000), a Rho GTPase that functions together with Rho3 in activation of formins, actin filament nucleators (Dong et al., 2003). Although further experimentation is necessary to clarify these results, they are intriguing because of the possible implications that the STRIPAK complex might be an upstream regulator of RhoA in controlling actin cytoskeleton organization and dynamics in mammalian cells.
4.1.3.2. STRIPAK-like complexes in Schizosaccharomyces pombe
Although functional conservation of some STRIPAK components has been demonstrated, there is also evidence for distinct functions of STRIPAK-like complexes in different organisms. Frost and colleagues have proposed that the STRIPAK-like complexes have undergone functional repurposing over the course of evolution (Frost et al., 2012). For example, although STRIPAK components in S. cerevisiae function together in the FAR complex to regulate cell cycle arrest in response to pheromone signaling, components of this complex (Far3 and Far7) appear to be lacking in Schizosaccharomyces pombe (S. pombe) and metazoa and new partners are found. In addition, comparison of the genetic interaction profiles of genes for STRIPAK component homologs demonstrated that in S. pombe, but not in S. cerevisiae, the genetic interaction patterns of genes encoding Far8, Far10, and Far11 are correlated with those of multiple genes involved in mitosis and cytokinesis. For example, strong synthetic genetic interactions are found between the S. pombe FAR genes and the gene for the mitotic exit phosphatase Cdc14 ortholog, Clp1, and mutation of S. pombe Far8, Far10, or Far11 together with Clp1 induces abnormal morphologies and increased ploidy not seen in the single mutants. In contrast, similar double mutants in S. cerevisiae did not show exacerbation of the cdc14 single mutant phenotype. In addition, the results from this study provided support for other functions for STRIPAK-like homologs in yeast. For example, in S. cerevisiae, genetic interactions were found between FAR complex genes and genes encoding the PP2A family catalytic subunit, Ppg1, the TORC2 kinase complex, and the endoplasmic reticulum-mitochondria encounter structure (ERMES), as well as lipid synthesis genes. In S. pombe, FAR complex genetic interactions correlated with those of genes encoding actomyosin contractile ring components, Pck1 kinase, Golgi components, and the PP2A catalytic subunit, Ppa3. Interestingly, strong synthetic genetic interactions of S. pombe with the PP2A B′ regulatory subunit, Par1, were found, prompting the authors to propose that Par1 (B′)-directed PP2A may compensate for FAR (STRIPAK)-directed PP2A. This possibility may have relevance to the question raised above in section 4.1.3.1 concerning PP2A B′ (Rts1) and FAR function in regulating TORC2 in S. cerevisiae. It is also consistent with the implication of both a STRIPAK-like complex and B′ in regulation of the septation initiation network (SIN) in S. pombe (see below).
In S. pombe, a STRIPAK-like complex has been implicated in the regulation of septation (Singh et al., 2011). The S. pombe septation initiation network (SIN) is critical for septum formation in cells at the completion of mitosis (Balasubramanian et al., 2004, Krapp and Simanis, 2008, Singh et al., 2011). Hyperactivity of the SIN (e.g., caused by mutation of negative regulators of the SIN or overexpression of certain positive SIN components) results in premature septum formation and, consequently, cellular compartments lacking nuclei. Conversely, inhibition of the SIN (e.g., caused by mutation of SIN components) reduces or prevents septation, resulting in multinucleated, elongated cells. Components of the SIN are anchored to the spindle pole body (SPB) via association with the scaffolding proteins, Cdc11 and Sid4, which in turn are tethered to the SPB protein, Ppc89. Central to the function of the SIN is the ras superfamily GTPase, Spg1, which is negatively regulated by a two-component GAP consisting of Cdc16 and Byr4. GTP-bound, active Spg1 on the SPB recruits Cdc7, the primary effector of Spg1 and the first of three kinases in the SIN. While Cdc7 initially localizes to both mother and daughter SPB in early mitosis, Cdc16-Byr4 is recruited to the mother SPB, presumably inactivating Spg1 in that location and consequently reducing Cdc7 binding to that SPB. Thus, one hallmark of a functioning SIN is the progression to an asymmetric localization of Cdc7 (on the daughter SPB) in mitosis.
Recently, a STRIPAK-like complex called the SIN-inhibitory PP2A complex (SIP) was identified that is a weak inhibitor of the SIN (Singh et al., 2011). Constituents of the SIP include homologs of the mammalian STRIPAK components, Csc1 (SLMAP homolog), Csc2 (STRIP1/2 homolog), Csc3 (striatin homolog), Paa1 (S. pombe PP2A A subunit), and Ppa3 (a novel, previously uncharacterized S. pombe PP2AC subunit), as well as Csc4, a protein of unknown function. Like components of the SIN, the SIP localizes to the SPB in a manner dependent on Ppc89 (Matsuyama et al., 2006, Singh et al., 2011). SIP localization to the SPB appears to be dependent on Cdk activity (Singh et al., 2011). Interestingly, while the SIP localizes to both SPB in early mitosis, as mitosis progresses it localizes only to the mother SPB, the SPB without Cdc7. Similar to loss of function mutations in the Spg1 GAP proteins, loss of SIP components abolishes the asymmetric localization of Cdc7 in mitosis and promotes septum formation during interphase. Moreover, the SIP promotes dephosphorylation of the SIN scaffolding protein, Cdc11, and loss of the SIP results in loss of Byr4 binding to the mother SPB. These and other lines of evidence suggest that one function of the SIP may be to promote the asymmetric localization of Cdc7 by dephosphorylating Cdc11 at the mother SPB, increasing recruitment of Byr4 (Singh et al., 2011), which binds hypophosphorylated Cdc11 (Krapp et al., 2003). However, another possibility is that SIP may indirectly regulate Cdc11 phosphorylation and/or may have additional targets in the SIN.
Of note, another PP2A catalytic subunit, Ppa2, also appears to be a negative regulator of the SIN (Singh et al., 2011), but whether it is present in the SIP is not known. Most likely Ppa3, Ppa2, or both participate in regulating the SIN in a separate PP2A complex since S. pombe PP2A B′ B-type regulatory/targeting subunits have also been shown to localize to SPB, to negatively regulate the SIN, and to be important for Cdc11 dephosphorylation (Jiang and Hallberg, 2000, 2001, Krapp et al., 2003, Le Goff et al., 2001, Tanabe et al., 2001). Interestingly, the multiseptation defect of a S. pombe B′ double deletion mutant was rescued by overexpression of a human B′ subunit, indicating remarkable functional conservation in this process (Tanabe et al., 2001). In addition, the S. pombe PP2A B B-type subunit has also been implicated as a negative regulator of the SIN (Lahoz et al., 2010). The involvement of multiple PP2A complexes in regulation of the SIN may explain why the STRIPAK-like SIP complex is only a weak negative regulator of the SIN. Thus, it will be important to determine the genetic and phenotypic interactions of these PP2A complexes in regulation of the SIN, and the specific targets in the SIN that they regulate. Finally, while components of the SIP localize to the SPB, Csc2 and Csc3 also localize to the nuclear envelope, consistent with reports that the Aspergillus nidulans and N. crassa striatin homologs are found in the nuclear envelope (Dettmann et al., 2013, Wang et al., 2010) and that mammalian SLMAP localizes primarily to the outer nuclear envelope and some endoplasmic reticulum structures, as well as in centrosomes (Frost et al., 2012, Guzzo et al., 2005, Guzzo et al., 2004a).
In summary, STRIPAK or STRIPAK-like complexes from yeast to humans regulate numerous cellular events including mitosis and cytokinesis. Some of these functions may be partially redundant with other PP2A holoenzyme forms, especially B′-directed holoenzymes, leading to masking of some of their roles. Thus, future studies should include investigations of the effects of co-targeting of B′- and striatin family-directed PP2A to increase our insight into the function of STRIPAK and STRIPAK-like complexes.
4.1.4. STRIPAK-like complexes in filamentous fungi
STRIPAK-like complexes have been implicated in sexual development and cell-cell fusion in filamentous fungi. Cell-cell fusion is a fundamental process involved in both vegetative growth and sexual development in filamentous fungi. Similar to cell fusion in higher eukaryotes, it involves communication between cells, cell adhesion, and fusion of cell membranes (Fleissner et al., 2008). Thus, understanding this process in fungi may provide important insights applicable to higher eukaryotes where cell-cell fusion is involved in processes such as fertilization, myoblast fusion, fusion of macrophages to form multinucleated giant cells, and placental development (Fleissner et al., 2008). Moreover, some fungi such as those of the genus Fusarium can be pathogenic in animals or plants, and understanding the biochemical basis for their virulence may help in the development of new anti-mycotics or increased resistance in plants. In this section, we will first provide a brief background on growth and sexual development of filamentous fungi and then discuss insights from studying STRIPAK-like complex function in filamentous fungi.
4.1.4.1. Growth and sexual development of filamentous fungi
Filamentous fungi such as Neurospora crassa (N. crassa) and Sordaria macrospora (S. macrospora) grow as mycelial colonies comprising a system of interconnected, multinucleate hyphal tubes (hyphae) (for reviews, see (Engh et al., 2010, Fleissner et al., 2008)). In N. crassa and S. macrospora, new colonies can be initiated when spores produced through sexual reproduction, called ascospores, germinate to produce a new colony. N. crassa, but not S. macrospora, can also reproduce asexually by producing asexual spores, called conidia, and the start of new N. crassa fungal colonies often involves fusion of multiple conidia (conidial germlings). After either type of germination, mycelial colonies are formed by vegetative growth involving hyphal tip growth and branching. During vegetative growth of both N. crassa and S. macrospora, hyphal fusion occurs to create the interconnected network of the colony, which allows the flow of organelles and cytoplasmic constituents between hyphae. Although septa are formed in the vegetative hyphae of many fungi, subdividing hyphae into chambers, these septa have pores that allow a similar movement of organelles and cytoplasmic constituents. In N. crassa, where mating occurs between two different mating types, conidia also function as male gametes. Sexual reproduction to produce ascospores involves production of specialized female reproductive hyphae called ascogonia. These ascogonia develop into female reproductive structures termed protoperithecia, and then male cells (microconidia or macroconidia) from a different mating type fuse with ascogonial hyphae emanating from protoperithecia. Finally, protoperithecia mature to form perithecia, fertile fruiting bodies containing ascospores that have been produced through meiosis. In S. macrospora, which is self-fertile, similar development from ascogonia to protoperithecia and then perithecia occurs, but with self-fertilization that does not involve conidia. As will become clear in the following subsections, STRIPAK-like complexes appear to play critical roles in growth, sexual development, and virulence of filamentous fungi.
4.1.4.2. STRIPAK-like complex function in Neurospora crassa growth and development
The N. crassa STRIP1/2 homolog, Ham-2 (Table 2), was originally identified as a protein required for sexual development and hyphal fusion (Xiang et al., 2002). Ham-2 loss or mutation resulted in slower growth, shorter aerial hyphae, female sterility, and hyphal fusion defects, despite normal vegetative hyphal architecture. Subsequently, Ham-2 was determined to be necessary for the chemotropic, polarized growth of conidial anastomosis (hyphal fusion) tubes required to facilitate fusion of conidial germlings (Roca et al., 2005). Moreover, Ham-2 mutant macroconidia lacked the ability to attract conidial anastomosis tubes from wild-type conidial germlings, indicating that Ham-2 was necessary for producing the yet unidentified chemoattractant. Because the Map kinase, Mak-2, and the Map kinase kinase, Nrc-1, also demonstrated these same defects, it was hypothesized that Ham-2 functions together with the Map kinase pathway to regulate chemotropic polarized growth in conidia and conidial germlings (Roca et al., 2005). In support of this possibility, a functional interaction between Mak-2 and Ham-2, Mob3, and some other STRIPAK-like complex components was recently reported (Dettmann et al., 2013). However, this functional interaction was not shown to affect tropic interactions of conidial germlings. Results from budding yeast also support the idea of cooperative functioning of STRIPAK-like complexes and Map kinases. Map kinase signaling is also important for chemotropic polarized growth in response to pheromone during mating in budding yeast (Arkowitz, 2009), where the FAR complex containing a Ham-2 homolog is necessary to maintain the G1 phase cell cycle arrest required for sexual polarized growth (Kemp and Sprague, 2003). Interestingly, another member of the FAR complex, Far9, a homolog of SLMAP, has been shown to be required for secretion of the yeast pheromone, α-factor (Bonangelino et al., 2002). Thus, STRIPAK-related complexes likely function together with Map kinases in regulating aspects of chemotropic polarized growth in both yeast and filamentous fungi.
Recently, Simonin and colleagues reported that N. crassa Ham-2, Ham-3 (striatin homolog), and Ham-4 (SLMAP homolog) mutants all display a similar reduction in growth rate, short aerial hyphae, dramatically reduced conidiation (production of conidia), enlarged vacuoles, and delayed formation of protoperithecia (Simonin et al., 2010). In addition, like Ham-2 mutants, loss of Ham-3 or Ham-4 caused loss of chemotropic interactions during germling fusion and these strains were highly defective in germling and hyphal fusion. While Ham-3 and Ham-4 were not required for chemotropic interactions during mating and sexual fusion (Ham-2 was not able to be evaluated), they were required for proper ascosporogenesis. Their loss resulted in defects in the morphology, number, and/or size of the ascospores produced and increased the osmotic fragility of the ascospores that were produced (Simonin et al., 2010). These data support the idea that Ham-2, Ham-3, and Ham-4 function together as a STRIPAK-like complex in these developmental processes.
Another N. crassa STRIPAK component homolog, Mob3, was implicated in sexual development and asexual cell fusion in a study examining the relationship between NDR kinases and Mob proteins in N. crassa (Maerz et al., 2009). While N. crassa Mob1 regulates the NDR kinase Dbf2, and Mob2a and Mob2b regulate the NDR kinase Cot1, Mob3 functions independently of NDR kinases (Maerz et al., 2009). Loss of Mob3 in N. crassa had little effect on growth and caused a weak conidiation defect, but dramatically reduced conidial germling and hyphal fusion and the development of protoperithecia. When ΔMob3 cells were used as the male in crosses with wild-type cells, development progressed further, but very few ascospores were produced, indicating that like Ham-2 and Ham-3, Mob3 has a role in ascosporogenesis (Maerz et al., 2009). Using a morphology-based screening protocol, another group recently identified 24 genes required for hyphal cell fusion in N. crassa (Fu et al., 2011). A number of these genes, including, but not limited to, Ham-2, Ham-3, and Mob3, had been previously implicated in hyphal fusion. Among the novel genes identified were the genes encoding the PP2A C subunit (Fu et al., 2011), another known component of STRIPAK complexes. Like Ham-2, Ham-3, and Ham-4, N. crassa PP2A C subunit is also critical for production of conidia (Nargang et al., 2012). Recently, similar results were obtained regarding the role of N. crassa STRIPAK-like components in vegetative hyphal fusion, except that while Ham-2, Ham-3, and the PP2A catalytic subunit, Ppg1, were found to be essential for hyphal fusion of conidial germlings, Ham-4 and Mob3 were found to be less important (Dettmann et al., 2013). Thus, five known homologs of STRIPAK components regulate hyphal fusion and conidiation in N. crassa, consistent with the idea that a N. crassa STRIPAK-like complex regulates these developmental processes and others in which these proteins have been implicated.
4.1.4.3. STRIPAK-like complex function in Sordaria macrospora growth and development
A STRIPAK-like complex also appears to regulate developmental processes in S. macrospora, a close relative of N. crassa. Mutation of S. macrospora Pro11 (Table 2), a striatin homolog found localized primarily in membranes, caused an increased density of aerial hyphae but no general growth defect (Poggeler and Kuck, 2004). It also resulted in a reduction in the number of protoperithecia, and a block in development from protoperithecia to perithecia. Strikingly, expression of mouse striatin fully restored the ability to develop fertile fruiting bodies and produce normal ascospores, although the frequency of abnormal asci with reduced numbers of ascospores was still modestly increased. Thus, striatin is a functional homolog of Pro11 (Poggeler and Kuck, 2004). A nonsense point mutation (Q437stop) in the S. macrospora STRIP1/2 homolog, Pro22, or deletion of its gene also caused defects in hyphal fusion and protoperithecia development, and a block in the developmental progression from protoperithecia to perithecia (Bloemendal et al., 2012, Bloemendal et al., 2010, Rech et al., 2007). Protoperithecia formed were clearly less differentiated and contained thin-walled hyphae (Bloemendal et al., 2010). As mentioned above, protoperithecia develop from specialized female hyphae called ascogonia. Interestingly, the ascogonia of a Pro22 mutant largely lacked intercalary septa (Bloemendal et al., 2010). The absence of septa was specific to these sexual reproductive structures as septation of vegetative hyphae was normal, suggesting that Pro22 may play a role in regulating septation specifically during sexual development. In contrast, a C-terminal truncation mutant of Pro11 that retains 545 out of the 845 amino acids found in the full-length protein did not show a similar defect (Bloemendal et al., 2010). However, we hypothesize that this difference may be due to dissimilarities in the relative severity of the Pro22 and Pro11 mutations. A more recent analysis shows that the Pro11 truncation mutant analyzed in this study has substantial residual activity in other assays for sexual developmental (Bloemendal et al., 2012). As evidence for the generality of results obtained in S. macrospora, multiple STRIPAK homologs, including a Pro22 homolog, Csc2, and a Pro11 homolog, Csc3, have been implicated in control of septation in S. pombe (Singh et al., 2011). Finally, a role for Pro22 in septation is consistent with the report that a S. cerevisiae Pro22 homolog, Far11, was found to bind to Rho4 (Uetz et al., 2000), a small GTPase whose N. crassa homolog is required for septation (Rasmussen and Glass, 2005).
S. macrospora Mob3 (SmMob3) is also essential for hyphal fusion and fruiting body development. Loss of SmMob3 expression caused slower growth, increased aerial hyphae density, greatly reduced numbers of protoperithecia, and a block in development from protoperithecia to perithecia (Bernhards and Poggeler, 2011), phenotypes which are similar to those caused by a Pro11 mutation in N. crassa (Poggeler and Kuck, 2004). RNAi knockdown of Pro11 in a ΔSmMob3 strain or deletion of Pro11 caused an even earlier block in sexual development, with almost no ascogonia production and complete loss of protoperithecia development (Bernhards and Poggeler, 2011, Bloemendal et al., 2012). Moreover, a mutant Pro11 strain was unable to complement a ΔSmMob3 strain (Bernhards and Poggeler, 2011). These results support the idea that S. macrospora striatin and Mob3 homologs function together, perhaps as a complex in these processes (Bernhards and Poggeler, 2011), but that Pro11 has Mob3-independent functions. However, unlike mammalian striatin, which was able to partially complement a Pro11 mutant strain (Poggeler and Kuck, 2004), expression of mouse Mob3 was unable to complement SmMob3 loss (Bernhards and Poggeler, 2011).
4.1.4.4. STRIPAK-like complex function in Fusarium verticillioides and Fusarium graminearum growth, development, and virulence
The striatin family homolog, Fsr1 (Table 2), plays a critical but undefined role in the virulence of the plant pathogenic filamentous fungi, Fusarium verticillioides (F. verticillioides) and Fusarium graminearum (F. graminearum) (Shim et al., 2006). F. verticillioides defective for Fsr1 display reduced radial growth on agar, fewer aerial mycelia, and inability to penetrate and grow in maize stalk in a stalk-rot assay (Shim et al., 2006, Yamamura and Shim, 2008). While Fsr1 is not critical for male fertility in F. verticillioides, it is required for female fertility and the development of perithecia (Shim et al., 2006). Similarly, deletion of Fsr1 in F. graminearu also leads to a block in perithecia development and significantly reduced head blight symptoms of this fungus on barley (Shim et al., 2006). In F. verticillioides, the growth and virulence defects of a ΔFsr1 strain could be rescued by expression of an Fsr1 mutant lacking the WD-repeat domain or by expression of a mutant lacking both the WD-repeat domain and the caveolin-binding domain (Yamamura and Shim, 2008). Given the importance of the WD-repeats for Mob3 binding in mammalian cells (Gordon et al., 2011), it will be interesting to determine if the F. verticillioides Mob3 homolog is dispensable for virulence. Also of note, caveolins do not appear to be expressed in fungi (Field et al., 2007) and some of the fungal striatin homologs lack a full consensus caveolin-binding domain (Couet et al., 1997); thus, the function of this stretch of amino acids in fungal striatin homologs is currently unclear. Interestingly, the growth and virulence defects of the ΔFsr1 strain could not be rescued by expression of a mutant lacking both the WD-repeat domain and the coiled-coil domain (Yamamura and Shim, 2008). Thus, at least in the absence of the WD-repeat domain, the coiled-coil domain is required for normal growth and virulence, while the WD-repeat domain appears to be dispensable for both.
4.1.4.5. STRIPAK-like complex function in Aspergillus nidulans growth and development
In A. nidulans, a fungus that produces cleistothecia (closed fruiting bodies) instead of perithecia, the striatin homolog, StrA (Table 2), is important for colony growth, conidiation, conidia germination, and sexual development (Wang et al., 2010). Loss of StrA expression reduces the number and size of cleistothecia. It also reduces the number of ascospores and increases the amount of abnormal (unseparated or improper shaped) ascospores, indicating a role for StrA in ascosporogenesis. On the other hand, overexpression of StrA decreased conidiation but increased the number of cleistothecia, demonstrating that StrA positively influences sexual development. All things considered, the roles for A. nidulans StrA are very similar to the roles described for STRIPAK component homologs from N. crassa, S. macrospora, F. verticillioides, and F. graminearu.
4.1.4.6. Structure-function analysis of the Sordaria macrospora STRIPAK-like complex
The functional similarities of the fungal STRIPAK component homologs described above suggest that they could function together in a STRIPAK-like complex in filamentous fungi. Recently, the first direct demonstration of a physical STRIPAK-like complex in fungi was reported for S. macrospora. The STRIP1/2 homolog, Pro22, forms a complex with the striatin homolog, Pro11, and the SmPP2A A and C subunits (Bloemendal et al., 2012). Pro11 also interacts with SmMob3, expanding the likely complex further. A similar STRIPAK-like complex was subsequently described in N. crassa that also included the N. crassa SLMAP homolog, Ham-4 (Dettmann et al., 2013). Results of a yeast two-hybrid analysis of deletion mutants from S. macrospora were also generally in good agreement with published results regarding mammalian STRIPAK structure-function (Bloemendal et al., 2012, Gordon et al., 2011, Kean et al., 2011). One exception was that the two-hybrid analysis provided evidence for an interaction of SmPP2AA with Pro11 residues 111–281, which includes only residues C-terminal to the Pro11 coiled-coil domain (Bloemendal et al., 2012). This is in apparent contrast to results from mammalian cells, which showed that the coiled-coil domain is necessary for PP2A association with striatin family members and that point mutants within this domain can dissociate PP2A without loss of oligomerization (Gordon et al., 2011, Kean et al., 2011). Moreover, purified mammalian PP2A A subunit directly interacts with SG2NA residues 57–169, which include the coiled-coil and a short stretch of amino acids after the coiled-coil domain (Gordon et al., 2011, Kean et al., 2011). Further experimentation will be necessary to determine whether these results arise from a difference between mammalian and fungal STRIPAK organization. Another interesting finding from the two-hybrid results was the discovery that Pro22 may interact with the C-terminal half of SmPP2AA independently of Pro11 (Bloemendal et al., 2012). While this result provides new insight into the organization of the fungal STRIPAK complex (Bloemendal et al., 2012), we speculate that it may have another important implication as well. If Pro22 indeed interacts with PP2A independently of striatin family members, then it may be capable of targeting other PP2A complexes (dimer or trimer) in addition to STRIPAK complexes. One prediction of this hypothesis is that STRIP1/2 homologs will be found to have cellular roles not wholly explained by STRIPAK function. A potential example of such a function may be found in FAR complex regulation of TORC2 signaling in yeast (discussed above in section 4.1.3.1).
