

Abstract
Trichloroethylene (TCE) is an organic solvent and common environmental contaminant. TCE exposure is associated with heart defects in humans and animal models. Primary metabolism of TCE in adult rodent models is by specific hepatic cytochrome P450 enzymes (Lash et al., 2000). As association of TCE exposure with cardiac defects is in exposed embryos prior to normal liver development, we investigated metabolism of TCE in the early embryo. Developing chick embryos were dosed in ovo with environmentally relevant doses of TCE (8 ppb and 800 ppb) and RNA was extracted from cardiac and extra-cardiac tissue (whole embryo without heart). Real time PCR showed upregulation of CYP2H1 transcripts in response to TCE exposure in the heart. No detectable cytochrome expression was found in extra-cardiac tissue. As seen previously, the dose response was non-monotonic and 8ppb elicited stronger upregulation than 800 ppb. Immunostaining for CYP2C subfamily expression confirmed protein expression and showed localization in both myocardium and endothelium. TCE exposure increased protein expression in both tissues. These data demonstrate that the earliest embryonic expression of phase I detoxification enzymes is in the developing heart. Expression of these CYPs is likely to be relevant to the susceptibility of the developing heart to environmental teratogens.
Introduction
Trichloroethylene (TCE; TRI; C2HCl3) is an organic solvent used primarily as an industrial degreasing agent but also found in consumer products (Page et al., 2001). Though its use has been phased out for many applications, TCE remains a common environmental contaminant. According to an EPA national groundwater survey, TCE is the most frequently detected organic solvent in groundwater supplies and is estimated to be in up to 34% of the nation’s drinking water supplies (Page et al., 2001). TCE is found in at least 852 of the 1,430 EPA Superfund sites around the country (ATSDR, 2003).
TCE was first linked to altered heart development in an epidemiological study that found an odds ratio of 3 for congenital heart disease in children living in an area of the Tucson Valley with TCE groundwater contamination in the range of 100–270 ppb (Goldberg et al., 1990). Defects found included both myocardial and valvular structures. Although this study was controversial for methodological reasons based upon the appropriateness of the selected controls, an independent reevaluation of the data validated the conclusion that TCE is cardio-teratogenic (Bove et al., 2002). Further, a Wisconsin epidemiological study shows a correlation between the proximity of maternal residence to TCE emitting sites and increased heart defects in their children (Yauck et al., 2004).
The connection between congenital heart disease and TCE has been a controversial issue for many years (see Boyer et al., 2000; Dugard, 2000). A review by Hardin et al. (2005) challenged the idea that TCE is a teratogen based largely upon inconsistent dose response data to various exposure protocols. However, an independent evaluation produced by the National Academy of Sciences (Research Council, 2006) concluded that TCE or one or more of its metabolites could cause cardiac teratogenesis. They suggested that additional studies of lowest-observed adverse effect level and mode of action are necessary (Research Council, 2006). Prevalence of TCE as a soil and water contaminant argues for a better understanding of the consequences of low-level exposures. For yet poorly understood reasons, TCE does not have a consistent dose response curve in the cardiovascular system. Early studies focused on moderate to high levels of exposure. However, recent studies of gene expression in H9C2 cells suggest that there are biphasic curves with maximum effects between 10–100 ppb and again at 10–100 ppm (Caldwell et al., 2008; Selmin et al., 2005). As described by Drake et al. (2006b), concentrations of TCE (8 ppb) close to the EPA maximum contaminant levels (5 ppb) are sufficient to alter cardiac hemodynamics. While non-monotonic dose response curves could reflect issues with metabolism, transport, compartmentalization or a combination of factors, we began to explore this issue by focusing on metabolism. Lash et al. (2000) demonstrated that in the murine model system, as well as in human liver microsomes, trichloroethylene undergoes cytochrome P450 mediated oxidative metabolism. As seen by Lash et al. (2000), the CYP2 family demonstrates both affinity and substrate specificity for TCE. CYP2E1 was implicated as the major TCE metabolizing CYP in both human liver microsomes and the murine model systems, while CYP2E1 is present in low levels in human fetal liver and brain (Brzezinski et al., 1999; Carpenter et al., 1996). We, and others, found that TCE affects cardiovascular development prior to both liver and brain development (Drake et al., 2006b; Makwana et al., 2010; Mishima et al., 2006). If metabolic activation of TCE is relevant to teratogenesis, then the existence and localization of cytochrome P450 enzymes in the early embryo is important to examine.