Finally, functional analysis of C-terminal deletion mutants of Pro11 provided some interesting correlative results. First, consistent with the requirement for SmMob3 for development from protoperithecia to perithecia (Bernhards and Poggeler, 2011), the Pro11 WD-repeat domain, the likely binding domain for SmMob3, is critical for development from protoperithecia to perithecia (Bloemendal et al., 2012). Second, wild-type ascogonial development, but not protoperithecia development, was supported by a mutant containing the coiled-coil domain, but lacking most sequences C-terminal to that domain. The ability of different Pro11 sequences to support different aspects of sexual development suggest that different Pro11-associated proteins may be more important for some stages of development than others.
In summary, functional studies of STRIPAK component homologs, the demonstration of physical interactions between these proteins, and the ability of mammalian striatin to functionally substitute for a fungal striatin homolog indicate the existence of a fungal STRIPAK-like complex that plays important roles in growth, conidiation, germling and hyphal fusion, sexual development, ascosporogenesis, and virulence. Much remains to be elucidated regarding the mechanisms by which the fungal STRIPAK-like complex contributes to these processes. Consideration of these events reveals that, among other roles, STRIPAK-like complexes function in cell-cell communication and polarized growth during sexual development, cell-cell fusion events, and meiosis.
Similarities between the roles of STRIPAK-like complexes in filamentous fungi and yeast provide additional insight. In budding yeast, the STRIPAK-like FAR complex also participates in regulation of polarized growth during mating (Kemp and Sprague, 2003). Interestingly, in neither yeast nor fungi do STRIPAK-like complexes appear to be critical for polarized growth unrelated to mating, such as budding in yeast or hyphal growth in fungi. This is consistent with the possibility that they play a regulatory role in this process during sexual development, although additional roles cannot be excluded. The role of Mob3 in these functions is intriguing. Fungal Mob3 is critical for cell fusion events, full sexual development and proper ascosporogenesis. Consistent with this, the striatin family WD-repeat domain shown to be important for Mob3 binding (Gordon et al., 2011) is also required for sexual development in filamentous fungi (Bloemendal et al., 2012). While Mob3 is related to other Mob proteins, including those in yeast, there is no direct Mob3 homolog in yeast. Moreover, the striatin homolog in budding yeast, Far8, lacks a WD-repeat domain (Goudreault et al., 2009). Thus, it appears that both Mob3 and its binding site on striatin homologs are lacking in yeast. Whether the absence of Mob3 in yeast indicates that its role in cell-cell fusion in yeast is played by a non-STRIPAK-like yeast component is unknown. Similarities in STRIPAK component homolog function in yeast and fungi include roles in mating factor/chemotropic agent secretion and polarized growth during sexual development and mating. Whether STRIPAK-like complex components play a role in the actual fusion events and what their role in ascosporogenesis might be remains to be determined, but their implication in cellular processes such as vesicular trafficking, microtubule organization, cell wall integrity pathway, cytokinesis and abscission in different organisms suggests possible directions that should be explored.
4.2. STRIPAK-like complexes and estrogen receptor signaling
Estrogens regulate cellular functions by signaling via their receptors, ERα and ERβ, through both genomic and non-genomic pathways (O’Lone et al., 2004, Raz et al., 2008). Genomic effects of estrogen refer to ligand-dependent activation of ERs as transcription factors in the nucleus to modulate gene expression. Nongenomic effects of estrogen are mediated by ER localized to the caveolae (Chambliss et al., 2000), and in endothelial cells involve rapid activation of several kinases, including Erks and Akt, and activation of eNOS (for reviews, see (Raz et al., 2008, Wu et al., 2011)). Ligand-independent regulation of ERs through the action of kinases and phosphatases has also been proposed (Lu et al., 2003).
Striatin family members serve as scaffolds for formation of separate PP2A/ERαcomplexes involved in genomic and non-genomic signaling by estrogens (Lu et al., 2004, Tan et al., 2008). Rat STRN3γ, a nuclear-localized splice variant of SG2NA, organizes an estrogen-inducible STRIPAK-like complex containing PP2A and ERα to facilitate downregulation of ERα transcriptional activity by PP2A-mediated dephosphorylation of ERα(Tan et al., 2008) (Fig. 2). On the other hand, striatin organizes an estrogen-enhanced STRIPAK-like complex for rapid, non-genomic ERαsignaling at the plasma membrane that contains PP2A, ERα, Gαi, and eNOS (Lu et al., 2004). Striatin binds ERα directly and targets it to the cell membrane. The first 203 amino acids of striatin and residues 183–253 of ERα are sufficient for formation of the striatin-ERα complex. Importantly, disruption of striatin binding to ERα using an ERαblocking peptide (aa176-253) inhibited striatin-ERα complex formation and estrogen-induced eNOS activation. However, the blocking peptide may have prevented other proteins like PP2A from interacting with ERα as well since the same peptide was previously shown to bind PP2A (Lu et al., 2003). More recently, striatin knockdown in endothelial cells was shown to block rapid activation of Akt and eNOS by estrogen, demonstrating clearly that striatin is essential for activation of these enzymes by non-genomic estrogen signaling (Bernelot Moens et al., 2012).
Nitric oxide produced by estrogen-activated eNOS has vasoprotective effects. These effects include vasodilation, enhanced growth and migration of endothelial cells, and inhibition of the growth and migration of vascular smooth muscle cells (Wu et al., 2011). A crucial role for the striatin-ERα complex in vasoprotection is supported by results from a mouse model in which striatin-ERα blocking peptide (aa176-253) was expressed (Bernelot Moens et al., 2012). The peptide disrupted the striatin-ERα complex in cells, inhibited estrogen-induced endothelial cell migration, and abolished estrogen inhibition of vascular smooth muscle cell growth. The relevance of these effects was strengthened by the fact that expression of the peptide prevented estrogen-mediated protection against vascular injury in a carotid artery injury mouse model (Bernelot Moens et al., 2012). These results represent a major advance in our understanding of non-genomic ERα signaling. However, an alternative approach such as generation of an endothelial-targeted striatin knockout mouse will be necessary to confirm striatin’s role because of the caveat mentioned above to experiments utilizing a blocking peptide. Nevertheless, as noted previously (Lu et al., 2004), the discovery of specific complexes that regulate genomic and non-genomic ERα signaling may facilitate the development of specific modulators for each pathway. Such modulators would represent potential therapeutic tools for combating diseases such as breast cancer and cardiovascular disease where differential modulation of these ERα signaling pathways could be beneficial. Also, given that disruption of the striatin-ERα complex was not lethal in mice, it is possible that mutations in striatin, ERα or other components of this assembly that disrupt these complexes will be found in humans. Thus, it would be interesting to look for such disrupting mutations in striatin, ERα, and other components of these complexes in cardiovascular patients, particularly among premenopausal women, since estrogen receptor signaling has been reported to have a protective function against cardiovascular diseases.
Interestingly, endothelial cell striatin was recently reported to complex with another steroid hormone receptor, the mineralocorticoid receptor (MR) (Pojoga et al., 2012). MR is activated by aldosterone, a mineralocorticoid that functions to enhance sodium uptake in cells that express the MR receptor. In rodent models, increasing aldosterone plasma concentrations increases the striatin protein level in heart tissue (Pojoga et al., 2012, Ricchiuti et al., 2011). Similarly, aldosterone treatment increases striatin protein and mRNA levels in endothelial cells in a MR-dependent manner (Pojoga et al., 2012). Together, these data suggest that aldosterone regulates striatin levels via activation of the MR. Given the role of striatin in targeting ERα to the cell membrane and orchestrating regulation of ERα non-genomic signaling by estrogen (Lu et al., 2004), aldosterone regulation of striatin levels via the MR has the potential to modulate estrogen non-genomic signaling (Pojoga et al., 2012). Additional studies will be necessary to further investigate the nature of the proposed striatin-MR complex and whether striatin family members form additional complexes with other steroid hormone receptors.
4.3. STRIPAK and vesicular trafficking
The fact that striatin family members have been found in all mammalian cells tested to date is consistent with the idea that they may participate in one or more functions common to all cell types. Multiple lines of evidence suggest that one such function might be their involvement in endocytosis and vesicular trafficking. Striatin, SG2NA, and the striatin family-associated protein Mob3 are located in the Golgi, cytoplasm, and plasma membrane (Baillat et al., 2001, Moreno et al., 2001). The association of SG2NA and Mob3 with the Golgi is rapidly altered by treatment of cells with brefeldin A (Baillat et al., 2001), which inhibits certain guanine nucleotide exchange factors for ARFs, small monomeric G proteins that regulate coatamer protein binding to membranes and thus vesicular trafficking (Donaldson et al., 1992). The fact that Mob3 has sequence homology with the σ light chain subunit of clathrin adaptor complexes led to the hypothesis that Mob3 might be a component of a new type of Arf-dependent coat protein involved in vesicular transport (Baillat et al., 2001). Additional evidence for a role of Mob3 (and potentially striatin family members) in endocytosis and vesicular transport came from the finding that Mob3 interacts with nucleoside-diphosphate kinase (NDPK), epidermal growth factor receptor substrate 15 (Eps15), and dynamin I, and also that Mob3 partially colocalizes with dynamin I in neurons (Baillat et al., 2002). NDPK, Eps15, and dynamin I are involved in membrane dynamics, including clathrin-dependent endocytosis. Eps15 (Fazioli et al., 1993) is an endocytic coat adaptor protein involved in ligand-induced receptor endocytosis of receptor tyrosine kinases (RTKs) like EGFR. This function is critical for homeostatic limiting of RTK signaling and down-regulation of RTK signaling after ligand-induced activation (for reviews, see (Marmor and Yarden, 2004, van Bergen En Henegouwen, 2009)). Mammalian Eps15 has been shown to interact directly with dynamin I and in C. elegans the two proteins interact genetically (Salcini et al., 2001). Dynamin I is a GTPase that functions in the fission of clathrin-coated vesicles from the plasma membrane during endocytosis. NDPK interacts directly with dynamin I (Baillat et al., 2002), and based on work in Drosophila, is thought to regulate endocytosis by providing a local pool of GTP for dynamin I (Krishnan et al., 2001). Thus, STRIPAK may regulate endocytosis and vesicular trafficking through Mob3 (Fig. 2).
Results from studies with dMob4, the Drosophila Mob3 functional homolog, also support a role for Mob3 in endocytosis and vesicular trafficking. Analysis of dMob4 mutants revealed that dMob4 is important for microtubule organization, axonal transport, synapse assembly, and neurite growth and branching (Schulte et al., 2010, Sepp et al., 2008). Moreover, dMob4 mutants displayed phenotypes similar to other known endocytic mutants, including Drosophila Eps15. One model that could explain many of the observed defects is that dMob4 is important for proper localization or activity of NDPK, which is needed to supply GTP for dynamin I function and mictrotubule polymerization.
Mammalian striatin and SG2NA are also known to associate with GIPC (GAIP-interacting protein, C terminus), a protein involved in receptor (e.g., VEGFR2 and TrkA) endocytosis and trafficking, further corroborating a possible role of striatin family members in vesicular trafficking (Varsano et al., 2006). Although the significance of the interaction between GIPC and striatin family members was not explored, GIPC and striatin family members might collaborate in endocytosis of cell surface receptors to regulate receptor-mediated signaling pathways. This model would be consistent with the association of Eps15 with Mob3 mentioned above.
In filamentous fungi, a possible role for STRIPAK component homologs in vesicular trafficking has also begun to be investigated. An overexpressed, functional GFP-fusion protein of the S. macrospora STRIP1/2 homolog, Pro22, localized to vesicular and tubular vacuolar structures near the tips of the growing hyphae and in ascogonia (Bloemendal et al., 2010), suggesting that it may be involved in some aspect of vacuolar function. Perhaps related, in N. crassa, mutations in Ham-2 (STRIP1/2 homolog), Ham-3 (striatin homolog), or Ham-4 (SLMAP homolog) caused an enlarged vacuole phenotype (Simonin et al., 2010). In A. nidulans, GFP-tagged striatin (StrA) was found mainly in the endoplasmic reticulum and nuclear envelope (Wang et al., 2010). Although the Mob3 proteins of filamentous fungi such as N. crassa and S. macrospora are much larger than Mob3 proteins from animals such as nematodes, fruit flies, and mammals, they share a conserved mob domain (Bernhards and Poggeler, 2011). As with Mob3 from animals, sequences homologous to the σ light chain subunit of clathrin adaptor complexes are found within their conserved mob domain (Baillat et al., 2001, Bernhards and Poggeler, 2011), suggesting that these Mob3 proteins may function in endocytosis. Loss of S. macrospora Mob3 (SmMob3) resulted in no detectable effect on endocytosis as measured by comparing internalization of the lipophilic dye, FM4-64 (Bernhards and Poggeler, 2011). Similarly, loss of the A. nidulans striatin homolog, StrA, had no effect on FM4-64 internalization (Wang et al., 2010). However, only certain endocytic mutants affect FM4-64 internalization in yeast (Vida and Emr, 1995). Thus, it is still possible that SmMob3 functions in one or more endocytic pathways that do not regulate FM4-64 internalization.
In yeast, members of the STRIPAK-related FAR complex have been implicated in vesicular trafficking. Based on experiments using fluorescent fusion proteins, the S. cerevisiae STRIP1/2 homolog, Far11, predominantly localizes to late Golgi vesicles at the cellular periphery, while the S. cerevisiae SLMAP homolog, Far9, mainly localizes to the endoplasmic reticulum-Golgi (Lisa-Santamaria et al., 2012). Far9, also called Vps64, is important for proper sorting of proteins to the vacuole and for secretion of α-factor, but not for its processing (Bonangelino et al., 2002). In addition, Far11 (Ynl127) has a weak effect on protein sorting to the vacuole (Bonangelino et al., 2002). Thus, the FAR complex may be involved in intracellular trafficking and possibly exocytosis. In addition, during a global analysis of protein localization in S. pombe, the S. pombe STRIP1/2 homolog (gene name SPBC27B12.04c) was found to be localized to the vacuole (Matsuyama et al., 2006).
In summary, substantial evidence from multiple organisms implicates a role for STRIPAK components in vesicular trafficking. However, much additional research is needed to determine the specific pathways in which they participate, the roles of the different STRIPAK components, and the mechanisms and regulation of their function.
5. The role of STRIPAK in diseases
STRIPAK complexes have been connected to a number of clinical conditions and diseases, including heart disease, diabetes, autism, cancer, and cerebral cavernous malformation. Evidence supporting these connections, and possible mechanistic involvement of STRIPAK complexes will be discussed below.
5.1. STRIPAK and heart disease
Several connections to cardiac disease have been reported. First, the striatin gene was discovered in one of twenty-two loci containing common variants associated with cardiac ventricular conduction and QRS interval length (Sotoodehnia et al., 2010). Prolonged QRS duration is associated with sudden cardiac death. Second, an 8 base-pair deletion in the 3′ untranslated region of striatin resulting in reduced striatin mRNA was recently implicated in a canine model of arrhythmogenic right ventricular cardiomyopathy (ARVC) (Meurs et al., 2010), a disease where prolonged QRS interval is a common electrocardiographic abnormality and can be predictive of an adverse outcome (Lemola et al., 2005, Steriotis et al., 2009, Zhao et al., 2011). Third, an association was found between dilated cardiomyopathy (DCM) in Boxer dogs and the same 8 base-pair striatin deletion, especially with the homozygous genotype (Meurs et al., 2013). As discussed previously, the STRIPAK component SLMAP has isoforms that appear to be specific for select muscle tissues (including cardiac tissue) and is located in the sarcolemma, T-tubules, and SR in cardiac muscle. SLMAP has therefore been postulated to play a structural role in the arrangement of the excitation-contraction coupling apparatus (Guzzo et al., 2005). However, SLMAP also has the potential to recruit STRIPAK components to these locations to regulate heart muscle function. Recently, transgenic overexpression of SLMAP in mice was found to induce pathologic cardiac remodeling, including apparent expansion of SR membranes, increased expression of cardiac fetal genes, and age-related impairment of contractility and relaxation (Nader et al., 2012). Intriguingly, transgenic overexpression of SLMAP greatly reduced the expression of some key SR Ca2+ handling proteins, including the SR Ca2+-ATPase, Serca2a, calsequestrin, and triadin. Reductions in each of these proteins have been linked to heart failure in humans (Chopra and Knollmann, 2009, Marks, 2013, Roux-Buisson et al., 2012). Most recently, two SLMAP mutations were identified that were associated with Brugada syndrome (Ishikawa et al., 2012), a cardiac channelopathy characterized by a high incidence of ventricular fibrillation and sudden cardiac death (Brugada and Brugada, 1992, Chen and Priori, 2008, Ishikawa et al., 2013). While the main disease-causing gene found to date for Brugada syndrome is SCN5A, which encodes the α-subunit of the cardiac sodium channel, hNav1.5 (Chen and Priori, 2008), mutations in the cardiac L-type calcium channel have also been found (Antzelevitch et al., 2007), and the genetic cause of many cases of Brugada syndrome is still not known. Expression of the newly identified SLMAP mutants in cardiomyocytes showed that they could act in a dominant-negative manner to reduce cell surface expression of hNav1.5 and, consequently, reduce hNav1.5 current (Ishikawa et al., 2012). The dominant-negative action of these mutants is consistent with their heterozygous status in Brugada syndrome patients. Considered together, these data link STRIPAK components to heart disease and justify further investigations into possible roles of STRIPAK complexes in heart function and disease.
5.2. STRIPAK and diabetes
Two components of STRIPAK have been implicated in diabetes through mouse models or patient studies. SLMAP is elevated in two different type 2 diabetes mouse models. Type 2 diabetes db/db mice are a monogenic mouse model with defective leptin receptors. Blood glucose and triglyceride levels are elevated in db/db mice as compared to heterozygous db/+ controls and acetylcholine-induced relaxation of small mesenteric arteries in homozygous db/db mice is significantly reduced as compared to db/+ controls, indicating vascular dysfunction (Ding et al., 2005). Levels of the 35kDa SLMAP1 protein and SLMAP mRNA in vascular tissue of these mice correlate with these changes. In particular, SLMAP expression in vasculature is increased in db/db mice but reduced by activation of peroxisome proliferator-activated receptor γ (PPARγ), which restores normal levels of blood glucose, triglycerides, and acetylcholine-induced relaxation in db/db mice. Moreover, upon removal of the PPARγ agonist, SLMAP expression, blood glucose and triglyceride levels revert to higher levels typical of db/db mice and the defect in acetylcholine-induced relaxation returns. Thus, SLMAP expression correlates with endothelial dysfunction and elevated glucose and triglycerides in this diabetic mouse model (Ding et al., 2005).
Although it was proposed that SLMAP expression might be regulated by PPARγ(Ding et al., 2005), additional evidence suggests that it may be regulated indirectly by the changes in glucose and/or triglyceride levels (Chen and Ding, 2011). In the polygenic Tally Ho type 2 diabetes mouse model, expression of SLMAP1 and SLMAP2 was reported to be elevated in adipose tissue of hyperglycemic mice and to be increased in Tally Ho adipocytes incubated in high glucose (Chen and Ding, 2011). Some evidence suggests an association of SLMAP2 with the glucose transporter, Glut-4. Consistent with this, downregulation of SLMAP in Tally Ho adipocytes reduced glucose uptake (Chen and Ding, 2011). Thus, expression of specific SLMAP isoforms appear to be regulated by glucose levels and, conversely, SLMAP may have a role in regulating glucose uptake.
Ysk1, another component of STRIPAK complexes, has also been implicated in control of glucose uptake (Nerstedt et al., 2012). Knockdown of Ysk1 in rat myoblasts enhances expression of the glucose transporters, Glut1 and Glut4, and hexokinase-2, enhances insulin-stimulated glucose uptake, increases the expression of uncoupling protein 3, and increases lipid oxidation (Nerstedt et al., 2012). Correspondingly, Ysk1 mRNA was found to be significantly elevated in skeletal muscles of type 2 diabetic patients (Nerstedt et al., 2012). Considered together, the results from these studies are consistent with the possibility that SLMAP-directed STRIPAK plays a role in regulation of glucose uptake in diabetes-relevant tissues and may therefore be a potential drug target for this disease.
5.3. STRIPAK and autism
The gene for the STRIPAK component CTTNBP2 (also called CORTBP2 or CBP90) was initially investigated as a possible autism susceptibility gene because it is located within the q31 band of human chromosome 7, which was a candidate site for an autism susceptibility locus (AUTS1) and contained the severe speech and language disorder locus, SPCH1 (Cheung et al., 2001). Although that initial study did not implicate CTTNBP2 in autism, a more recent study found a male autism patient with a de novo 2 base pair frame shift deletion in CTTNBP2 that is likely to disrupt protein function, providing the first evidence that mutations in CTTNBP2 may cause autism (Iossifov et al., 2012). Consistent with the finding of increased spine density in autism spectrum disorders (Hutsler and Zhang, 2010, Penzes et al., 2011), CTTNBP2 is stably localized to the spines of neuronal dendrites, is important for targeting STRIPAK to dendritic spines, and regulates dendritic spine density (Chen et al., 2012). However, since CTTNBP2 is important for spinogenesis and/or maintenance of spine density and spine density increases in autism spectrum disorders, the autism patient frame shift mutation would be anticipated to cause enhanced CTTNBP2 function. The mutation is predicted to alter the CTTNBP2 coding sequence after amino acid 759 of the 1663 amino acid form of the protein (Iossifov et al., 2012), retaining N-terminal sequences shown to mediate complex formation with striatin (and presumably STRIPAK). Studies to date have only examined the role of a short 630 amino acid splice variant form of CTTNBP2 that is the major form of the protein found in mouse and rat brain (Chen and Hsueh, 2012). Thus, to elucidate the effects of the autism patient CTTNBP2 mutation on CTTNBP2 function, it will be important to determine the forms of CTTNBP2 expressed in human brain, the function of the longer form of the protein, and the effect of the autism patient frameshift mutation on the levels and function of all the forms of CTTNBP2, including the effect on the targeting of the STRIPAK complex to dendritic spines.
5.4. STRIPAK and cancer
The striatin gene was identified as a novel fusion partner for platelet derived growth factor receptor alpha (PDGFRA), leading to PDGFRA activation and contributing to the development of chronic eosinophilic leukaemia (Weber et al., 2001). The fusion contributes the striatin oligomerization domain to PDGFRA and also disrupts the negative regulatory WW-like domain of PDGFRA. While not formally demonstrated as of yet, both of these alterations could contribute to development of the disease.
In a separate study, the gene for the STRIPAK component, FGFR1OP2, was identified as one of a number of partner genes found fused to the FGFR1 in 8p11 myeloproliferative syndrome (EMS) (Grand et al., 2004). The partner proteins, including FGFR1OP2, promote dimerization and ligand-independent activation of FGFR1. In the case of FGFR1OP2, this probably occurs by dimerization via the FGFR1OP2 coiled-coil domains (Grand et al., 2004, Lin et al., 2010). Because of the number of unrelated proteins that can mediate the dimerization of FGFR1 in EMS, it is likely that there may not be any additional contribution of FGFR1OP2 (and thus STRIPAK) to the pathogenesis of EMS beyond mediating dimerization; however possible effects of reduction in cellular FGFR1OP2 on STRIPAK function remain to be determined.
5.5. STRIPAK and cerebral cavernous malformation (CCM)
The identification of Ccm3 as a component of the STRIPAK complex implicated striatin family complexes in a common vascular disease called cerebral cavernous malformation (CCM) (Goudreault et al., 2009), which can be caused by mutation of Ccm3 (Bergametti et al., 2005). Cerebral cavernous malformations are characterized by abnormally enlarged capillary cavities lacking supportive smooth muscle and are predominantly found in the central nervous system, especially the brain (for reviews on CCM, see (Chan et al., 2010, Faurobert and Albiges-Rizo, 2010, Labauge et al., 2007, Riant et al., 2010). Symptoms of this disease include headache, recurrent hemorrhages, stroke, seizure, or even death, while many carriers are asymptomatic.
Functions of Ccm3 as a part of, and independent of, STRIPAK are under active investigation in an effort to solve the molecular mechanisms underlying CCM pathogenesis. Ccm3 is critical for tethering GCKIII kinases to striatin family members in STRIPAK complexes (Kean et al., 2011), and GCKIII kinases have been implicated in Ccm3 function and thus CCM (Chan et al., 2011, Fidalgo et al., 2010, Fidalgo et al., 2012, Ma et al., 2007, Preisinger et al., 2004, Voss et al., 2009, Yoruk et al., 2012, Zhang, 2012, Zheng et al., 2010). Therefore, STRIPAK may regulate Ccm3-GCKIII function in CCM-relevant pathways. Because excellent reviews on CCM exist and a full assessment of CCM literature is beyond the scope of this review, we will not present a comprehensive analysis of the disease. Rather, we will provide background important for discussing possible functions of STRIPAK in the context of CCM pathogenesis, with a special focus on what is known about Ccm3 since it is the only CCM disease protein known to associate with STRIPAK. To do this, in the sections that follow we will first provide a brief background on CCM and then discuss what is known regarding Ccm3 functions, Ccm3 regulation of GCKIII kinases, Ccm3, GCKIII kinase, and STRIPAK regulation of RhoA signaling, and how mutations in Ccm3, GCKIII kinases, or STRIPAK components might contribute to CCM.
5.5.1. CCM disease
Approximately one in 200–250 people in the general population is affected by CCM, among which sporadic cases account for the majority of cases (Del Curling et al., 1991, Robinson et al., 1991). The rest consist of autosomal-dominantly inherited (familial) forms with incomplete penetrance. Most of the inherited forms are likely recessive at the level of the cell, with a secondary somatic mutation being required to cause a malformation (see discussion of the two-hit hypothesis below). The bulk of familial CCM cases arise from loss-of-function mutations in one of three CCM loci: Ccm1 (K-Rev interaction trapped 1; Krit1)(Laberge-le Couteulx et al., 1999, Sahoo et al., 1999), Ccm2 (Osmosensing Scaffold for MEKK3; OSM; Malcavernin)(Denier et al., 2004, Liquori et al., 2003), or Ccm3 (Bergametti et al., 2005). However, there are still cases without detectable mutations in these genes, suggesting that additional CCM genes may exist (Riant et al., 2010). Epigenetic silencing of one of the CCM genes might explain some CCM cases lacking Ccm1-3 protein mutations, but there are other possibilities as well (Akers et al., 2009).
CCM lesions have clusters of dilated blood vessels surrounded by a thin layer of endothelium without supportive smooth muscle cells, making them susceptible to recurrent hemorrhages. The dilated vessels in CCM lesions are characterized by compromised or absent endothelial cell-cell junctions, gaps between endothelial cells, and lack of normal ensheathing cells (Burkhardt et al., 2010, Clatterbuck et al., 2001, Schneider et al., 2011, Tu et al., 2005, Wong et al., 2000). This compromised blood-brain barrier in turn is thought to allow chronic low-level leakage of red blood cells and to help predispose these lesions to hemorrhage, resulting in the hemosiderin deposits often seen in CCM. Consistent with the observed defects in endothelial cell-cell junctions, altered distribution and expression of the vascular adhesion molecule, CD31, and the major TJ and AJ proteins, occludin and VE-cadherin are seen, including detection of their expression beyond the intima of small vessels and capillaries and in surrounding brain tissues of CCM lesions (Burkhardt et al., 2010). Similarly, another study reported altered expression and redistribution of the tight junction proteins occludin, claudin-5, and ZO-1 as well as down-regulation of the major endothelial cell glucose transporter, GLUT-1 (Schneider et al., 2011). Moreover, infiltration of a variety of immune cells, likely due to defective tight junctions and blood brain barrier, is also detected in lesions (Shi et al., 2009). However, it is unclear whether this inflammatory response contributes to CCM lesion genesis. These and other similar observations have led to the conclusion that the Ccm1, Ccm2, and Ccm3 proteins are important for the proper assembly and/or stability of endothelial cell-cell junctions.
The symptoms arising from mutations in any of the three CCM genes cannot be distinguished and Ccm1, Ccm2, and Ccm3 are known to exist in complex with each other, suggesting that Ccm proteins are engaged in similar pathways (Hilder et al., 2007, Labauge et al., 2007, Stahl et al., 2008, Voss et al., 2007). However, several lines of evidence suggest that Ccm3 may have roles that overlap, but are not identical to those of Ccm1 and Ccm2. For example, fewer individuals in Ccm3-affected families present with disease; onset of symptoms tends to be earlier in patients with Ccm3 mutations; and individuals with Ccm3 mutations may have a higher risk of cerebral hemorrhage as compared to those with Ccm1 or Ccm2 mutations (Denier et al., 2006). Similarly, endothelial-specific knockout of Ccm3 in newborn mice caused earlier onset of CCM lesions and a more severe phenotype than the analogous knockout of Ccm2 (Chan et al., 2011). Also, global Ccm3 knockout embryos die earlier than Ccm1 and Ccm2 disrupted embyros (Chan et al., 2011, He et al., 2010), indicating that Ccm3 has a more critical function earlier in embryonic development than Ccm1 and Ccm2. Moreover, unlike Ccm1 and Ccm2, Ccm3 has been reported to not be important for the establishment of circulation (Chan et al., 2011). Thus, Ccm3 may have roles that overlap with, but are not identical to, those of Ccm1 and Ccm2 in CCM-relevant pathways (Fig. 4A). As will be discussed in the next section, the functions of Ccm3 relevant to CCM are still being elucidated and much remains to be determined. Even less is known about how STRIPAK might regulate Ccm3 in one or more of these functions, although this too is currently under investigation.
Fig. 4. Alternative models for Ccm protein function in vascular development and integrity.
(A) Based on the evidence discussed in the text, we propose a model in which Ccm3/GCKIII has overlapping functions with Ccm1/Ccm2. This model is consistent with the common function of these proteins in vascular development and integrity, the fact that Ccm3, Ccm2, and Ccm1 form a complex together, and the differences reported for Ccm3 versus Ccm1 and Ccm2 in CCM mouse models, zebrafish, and human CCM. The arrow from Ccm3 to Ccm2/Ccm1 indicates that Ccm3 functions together with these proteins, not necessarily upstream of them, while the arrow from Ccm3 directly to vascular development and integrity represents a Ccm2/Ccm1-independent pathway. (B) In an alternative model, Ccm3/GCKIII acts only through a distinct pathway from Ccm1 and Ccm2 in vascular development and in promoting vascular integrity (for examples, see (Chan et al., 2011, Yoruk et al., 2012)). (C) In a third model, Ccm3/GCKIII signals together with Ccm1/Ccm2 in a common pathway in vascular development and in promoting vascular integrity (for example, see (Zheng et al., 2010)). Each of these models is consistent with mutation in any one of CCM genes leading to development of CCM, but the first model is the most compatible with existing data.
Development of CCM lesions seems to follow a two-hit mechanism, which means that somatic mutations need to be induced in the background of heterozygous germline mutations to achieve a biallelic mutation of one of the three CCM genes and ultimately develop lesions (Akers et al., 2009, Gault et al., 2009, Gault et al., 2005, Kehrer-Sawatzki et al., 2002, Pagenstecher et al., 2009). This biallelic mutation hypothesis is consistent with the occurrence of multiple lesions in familial cases and a single lesion in sporadic cases (Akers et al., 2009, Pagenstecher et al., 2009, Rigamonti et al., 1988). Studies using Ccm1 and Ccm2 heterozygous mouse models also support the two-hit mutation theory since most of these mice do not develop CCM lesions during the short lifespan of the mouse unless the rate of somatic mutations is induced by loss of Trp53, the mouse gene for the p53 tumor suppressor, or by loss of the gene encoding Msh2, a key mismatch repair protein (McDonald et al., 2011, Plummer et al., 2004, Shenkar et al., 2008).
CCM lesions are found most typically in the brain even though expression of Ccm proteins has been detected in various tissues, leading to a question of what cell type underlies disease pathology. Immunohistochemical analyses and sequencing of amplified genes from lesions made up of different cell types showed that the second, disease-causing somatic mutation in a CCM gene is exclusively found in endothelial cells in all three forms of inherited CCM, indicating that mutations in endothelial cells lead to formation of lesions (Akers et al., 2009, Pagenstecher et al., 2009). Most evidence from CCM animal models supports this conclusion as well (Boulday et al., 2009, Boulday et al., 2011, Chan et al., 2011, He et al., 2010, Kleaveland et al., 2009, McDonald et al., 2011, Whitehead et al., 2009), although results from one study suggest that Ccm3 loss in neuroglial cells could contribute as well (Louvi et al., 2011). In addition, only a subset of germline mutation-carrying endothelial cells in human familial CCM lesions shows the additional somatic mutation in one of three CCM genes, demonstrating endothelial cell mosaicism within lesions (Pagenstecher et al., 2009). This result suggests that Ccm protein-deficient endothelial cells may have dominant effects within lesions. Given that cell-cell adhesion involves the cooperative effort of neighboring cells, defects in cell junctions could conceivably cause such a dominant effect.
5.5.2. Cerebral cavernous malformation 3 (Ccm3) functions
An important question is how Ccm3 loss causes CCM. In this section we will discuss reported functions of Ccm3 that may be relevant to CCM, and in the following sections we will relate these functions to regulation of GCKIII kinases by Ccm3 and STRIPAK. Ccm3 has been implicated in control of apoptosis, cell proliferation, cell migration, cell-cell adhesion, cell polarity, and vascular development. Ccm3 was originally discovered as a gene termed TF-1 cell apoptosis related gene-15 (TFAR15) whose mRNA and protein products increased in the TF-1 myeloid cell upon GM-CSF deprivation-induced apoptosis (Wang et al., 1999). Consistent with this initial finding, Ccm3 expression is induced in serum-starved human umbilical vein endothelial cells (HUVECs) undergoing apoptosis (Chen et al., 2009). However, the role that Ccm3 plays in apoptosis is more controversial. Results from several studies support a proapoptotic role for Ccm3. For example, Ccm3 knockdown reduced apoptosis in HUVECs (Chen et al., 2009, Zhu et al., 2010), human brain microvascular endothelial cells (HBMECs) (Zhu et al., 2010), and CCM patient endothelial cells (CCM-ECs) (Zhu et al., 2010). Correspondingly, overexpression of wild-type Ccm3, but not CCM patient-derived Ccm3 mutants, induced apoptosis in HeLa cells, suggesting that aberrant apoptosis due to loss of pro-apoptotic Ccm3 might contribute to CCM pathology (Chen et al., 2009). In contrast, others have reported an anti-apoptotic role for Ccm3. For example, overexpression of Ccm3 was found to reduce apoptosis in HUVECs (Schleider et al., 2011). Also, Ccm3 knockdown induced apoptosis in some malignant T cell lines while in other lines it did not (Lauenborg et al., 2010). Discrepancies in the observed role of Ccm3 in apoptosis may stem from the use of different cell lines and approaches. Thus, it is difficult to make generalizations regarding Ccm3 function in apoptosis. Moreover, whether Ccm3 function in apoptosis plays an important role in CCM pathogenesis is still questionable, and further evidence is necessary to address this hypothesis.
Roles for Ccm3 in cell proliferation and migration have also been described. Ccm3 knockdown increased proliferation in HUVECs, HBMECs, and CCM-ECs (Zhu et al., 2010). Consistent with those results, overexpression of Ccm3 reduced cell proliferation in HUVECs (Schleider et al., 2011). In addition, one study reported that Ccm3 knockout in neuroglial cells in a mouse model caused increased proliferation, survival, and activation of astrocytes (Louvi et al., 2011). However, Ccm3 knockdown inhibited proliferation in some malignant T cell lines while in other lines it did not (Lauenborg et al., 2010). Thus, while Ccm3 may suppress cell proliferation in endothelial cells and astrocytes, there may also be cell type-specific effects. Regarding Ccm3 effects on migration, Ccm3 knockdown was reported to specifically increase migration of CCM-ECs but not HUVECs or HBMECs (Zhu et al., 2010), while overexpression of Ccm3 reduced migration of HUVECs (Schleider et al., 2011). Thus, Ccm3 can negatively regulate migration of endothelial cells in vitro and it is possible that aberrant migration might contribute to CCM.
Ccm3 is critical for vascular development. As has been reported for Ccm2 (Kleaveland et al., 2009, Whitehead et al., 2009), depletion of Ccm3 in HUVECs caused increased angiogenic sprouting but defective lumen formation (Chan et al., 2011, Zhu et al., 2010). Cranial vasculature lumenization defects are also caused by morpholino knockdown of Ccm3 in zebrafish (Yoruk et al., 2012). Ccm3 overexpression reduces the ability of HUVECs to form capillary-like structures (Schleider et al., 2011), suggesting that either too much or too little Ccm3 may interfere with angiogenesis. Similar to deletion of Ccm1 and Ccm2 (Boulday et al., 2009, Cunningham et al., 2011, Kleaveland et al., 2009, Whitehead et al., 2009, Whitehead et al., 2004), loss of Ccm3 in mice was also reported to cause defects in cell-cell junctions and vascular integrity (He et al., 2010). Endothelial cell-specific inactivation caused increased vessel size, decreased vessel density, compromised vessel integrity, and disruption of TJs and AJs (He et al., 2010). Correspondingly, human dermal microvascular endothelial cells (HMVECs) knocked down for Ccm3 also displayed disruption of TJs and AJs. A second study also reported vascular defects in Ccm3 knockout mice, including increased venous size and venous rupture, but reported no effects on TJs (Chan et al., 2011). These studies additionally demonstrated that endothelial cell-specific knockout of Ccm3 in mice causes vascular defects not observed when Ccm3 is inactivated specifically in neuronal or smooth muscle cells (Chan et al., 2011, He et al., 2010), suggesting that, as for Ccm1 and Ccm2 (Boulday et al., 2009, Boulday et al., 2011, Chan et al., 2011, Cunningham et al., 2011), endothelial cells are the most important tissue in which Ccm3 function is required for development of normal vasculature.
On the molecular level, Ccm3 was reported to regulate the homeostatic level of total and plasma membrane-localized VEGFR2 in endothelial cells and, consequently, the ability of endothelial cells to respond appropriately to VEGF (He et al., 2010). Ccm3 appeared to stabilize VEGFR2 by binding via its N-terminal region to VEGFR2 and blocking endocytosis and degradation of VEGFR2 (Fig. 2). Conversely, expression of C-terminally truncated Ccm3 mutants found in patients caused destabilization of VEGFR2 by enhancing its internalization and degradation (He et al., 2010). However, controversy exists since another report failed to find a connection between Ccm3 and VEGF signaling (Chan et al., 2011). In addition, Ccm2 overexpression did not stabilize VEGFR2 (He et al., 2010) and Ccm2 loss caused defects in endothelial cell junctions and increased VEGF-induced vascular permeability rather than decreasing it (Kleaveland et al., 2009, Whitehead et al., 2009). However, given the connection of striatin family members and Mob3 to endocytosis, the known association of Ccm3 with striatin family complexes, and the fact that mammalian striatin and SG2NA associate with GIPC, a protein involved in VEGFR2 endocytosis and trafficking (Varsano et al., 2006), it will be important to investigate the possibility that STRIPAK may orchestrate Ccm3 regulation of VEGFR2.
5.5.3. Ccm3 as a regulatory adaptor of germinal center kinase III (GCKIII) kinases
Emerging evidence suggests that Ccm3 is critical for STRIPAK regulation of GCKIII kinases by PP2A and that misregulation of GCKIII kinases by mutant Ccm3 contributes to CCM pathogenesis. Mammalian GCKIII kinases regulate diverse cellular processes including cell polarization, cell migration, neuronal differentiation, cell cycle, cell growth and transformation, and cell survival and death (for reviews, see (Ling et al., 2008, Sugden et al., 2013)). Ccm3 binds GCKIII kinases (Ma, 2007, Rual et al., 2005, Voss et al., 2007) and has been implicated in many of these same processes, likely because most functions attributed to Ccm3 are mediated through the associated GCKIII kinases. Ccm3 has two domains, an N-terminal dimerization domain and a C-terminal focal adhesion targeting (FAT)-homology domain (Li et al., 2010) (Fig. 5). Most Ccm3 mutations found in CCM patients affect the FAT-homology domain; therefore, disruption of complexes formed through this domain may be the central cause of CCM development due to Ccm3 mutations (Li et al., 2010). However, since Ccm3 mutants from patients often express poorly, it is difficult to rule out a contribution from loss of associations mediated by the N-terminal region of Ccm3 as well.
Fig. 5. Ccm3 forms different complexes by associating with alternative binding partners through its N- and C-terminal domains.
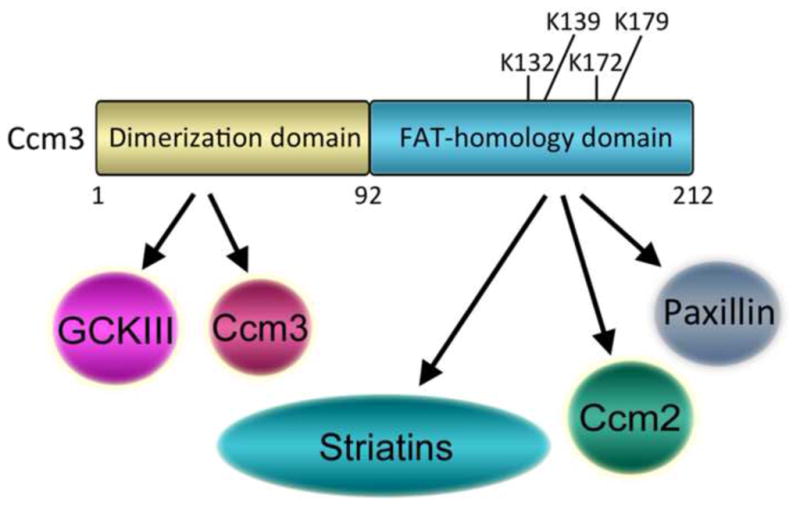
Ccm3 has a dimerization domain at its N-terminus and a FAT-homology domain at its C-terminus. The Ccm3 dimerization domain mediates homodimerization of Ccm3 proteins and binding of GCKIII kinases, two mutually exclusive events. The Ccm3 FAT-homology domain mediates binding of Ccm2, Paxillin, or striatin family members (striatins) to form multiple, independent complexes. Ccm3 may recruit GCKIII kinases to each of these complexes. Four lysines in the FAT-homology domain known to be important for complex formation with its binding partners are indicated.
Ccm3 forms multiple independent complexes in cells through associations mediated by these domains (Fig. 5). For example, Ccm3 binds GCKIII kinases via its N-terminal dimerization domain (Ceccarelli, 2011). In addition, via its FAT-homology domain, Ccm3 binds directly to striatin family members in STRIPAK complexes (Goudreault et al., 2009, Kean et al., 2011), binds Ccm2 as a component of a Ccm1-Ccm2-Ccm3 complex (Hilder et al., 2007, Li et al., 2010, Stahl et al., 2008, Voss et al., 2007), and binds paxillin to form yet another complex (Li et al., 2010). Thus, Ccm3 may bring GCKIII kinases to each of these complexes. Ccm3 has been clearly demonstrated to recruit GCKIII kinases to the STRIPAK complex (Kean et al., 2011). In addition, all three GCKIII kinases associate with Ccm2 (Costa et al., 2012, Voss et al., 2007), consistent with the possibility that Ccm3 brings GCKIII kinases to the Ccm1-Ccm2-Ccm3 complex (Fig. 6, top right). However, it remains to be determined whether GCKIII kinases are present in the Ccm3-paxillin complex. Based on mass spectrometry data, the majority of Ccm3 has been suggested to be in STRIPAK complexes (Kean et al., 2011). By tethering GCKIII kinases to striatin family members in these complexes, Ccm3 brings these kinases in proximity to STRIPAK-bound PP2A, which appears to modulate GCKIII function by downregulating their kinase activity (Gordon et al., 2011). Conversely, GCKIII kinases may regulate STRIPAK function by phosphorylating its components (Fig. 6, top left), many of which are phosphorylated (see, for example (Moreno et al., 2001)); however, no direct evidence for a GCKIII kinase substrate in STRIPAK has been reported.
Fig. 6. Proposed model of CCM pathology.