In this study, avian embryos were injected in ovo (Drake et al., 2006b) with low levels (8 and 800 ppb) of TCE, and cardiac and extra-cardiac tissue was analyzed for expression of avian homologues of TCE-metabolizing cytochromes. The data show that cytochromes 1A4, 2C45 and 2H1 are present in the early embryonic heart and that expression of mRNA and protein for CYP2H1 is increased by TCE exposure. These enzymes are unique to the heart at an early stage of development as little signal is found in the rest of the embryo (extra-cardiac tissue). The expression data recapitulate the non-monotonic dose response seen previously (Caldwell et al., 2008; Drake et al., 2006b; Makwana et al., 2010; Rufer et al., 2010). These data suggest that the developing heart, a target of TCE toxicity, has a unique ability to metabolize TCE at an early stage in development. The localized expression of cytochromes at this early time may be relevant to the frequency of environmentally caused defects found in this organ.
Material and Methods
Trichloroethylene ACS reagent, ≥99.5% (TCE) was obtained from Sigma-Aldrich Co. (St. Louis, MO) Catalog #251402.
Dosing
TCE was dosed at 8 ppb (60 nM) and 800 ppb (6000 nM) through injection into stage Hamburger Hamilton (HH)13 eggs using Hamilton Co. (Reno, NV) 800 Series Syringes (Part #7646-01) paired with custom needles (Part #7806-02) with the following specifications: RN NDL 6/PK (22s/1″/4)L. Methods were as described by Drake et al. (2006b) and Makwana et al. (2010).
Quantitative real-time PCR
After dosing embryos were allowed to develop for approximately 24 hrs until reaching stage HH17. Approximately two to three HH17 whole embryos with hearts removed, defined henceforth as “extra-cardiac tissue,” were pooled for homogenization in Trizol (#15596-018, Invitrogen) and processed for RNA isolation using PureLink™ Micro-to-Midi™ Total RNA Purification System (# 12183-018, Invitrogen). Additionally, this protocol was repeated for pooled samples of approximately twenty Hamburger Hamilton (HH) 17 hearts. Data shown are a representation of three experimental replicates representing a total of 9 extracardiac tissues and 60 hearts per treatment for a 24 hour exposure period. Concentration of cDNA was measured using fluorometry (Turner Biosystems) after staining with Quant-iT™ OliGreenR®ssDNA Assay Kit (# O11492, Molecular Probes) and equal aliquots of control and experimental cDNA samples were added to triplicate reaction mixtures. Real-time PCR reactions were carried out using primers in Table 1, FastStart SYBR Green Master (Roche) and the Rotor Gene 3000 System from Corbett Research. Analysis of the data was carried out using the Rotor Gene 6 software. All real-time PCR results were then normalized to the housekeeping gene GAPDH.
Table 1.
Primers used for detection of avian cytochromes
| Primers | Forward Sequence | Reverse Sequence |
|---|---|---|
| CYP1A4 | 5′-GTCAATGCTCGTTTCAGTGCCT-3′ | 5′-ATCCTCCCCTGTCCTTTTCTCC-3′ |
| CYP2C45 | 5′-GGTTTGTGTTGCTTGCCTGC-3′ | 5′-TTCACCTCCAGTATGTTCCCTACG-3′ |
| CYP2H1 | 5′-TGGCTTGAAAGGCAACCTACG-3′ | 5′-TTGTCTGCTCAGTATGGAGGAAGG-3′ |
| CYP3A37 | 5′–CCTGGAATACCGCAAAGGCTT-3′ | 5′-CCACTGGTGAAGGTTGGAGAGA-′ |
| UGT1A1 | 5′-ACTCAATGTCCCAATCCCCCT-3′ | 5′-TCGGTATGGTCTGTAAATGCCCT-3′ |
| GAPDH | 5′-GTGTGCCAACCCCCAATGTCT-3′ | 5′-CCCATCAGCAGCCTTCA-3′ |
Statistical Analysis
One-way ANOVA was performed using GraphPad Prism version 5.0b for Mac OS X, GraphPad Software, San Diego California USA, www.graphpad.com
Immunofluorescent Microscopy
Embryos were collected in Tyrode’s solution and the thorax region dissected from the embryo. Thoraxes were fixed in a solution of 20°C 80% methanol/20% DMSO and cryosubstituted in 100% ethanol at 20°C for one week. The tissues were then embedded in paraffin and sectioned. After deparaffinization, sections were rinsed in phosphate buffered saline (PBS) for 10 min, blocked for 1 hr at room temperature with a blocking solution containing 1% bovine serum albumin and 0.1% Tween 20 in PBS. Sections containing the heart were processed using indirect immunofluorescence. Sections were incubated overnight with primary antibody (goat anti-CYP2C8/9/18/19 K-21, Santa Cruz Biotechnology, no. sc-23435) at 4°C in a moist chamber at 1:50 dilution. The immunogen used to make this antibody is an epitope from the C-terminus of human CYP2C9. This region is highly conserved with the C-terminus of chick CYP2H1. After several rinses in PBS, Alexa fluor 488 or 546-conjugated rabbit anti-goat secondary antibody (Molecular Probes, Eugene, OR; A-11008, A-11010) was incubated for 1 hr at room temperature in a moist chamber at 1:200 dilution. After rinsing in PBS, the nuclei were stained with TO-PRO-3 or DAPI (Molecular Probes) and the sections mounted using Prolong Gold mounting media (Molecular Probes). Sections were analyzed using a Zeiss 510 Meta confocal microscope or with a Deltavision deconvolution microscope.