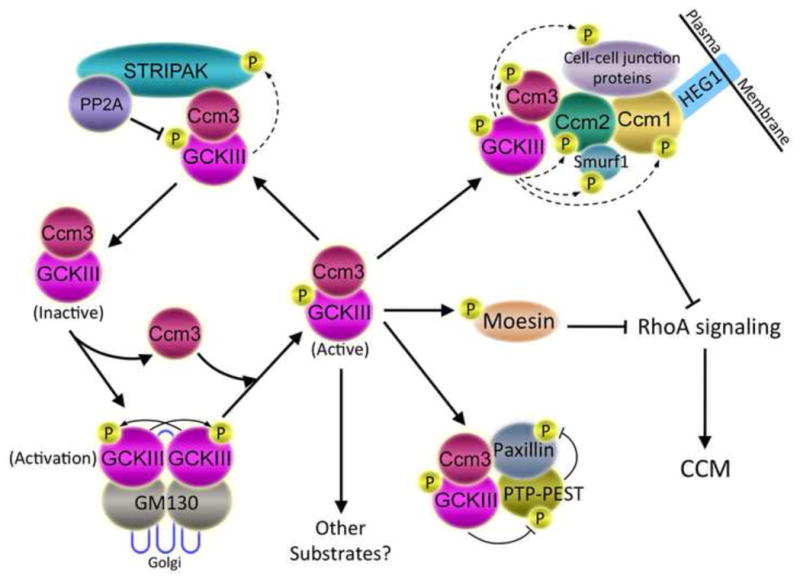
Binding of GCKIII monomers to GM130 in the Golgi induces GCKIII dimerization, autophosphorylation, and activation (lower left). Activated GCKIII kinases are bound and stabilized by Ccm3 (center) and targeted to the STRIPAK complex (upper left), the Ccm protein complex (upper right), the PTP-PEST-Paxillin complex (lower right), moesin (center right), and potentially to other unknown pathways. By binding to Ccm2 protein, Ccm3 is thought to bring GCKIII kinases to Ccm2 and Ccm1, forming a complex that regulates cell-cell junction formation, disassembly, and stability (upper right). Dashed arrows indicate potential phosphorylation of proteins by GCKIII kinases. For example, GCKIII kinases might phosphorylate STRIPAK components (upper left), Ccm proteins (upper right), Ccm protein-associated proteins (represented in the diagram by Smurf1, which is regulated by phosphorylation, but not currently known to be a substrate of GCKIII kinases), or neighboring junctional proteins. Both the Ccm1-Ccm2-Ccm3 complex and GCKIII kinase-phosphorylated moesin inhibit RhoA signaling (right), maintaining junctional homeostasis. Depletion of Ccm1, Ccm2, Ccm3, or GCKIII kinases leads to enhanced RhoA activity, junctional instability, and onset of CCM. Since STRIPAK negatively regulates Ccm3-GCKIII kinases, loss of STRIPAK function is expected to inhibit CCM-relevant pathways, and not induce CCM.
Ccm3 not only participates in the negative regulation of GCKIII kinases by tethering them to STRIPAK complexes (Gordon et al., 2011, Kean et al., 2011) but it also positively regulates these kinases by promoting their stability (Fidalgo et al., 2010). Consequently, GCKIII function is critically linked to the physical integrity of Ccm3. For example, Ccm3 regulates Golgi assembly, Golgi and centrosome polarization, and cell migration by stabilizing GCKIII kinases (Fidalgo et al., 2010) (Fig. 2). Ccm3 interacts with both GM130, a Golgi-resident protein that faces the cytoplasm, and GCKIII kinases and localizes to the Golgi apparatus. Similarly, Mst4 and Ysk1 are targeted to the Golgi via GM130 where they are activated to regulate cell polarization and migration (Preisinger et al., 2004). Depletion of Ccm3 induces ubiquitination and proteasome-mediated degradation of GCKIII kinases and causes disassembly of the Golgi, defective Golgi and centrosome reorientation in wound healing assays, and reduced cell migration (Fidalgo et al., 2010). Ccm3’s role in Golgi assembly is mediated in part by Ysk1 phosphorylation of its downstream target, 14-3-3ζ (Preisinger et al., 2004), since loss of Ccm3 decreases the amount of phosphorylated 14-3-3ζ, and a 14-3-3ζ phosphomimetic mutant rescues the dispersed Golgi phenotype caused by Ccm3 depletion (Fidalgo et al., 2010). Importantly, clinically relevant Ccm3 mutants that do not interact with Ysk1, Mst3, or Mst4 are unable to rescue the effects of Ccm3 depletion, suggesting that interaction of Ccm3 with these GCKIII kinases is important for Golgi function and cell polarity and migration, and may play a critical role in CCM pathology (Fidalgo et al., 2010).
In the experiments just described, Ccm3 protein level was shown to positively influence cell migration in SaOS2 and HeLa cells (Fidalgo et al., 2010). However, Ccm3 is also able to negatively regulate migration of endothelial cells (see section 5.5.2). One possible mechanism for Ccm3 negative regulation of cell migration may be through Ccm3 regulation of Mst3, which has been shown to inhibit cell migration by phosphorylating, and thus inhibiting, the paxillin phosphatase, protein-tyrosine phosphatase (PTP)-PEST, leading to increased paxillin tyrosine phosphorylation (Lu et al., 2006). As Ccm3 reduction leads to concomitant reduction in Mst3, Ccm3 might negatively regulate endothelial cell migration in part via this indirect regulation of paxillin phosphorylation by Mst3. In this regard it is interesting that Ccm3 directly binds paxillin (Li et al., 2010), although whether Ccm3, Mst3, paxillin, and PTP-PEST form a quaternary complex is not yet known (Fig. 6, bottom right).
GCKIII kinases have been implicated in most of the other functions of Ccm3 as well. Ccm3 works together with Mst4 to promote cell growth and transformation by modulating the Erk pathway (Ma et al., 2007). Furthermore, the ability of Ccm3 to promote either cell survival or death may be determined in part by the GCKIII kinase with which it interacts. For example, Ccm3 enhances the phosphorylation of the ERM family of proteins by Mst4 to protect cells from death induced by oxidative stress (Fidalgo et al., 2012) while, in contrast, Ccm3 interacts with Ysk1 to promote apoptosis under oxidative stress (Zhang, 2012). In addition, GCKIII kinases have been connected to the function of Ccm3 in vascular lumenization, because depletion of Ysk1 and either Mst3 or Mst4 in human endothelial cells replicates the lumen formation defects seen with Ccm3 knockdown (Chan et al., 2011). In Drosophila, which has no Ccm1 or Ccm2 orthologs or vascular system, loss of Ccm3 or the single dGckIII kinase results in tracheal tube lumenization failure. The tracheal tube lumenization defect caused by loss of Ccm3 cannot be rescued by a Ccm3 mutant that cannot bind to dGckIII, indicating that Ccm3 functions through dGckIII in this process. These and the other studies mentioned above clearly demonstrate both physical and functional connections between Ccm3 and GCKIII kinases in cellular processes that may be relevant to development of CCM.
In addition, results of several studies in zebrafish suggest that the interaction between Ccm3 and GCKIII kinases may be critical for CCM pathogenesis. A small N-terminal Ccm3 in-frame deletion mutant found in a CCM patient family and/or the equivalent deletion in zebrafish is defective in binding Ysk1, Mst3, and Mst4, but still forms a ternary complex with Ccm1 and Ccm2 (Voss et al., 2009, Zheng et al., 2010). This finding not only revealed a short stretch of residues in Ccm3 important for binding GCKIII kinases but also implicated Ccm3 signaling through GCKIII kinases in CCM. Interestingly, Ysk1 phosphorylated two residues within this binding site in vitro (Voss et al., 2009, Voss et al., 2007). In zebrafish, simultaneous inactivation of its two redundant Ccm3 genes (Ccm3a and Ccm3b) or morpholino-induced in-frame skipping of the newly identified GCKIII binding site in Ccm3a combined with repression of Ccm3b resulted in severe cardiac dilation and pericardial edema, similar to Ccm1 or Ccm2 depletion (Voss et al., 2009, Zheng et al., 2010). In addition, low level depletion of all three GCKIII kinases or of a combination of Ccm3 and either Mst3 or Ysk1, but not any single GCKIII kinase by itself, led to the similar cardiovascular phenotypes, again supporting the importance of the interaction between Ccm3 and GCKIII kinases in cardiovascular development and disease (Zheng et al., 2010). These results indicate that Ccm3 and the interaction of Ccm3 with multiple GCKIII kinases are necessary for proper cardiovascular development in zebrafish. They also support the idea that loss of Ccm3 interaction with GCKIII kinases contributes to CCM development (Voss et al., 2009, Zheng et al., 2010). Yet another study using zebrafish provides additional evidence for the cooperative work between Ccm3 and GCKIII kinases in vascular development (Yoruk et al., 2012). Although morpholino knockdown of Ccm3 did not cause the previously reported dilated heart phenotype, marked pericardial edema and lack of circulation were seen. In addition, this study showed for the first time cranial vasculature defects due to Ccm3 depletion. These defects developed rapidly and did not seem to be due to overproliferation or apoptosis (Yoruk et al., 2012), consistent with previous results from a CCM mouse model system targeting Ccm2 (Boulday et al., 2011). Notably, Ysk1 depletion also caused cranial vasculature defects and overexpression of Ysk1 rescued the cranial vascular defects in Ccm3-depleted zebrafish, but not the dilated heart phenotype or circulation defects of in Ccm2-depleted zebrafish (Yoruk et al., 2012). These results indicate that Ysk1 works downstream of Ccm3 and that Ccm3 positively modulates Ysk1 function in cranial vasculature morphogenesis. Based on obtaining a more severe defect upon reduction of both Ccm2 and Ccm3 and the lack of rescue of Ccm2 by Ysk1 overexpression, the authors suggested that Ccm2 and Ccm3 act through distinct pathways in cranial vascular morphogenesis (Fig. 4B). This hypothesis was in agreement with a similar hypothesis previously put forward by others based on mouse model and mammalian cell data (Chan et al., 2011). However, as illustrated in Figure 4A and discussed below in section 5.5.5, we propose that these results and those of others could be better explained by a model in which Ccm3 and GCKIII kinases function together with Ccm2-Ccm1, but also have at least one independent function. In summary, results from multiple organisms strongly suggest that the primary role of Ccm3 is as an adapter that regulates GCKIII kinase function, that CCM-relevant Ccm3 functions are largely if not wholly carried out through these kinases, and that loss of GCKIII function contributes to the development of CCM.
5.5.4. STRIPAK regulation of Ccm3 and GCKIII kinases
Despite the identification of Ccm3 as a component of the STRIPAK complex in 2009, to date little research has been done to investigate how STRIPAK might regulate Ccm3-GCKIII kinase function. One study recently showed that STRIPAK-associated PP2A negatively regulates the activation state of STRIPAK-bound Mst3 and likely the other STRIPAK-associated GCKIII kinases as well (Gordon et al., 2011). This regulation affects a substantial portion of cellular Mst3, indicating that STRIPAK may regulate the majority of cellular GCKIII kinases. Thus, a major function of STRIPAK may be to employ PP2A to negatively regulate GCKIII kinase function (Gordon et al., 2011). Since loss of function of Ccm3 and GCKIII kinases activates CCM-relevant pathways, mutations that enhance the ability of STRIPAK to downregulate GCKIII kinases or cause increased expression of striatin family members might contribute to CCM. Thus it would be interesting to examine the status of STRIPAK components in CCM patients. Conversely, loss of STRIPAK function might activate Ccm3-GCKIII kinase function and not lead to CCM. Consistent with this rationale, depletion of striatin family members enhances cell polarization (Kean et al., 2011), while depletion of Ccm3 or GCKIII kinases impairs it (Fidalgo et al., 2010, Kean et al., 2011). Considering the fact that Ccm3 and GCKIII kinases are downstream of STRIPAK, it seems unlikely that inhibiting STRIPAK therapeutically would be of benefit for CCM disease caused by Ccm3 mutations that abrogate binding to STRIPAK or GCKIII kinases. However, in cases of CCM where Ccm3-GCKIII signaling is at least partially intact this approach may hold promise.
Ccm3 and striatin family members not only have opposing effects on cell polarization (as assayed by Golgi reorientation during wound healing) but also on Mst4 localization to the Golgi (Kean et al., 2011). Depletion of Ccm3 enhances Mst4 localization to the Golgi while, conversely, depletion of striatin family members reduces Mst4 Golgi localization. Mst4 and Ysk1 interact with, and are targeted to the Golgi by, the cis-Golgi matrix protein, GM130 (Preisinger et al., 2004). GM130 activates both Ysk1 and Mst4 in vitro, presumably by promoting their dimerization (GM130 itself forms dimers) and thus autophosphorylation (Preisinger et al., 2004). Since Ccm3 and GCKIII kinases heterodimerize with each other via their highly similar homodimerization domains (Ceccarelli, 2011, Zhang et al., 2013), we speculate that depletion of Ccm3 may enhance interaction of Mst4 with GM130, Mst4 dimerization and thus Mst4 Golgi localization. Consistent with this possibility, Mst4-GM130 interaction was indeed increased upon Ccm3 knockdown in some experiments (Kean et al., 2011). These results are consistent with a model (Fig. 6, left half) in which GCKIII kinases (at least Mst4 and Ysk1) go through a cycle of dimerization (with concomitant dissociation from Ccm3) and activation promoted by GM130, followed by re-heterodimerization with Ccm3, and then inactivation by STRIPAK-associated PP2A. Since STRIPAK-GCKIII complexes are found in the Golgi as well as other membranes but GM130 is only found in the Golgi, one prediction of this model is that GCKIII kinases in the Golgi might be more activated than those in other membranes. This has been demonstrated in the case of Ysk1 (Preisinger et al., 2004).
Since STRIPAK regulation of GCKIII kinases likely depends on tethering of these kinases to striatin family members by Ccm3, modulation of Ccm3 association with STRIPAK may regulate GCKIII function. It is interesting that of the four lysines in Ccm3 important for binding to STRIPAK (Kean et al., 2011) (Fig. 5), two are modified by potentially reversible covalent modifications. Both lysine 132 and lysine 179 can be ubiquitinated (Wagner et al., 2011), although whether this plays a role in regulating Ccm3 stability or instead a non-proteasomal role is not known. Since these lysines are important for association of Ccm3 with Ccm2, paxillin, and striatin family members (Kean et al., 2011, Li et al., 2010), it is possible that ubiquitination occurs on these sites when they are exposed as a means of degrading excess Ccm3 not in one of these complexes. However, Lysine 179 can also be acetylated (Choudhary et al., 2009). While it is possible that this modification might regulate ubiquitination on this lysine, it is also possible that one or more of these modifications could serve the purpose of reversibly regulating Ccm3 complex formation with STRIPAK and other binding partners such as Ccm2 and paxillin. Further experimentation will be required to investigate these attractive possibilities.
5.5.5. Ccm3 and RhoA signaling
RhoA is a small GTPase that acts through its effectors, Rho-associated coiled coil-forming kinase (ROCK) and mammalian homolog of Drosophila diaphanous (mDia), to regulate actin polymerization and stress fiber formation (Watanabe et al., 1999). In addition, Rho GTPases regulate cell-cell junctions, cell-cell adhesion, and cell adhesion to extracellular matrix (for review, see (Beckers et al., 2010)). Inappropriate activation of the RhoA pathway disrupts cell junction integrity and cell adhesion, and consequently reduces vascular integrity. Recently, Ccm proteins and GCKIII kinases were implicated in the negative regulation of RhoA GTPase signaling (Borikova et al., 2010, Crose et al., 2009, Stockton et al., 2010, Whitehead et al., 2009, Zheng et al., 2010) and the RhoA pathway was found to be activated in human CCM (Stockton et al., 2010). Thus, it was postulated that impaired function of Ccm proteins leads to vascular defects at least in part because of increased RhoA signaling. In the next two sections, we will discuss some of the key evidence for Ccm protein and GCKIII kinase regulation of RhoA signaling, as well as possible connections to STRIPAK.
The evidence for Ccm protein involvement in RhoA pathway regulation is clear for Ccm1 and Ccm2. For example, depletion of either Ccm1 or Ccm2 in human endothelial cells increases the amount of activated (GTP-bound) RhoA, increases stress fibers and monolayer permeability in a RhoA-dependent manner, and increases ROCK-dependent phosphorylation of myosin light chain (MLC) and myosin phosphatase binding protein (Stockton et al., 2010, Whitehead et al., 2009). In murine embryonic endothelial cells, depletion of Ccm1 or Ccm2 also activated RhoA, increased ROCK-dependent phosphorylation of MLC, enhanced RhoA protein levels, and inhibited endothelial cell invasion of extracellular matrix and vessel-like tube formation in a ROCK-dependent manner in vitro (Borikova et al., 2010). In vivo data also support the idea that RhoA activation due to reduced Ccm1 or Ccm2 function likely contributes to CCM development. First, endothelium in both Ccm1-and Ccm2-deficient sporadic and familial human CCM showed evidence of activated ROCK, increasing the relevance of the previous findings to the human disease (Stockton et al., 2010). Second, treatment with simvastatin, a statin drug that indirectly blocks RhoA isoprenylation and thus RhoA function, rescued the compromised vascular permeability response in Ccm2 heterozygous mice (Whitehead et al., 2009). Third, treatment with fasudil, a ROCK inhibitor, rescued vascular leak in Ccm1 and Ccm2 heterozygous mice (Stockton et al., 2010) and reversed higher vascular leak, reduced lesion genesis, and prevented lesion maturation in Ccm1+/−Msh2−/− mice (McDonald et al., 2012). Fasudil treatment also reduced CCM lesion burden in Ccm2+/−Trp53−/− mice (McDonald et al., 2012). Excitingly, these results suggest that that targeting RhoA-ROCK signaling may be a novel pharmacological approach to prevent CCM lesion development and to reduce the potential complications of CCM. At least one clinical trial addressing the efficacy of simvastatin to reduce vascular leakage is in progress (http://clinicaltrials.gov/ct2/show/NCT01764451).
While the evidence that Ccm1 and Ccm2 function together to down-regulate RhoA signaling is clear, a role for Ccm3 in the regulation of RhoA signaling has been more controversial. In SaOS2 osteosarcoma cells, neither stress fiber formation nor RhoA activity was affected by Ccm3 knockdown either in the presence of serum or in serum-starved conditions (Fidalgo et al., 2010). In another study, depletion of Ccm3 in HMVECs did not increase stress fiber formation or enhance phosphorylation of MLC (Chan et al., 2011). These results and the identification of some distinct roles for Ccm3 in development and signaling led to the proposal that Ccm3 likely did not signal through RhoA (Chan et al., 2011, Fidalgo et al., 2010).
In contrast, results of other studies suggest that Ccm3 and its associated GCKIII kinases may indeed negatively regulate RhoA signaling. For example, using murine embryonic endothelial cells, Ccm3 depletion was found, like Ccm1 or Ccm2 depletion, to activate RhoA, to enhance total RhoA protein levels, to increase activated RhoA- and ROCK-dependent phosphorylation of MLC, and to inhibit endothelial cell invasion of extracellular matrix and vessel-like tube formation in a ROCK-dependent manner in vitro (Borikova et al., 2010). Although studies by other groups using HUVECs reported that depletion of Ccm1, Ccm2, or Ccm3 in HUVECs did not affect the total level of RhoA protein (Stockton et al., 2010, Zheng et al., 2010), this discrepancy might be due to differences between mouse and human endothelial cells as Ccm2 depletion was also found to increase total RhoA levels in murine brain endothelial (bEND.3) cells (Crose et al., 2009). Additionally, in a separate study, knockdown of Ccm3, Mst3, or Ysk1 in HUVECs increased stress fiber formation and RhoA activity similarly to depletion of Ccm2 (Zheng et al., 2010). In vitro, Ysk1, and to a lesser extent, Mst3, phosphorylated and activated moesin (Zheng et al., 2010), a negative regulator of RhoA signaling in epithelial and endothelial cells (Speck et al., 2003, Zheng et al., 2010). Also, Ysk1 overexpression in bovine aortic endothelial cells activated moesin while depletion of Ccm3 and/or Ysk1 in HMVECs reduced the amount of activated moesin at cell borders. Thus, Ccm3 may signal through GCKIII kinases to negatively regulate RhoA signaling in endothelial cells by phosphorylating and activating moesin (Zheng et al., 2010) (Fig. 6, center right). Combined with the finding that Ccm3 and Ysk1 regulate endothelial barrier function similar to Ccm2 in vitro, these results led to the postulate that Ccm1, Ccm2, Ccm3, and GCKIII kinases may function in a linear pathway to regulate endothelial cell junctions and barrier function (Zheng et al., 2010) (Fig. 4C). Although the reasons for the negative results in some Ccm3 depletion studies are unclear, the results of the last two studies support the hypothesis that Ccm3, like Ccm1 and Ccm2, negatively regulates RhoA signaling. However, it still needs to be determined whether, as is the case for Ccm1 and Ccm2, inhibiting RhoA-ROCK pathway signaling will provide a therapeutic benefit with a Ccm3 mouse model and whether the RhoA pathway is activated in human CCM resulting from Ccm3 mutations. Moreover, additional critical insight may be gained if, in the clinical trial mentioned above, the efficacy of simvastatin is analyzed on patients with CCM caused by mutation of Ccm3.
The purpose of Ccm1-Ccm2-Ccm3 complex formation has only begun to be elucidated. A simplistic view is that it functions to recruit, scaffold, and target modulators of Rho GTPase signaling to cellular junctions to inhibit RhoA signaling and positively regulate junctional integrity. For instance, the Ccm1-binding protein, ICAP-1 (Integrin Cytoplasmic Associated Protein-1), mediates the interaction of β1 integrin with ROCK (Stroeken et al., 2006), while Ccm2 binds RhoA, at least when the proteins are overexpressed (Whitehead et al., 2009), and interacts with the E3 ubiquitin ligase Smurf1 to control RhoA degradation (Crose et al., 2009). In addition, Ccm3 recruits GCKIII kinases, which likely negatively regulate RhoA signaling via phosphorylation of moesin (Zheng et al., 2010).
Ccm1 is an effector of the small GTPase, Rap1, which promotes junction stability in endothelial cells by inhibiting RhoA signaling (Cullere et al., 2005, Glading et al., 2007, Liu et al., 2011). Activation of Rap1 triggers its binding to Ccm1, releasing Ccm1 from cytoplasmic microtubules and allowing Ccm1 to localize to cell-cell junctions (Glading et al., 2007, Liu et al., 2011), where it is anchored by binding to HEG1, the mammalian homolog of the zebrafish heart of glass protein (Gingras et al., 2012, Kleaveland et al., 2009). Consistent with this localization, Ccm1 binds junction proteins including α/β-catenin, p120, AF-6, and VE-cadherin (Glading et al., 2007). Presumably, junctional localization of Ccm1 facilitates junctional localization of the entire Ccm1-Ccm2-Ccm3 complex, although this has only been directly demonstrated for Ccm2 (Stockton et al., 2010). If that were the case, then the entire Ccm protein complex may be an effector of Rap1, functioning to stabilize cell-cell junctions upon Rap1 activation. It is clear that Ccm1 and Ccm2 act as a complex to negatively regulate RhoA because expression of a Ccm2 point mutant that no longer binds Ccm1 does not rescue phenotypes caused by Ccm2 depletion, including activated RhoA-ROCK signaling, increased RhoA-induced stress fibers, and RhoA-pathway-dependent defects in endothelial monolayer permeability (Stockton et al., 2010). Moreover, the Ccm2 interaction with Ccm1 is required for localization of either Ccm1 or Ccm2 to cell-cell junctions (Stockton et al., 2010). However, a requirement for Ccm3 to associate with Ccm1-Ccm2 for negative regulation of RhoA signaling and junction stability has not been established and needs to be investigated. The fact that all three Ccm proteins regulate total RhoA levels in mouse endothelial cells and are known to associate suggests that the three proteins may indeed work together as a complex to carry out at least this function (Borikova et al., 2010).
We speculate that Ccm3-GCKIII is recruited to Ccm2-Ccm1 and thus potentially cell-cell junctions to facilitate GCKIII regulation of proteins in the Ccm1-Ccm2-Ccm3 complex and proteins in proximity to this complex that participate in the regulation of RhoA signaling and junction formation, disassembly, and stability (Fig. 6). To test this idea, it will be important to search for GCKIII kinase substrates in the Ccm protein complex, junctional complexes, and among Rho regulatory proteins. For a number of the prospective targets, phosphorylation is known to negatively regulate RhoA signaling, although GCKIII kinases have not been implicated in most cases. For example, serine phosphorylation of RhoA inhibits its activity in vivo (Ellerbroek et al., 2003), phosphorylation of Smurf1 by protein kinase A increases the degradation of RhoA (Cheng et al., 2011), phosphorylation of the junctional RhoGEF, Syx, reduces its junctional localization and ability to activate RhoA-mDia signaling (Ngok et al., 2013, Ngok et al., 2012), and both Mst3 and Mst4 are important for phosphorylation and activation of ERM proteins, at least in the case of Mst4 in a Ccm3-dependent manner (Fidalgo et al., 2012). Moreover, Mst3 was found to interact with the Rho family GEF, Vav2 in a two-hybrid interaction assay (Thalappilly et al., 2008). All things considered, however, the field is in great need of more insight into the pathways by which Ccm3 and GCKIII bolster vascular integrity.
In summary, based on the data discussed to this point, we propose a model in which Ccm3 brings GCKIII kinases to Ccm1-Ccm2 to regulate RhoA signaling and cell-cell junction assembly and stability via phosphorylation of proteins in or proximal to the Ccm protein complex (Fig. 6). In this model, Ccm3-GCKIII would regulate select aspects of Ccm2-Ccm1 function while also signaling via an additional pathway(s) (Fig. 4A). While many predictions of this model remain to be tested, the model is consistent with 1) the existence of a Ccm1-Ccm2-Ccm3 complex; 2) the additional Ccm1-Ccm2-independent localization of Ccm3; 3) partially overlapping signaling pathways for Ccm3 vs Ccm2-Ccm1; 4) regulation of RhoA function by the three CCM proteins; 5) regulation of RhoA signaling by Ccm3 through its associated GCKIII kinases; and 6) the known inhibition of RhoA function at many levels by phosphorylation.
5.5.6. STRIPAK and RhoA signaling
The connections between Ccm3, GCKIII kinases and RhoA suggest strongly that STRIPAK complexes function on some level to regulate RhoA signaling and function. Since STRIPAK appears to be a negative regulator of GCKIII kinases (Gordon et al., 2011), which in turn negatively regulate RhoA signaling, STRIPAK may be a positive regulator of RhoA signaling. STRIPAK may also compete with Ccm2-Ccm1 for binding to Ccm3-GCKIII kinases (Kean et al., 2011), and therefore increases in STRIPAK levels due to enhanced striatin family member expression or stability might inhibit Ccm3-GCKIII function through the Ccm protein complex, which again would be predicted to activate RhoA signaling. Because STRIPAK is multifunctional, it may also regulate RhoA in other ways and/or impinge on pathways downstream of RhoA in positive or negative directions. Consistent with these ideas, Cka, the Drosophila striatin homolog, and both Drosophila and human RhoA all positively regulate the Jnk pathway (Chen et al., 2002, Neisch et al., 2010, Whitehead et al., 2009). Moreover, the synthetic genetic interaction found for Cka and dRhoA (Rho1) (Horn et al., 2011) (discussed in section 4.1.2.2) supports the idea that Cka and dRhoA function in the same direction to regulate proliferation. As discussed in section 4.1.3.1, Far11, the budding yeast homolog of the STRIPAK components STRIP1/2, functions downstream of TORC2 and apparently upstream of Rho to regulate actin polarization (Baryshnikova et al., 2010, Fadri et al., 2005, Pracheil et al., 2012). While this regulation appears to be negative, it is important to note that yeast lack an obvious Ccm3 homolog, suggesting that the regulation might be through modulation of phosphorylation by Far complex-associated PP2A. Perhaps related, STRIP1 itself has been implicated in regulation of the actin cytoskeleton, cell spreading, and cell morphology in mammalian cells (Bai et al., 2011). Together, these findings suggest that STRIPAK is capable of regulating pathways both upstream and downstream of RhoA.
The identification in a high-throughput screen of an interaction between Far11 and Rho4 (Uetz et al., 2000), a Rho GTPase that functions together with Rho3 in activation of formins, actin filament nucleators (Dong et al., 2003) suggested that the yeast STRIPAK-like FAR complex might interact with Rho proteins to modulate their function and/or be regulated themselves by Rho. Consistent with this possibility, mammalian SG2NA was recently identified as a novel RhoA effector candidate because it interacts specifically with GTPγ-S-activated mammalian RhoA and a constitutively active RhoA mutant, but not with inactive GDP-RhoA or a dominant negative RhoA mutant (Kato et al., 2012). While SG2NA may indeed be a RhoA effector, these results are also consonant with the idea that SG2NA may be involved in modulating RhoA activity once it is active, or, based on the clear role of STRIPAK in vesicular transport, in transporting active RhoA to or from subcellular sites important for its function. Interestingly, striatin has been reported to localize to cell-cell junctions and to be important for normal organization of filamentous actin and proper localization of ZO-1, suggesting that it may regulate TJ formation (Breitman et al., 2008). Moreover, the STRIPAK components CTTNBP2 and CTTNBP2NL are cortactin binding proteins that preferentially localize to the cell cortex and stress fibers, respectively (discusssed in section 4.1.1.3). Although further experimentation is necessary to investigate and clarify these results, they are intriguing because of the possible implications that the STRIPAK complex might be a significant regulator and/or effector of RhoA in controlling actin cytoskeleton organization and dynamics; cell junction assembly, disassembly, and stability; and vascular integrity in mammalian cells. Therefore, it is of great interest to determine if modulation of STRIPAK function can indeed regulate RhoA signaling in endothelial cells and potentially the development of CCM.
6. Conclusions and future directions
In summary, STRIPAK and STRIPAK-like complexes are large, multifunctional protein assemblies that participate in a wide variety of cellular processes. The roles of these complexes range from regulation of signal transduction pathways shared by many cell types to regulation of more specialized signaling pathways and events. Common themes emerging from studies of STRIPAK and STRIPAK-like complexes in multiple organisms include STRIPAK regulation of signal transduction pathways, mitosis and cytokinesis, cell polarity, and protein trafficking, to name a few.
Consistent with the range of functions they possess, there is enormous variation possible in the composition of STRIPAK complexes, with well over 100 variants predicted. One challenge in the future will be to understand how cells independently regulate different STRIPAK complexes to carry out distinct functions. Differential expression of both striatin family members and some other STRIPAK components in various cell types has been demonstrated and likely contributes to helping cells regulate these complexes by reducing the complexity of STRIPAK complexes within any one cell type. However, multiple STRIPAK complexes are clearly present in the same cell. Based on subcellular localization studies, accessory targeting proteins like SLMAPs and CTTNBP2 probably localize STRIPAK complexes to different subcellular locations, providing for spatial separation of distinct STRIPAK complexes that facilitates selective regulation. However, many questions remain in regard to how this is achieved and modulated in response to extrinsic and intrinsic cues, and other mechanisms likely contribute and remain to be elucidated.
In addition to understanding the molecular details of how multiple STRIPAK complexes are orchestrated within a single cell, there are a number of key questions raised in this review about function, regulation, and contributions of STRIPAK and STRIPAK components to human disease. We highlight a number of these key questions here but many additional areas of investigation are mentioned throughout the review. Moreover, our model shown in Fig. 6 summarizes several testable hypotheses regarding STRIPAK, Ccm3, and GCKIII kinase function and regulation that need to be addressed in the future.
An important question to resolve is whether STRIPAK components have STRIPAK-independent functions. This question is significant because, in a number of studies, only one STRIPAK component was implicated in a particular function or connected to a particular disease. Thus, it will be important to assess the involvement of other members of the STRIPAK complex in these functions or diseases to rule out the possibility that the reported findings represent STRIPAK-independent functions of individual STRIPAK components.
An additional pertinent area of investigation in the future will be to elucidate the role of covalent modification of STRIPAK components in STRIPAK function and regulation. The presence of PP2A and various kinases in STRIPAK complexes, the knowledge that many STRIPAK components are phosphorylated and become hyperphosphorylated in the presence of an inhibitor of PP2A family phosphatases, and the finding that STRIPAK-associated PP2A appears to regulate GCKIII kinase phosphorylation and activity suggest that STRIPAK functions are intricately regulated by reversible phosphorylation. Moreover, these same findings suggest that a major mechanism of STRIPAK function is to direct PP2A to key substrates, many of which are likely kinases. Similar targeting of PP2A to substrates by STRIPAK-like complexes in other organisms suggests that this is a general theme in STRIPAK function.
Yet another area of future inquiry stems from the likelihood that STRIPAK complexes play critical roles in mammalian nervous system development and function. STRIPAK components have been broadly implicated in neuronal function and multiple findings support the idea that STRIPAK function in neurons may be regulated by Ca2+ signaling. In addition, the localization of all three striatin family members to dendritic spines and the identification of a CTTNBP2 mutant that may contribute to the development of autism provide further impetus for dissecting the roles STRIPAK complexes play in nervous system development and function. Development of mouse models for select STRIPAK components would greatly facilitate these and other investigations of STRIPAK function in mammalian cells.
Studies of STRIPAK-like complexes in lower eukaryotes have made substantial contributions to elucidating mammalian STRIPAK functions and have provided direction for future experimentation in mammalian systems. For example, investigations of STRIPAK homologs in lower eukaryotes have uncovered some novel functions not yet studied in mammalian cells. Examples of these functions include roles for STRIPAK complexes in cell-cell communication, sexual development, and protein secretion. Although all of these functions might not be conserved in mammals, their potential significance justifies making a substantial effort to determine if they are indeed conserved. In other cases, the evidence supporting certain roles for STRIPAK is much stronger in lower eukaryotic model systems, providing incentive for further study of these functions in mammalian cells. For instance, while STRIPAK-like complexes are clearly important for cell-cell fusion events in filamentous fungi, in mammals the STRIPAK components SLMAP and Map4k4 have been connected to myoblast fusion but it is not known whether a STRIPAK complex is involved. The results with filamentous fungi support the idea that STRIPAK may be important for cell-cell fusion events in general. Thus, future studies should address the possible role of STRIPAK not only in myoblast fusion, but also fertilization and other mammalian cell-cell fusion events.
Related to the above-mentioned area of inquiry, it will be also be important to sort out possible functional and mechanistic differences between STRIPAK and STRIPAK-like complexes in different organisms. For example, Ccm3 is only found in metazoans. Therefore, the general theme of targeting GCKIII (and perhaps other) kinases to STRIPAK via Ccm3 must be unique to metazoan organisms. In theory, this might imply possible differences in function for STRIPAK and STRIPAK-like complexes in metazoan and non-metazoan organisms. Consistent with this possibility, while GCKIII homologs have been described in non-metazoans (for examples, see (Leonhard and Nurse, 2005, Pombo et al., 2007, Rohlfs, 2007, Seiler et al., 2006)), none have been reported as components of STRIPAK-like complexes. Whether this signifies a true difference in targets or simply reflects limitations in the current sampling of STRIPAK and STRIPAK-like complexes in non-metazoans needs to be determined. Even within metazoans, it is unclear how GCKII, GCKIV, and other kinases such as Jnk are targeted to STRIPAK and STRIPAK-like complexes. Addressing these gaps in knowledge will be important to understanding both common and unique themes of STRIPAK and STRIPAK-like function in different organisms.
One of the most exciting areas of inquiry will be elucidating the roles of STRIPAK complexes in CCM. Given the demonstrated importance of dysregulated RhoA signaling for the development of CCM and the evidence connecting STRIPAK to RhoA signaling, it will be key to investigate the role(s) of STRIPAK in RhoA activation and signaling, in regulation of actin cytoskeleton, and in the assembly, disassembly, and stability of cell-cell junctions, as well as the possibility of targeting STRIPAK therapeutically in some cases of CCM. In addition, because GCKIII kinases negatively regulate RhoA signaling, their genes are candidate CCM genes. While functional redundancy among these kinases might reduce the likelihood of finding their genes mutated in CCM cases, the studies described in this review suggest that partial loss of multiple GCKIII kinases or a GCKIII kinase and Ccm3 might promote the development of CCM.
This review presents a summary of our current knowledge of the many forms and functions of STRIPAK and STRIPAK-like complexes. Clearly, much additional work will be required to fully define the intricate functions of these complexes and, importantly, to uncover the molecular basis underlying the growing number of links between STRIPAK and a variety of human diseases.
Acknowledgments
This work was supported by the National Cancer Institute of the National Institutes of Health under award number R01CA057327 to D.C.P. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank Anita Corbett and Jennifer Jackson for critical reading of the manuscript, and Michael Konomos for advice on graphic design. Dr. David Pallas is entitled to royalty from the sale of products related to the research described in this paper by Millipore Inc., Santa Cruz Biotechnologies Inc., Invitrogen Corp., and Novus Biologicals Inc. In addition, this same author serves as a consultant to Millipore. The terms of these arrangements have been reviewed and approved by Emory University in accordance with its conflict of interest policies.
Abbreviations
- AJ
adherens junction
- APC
adenomatous polyposis coli
- ARD
armadillo repeat domain
- ARVC
arrhythmogenic right ventricular cardiomyopathy
- Bsk
Basket
- CaM
calmodulin
- Cav-1
caveolin-1
- CCM
cerebral cavernous malformations
- Ccm3
cerebral cavernous malformation 3
- CCM-ECs
CCM patient endothelial cells
- CCT/TRiC
chaperonin containing TCP-1/TCP-1 Ring Complex
- Cka
connector of kinase to AP-1
- CTTNBP2/NL
cortactin-binding protein 2/Cortactin-binding protein 2, N-terminal-like
- DCM
dilated cardiomyopathy
- dErk
Drosophila extracellular signal-regulated kinase
- dJnk
Drosophila Jun N-terminal kinase
- dMob4
Drosophila Mob3 functional homolog
- dRassf
Drosophila ras association domain family protein
- DUB
deubiquitinase
- EGFR
epidermal growth factor receptor
- EMS
8p11 myeloproliferative syndrome
- eNOS
endothelial nitric oxide synthase
- Eps15
epidermal growth factor receptor substrate 15
- ERα
Estrogen receptor alpha
- Erk
extracellular signal-regulated kinase
- ERM
ezrin/radixin/moesin
- ERMES
endoplasmic reticulum-mitochondria encounter structure
- FAR
factor arrest
- FAT
focal adhesion targeting
- FGFR1OP2
fibroblast growth factor receptor 1 oncogene partner 2
- GAP
GTPase activating protein
- GCKIII
germinal center kinase III
- GH
glycine-histidine
- GIPC
GAIP-interacting protein, C terminus
- HBMECs
human brain microvascular endothelial cells
- HEG1
heart of glass 1
- Hep
Hemipterous
- HGNC
HUGO Gene Nomenclature Committee
- HMVECs
human dermal microvascular endothelial cells
- Hpo
Hippo
- HUVECs
human umbilical vein endothelial cells
- ICAP-1
integrin cytoplasmic associated protein-1
- IRF-3
interferon regulatory factor 3
- Jra
Jun-related antigen
- Kay
kayak
- Krit1
K-Rev interaction trapped 1
- Map4k4
Mitogen-activated protein kinase kinase kinase kinase 4
- MASK
Mst3 and Sok1-related kinase
- mDia
mammalian homolog of Drosophila diaphanous
- MEKK3
mitogen-activated protein kinase kinase kinase 3
- Mink1
Msn-like kinase 1
- MLC
myosin light chain
- Mob3
monopolar spindle-one-binder family 3
- MR
mineralocorticoid receptor
- Msn
Misshapen
- Mst
mammalian sterile 20-like
- NDPK
nucleoside-diphosphate kinase
- NLS
nuclear localization signal
- NMDA
N-methyl-D-aspartate
- NMDARs
N-methyl-D-aspartate receptors
- OSM
osmosensing scaffold for MEKK3
- PDCD10
programmed cell death 10
- PDGFRA
platelet derived growth factor receptor alpha
- PPARγ
peroxisome proliferator-activated receptor γ
- PP2A
protein phosphatase 2A
- PTB
phosphotyrosine-binding
- PTP
protein-tyrosine phosphatase
- RhoA
ras homolog gene family member A
- ROCK
Rho-associated coiled coil-forming kinase
- RTKs
receptor tyrosine kinases
- SG2NA
S/G2 nuclear autoantigen
- SIKE
suppressor of IKKε
- SIN
septation initiation network
- SIP
SIN-inhibitory PP2A complex
- SLMAP
sarcolemmal membrane-associated protein
- Sok1
Sterile 20/oxidant stress-response kinase 1
- SPB
spindle pole body
- SR
sarcoplasmic reticulum
- STRIPAK
striatin-interacting phosphatase and kinase
- STRIP1/2
striatin interacting proteins 1 and 2
- TFAR15
TF-1 cell apoptosis related gene-15
- TJ
tight junction
- TLR3
toll-like receptor 3
- Tnf-α
tumor necrosis factor alpha
- Tnik
TRAF2- and NCK-interacting kinase
- TORC1
target of rapamycin complex 1
- WD
tryptophan-aspartate
- Ysk1
yeast Sps1/Sterile-20-related kinase 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Juyeon Hwang, Email: jhwang8231@gmail.com.
David C. Pallas, Email: dpallas@emory.edu.
References
- Akers AL, Johnson E, Steinberg GK, Zabramski JM, Marchuk DA. Biallelic somatic and germline mutations in cerebral cavernous malformations (CCMs): evidence for a two-hit mechanism of CCM pathogenesis. Human molecular genetics. 2009;18:919–30. doi: 10.1093/hmg/ddn430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antzelevitch C, Pollevick GD, Cordeiro JM, Casis O, Sanguinetti MC, Aizawa Y, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007;115:442–9. doi: 10.1161/CIRCULATIONAHA.106.668392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aouadi M, Tesz GJ, Nicoloro SM, Wang M, Chouinard M, Soto E, et al. Orally delivered siRNA targeting macrophage Map4k4 suppresses systemic inflammation. Nature. 2009;458:1180–4. doi: 10.1038/nature07774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkowitz RA. Chemical gradients and chemotropism in yeast. Cold Spring Harbor perspectives in biology. 2009;1:a001958. doi: 10.1101/cshperspect.a001958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong CT, Vincent TL, Green PJ, Woolfson DN. SCORER 2.0: an algorithm for distinguishing parallel dimeric and trimeric coiled-coil sequences. Bioinformatics. 2011;27:1908–14. doi: 10.1093/bioinformatics/btr299. [DOI] [PubMed] [Google Scholar]
- Audhya A, Loewith R, Parsons AB, Gao L, Tabuchi M, Zhou H, et al. Genome-wide lethality screen identifies new PI4,5P2 effectors that regulate the actin cytoskeleton. Embo J. 2004;23:3747–57. doi: 10.1038/sj.emboj.7600384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai SW, Herrera-Abreu MT, Rohn JL, Racine V, Tajadura V, Suryavanshi N, et al. Identification and characterization of a set of conserved and new regulators of cytoskeletal organization, cell morphology and migration. BMC biology. 2011;9:54. doi: 10.1186/1741-7007-9-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baillat G, Gaillard S, Castets F, Monneron A. Interactions of Phocein with Nucleoside-Diphosphate Kinase, Eps15, and Dynamin I. J Biol Chem. 2002;277:18961–6. doi: 10.1074/jbc.M108818200. [DOI] [PubMed] [Google Scholar]
- Baillat G, Moqrich A, Castets F, Baude A, Bailly Y, Benmerah A, et al. Molecular cloning and characterization of phocein, a protein found from the golgi complex to dendritic spines. Mol Biol Cell. 2001;12:663–73. doi: 10.1091/mbc.12.3.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian MK, Bi E, Glotzer M. Comparative analysis of cytokinesis in budding yeast, fission yeast and animal cells. Current biology: CB. 2004;14:R806–18. doi: 10.1016/j.cub.2004.09.022. [DOI] [PubMed] [Google Scholar]
- Bartoli M, Monneron A, Ladant D. Interaction of calmodulin with striatin, a WD-repeat protein present in neuronal dendritic spines. J Biol Chem. 1998;273:22248–53. doi: 10.1074/jbc.273.35.22248. [DOI] [PubMed] [Google Scholar]
- Bartoli M, Ternaux JP, Forni C, Portalier P, Salin P, Amalric M, et al. Down-regulation of striatin, a neuronal calmodulin-binding protein, impairs rat locomotor activity. Journal of neurobiology. 1999;40:234–43. [PubMed] [Google Scholar]
- Baryshnikova A, Costanzo M, Kim Y, Ding H, Koh J, Toufighi K, et al. Quantitative analysis of fitness and genetic interactions in yeast on a genome scale. Nature methods. 2010;7:1017–24. doi: 10.1038/nmeth.1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckers CM, van Hinsbergh VW, van Nieuw Amerongen GP. Driving Rho GTPase activity in endothelial cells regulates barrier integrity. Thrombosis and haemostasis. 2010;103:40–55. doi: 10.1160/TH09-06-0403. [DOI] [PubMed] [Google Scholar]
- Benoist M, Baude A, Tasmadjian A, Dargent B, Kessler JP, Castets F. Distribution of zinedin in the rat brain. Journal of neurochemistry. 2008;106:969–77. doi: 10.1111/j.1471-4159.2008.05448.x. [DOI] [PubMed] [Google Scholar]
- Benoist M, Gaillard S, Castets F. The striatin family: A new signaling platform in dendritic spines. J Physiol Paris. 2006;99:146–53. doi: 10.1016/j.jphysparis.2005.12.006. [DOI] [PubMed] [Google Scholar]
- Bergametti F, Denier C, Labauge P, Arnoult M, Boetto S, Clanet M, et al. Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. Am J Hum Genet. 2005;76:42–51. doi: 10.1086/426952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernelot Moens SJ, Schnitzler GR, Nickerson M, Guo H, Ueda K, Lu Q, et al. Rapid estrogen receptor signaling is essential for the protective effects of estrogen against vascular injury. Circulation. 2012;126:1993–2004. doi: 10.1161/CIRCULATIONAHA.112.124529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhards Y, Poggeler S. The phocein homologue SmMOB3 is essential for vegetative cell fusion and sexual development in the filamentous ascomycete Sordaria macrospora. Current genetics. 2011;57:133–49. doi: 10.1007/s00294-010-0333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloemendal S, Bernhards Y, Bartho K, Dettmann A, Voigt O, Teichert I, et al. A homologue of the human STRIPAK complex controls sexual development in fungi. Molecular microbiology. 2012;84:310–23. doi: 10.1111/j.1365-2958.2012.08024.x. [DOI] [PubMed] [Google Scholar]
- Bloemendal S, Lord KM, Rech C, Hoff B, Engh I, Read ND, et al. A mutant defective in sexual development produces aseptate ascogonia. Eukaryotic cell. 2010;9:1856–66. doi: 10.1128/EC.00186-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonangelino CJ, Chavez EM, Bonifacino JS. Genomic screen for vacuolar protein sorting genes in Saccharomyces cerevisiae. Mol Biol Cell. 2002;13:2486–501. doi: 10.1091/mbc.02-01-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borikova AL, Dibble CF, Sciaky N, Welch CM, Abell AN, Bencharit S, et al. Rho kinase inhibition rescues the endothelial cell cerebral cavernous malformation phenotype. J Biol Chem. 2010;285:11760–4. doi: 10.1074/jbc.C109.097220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boscher C, Nabi IR. Caveolin-1: role in cell signaling. Advances in experimental medicine and biology. 2012;729:29–50. doi: 10.1007/978-1-4614-1222-9_3. [DOI] [PubMed] [Google Scholar]
- Boulday G, Blecon A, Petit N, Chareyre F, Garcia LA, Niwa-Kawakita M, et al. Tissue-specific conditional CCM2 knockout mice establish the essential role of endothelial CCM2 in angiogenesis: implications for human cerebral cavernous malformations. Disease models & mechanisms. 2009;2:168–77. doi: 10.1242/dmm.001263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulday G, Rudini N, Maddaluno L, Blecon A, Arnould M, Gaudric A, et al. Developmental timing of CCM2 loss influences cerebral cavernous malformations in mice. J Exp Med. 2011;208:1835–47. doi: 10.1084/jem.20110571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitman M, Zilberberg A, Caspi M, Rosin-Arbesfeld R. The armadillo repeat domain of the APC tumor suppressor protein interacts with Striatin family members. Biochim Biophys Acta. 2008;1783:1792–802. doi: 10.1016/j.bbamcr.2008.04.017. [DOI] [PubMed] [Google Scholar]
- Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. Journal of the American College of Cardiology. 1992;20:1391–6. doi: 10.1016/0735-1097(92)90253-j. [DOI] [PubMed] [Google Scholar]
- Buda A, Pignatelli M. E-cadherin and the cytoskeletal network in colorectal cancer development and metastasis. Cell communication & adhesion. 2011;18:133–43. doi: 10.3109/15419061.2011.636465. [DOI] [PubMed] [Google Scholar]
- Bultynck G, Heath VL, Majeed AP, Galan JM, Haguenauer-Tsapis R, Cyert MS. Slm1 and slm2 are novel substrates of the calcineurin phosphatase required for heat stress-induced endocytosis of the yeast uracil permease. Mol Cell Biol. 2006;26:4729–45. doi: 10.1128/MCB.01973-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhard P, Stetefeld J, Strelkov SV. Coiled coils: a highly versatile protein folding motif. Trends in cell biology. 2001;11:82–8. doi: 10.1016/s0962-8924(00)01898-5. [DOI] [PubMed] [Google Scholar]
- Burkhardt JK, Schmidt D, Schoenauer R, Brokopp C, Agarkova I, Bozinov O, et al. Upregulation of transmembrane endothelial junction proteins in human cerebral cavernous malformations. Neurosurgical focus. 2010;29:E3. doi: 10.3171/2010.6.FOCUS10125. [DOI] [PubMed] [Google Scholar]
- Byers JT, Guzzo RM, Salih M, Tuana BS. Hydrophobic profiles of the tail anchors in SLMAP dictate subcellular targeting. BMC cell biology. 2009;10:48. doi: 10.1186/1471-2121-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castets F, Bartoli M, Barnier JV, Baillat G, Salin P, Moqrich A, et al. A novel calmodulin-binding protein, belonging to the WD-repeat family, is localized in dendrites of a subset of CNS neurons. J Cell Biol. 1996;134:1051–62. doi: 10.1083/jcb.134.4.1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castets F, Rakitina T, Gaillard S, Moqrich A, Mattei MG, Monneron A. Zinedin, SG2NA, and striatin are calmodulin-binding, WD repeat proteins principally expressed in the brain. J Biol Chem. 2000;275:19970–7. doi: 10.1074/jbc.M909782199. [DOI] [PubMed] [Google Scholar]
- Ceccarelli DF. CCM3/PDCD10 Heterodimerizes with Germinal Center Kinase III (GCKIII) Proteins Using a Mechanism Analogous to CCM3 Homodimerization. The Journal of Biological Chemistry. 2011;286:25056–64. doi: 10.1074/jbc.M110.213777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambliss KL, Shaul PW. Rapid activation of endothelial NO synthase by estrogen: evidence for a steroid receptor fast-action complex (SRFC) in caveolae. Steroids. 2002;67:413–9. doi: 10.1016/s0039-128x(01)00177-5. [DOI] [PubMed] [Google Scholar]
- Chambliss KL, Yuhanna IS, Mineo C, Liu P, German Z, Sherman TS, et al. Estrogen receptor alpha and endothelial nitric oxide synthase are organized into a functional signaling module in caveolae. Circulation research. 2000;87:E44–52. doi: 10.1161/01.res.87.11.e44. [DOI] [PubMed] [Google Scholar]
- Chan AC, Drakos SG, Ruiz OE, Smith AC, Gibson CC, Ling J, et al. Mutations in 2 distinct genetic pathways result in cerebral cavernous malformations in mice. The Journal of clinical investigation. 2011;121:1871–81. doi: 10.1172/JCI44393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan AC, Li DY, Berg MJ, Whitehead KJ. Recent insights into cerebral cavernous malformations: animal models of CCM and the human phenotype. The FEBS journal. 2010;277:1076–83. doi: 10.1111/j.1742-4658.2009.07536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HW, Marinissen MJ, Oh SW, Chen X, Melnick M, Perrimon N, et al. CKA, a novel multidomain protein, regulates the JUN N-terminal kinase signal transduction pathway in Drosophila. Mol Cell Biol. 2002;22:1792–803. doi: 10.1128/MCB.22.6.1792-1803.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Tanriover G, Yano H, Friedlander R, Louvi A, Gunel M. Apoptotic functions of PDCD10/CCM3, the gene mutated in cerebral cavernous malformation 3. Stroke; a journal of cerebral circulation. 2009;40:1474–81. doi: 10.1161/STROKEAHA.108.527135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PS, Priori SG. The Brugada syndrome. Journal of the American College of Cardiology. 2008;51:1176–80. doi: 10.1016/j.jacc.2007.12.006. [DOI] [PubMed] [Google Scholar]
- Chen X, Ding H. Increased expression of the tail-anchored membrane protein SLMAP in adipose tissue from type 2 Tally Ho diabetic mice. Experimental diabetes research. 2011;2011:421982. doi: 10.1155/2011/421982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YK, Chen CY, Hu HT, Hsueh YP. CTTNBP2, but not CTTNBP2NL, regulates dendritic spinogenesis and synaptic distribution of the striatin-PP2A complex. Mol Biol Cell. 2012 doi: 10.1091/mbc.E12-05-0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YK, Hsueh YP. Cortactin-binding protein 2 modulates the mobility of cortactin and regulates dendritic spine formation and maintenance. J Neurosci. 2012;32:1043–55. doi: 10.1523/JNEUROSCI.4405-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng PL, Lu H, Shelly M, Gao H, Poo MM. Phosphorylation of E3 ligase Smurf1 switches its substrate preference in support of axon development. Neuron. 2011;69:231–43. doi: 10.1016/j.neuron.2010.12.021. [DOI] [PubMed] [Google Scholar]
- Cheng W, Nishimura I. High throughput screening of biologically functional small molecules for modulating the expression of FGFR1OP2/wit3.0 in fibroblasts. Journal of the California Dental Association. 2012;40:929–31. 34–7. [PubMed] [Google Scholar]
- Cheung J, Petek E, Nakabayashi K, Tsui LC, Vincent JB, Scherer SW. Identification of the human cortactin-binding protein-2 gene from the autism candidate region at 7q31. Genomics. 2001;78:7–11. doi: 10.1006/geno.2001.6651. [DOI] [PubMed] [Google Scholar]
- Chidlow JH, Jr, Sessa WC. Caveolae, caveolins, and cavins: complex control of cellular signalling and inflammation. Cardiovascular research. 2010;86:219–25. doi: 10.1093/cvr/cvq075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopra N, Knollmann BC. Cardiac calsequestrin: the new kid on the block in arrhythmias. Journal of cardiovascular electrophysiology. 2009;20:1179–85. doi: 10.1111/j.1540-8167.2009.01531.x. [DOI] [PubMed] [Google Scholar]
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–40. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- Ciani B, Bjelic S, Honnappa S, Jawhari H, Jaussi R, Payapilly A, et al. Molecular basis of coiled-coil oligomerization-state specificity. Proc Natl Acad Sci U S A. 2010;107:19850–5. doi: 10.1073/pnas.1008502107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clatterbuck RE, Eberhart CG, Crain BJ, Rigamonti D. Ultrastructural and immunocytochemical evidence that an incompetent blood-brain barrier is related to the pathophysiology of cavernous malformations. Journal of neurology, neurosurgery, and psychiatry. 2001;71:188–92. doi: 10.1136/jnnp.71.2.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins CS, Hong J, Sapinoso L, Zhou Y, Liu Z, Micklash K, et al. A small interfering RNA screen for modulators of tumor cell motility identifies MAP4K4 as a promigratory kinase. Proc Natl Acad Sci U S A. 2006;103:3775–80. doi: 10.1073/pnas.0600040103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor JA, Miller LD, Petrozzino J, Muller W. Calcium signaling in dendritic spines of hippocampal neurons. Journal of neurobiology. 1994;25:234–42. doi: 10.1002/neu.480250304. [DOI] [PubMed] [Google Scholar]
- Costa B, Kean MJ, Ast V, Knight JD, Mett A, Levy Z, et al. STK25 protein mediates TrkA and CCM2 protein-dependent death in pediatric tumor cells of neural origin. J Biol Chem. 2012;287:29285–9. doi: 10.1074/jbc.C112.345397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couet J, Li S, Okamoto T, Ikezu T, Lisanti MP. Identification of peptide and protein ligands for the caveolin-scaffolding domain. Implications for the interaction of caveolin with caveolae-associated proteins. J Biol Chem. 1997;272:6525–33. doi: 10.1074/jbc.272.10.6525. [DOI] [PubMed] [Google Scholar]
- Crose LE, Hilder TL, Sciaky N, Johnson GL. Cerebral cavernous malformation 2 protein promotes smad ubiquitin regulatory factor 1-mediated RhoA degradation in endothelial cells. J Biol Chem. 2009;284:13301–5. doi: 10.1074/jbc.C900009200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullere X, Shaw SK, Andersson L, Hirahashi J, Luscinskas FW, Mayadas TN. Regulation of vascular endothelial barrier function by Epac, a cAMP-activated exchange factor for Rap GTPase. Blood. 2005;105:1950–5. doi: 10.1182/blood-2004-05-1987. [DOI] [PubMed] [Google Scholar]
- Cunningham K, Uchida Y, O’Donnell E, Claudio E, Li W, Soneji K, et al. Conditional deletion of Ccm2 causes hemorrhage in the adult brain: a mouse model of human cerebral cavernous malformations. Human molecular genetics. 2011;20:3198–206. doi: 10.1093/hmg/ddr225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daquinag A, Fadri M, Jung SY, Qin J, Kunz J. The yeast PH domain proteins Slm1 and Slm2 are targets of sphingolipid signaling during the response to heat stress. Mol Cell Biol. 2007;27:633–50. doi: 10.1128/MCB.00461-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Curling O, Jr, Kelly DL, Jr, Elster AD, Craven TE. An analysis of the natural history of cavernous angiomas. J Neurosurg. 1991;75:702–8. doi: 10.3171/jns.1991.75.5.0702. [DOI] [PubMed] [Google Scholar]
- Denier C, Goutagny S, Labauge P, Krivosic V, Arnoult M, Cousin A, et al. Mutations within the MGC4607 gene cause cerebral cavernous malformations. Am J Hum Genet. 2004;74:326–37. doi: 10.1086/381718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denier C, Labauge P, Bergametti F, Marchelli F, Riant F, Arnoult M, et al. Genotype-phenotype correlations in cerebral cavernous malformations patients. Annals of neurology. 2006;60:550–6. doi: 10.1002/ana.20947. [DOI] [PubMed] [Google Scholar]
- Dettmann A, Heilig Y, Ludwig S, Schmitt K, Illgen J, Fleissner A, et al. HAM-2 and HAM-3 are central for the assembly of the Neurospora STRIPAK complex at the nuclear envelope and regulate nuclear accumulation of the MAP kinase MAK-1 in a MAK-2-dependent manner. Molecular microbiology. 2013 doi: 10.1111/mmi.12399. [DOI] [PubMed] [Google Scholar]
- Ding H, Howarth AG, Pannirselvam M, Anderson TJ, Severson DL, Wiehler WB, et al. Endothelial dysfunction in Type 2 diabetes correlates with deregulated expression of the tail-anchored membrane protein SLMAP. American journal of physiology Heart and circulatory physiology. 2005;289:H206–11. doi: 10.1152/ajpheart.00037.2005. [DOI] [PubMed] [Google Scholar]
- Donaldson JG, Finazzi D, Klausner RD. Brefeldin A inhibits Golgi membrane-catalysed exchange of guanine nucleotide onto ARF protein. Nature. 1992;360:350–2. doi: 10.1038/360350a0. [DOI] [PubMed] [Google Scholar]
- Dong Y, Pruyne D, Bretscher A. Formin-dependent actin assembly is regulated by distinct modes of Rho signaling in yeast. J Cell Biol. 2003;161:1081–92. doi: 10.1083/jcb.200212040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellerbroek SM, Wennerberg K, Burridge K. Serine phosphorylation negatively regulates RhoA in vivo. J Biol Chem. 2003;278:19023–31. doi: 10.1074/jbc.M213066200. [DOI] [PubMed] [Google Scholar]
- Engh I, Nowrousian M, Kuck U. Sordaria macrospora, a model organism to study fungal cellular development. European journal of cell biology. 2010;89:864–72. doi: 10.1016/j.ejcb.2010.07.002. [DOI] [PubMed] [Google Scholar]
- Fadri M, Daquinag A, Wang S, Xue T, Kunz J. The pleckstrin homology domain proteins Slm1 and Slm2 are required for actin cytoskeleton organization in yeast and bind phosphatidylinositol-4,5-bisphosphate and TORC2. Mol Biol Cell. 2005;16:1883–900. doi: 10.1091/mbc.E04-07-0564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faurobert E, Albiges-Rizo C. Recent insights into cerebral cavernous malformations: a complex jigsaw puzzle under construction. The FEBS journal. 2010;277:1084–96. doi: 10.1111/j.1742-4658.2009.07537.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazioli F, Minichiello L, Matoskova B, Wong WT, Di Fiore PP. eps15, a novel tyrosine kinase substrate, exhibits transforming activity. Mol Cell Biol. 1993;13:5814–28. doi: 10.1128/mcb.13.9.5814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidalgo M, Fraile M, Pires A, Force T, Pombo C, Zalvide J. CCM3/PDCD10 stabilizes GCKIII proteins to promote Golgi assembly and cell orientation. J Cell Sci. 2010;123:1274–84. doi: 10.1242/jcs.061341. [DOI] [PubMed] [Google Scholar]
- Fidalgo M, Guerrero A, Fraile M, Iglesias C, Pombo CM, Zalvide J. Adaptor protein cerebral cavernous malformation 3 (CCM3) mediates phosphorylation of the cytoskeletal proteins ezrin/radixin/moesin by mammalian Ste20–4 to protect cells from oxidative stress. J Biol Chem. 2012;287:11556–65. doi: 10.1074/jbc.M111.320259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field MC, Gabernet-Castello C, Dacks JB. Reconstructing the evolution of the endocytic system: insights from genomics and molecular cell biology. Advances in experimental medicine and biology. 2007;607:84–96. doi: 10.1007/978-0-387-74021-8_7. [DOI] [PubMed] [Google Scholar]
- Fleissner A, Simonin AR, Glass NL. Cell fusion in the filamentous fungus, Neurospora crassa. Methods Mol Biol. 2008;475:21–38. doi: 10.1007/978-1-59745-250-2_2. [DOI] [PubMed] [Google Scholar]
- Friedman A, Perrimon N. A functional RNAi screen for regulators of receptor tyrosine kinase and ERK signalling. Nature. 2006;444:230–4. doi: 10.1038/nature05280. [DOI] [PubMed] [Google Scholar]
- Frost A, Elgort MG, Brandman O, Ives C, Collins SR, Miller-Vedam L, et al. Functional repurposing revealed by comparing S. pombe and S. cerevisiae genetic interactions. Cell. 2012;149:1339–52. doi: 10.1016/j.cell.2012.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu C, Iyer P, Herkal A, Abdullah J, Stout A, Free SJ. Identification and characterization of genes required for cell-to-cell fusion in Neurospora crassa. Eukaryotic cell. 2011;10:1100–9. doi: 10.1128/EC.05003-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaillard S, Bailly Y, Benoist M, Rakitina T, Kessler JP, Fronzaroli-Molinieres L, et al. Targeting of proteins of the striatin family to dendritic spines: role of the coiled-coil domain. Traffic. 2006;7:74–84. doi: 10.1111/j.1600-0854.2005.00363.x. [DOI] [PubMed] [Google Scholar]
- Gaillard S, Bartoli M, Castets F, Monneron A. Striatin, a calmodulin-dependent scaffolding protein, directly binds caveolin-1. FEBS Lett. 2001;508:49–52. doi: 10.1016/s0014-5793(01)03020-4. [DOI] [PubMed] [Google Scholar]
- Garcia-Cardena G, Martasek P, Masters BS, Skidd PM, Couet J, Li S, et al. Dissecting the interaction between nitric oxide synthase (NOS) and caveolin. Functional significance of the nos caveolin binding domain in vivo. J Biol Chem. 1997;272:25437–40. doi: 10.1074/jbc.272.41.25437. [DOI] [PubMed] [Google Scholar]
- Gault J, Awad IA, Recksiek P, Shenkar R, Breeze R, Handler M, et al. Cerebral cavernous malformations: somatic mutations in vascular endothelial cells. Neurosurgery. 2009;65:138–44. doi: 10.1227/01.NEU.0000348049.81121.C1. discussion 44–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gault J, Shenkar R, Recksiek P, Awad IA. Biallelic somatic and germ line CCM1 truncating mutations in a cerebral cavernous malformation lesion. Stroke; a journal of cerebral circulation. 2005;36:872–4. doi: 10.1161/01.STR.0000157586.20479.fd. [DOI] [PubMed] [Google Scholar]
- Gingras AR, Liu JJ, Ginsberg MH. Structural basis of the junctional anchorage of the cerebral cavernous malformations complex. J Cell Biol. 2012;199:39–48. doi: 10.1083/jcb.201205109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glading A, Han J, Stockton RA, Ginsberg MH. KRIT-1/CCM1 is a Rap1 effector that regulates endothelial cell cell junctions. J Cell Biol. 2007;179:247–54. doi: 10.1083/jcb.200705175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glatter T, Wepf A, Aebersold R, Gstaiger M. An integrated workflow for charting the human interaction proteome: insights into the PP2A system. Mol Syst Biol. 2009;5:237. doi: 10.1038/msb.2008.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloerich M, ten Klooster JP, Vliem MJ, Koorman T, Zwartkruis FJ, Clevers H, et al. Rap2A links intestinal cell polarity to brush border formation. Nat Cell Biol. 2012;14:793–801. doi: 10.1038/ncb2537. [DOI] [PubMed] [Google Scholar]
- Goetz RM, Thatte HS, Prabhakar P, Cho MR, Michel T, Golan DE. Estradiol induces the calcium-dependent translocation of endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 1999;96:2788–93. doi: 10.1073/pnas.96.6.2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon J, Hwang J, Carrier KJ, Jones CA, Kern QL, Moreno CS, et al. Protein phosphatase 2a (PP2A) binds within the oligomerization domain of striatin and regulates the phosphorylation and activation of the mammalian Ste20-Like kinase Mst3. BMC biochemistry. 2011;12:54. doi: 10.1186/1471-2091-12-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudreault M, D’Ambrosio LM, Kean MJ, Mullin MJ, Larsen BG, Sanchez A, et al. A PP2A phosphatase high density interaction network identifies a novel striatin-interacting phosphatase and kinase complex linked to the cerebral cavernous malformation 3 (CCM3) protein. Mol Cell Proteomics. 2009;8:157–71. doi: 10.1074/mcp.M800266-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grand EK, Grand FH, Chase AJ, Ross FM, Corcoran MM, Oscier DG, et al. Identification of a novel gene, FGFR1OP2, fused to FGFR1 in 8p11 myeloproliferative syndrome. Genes, chromosomes & cancer. 2004;40:78–83. doi: 10.1002/gcc.20023. [DOI] [PubMed] [Google Scholar]
- Guzzo RM, Salih M, Moore ED, Tuana BS. Molecular properties of cardiac tail-anchored membrane protein SLMAP are consistent with structural role in arrangement of excitation-contraction coupling apparatus. American journal of physiology Heart and circulatory physiology. 2005;288:H1810–9. doi: 10.1152/ajpheart.01015.2004. [DOI] [PubMed] [Google Scholar]
- Guzzo RM, Sevinc S, Salih M, Tuana BS. A novel isoform of sarcolemmal membrane-associated protein (SLMAP) is a component of the microtubule organizing centre. J Cell Sci. 2004a;117:2271–81. doi: 10.1242/jcs.01079. [DOI] [PubMed] [Google Scholar]
- Guzzo RM, Wigle J, Salih M, Moore ED, Tuana BS. Regulated expression and temporal induction of the tail-anchored sarcolemmal-membrane-associated protein is critical for myoblast fusion. The Biochemical journal. 2004b;381:599–608. doi: 10.1042/BJ20031723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayer A, Stoeber M, Bissig C, Helenius A. Biogenesis of caveolae: stepwise assembly of large caveolin and cavin complexes. Traffic. 2010;11:361–82. doi: 10.1111/j.1600-0854.2009.01023.x. [DOI] [PubMed] [Google Scholar]
- He Y, Zhang H, Yu L, Gunel M, Boggon TJ, Chen H, et al. Stabilization of VEGFR2 signaling by cerebral cavernous malformation 3 is critical for vascular development. Sci Signal. 2010;3:ra26. doi: 10.1126/scisignal.2000722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hering H, Sheng M. Activity-dependent redistribution and essential role of cortactin in dendritic spine morphogenesis. J Neurosci. 2003;23:11759–69. doi: 10.1523/JNEUROSCI.23-37-11759.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog F, Kahraman A, Boehringer D, Mak R, Bracher A, Walzthoeni T, et al. Structural probing of a protein phosphatase 2A network by chemical cross-linking and mass spectrometry. Science. 2012;337:1348–52. doi: 10.1126/science.1221483. [DOI] [PubMed] [Google Scholar]
- Hilder TL, Malone MH, Bencharit S, Colicelli J, Haystead TA, Johnson GL, et al. Proteomic identification of the cerebral cavernous malformation signaling complex. Journal of proteome research. 2007;6:4343–55. doi: 10.1021/pr0704276. [DOI] [PubMed] [Google Scholar]
- Horn T, Sandmann T, Fischer B, Axelsson E, Huber W, Boutros M. Mapping of signaling networks through synthetic genetic interaction analysis by RNAi. Nature methods. 2011;8:341–6. doi: 10.1038/nmeth.1581. [DOI] [PubMed] [Google Scholar]
- Huang J, Liu T, Xu LG, Chen D, Zhai Z, Shu HB. SIKE is an IKK epsilon/TBK1-associated suppressor of TLR3- and virus-triggered IRF-3 activation pathways. Embo J. 2005;24:4018–28. doi: 10.1038/sj.emboj.7600863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutsler JJ, Zhang H. Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 2010;1309:83–94. doi: 10.1016/j.brainres.2009.09.120. [DOI] [PubMed] [Google Scholar]
- Hyodo T, Ito S, Hasegawa H, Asano E, Maeda M, Urano T, et al. Misshapen-like kinase 1 (MINK1) is a novel component of striatin-interacting phosphatase and kinase (STRIPAK) and is required for the completion of cytokinesis. J Biol Chem. 2012;287:25019–29. doi: 10.1074/jbc.M112.372342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–99. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa T, Sato A, Marcou CA, Tester DJ, Ackerman MJ, Crotti L, et al. A novel disease gene for Brugada syndrome: sarcolemmal membrane-associated protein gene mutations impair intracellular trafficking of hNav1.5. Circulation Arrhythmia and electrophysiology. 2012;5:1098–107. doi: 10.1161/CIRCEP.111.969972. [DOI] [PubMed] [Google Scholar]
- Ishikawa T, Takahashi N, Ohno S, Sakurada H, Nakamura K, On YK, et al. Novel SCN3B Mutation Associated With Brugada Syndrome Affects Intracellular Trafficking and Function of Nav1.5. Circulation journal: official journal of the Japanese Circulation Society. 2013;77:959–67. doi: 10.1253/circj.cj-12-0995. [DOI] [PubMed] [Google Scholar]
- Jiang W, Hallberg RL. Isolation and characterization of par1(+) and par2(+): two Schizosaccharomyces pombe genes encoding B′ subunits of protein phosphatase 2A. Genetics. 2000;154:1025–38. doi: 10.1093/genetics/154.3.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Hallberg RL. Correct regulation of the septation initiation network in Schizosaccharomyces pombe requires the activities of par1 and par2. Genetics. 2001;158:1413–29. doi: 10.1093/genetics/158.4.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju H, Zou R, Venema VJ, Venema RC. Direct interaction of endothelial nitric-oxide synthase and caveolin-1 inhibits synthase activity. J Biol Chem. 1997;272:18522–5. doi: 10.1074/jbc.272.30.18522. [DOI] [PubMed] [Google Scholar]
- Kammerer RA, Kostrewa D, Progias P, Honnappa S, Avila D, Lustig A, et al. A conserved trimerization motif controls the topology of short coiled coils. Proc Natl Acad Sci U S A. 2005;102:13891–6. doi: 10.1073/pnas.0502390102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato K, Yazawa T, Taki K, Mori K, Wang S, Nishioka T, et al. The inositol 5-phosphatase SHIP2 is an effector of RhoA and is involved in cell polarity and migration. Mol Biol Cell. 2012;23:2593–604. doi: 10.1091/mbc.E11-11-0958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kean MJ, Ceccarelli DF, Goudreault M, Sanches M, Tate S, Larsen B, et al. Structure-function analysis of core STRIPAK Proteins: a signaling complex implicated in Golgi polarization. J Biol Chem. 2011;286:25065–75. doi: 10.1074/jbc.M110.214486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehrer-Sawatzki H, Wilda M, Braun VM, Richter HP, Hameister H. Mutation and expression analysis of the KRIT1 gene associated with cerebral cavernous malformations (CCM1) Acta neuropathologica. 2002;104:231–40. doi: 10.1007/s00401-002-0552-6. [DOI] [PubMed] [Google Scholar]
- Kemp HA, Sprague GF., Jr Far3 and five interacting proteins prevent premature recovery from pheromone arrest in the budding yeast Saccharomyces cerevisiae. Mol Cell Biol. 2003;23:1750–63. doi: 10.1128/MCB.23.5.1750-1763.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HP, Lee JY, Jeong JK, Bae SW, Lee HK, Jo I. Nongenomic stimulation of nitric oxide release by estrogen is mediated by estrogen receptor alpha localized in caveolae. Biochem Biophys Res Commun. 1999;263:257–62. doi: 10.1006/bbrc.1999.1348. [DOI] [PubMed] [Google Scholar]
- Kim JH, Oh MY, Paek J, Lee J. Association between FGFR1OP2/wit3.0 polymorphisms and residual ridge resorption of mandible in Korean population. PLoS One. 2012;7:e42734. doi: 10.1371/journal.pone.0042734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleaveland B, Zheng X, Liu JJ, Blum Y, Tung JJ, Zou Z, et al. Regulation of cardiovascular development and integrity by the heart of glass-cerebral cavernous malformation protein pathway. Nature medicine. 2009;15:169–76. doi: 10.1038/nm.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krapp A, Cano E, Simanis V. Mitotic hyperphosphorylation of the fission yeast SIN scaffold protein cdc11p is regulated by the protein kinase cdc7p. Current biology: CB. 2003;13:168–72. doi: 10.1016/s0960-9822(02)01417-3. [DOI] [PubMed] [Google Scholar]
- Krapp A, Simanis V. An overview of the fission yeast septation initiation network (SIN) Biochemical Society transactions. 2008;36:411–5. doi: 10.1042/BST0360411. [DOI] [PubMed] [Google Scholar]
- Krishnan KS, Rikhy R, Rao S, Shivalkar M, Mosko M, Narayanan R, et al. Nucleoside diphosphate kinase, a source of GTP, is required for dynamin-dependent synaptic vesicle recycling. Neuron. 2001;30:197–210. doi: 10.1016/s0896-6273(01)00273-2. [DOI] [PubMed] [Google Scholar]
- Labauge P, Denier C, Bergametti F, Tournier-Lasserve E. Genetics of cavernous angiomas. Lancet neurology. 2007;6:237–44. doi: 10.1016/S1474-4422(07)70053-4. [DOI] [PubMed] [Google Scholar]
- Laberge-le Couteulx S, Jung HH, Labauge P, Houtteville JP, Lescoat C, Cecillon M, et al. Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas. Nat Genet. 1999;23:189–93. doi: 10.1038/13815. [DOI] [PubMed] [Google Scholar]
- Lahoz A, Alcaide-Gavilan M, Daga RR, Jimenez J. Antagonistic roles of PP2A-Pab1 and Etd1 in the control of cytokinesis in fission yeast. Genetics. 2010;186:1261–70. doi: 10.1534/genetics.110.121368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lajoie P, Goetz JG, Dennis JW, Nabi IR. Lattices, rafts, and scaffolds: domain regulation of receptor signaling at the plasma membrane. J Cell Biol. 2009;185:381–5. doi: 10.1083/jcb.200811059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauenborg B, Kopp K, Krejsgaard T, Eriksen KW, Geisler C, Dabelsteen S, et al. Programmed cell death-10 enhances proliferation and protects malignant T cells from apoptosis. APMIS: acta pathologica, microbiologica, et immunologica Scandinavica. 2010;118:719–28. doi: 10.1111/j.1600-0463.2010.02669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Goff X, Buvelot S, Salimova E, Guerry F, Schmidt S, Cueille N, et al. The protein phosphatase 2A B′-regulatory subunit par1p is implicated in regulation of the S. pombe septation initiation network. FEBS Lett. 2001;508:136–42. doi: 10.1016/s0014-5793(01)03047-2. [DOI] [PubMed] [Google Scholar]
- Le Lay S, Kurzchalia TV. Getting rid of caveolins: phenotypes of caveolin-deficient animals. Biochim Biophys Acta. 2005;1746:322–33. doi: 10.1016/j.bbamcr.2005.06.001. [DOI] [PubMed] [Google Scholar]
- Lemola K, Brunckhorst C, Helfenstein U, Oechslin E, Jenni R, Duru F. Predictors of adverse outcome in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy: long term experience of a tertiary care centre. Heart. 2005;91:1167–72. doi: 10.1136/hrt.2004.038620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonhard K, Nurse P. Ste20/GCK kinase Nak1/Orb3 polarizes the actin cytoskeleton in fission yeast during the cell cycle. J Cell Sci. 2005;118:1033–44. doi: 10.1242/jcs.01690. [DOI] [PubMed] [Google Scholar]
- Li D, Roberts R. WD-repeat proteins: structure characteristics, biological function, and their involvement in human diseases. Cellular and molecular life sciences: CMLS. 2001;58:2085–97. doi: 10.1007/PL00000838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Couet J, Lisanti MP. Src tyrosine kinases, Galpha subunits, and H-Ras share a common membrane-anchored scaffolding protein, caveolin. Caveolin binding negatively regulates the auto-activation of Src tyrosine kinases. J Biol Chem. 1996;271:29182–90. doi: 10.1074/jbc.271.46.29182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zhang R, Zhang H, He Y, Ji W, Min W, et al. Crystal structure of CCM3, a cerebral cavernous malformation protein critical for vascular integrity. J Biol Chem. 2010;285:24099–107. doi: 10.1074/jbc.M110.128470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin A, Hokugo A, Choi J, Nishimura I. Small cytoskeleton-associated molecule, fibroblast growth factor receptor 1 oncogene partner 2/wound inducible transcript-3.0 (FGFR1OP2/wit3.0), facilitates fibroblast-driven wound closure. Am J Pathol. 2010;176:108–21. doi: 10.2353/ajpath.2010.090256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling P, Lu TJ, Yuan CJ, Lai MD. Biosignaling of mammalian Ste20-related kinases. Cell Signal. 2008;20:1237–47. doi: 10.1016/j.cellsig.2007.12.019. [DOI] [PubMed] [Google Scholar]
- Liquori CL, Berg MJ, Siegel AM, Huang E, Zawistowski JS, Stoffer T, et al. Mutations in a gene encoding a novel protein containing a phosphotyrosine-binding domain cause type 2 cerebral cavernous malformations. Am J Hum Genet. 2003;73:1459–64. doi: 10.1086/380314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisa-Santamaria P, Jimenez A, Revuelta JL. The protein factor-arrest 11 (Far11) is essential for the toxicity of human caspase-10 in yeast and participates in the regulation of autophagy and the DNA damage signaling. J Biol Chem. 2012;287:29636–47. doi: 10.1074/jbc.M112.344192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Su YC, Becker E, Treisman J, Skolnik EY. A Drosophila TNF-receptor-associated factor (TRAF) binds the ste20 kinase Misshapen and activates Jun kinase. Current biology: CB. 1999;9:101–4. doi: 10.1016/s0960-9822(99)80023-2. [DOI] [PubMed] [Google Scholar]
- Liu JJ, Stockton RA, Gingras AR, Ablooglu AJ, Han J, Bobkov AA, et al. A mechanism of Rap1-induced stabilization of endothelial cell--cell junctions. Mol Biol Cell. 2011;22:2509–19. doi: 10.1091/mbc.E11-02-0157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louvi A, Chen L, Two AM, Zhang H, Min W, Gunel M. Loss of cerebral cavernous malformation 3 (Ccm3) in neuroglia leads to CCM and vascular pathology. Proc Natl Acad Sci U S A. 2011;108:3737–42. doi: 10.1073/pnas.1012617108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Q, Pallas DC, Surks HK, Baur WE, Mendelsohn ME, Karas RH. Striatin assembles a membrane signaling complex necessary for rapid, nongenomic activation of endothelial NO synthase by estrogen receptor alpha. Proc Natl Acad Sci U S A. 2004;101:17126–31. doi: 10.1073/pnas.0407492101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Q, Surks HK, Ebling H, Baur WE, Brown D, Pallas DC, et al. Regulation of estrogen receptor alpha-mediated transcription by a direct interaction with protein phosphatase 2A. J Biol Chem. 2003;278:4639–45. doi: 10.1074/jbc.M210949200. [DOI] [PubMed] [Google Scholar]
- Lu TJ, Lai WY, Huang CY, Hsieh WJ, Yu JS, Hsieh YJ, et al. Inhibition of cell migration by autophosphorylated mammalian sterile 20-like kinase 3 (MST3) involves paxillin and protein-tyrosine phosphatase-PEST. J Biol Chem. 2006;281:38405–17. doi: 10.1074/jbc.M605035200. [DOI] [PubMed] [Google Scholar]
- Ma X. PDCD10 Interacts with Ste20-related Kinase MST4 to Promote Cell Growth and Transformation via Modulation of the ERK Pathway. Mol Biol Cell. 2007;18:1965–78. doi: 10.1091/mbc.E06-07-0608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Zhao H, Shan J, Long F, Chen Y, Chen Y, et al. PDCD10 interacts with Ste20-related kinase MST4 to promote cell growth and transformation via modulation of the ERK pathway. Mol Biol Cell. 2007;18:1965–78. doi: 10.1091/mbc.E06-07-0608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maerz S, Dettmann A, Ziv C, Liu Y, Valerius O, Yarden O, et al. Two NDR kinase-MOB complexes function as distinct modules during septum formation and tip extension in Neurospora crassa. Molecular microbiology. 2009;74:707–23. doi: 10.1111/j.1365-2958.2009.06896.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahrenholz CC, Abfalter IG, Bodenhofer U, Volkmer R, Hochreiter S. Complex networks govern coiled-coil oligomerization--predicting and profiling by means of a machine learning approach. Mol Cell Proteomics. 2011;10:M110 004994. doi: 10.1074/mcp.M110.004994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks AR. Calcium cycling proteins and heart failure: mechanisms and therapeutics. The Journal of clinical investigation. 2013;123:46–52. doi: 10.1172/JCI62834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmor MD, Yarden Y. Role of protein ubiquitylation in regulating endocytosis of receptor tyrosine kinases. Oncogene. 2004;23:2057–70. doi: 10.1038/sj.onc.1207390. [DOI] [PubMed] [Google Scholar]
- Matsuyama A, Arai R, Yashiroda Y, Shirai A, Kamata A, Sekido S, et al. ORFeome cloning and global analysis of protein localization in the fission yeast Schizosaccharomyces pombe. Nat Biotechnol. 2006;24:841–7. doi: 10.1038/nbt1222. [DOI] [PubMed] [Google Scholar]
- McDonald DA, Shenkar R, Shi C, Stockton RA, Akers AL, Kucherlapati MH, et al. A novel mouse model of cerebral cavernous malformations based on the two-hit mutation hypothesis recapitulates the human disease. Human molecular genetics. 2011;20:211–22. doi: 10.1093/hmg/ddq433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald DA, Shi C, Shenkar R, Stockton RA, Liu F, Ginsberg MH, et al. Fasudil decreases lesion burden in a murine model of cerebral cavernous malformation disease. Stroke; a journal of cerebral circulation. 2012;43:571–4. doi: 10.1161/STROKEAHA.111.625467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meurs KM, Mauceli E, Lahmers S, Acland GM, White SN, Lindblad-Toh K. Genome-wide association identifies a deletion in the 3′ untranslated region of striatin in a canine model of arrhythmogenic right ventricular cardiomyopathy. Hum Genet. 2010;128:315–24. doi: 10.1007/s00439-010-0855-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meurs KM, Stern JA, Sisson DD, Kittleson MD, Cunningham SM, Ames MK, et al. Association of Dilated Cardiomyopathy with the Striatin Mutation Genotype in Boxer Dogs. Journal of veterinary internal medicine/American College of Veterinary Internal Medicine. 2013 doi: 10.1111/jvim.12163. [DOI] [PubMed] [Google Scholar]
- Michel JB, Feron O, Sacks D, Michel T. Reciprocal regulation of endothelial nitric-oxide synthase by Ca2+-calmodulin and caveolin. J Biol Chem. 1997;272:15583–6. doi: 10.1074/jbc.272.25.15583. [DOI] [PubMed] [Google Scholar]
- Mineo C, Shaul PW. Regulation of eNOS in caveolae. Advances in experimental medicine and biology. 2012;729:51–62. doi: 10.1007/978-1-4614-1222-9_4. [DOI] [PubMed] [Google Scholar]
- Moqrich A, Mattei MG, Bartoli M, Rakitina T, Baillat G, Monneron A, et al. Cloning of human striatin cDNA (STRN), gene mapping to 2p22-p21, and preferential expression in brain. Genomics. 1998;51:136–9. doi: 10.1006/geno.1998.5342. [DOI] [PubMed] [Google Scholar]
- Moreno CS, Lane WS, Pallas DC. A mammalian homolog of yeast MOB1 is both a member and a putative substrate of striatin family-protein phosphatase 2A complexes. J Biol Chem. 2001;276:24253–60. doi: 10.1074/jbc.M102398200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno CS, Park S, Nelson K, Ashby D, Hubalek F, Lane WS, et al. WD40 repeat proteins striatin and S/G(2) nuclear autoantigen are members of a novel family of calmodulin-binding proteins that associate with protein phosphatase 2A. J Biol Chem. 2000;275:5257–63. doi: 10.1074/jbc.275.8.5257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller W, Connor JA. Dendritic spines as individual neuronal compartments for synaptic Ca2+ responses. Nature. 1991;354:73–6. doi: 10.1038/354073a0. [DOI] [PubMed] [Google Scholar]
- Muro Y, Chan EK, Landberg G, Tan EM. A cell-cycle nuclear autoantigen containing WD-40 motifs expressed mainly in S and G2 phase cells. Biochem Biophys Res Commun. 1995;207:1029–37. doi: 10.1006/bbrc.1995.1288. [DOI] [PubMed] [Google Scholar]
- Nader M, Westendorp B, Hawari O, Salih M, Stewart AF, Leenen FH, et al. Tail-anchored membrane protein SLMAP is a novel regulator of cardiac function at the sarcoplasmic reticulum. American journal of physiology Heart and circulatory physiology. 2012;302:H1138–45. doi: 10.1152/ajpheart.00872.2011. [DOI] [PubMed] [Google Scholar]
- Nargang FE, Adames K, Rub C, Cheung S, Easton N, Nargang CE, et al. Identification of genes required for alternative oxidase production in the Neurospora crassa gene knockout library. G3 (Bethesda) 2012;2:1345–56. doi: 10.1534/g3.112.004218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neisch AL, Speck O, Stronach B, Fehon RG. Rho1 regulates apoptosis via activation of the JNK signaling pathway at the plasma membrane. J Cell Biol. 2010;189:311–23. doi: 10.1083/jcb.200912010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerstedt A, Cansby E, Andersson CX, Laakso M, Stancakova A, Bluher M, et al. Serine/threonine protein kinase 25 (STK25): a novel negative regulator of lipid and glucose metabolism in rodent and human skeletal muscle. Diabetologia. 2012;55:1797–807. doi: 10.1007/s00125-012-2511-7. [DOI] [PubMed] [Google Scholar]
- Neumann B, Walter T, Heriche JK, Bulkescher J, Erfle H, Conrad C, et al. Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature. 2010;464:721–7. doi: 10.1038/nature08869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngok SP, Geyer R, Kourtidis A, Storz P, Anastasiadis PZ. Phosphorylation-mediated 14-3-3 protein binding regulates the function of the rho-specific guanine nucleotide exchange factor (RhoGEF) Syx. J Biol Chem. 2013;288:6640–50. doi: 10.1074/jbc.M112.432682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngok SP, Geyer R, Liu M, Kourtidis A, Agrawal S, Wu C, et al. VEGF and Angiopoietin-1 exert opposing effects on cell junctions by regulating the Rho GEF Syx. J Cell Biol. 2012;199:1103–15. doi: 10.1083/jcb.201207009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Lone R, Frith MC, Karlsson EK, Hansen U. Genomic targets of nuclear estrogen receptors. Mol Endocrinol. 2004;18:1859–75. doi: 10.1210/me.2003-0044. [DOI] [PubMed] [Google Scholar]
- Pagenstecher A, Stahl S, Sure U, Felbor U. A two-hit mechanism causes cerebral cavernous malformations: complete inactivation of CCM1, CCM2 or CCM3 in affected endothelial cells. Human molecular genetics. 2009;18:911–8. doi: 10.1093/hmg/ddn420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parton RG, Simons K. The multiple faces of caveolae. Nature reviews Molecular cell biology. 2007;8:185–94. doi: 10.1038/nrm2122. [DOI] [PubMed] [Google Scholar]
- Penzes P, Cahill ME, Jones KA, VanLeeuwen JE, Woolfrey KM. Dendritic spine pathology in neuropsychiatric disorders. Nature neuroscience. 2011;14:285–93. doi: 10.1038/nn.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plummer NW, Gallione CJ, Srinivasan S, Zawistowski JS, Louis DN, Marchuk DA. Loss of p53 sensitizes mice with a mutation in Ccm1 (KRIT1) to development of cerebral vascular malformations. Am J Pathol. 2004;165:1509–18. doi: 10.1016/S0002-9440(10)63409-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poggeler S, Kuck U. A WD40 repeat protein regulates fungal cell differentiation and can be replaced functionally by the mammalian homologue striatin. Eukaryotic cell. 2004;3:232–40. doi: 10.1128/EC.3.1.232-240.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pojoga LH, Coutinho P, Rivera A, Yao TM, Maldonado ER, Youte R, et al. Activation of the mineralocorticoid receptor increases striatin levels. American journal of hypertension. 2012;25:243–9. doi: 10.1038/ajh.2011.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pombo CM, Bonventre JV, Molnar A, Kyriakis J, Force T. Activation of a human Ste20-like kinase by oxidant stress defines a novel stress response pathway. Embo J. 1996;15:4537–46. [PMC free article] [PubMed] [Google Scholar]
- Pombo CM, Force T, Kyriakis J, Nogueira E, Fidalgo M, Zalvide J. The GCK II and III subfamilies of the STE20 group kinases. Front Biosci. 2007;12:850–9. doi: 10.2741/2107. [DOI] [PubMed] [Google Scholar]
- Pracheil T, Thornton J, Liu Z. TORC2 signaling is antagonized by protein phosphatase 2A and the Far complex in Saccharomyces cerevisiae. Genetics. 2012;190:1325–39. doi: 10.1534/genetics.111.138305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preisinger C, Short B, De Corte V, Bruyneel E, Haas A, Kopajtich R, et al. YSK1 is activated by the Golgi matrix protein GM130 and plays a role in cell migration through its substrate 14-3-3zeta. J Cell Biol. 2004;164:1009–20. doi: 10.1083/jcb.200310061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen CG, Glass NL. A Rho-type GTPase, rho-4, is required for septation in Neurospora crassa. Eukaryotic cell. 2005;4:1913–25. doi: 10.1128/EC.4.11.1913-1925.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raz L, Khan MM, Mahesh VB, Vadlamudi RK, Brann DW. Rapid estrogen signaling in the brain. Neuro-Signals. 2008;16:140–53. doi: 10.1159/000111559. [DOI] [PubMed] [Google Scholar]
- Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, et al. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem. 2001;276:38121–38. doi: 10.1074/jbc.M105408200. [DOI] [PubMed] [Google Scholar]
- Rech C, Engh I, Kuck U. Detection of hyphal fusion in filamentous fungi using differently fluorescence-labeled histones. Current genetics. 2007;52:259–66. doi: 10.1007/s00294-007-0158-6. [DOI] [PubMed] [Google Scholar]
- Riant F, Bergametti F, Ayrignac X, Boulday G, Tournier-Lasserve E. Recent insights into cerebral cavernous malformations: the molecular genetics of CCM. The FEBS journal. 2010;277:1070–5. doi: 10.1111/j.1742-4658.2009.07535.x. [DOI] [PubMed] [Google Scholar]
- Ribeiro PS, Josue F, Wepf A, Wehr MC, Rinner O, Kelly G, et al. Combined functional genomic and proteomic approaches identify a PP2A complex as a negative regulator of Hippo signaling. Mol Cell. 2010;39:521–34. doi: 10.1016/j.molcel.2010.08.002. [DOI] [PubMed] [Google Scholar]
- Ricchiuti V, Lapointe N, Pojoga L, Yao T, Tran L, Williams GH, et al. Dietary sodium intake regulates angiotensin II type 1, mineralocorticoid receptor, and associated signaling proteins in heart. The Journal of endocrinology. 2011;211:47–54. doi: 10.1530/JOE-10-0458. [DOI] [PubMed] [Google Scholar]
- Rigamonti D, Hadley MN, Drayer BP, Johnson PC, Hoenig-Rigamonti K, Knight JT, et al. Cerebral cavernous malformations. Incidence and familial occurrence. The New England journal of medicine. 1988;319:343–7. doi: 10.1056/NEJM198808113190605. [DOI] [PubMed] [Google Scholar]
- Robinson JR, Awad IA, Little JR. Natural history of the cavernous angioma. J Neurosurg. 1991;75:709–14. doi: 10.3171/jns.1991.75.5.0709. [DOI] [PubMed] [Google Scholar]
- Roca MG, Arlt J, Jeffree CE, Read ND. Cell biology of conidial anastomosis tubes in Neurospora crassa. Eukaryotic cell. 2005;4:911–9. doi: 10.1128/EC.4.5.911-919.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohlfs M. The Ste20-like kinase SvkA of Dictyostelium discoideum is essential for late stages of cytokinesis. Journal of Cell Science. 2007;120:4345–54. doi: 10.1242/jcs.012179. [DOI] [PubMed] [Google Scholar]
- Roux-Buisson N, Cacheux M, Fourest-Lieuvin A, Fauconnier J, Brocard J, Denjoy I, et al. Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human. Human molecular genetics. 2012;21:2759–67. doi: 10.1093/hmg/dds104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A, Li N, et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437:1173–8. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- Sahoo T, Johnson EW, Thomas JW, Kuehl PM, Jones TL, Dokken CG, et al. Mutations in the gene encoding KRIT1, a Krev-1/rap1a binding protein, cause cerebral cavernous malformations (CCM1) Human molecular genetics. 1999;8:2325–33. doi: 10.1093/hmg/8.12.2325. [DOI] [PubMed] [Google Scholar]
- Salcini AE, Hilliard MA, Croce A, Arbucci S, Luzzi P, Tacchetti C, et al. The Eps15 C. elegans homologue EHS-1 is implicated in synaptic vesicle recycling. Nat Cell Biol. 2001;3:755–60. doi: 10.1038/35087075. [DOI] [PubMed] [Google Scholar]
- Salin P, Kachidian P, Bartoli M, Castets F. Distribution of striatin, a newly identified calmodulin-binding protein in the rat brain: an in situ hybridization and immunocytochemical study. J Comp Neurol. 1998;397:41–59. [PubMed] [Google Scholar]
- Sanghamitra M, Talukder I, Singarapu N, Sindhu KV, Kateriya S, Goswami SK. WD-40 repeat protein SG2NA has multiple splice variants with tissue restricted and growth responsive properties. Gene. 2008;420:48–56. doi: 10.1016/j.gene.2008.04.016. [DOI] [PubMed] [Google Scholar]
- Schleider E, Stahl S, Wustehube J, Walter U, Fischer A, Felbor U. Evidence for anti-angiogenic and pro-survival functions of the cerebral cavernous malformation protein 3. Neurogenetics. 2011;12:83–6. doi: 10.1007/s10048-010-0261-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider H, Errede M, Ulrich NH, Virgintino D, Frei K, Bertalanffy H. Impairment of tight junctions and glucose transport in endothelial cells of human cerebral cavernous malformations. Journal of neuropathology and experimental neurology. 2011;70:417–29. doi: 10.1097/NEN.0b013e31821bc40e. [DOI] [PubMed] [Google Scholar]
- Schulte J, Sepp KJ, Jorquera RA, Wu C, Song Y, Hong P, et al. DMob4/Phocein regulates synapse formation, axonal transport, and microtubule organization. J Neurosci. 2010;30:5189–203. doi: 10.1523/JNEUROSCI.5823-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeling JM, JR, Miller R, Gil R, Moon T, White R, Virshup DM. Regulation of b-Catenin signaling by the B56 subunit of Protein Phosphatase 2A. Science. 1999;283:2089–91. doi: 10.1126/science.283.5410.2089. [DOI] [PubMed] [Google Scholar]
- Seiler S, Vogt N, Ziv C, Gorovits R, Yarden O. The STE20/germinal center kinase POD6 interacts with the NDR kinase COT1 and is involved in polar tip extension in Neurospora crassa. Mol Biol Cell. 2006;17:4080–92. doi: 10.1091/mbc.E06-01-0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepp KJ, Hong P, Lizarraga SB, Liu JS, Mejia LA, Walsh CA, et al. Identification of neural outgrowth genes using genome-wide RNAi. PLoS genetics. 2008;4:e1000111. doi: 10.1371/journal.pgen.1000111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenkar R, Venkatasubramanian PN, Wyrwicz AM, Zhao JC, Shi C, Akers A, et al. Advanced magnetic resonance imaging of cerebral cavernous malformations: part II. Imaging of lesions in murine models. Neurosurgery. 2008;63:790–7. doi: 10.1227/01.NEU.0000315862.24920.49. discussion 7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C, Shenkar R, Du H, Duckworth E, Raja H, Batjer HH, et al. Immune response in human cerebral cavernous malformations. Stroke; a journal of cerebral circulation. 2009;40:1659–65. doi: 10.1161/STROKEAHA.108.538769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim WB, Sagaram US, Choi YE, So J, Wilkinson HH, Lee YW. FSR1 is essential for virulence and female fertility in Fusarium verticillioides and F. graminearum. Molecular plant-microbe interactions: MPMI. 2006;19:725–33. doi: 10.1094/MPMI-19-0725. [DOI] [PubMed] [Google Scholar]
- Simonin AR, Rasmussen CG, Yang M, Glass NL. Genes encoding a striatin-like protein (ham-3) and a forkhead associated protein (ham-4) are required for hyphal fusion in Neurospora crassa. Fungal genetics and biology: FG & B. 2010;47:855–68. doi: 10.1016/j.fgb.2010.06.010. [DOI] [PubMed] [Google Scholar]
- Singh NS, Shao N, McLean JR, Sevugan M, Ren L, Chew TG, et al. SIN-inhibitory phosphatase complex promotes Cdc11p dephosphorylation and propagates SIN asymmetry in fission yeast. Current biology: CB. 2011;21:1968–78. doi: 10.1016/j.cub.2011.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TF. Diversity of WD-repeat proteins. Sub-cellular biochemistry. 2008;48:20–30. doi: 10.1007/978-0-387-09595-0_3. [DOI] [PubMed] [Google Scholar]
- Sotoodehnia N, Isaacs A, de Bakker PI, Dorr M, Newton-Cheh C, Nolte IM, et al. Common variants in 22 loci are associated with QRS duration and cardiac ventricular conduction. Nat Genet. 2010;42:1068–76. doi: 10.1038/ng.716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speck O, Hughes SC, Noren NK, Kulikauskas RM, Fehon RG. Moesin functions antagonistically to the Rho pathway to maintain epithelial integrity. Nature. 2003;421:83–7. doi: 10.1038/nature01295. [DOI] [PubMed] [Google Scholar]
- Stahl S, Gaetzner S, Voss K, Brackertz B, Schleider E, Surucu O, et al. Novel CCM1, CCM2, and CCM3 mutations in patients with cerebral cavernous malformations: in-frame deletion in CCM2 prevents formation of a CCM1/CCM2/CCM3 protein complex. Human mutation. 2008;29:709–17. doi: 10.1002/humu.20712. [DOI] [PubMed] [Google Scholar]
- Steriotis AK, Bauce B, Daliento L, Rigato I, Mazzotti E, Folino AF, et al. Electrocardiographic pattern in arrhythmogenic right ventricular cardiomyopathy. The American journal of cardiology. 2009;103:1302–8. doi: 10.1016/j.amjcard.2009.01.017. [DOI] [PubMed] [Google Scholar]
- Stockton RA, Shenkar R, Awad IA, Ginsberg MH. Cerebral cavernous malformations proteins inhibit Rho kinase to stabilize vascular integrity. J Exp Med. 2010;207:881–96. doi: 10.1084/jem.20091258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroeken PJ, Alvarez B, Van Rheenen J, Wijnands YM, Geerts D, Jalink K, et al. Integrin cytoplasmic domain-associated protein-1 (ICAP-1) interacts with the ROCK-I kinase at the plasma membrane. Journal of cellular physiology. 2006;208:620–8. doi: 10.1002/jcp.20699. [DOI] [PubMed] [Google Scholar]
- Su YC, Treisman JE, Skolnik EY. The Drosophila Ste20-related kinase misshapen is required for embryonic dorsal closure and acts through a JNK MAPK module on an evolutionarily conserved signaling pathway. Genes & development. 1998;12:2371–80. doi: 10.1101/gad.12.15.2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugden PH, McGuffin LJ, Clerk A. SOcK, MiSTs, MASK and STicKs: the GCKIII (germinal centre kinase III) kinases and their heterologous protein-protein interactions. The Biochemical journal. 2013;454:13–30. doi: 10.1042/BJ20130219. [DOI] [PubMed] [Google Scholar]
- Sukotjo C, Abanmy AA, Ogawa T, Nishimura I. Molecular cloning of wound inducible transcript (wit 3.0) differentially expressed in edentulous oral mucosa undergoing tooth extraction wound-healing. Journal of dental research. 2002;81:229–35. doi: 10.1177/154405910208100402. [DOI] [PubMed] [Google Scholar]
- Sukotjo C, Lin A, Song K, Ogawa T, Wu B, Nishimura I. Oral fibroblast expression of wound-inducible transcript 3.0 (wit3.0) accelerates the collagen gel contraction in vitro. J Biol Chem. 2003;278:51527–34. doi: 10.1074/jbc.M309616200. [DOI] [PubMed] [Google Scholar]
- Suwanwela J, Lee J, Lin A, Ucer TC, Devlin H, Sinsheimer J, et al. A genetic association study of single nucleotide polymorphisms in FGFR1OP2/wit3.0 and long-term atrophy of edentulous mandible. PLoS One. 2011;6:e16204. doi: 10.1371/journal.pone.0016204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan B, Long X, Nakshatri H, Nephew KP, Bigsby RM. Striatin-3 gamma inhibits estrogen receptor activity by recruiting a protein phosphatase. Journal of molecular endocrinology. 2008;40:199–210. doi: 10.1677/JME-07-0132. [DOI] [PubMed] [Google Scholar]
- Tanabe O, Hirata D, Usui H, Nishito Y, Miyakawa T, Igarashi K, et al. Fission yeast homologues of the B′ subunit of protein phosphatase 2A: multiple roles in mitotic cell division and functional interaction with calcineurin. Genes to cells: devoted to molecular & cellular mechanisms. 2001;6:455–73. doi: 10.1046/j.1365-2443.2001.00429.x. [DOI] [PubMed] [Google Scholar]
- Thalappilly S, Suliman M, Gayet O, Soubeyran P, Hermant A, Lecine P, et al. Identification of multi-SH3 domain-containing protein interactome in pancreatic cancer: a yeast two-hybrid approach. Proteomics. 2008;8:3071–81. doi: 10.1002/pmic.200701157. [DOI] [PubMed] [Google Scholar]
- Trammell MA, Mahoney NM, Agard DA, Vale RD. Mob4 plays a role in spindle focusing in Drosophila S2 cells. J Cell Sci. 2008;121:1284–92. doi: 10.1242/jcs.017210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran H, Bustos D, Yeh R, Rubinfeld B, Lam C, Shriver S, et al. HectD1 E3 Ligase Modifies Adenomatous Polyposis Coli (APC) with Polyubiquitin to Promote the APC-Axin Interaction. J Biol Chem. 2013;288:3753–67. doi: 10.1074/jbc.M112.415240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran H, Hamada F, Schwarz-Romond T, Bienz M. Trabid, a new positive regulator of Wnt-induced transcription with preference for binding and cleaving K63-linked ubiquitin chains. Genes & development. 2008;22:528–42. doi: 10.1101/gad.463208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu J, Stoodley MA, Morgan MK, Storer KP. Ultrastructural characteristics of hemorrhagic, nonhemorrhagic, and recurrent cavernous malformations. J Neurosurg. 2005;103:903–9. doi: 10.3171/jns.2005.103.5.0903. [DOI] [PubMed] [Google Scholar]
- Uetz P, Giot L, Cagney G, Mansfield TA, Judson RS, Knight JR, et al. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae [see comments] Nature. 2000;403:623–7. doi: 10.1038/35001009. [DOI] [PubMed] [Google Scholar]
- Valpuesta JM, Martin-Benito J, Gomez-Puertas P, Carrascosa JL, Willison KR. Structure and function of a protein folding machine: the eukaryotic cytosolic chaperonin CCT. FEBS Lett. 2002;529:11–6. doi: 10.1016/s0014-5793(02)03180-0. [DOI] [PubMed] [Google Scholar]
- van Bergen En Henegouwen PM. Eps15: a multifunctional adaptor protein regulating intracellular trafficking. Cell communication and signaling: CCS. 2009;7:24. doi: 10.1186/1478-811X-7-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varsano T, Dong MQ, Niesman I, Gacula H, Lou X, Ma T, et al. GIPC is recruited by APPL to peripheral TrkA endosomes and regulates TrkA trafficking and signaling. Mol Cell Biol. 2006;26:8942–52. doi: 10.1128/MCB.00305-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vida TA, Emr SD. A new vital stain for visualizing vacuolar membrane dynamics and endocytosis in yeast. J Cell Biol. 1995;128:779–92. doi: 10.1083/jcb.128.5.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent TL, Green PJ, Woolfson DN. LOGICOIL--multi-state prediction of coiled-coil oligomeric state. Bioinformatics. 2013;29:69–76. doi: 10.1093/bioinformatics/bts648. [DOI] [PubMed] [Google Scholar]
- Voss K, Stahl S, Hogan BM, Reinders J, Schleider E, Schulte-Merker S, et al. Functional analyses of human and zebrafish 18-amino acid in-frame deletion pave the way for domain mapping of the cerebral cavernous malformation 3 protein. Human mutation. 2009;30:1003–11. doi: 10.1002/humu.20996. [DOI] [PubMed] [Google Scholar]
- Voss K, Stahl S, Schleider E, Ullrich S, Nickel J, Mueller TD, et al. CCM3 interacts with CCM2 indicating common pathogenesis for cerebral cavernous malformations. Neurogenetics. 2007;8:249–56. doi: 10.1007/s10048-007-0098-9. [DOI] [PubMed] [Google Scholar]
- Wagner SA, Beli P, Weinert BT, Nielsen ML, Cox J, Mann M, et al. A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol Cell Proteomics. 2011;10:M111 013284. doi: 10.1074/mcp.M111.013284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CL, Shim WB, Shaw BD. Aspergillus nidulans striatin (StrA) mediates sexual development and localizes to the endoplasmic reticulum. Fungal genetics and biology: FG & B. 2010;47:789–99. doi: 10.1016/j.fgb.2010.06.007. [DOI] [PubMed] [Google Scholar]
- Wang M, Amano SU, Flach RJ, Chawla A, Aouadi M, Czech MP. Identification of Map4k4 as a novel suppressor of skeletal muscle differentiation. Mol Cell Biol. 2013;33:678–87. doi: 10.1128/MCB.00618-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Liu H, Zhang Y, Ma D. cDNA cloning and expression of an apoptosis-related gene, humanTFAR15 gene. Science in China Series C, Life sciences/Chinese Academy of Sciences. 1999;42:323–9. doi: 10.1007/BF03183610. [DOI] [PubMed] [Google Scholar]
- Watanabe N, Kato T, Fujita A, Ishizaki T, Narumiya S. Cooperation between mDia1 and ROCK in Rho-induced actin reorganization. Nat Cell Biol. 1999;1:136–43. doi: 10.1038/11056. [DOI] [PubMed] [Google Scholar]
- Weber A, Engers R, Nockemann S, Gohr LL, Zur Hausen A, Gabbert HE. Differentially expressed genes in association with in vitro invasiveness of human epithelioid sarcoma. Molecular pathology: MP. 2001;54:324–30. doi: 10.1136/mp.54.5.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehead KJ, Chan AC, Navankasattusas S, Koh W, London NR, Ling J, et al. The cerebral cavernous malformation signaling pathway promotes vascular integrity via Rho GTPases. Nature medicine. 2009;15:177–84. doi: 10.1038/nm.1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehead KJ, Plummer NW, Adams JA, Marchuk DA, Li DY. Ccm1 is required for arterial morphogenesis: implications for the etiology of human cavernous malformations. Development. 2004;131:1437–48. doi: 10.1242/dev.01036. [DOI] [PubMed] [Google Scholar]
- Wielowieyski PA, Sevinc S, Guzzo R, Salih M, Wigle JT, Tuana BS. Alternative splicing, expression, and genomic structure of the 3′ region of the gene encoding the sarcolemmal-associated proteins (SLAPs) defines a novel class of coiled-coil tail-anchored membrane proteins. J Biol Chem. 2000;275:38474–81. doi: 10.1074/jbc.M007682200. [DOI] [PubMed] [Google Scholar]
- Wigle JT, Demchyshyn L, Pratt MA, Staines WA, Salih M, Tuana BS. Molecular cloning, expression, and chromosomal assignment of sarcolemmal-associated proteins. A family of acidic amphipathic alpha-helical proteins associated with the membrane. J Biol Chem. 1997;272:32384–94. doi: 10.1074/jbc.272.51.32384. [DOI] [PubMed] [Google Scholar]
- Wolf E, Kim PS, Berger B. MultiCoil: a program for predicting two- and three-stranded coiled coils. Protein science: a publication of the Protein Society. 1997;6:1179–89. doi: 10.1002/pro.5560060606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong JH, Awad IA, Kim JH. Ultrastructural pathological features of cerebrovascular malformations: a preliminary report. Neurosurgery. 2000;46:1454–9. doi: 10.1097/00006123-200006000-00027. [DOI] [PubMed] [Google Scholar]
- Wu Q, Chambliss K, Umetani M, Mineo C, Shaul PW. Non-nuclear estrogen receptor signaling in the endothelium. J Biol Chem. 2011;286:14737–43. doi: 10.1074/jbc.R110.191791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Huang J, Dong J, Pan D. hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell. 2003;114:445–56. doi: 10.1016/s0092-8674(03)00549-x. [DOI] [PubMed] [Google Scholar]
- Wullschleger S, Loewith R, Oppliger W, Hall MN. Molecular organization of target of rapamycin complex 2. J Biol Chem. 2005;280:30697–704. doi: 10.1074/jbc.M505553200. [DOI] [PubMed] [Google Scholar]
- Wurzenberger C, Gerlich DW. Phosphatases: providing safe passage through mitotic exit. Nature reviews Molecular cell biology. 2011;12:469–82. doi: 10.1038/nrm3149. [DOI] [PubMed] [Google Scholar]
- Xiang Q, Rasmussen C, Glass NL. The ham-2 locus, encoding a putative transmembrane protein, is required for hyphal fusion in Neurospora crassa. Genetics. 2002;160:169–80. doi: 10.1093/genetics/160.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamura Y, Shim WB. The coiled-coil protein-binding motif in Fusarium verticillioides Fsr1 is essential for maize stalk rot virulence. Microbiology. 2008;154:1637–45. doi: 10.1099/mic.0.2008/016782-0. [DOI] [PubMed] [Google Scholar]
- Yoruk B, Gillers BS, Chi NC, Scott IC. Ccm3 functions in a manner distinct from Ccm1 and Ccm2 in a zebrafish model of CCM vascular disease. Developmental biology. 2012;362:121–31. doi: 10.1016/j.ydbio.2011.12.006. [DOI] [PubMed] [Google Scholar]
- Zhang H. PDCD10 interacts with STK25 to accelerate cell apoptosis under oxidative stress. Front Biosci. 2012;17:2295–305. doi: 10.2741/4053. [DOI] [PubMed] [Google Scholar]
- Zhang M, Dong L, Shi Z, Jiao S, Zhang Z, Zhang W, et al. Structural mechanism of CCM3 heterodimerization with GCKIII kinases. Structure. 2013;21:680–8. doi: 10.1016/j.str.2013.02.015. [DOI] [PubMed] [Google Scholar]
- Zhao YJ, Jia YH, Guan LK, Wei W, Wang J, Mao KX, et al. Association between abnormal electrocardiographic features and disease severity in patients with arrhythmogenic right ventricular cardiomyopathy. Zhonghua xin xue guan bing za zhi. 2011;39:734–8. [PubMed] [Google Scholar]
- Zheng X, Xu C, Di Lorenzo A, Kleaveland B, Zou Z, Seiler C, et al. CCM3 signaling through sterile 20-like kinases plays an essential role during zebrafish cardiovascular development and cerebral cavernous malformations. The Journal of clinical investigation. 2010;120:2795–804. doi: 10.1172/JCI39679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Chan EK, Li J, Hemmerich P, Tan EM. Transcription activating property of autoantigen SG2NA and modulating effect of WD-40 repeats. Experimental cell research. 2001;269:312–21. doi: 10.1006/excr.2001.5320. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Wu Q, Xu JF, Miller D, Sandalcioglu IE, Zhang JM, et al. Differential angiogenesis function of CCM2 and CCM3 in cerebral cavernous malformations. Neurosurgical focus. 2010;29:E1. doi: 10.3171/2010.5.FOCUS1090. [DOI] [PubMed] [Google Scholar]