Results
Avian Cytochrome P450s
We identified the avian homologues of mouse and human cytochromes associated with TCE metabolism using NCBI homologene (http://www.ncbi.nlm.nih.gov/homologene) (Table 2a) (Lash et al., 2000). The major TCE metabolizing enzyme in adult organisms (mice and rats) and adult human liver microsomes indicated by Lash et al. (2000) was CYP2E1. Other minor enzymes indentified by Lash et al. (2000) were, CYP1A1, CYP2C9 and CYP2B6. While the chicken does not express either CYP2E1 or CYP2B6, it does express CYP2 family members: CYP2H1 and CYP2C45 whose respective human homologues are CYP2C18 and CYP2C9 (Table 2). Of these two CYPs Thum et al. (2000) report CYP2C18 expression in adult human heart. Additionally, the avian homologue of CYP1A1 is CYP1A4 (Table 2a). CYP2H1 was characterized as being closely related to the human CYP2C subfamily (63.4 % DNA Identity with human) (Mattschoss et al., 1986). We designed PCR primers for CYP1A4, CYP2C45 and CYP2H1 (Table 1) and evaluated expression in both cardiac and extra-cardiac tissue during development with or without TCE exposure.
Table 2.
Cytochrome p450 homologues corresponding to TCE metabolizing enzymes identified by Lash et al., 2000. No CYP2E1 or CYP2B6 homologs have been identified in the chick. CYP2C18 was identified in the human heart its avian homolog is CYP2H1
| P450 | Human | Murine | Chick (% DNA Identity) |
|---|---|---|---|
| Major | CYP2E1 | CYP2E1 | - |
| Minor | CYP1A1 | CYP1A1 | CYP1A4 (66.9) |
| Minor | CYP2C9 | CYP2C | CYP2C45 (61.4) |
| Minor | CYP2B6 | CYP2B2 | - |
|
| |||
| Cardiac | CYP2C18 | CYP2C80 | CYP2H1 (63.4) |
CYP Gene Expression after TCE Exposure
The developing heart of the embryo is seen as a looped structure on the anterior thorax during the organogenesis stage of the development. While the heart initially forms as a linear heart tube, developmental movements during morphogenesis produce a loop with concomitant internal processes of septation to produce a 4 chambered heart. Previous studies in animal models have shown that the window of sensitivity to TCE in chick and mouse models coincides with looping and septation (Boyer et al., 2000; Collier et al., 2003; Drake et al., 2006b; Johnson et al., 2003; Makwana et al., 2010; Mishima et al., 2006; Rufer et al., 2010). In the interval between HH13 (exposure) and HH17 (collection), the heart is comprised of endothelial and myocardial cell layers with an intervening extracellular matrix. At HH17, some mesenchymal cells of endothelial origin arise in the atrioventricular (AV) canal between the single atrial and ventricular chambers (Boyer et al., 2000). These cells are the earliest precursors of the mitral and tricuspid valves. Embryos were collected at this stage of development for the analyses described here.
As the major cytochrome for TCE in mouse and humans is not expressed in the chick, we examined the minor cytochromes. CYP1A4 was expressed in isolated chick heart myocytes after TCDD exposure (Gannon et al., 2000). Here, real-time PCR was used to examine CYP1A4 expression after TCE exposure in ovo. Incubated avian embryos were exposed to TCE by injection into the yolk at 48 hrs of development (HH Stage 13), and embryos were allowed to develop for another 24 hrs (to reach HH 17) at which time extra-cardiac tissue and heart tissue was extracted for RNA isolation. Real-time PCR data were normalized to GAPDH expression within that tissue. We verified that GAPDH expression was not sensitive to TCE exposure by evaluating GAPDH levels in equal aliquots of cDNA from treated and untreated samples. Results shown in Fig. 1 (A&B) illustrate that CYP1A4 was detectable but does not demonstrate significantly altered expression at 8 or 800 ppb TCE exposure in either extra-cardiac or heart tissue.
Figure 1.

Real-time PCR measurement of CYP expression. Data are normalized to GAPDH and PBS injection into the yolk. A. CYP1A4 expression in HH17 hearts. B. CYP1A4 expression in HH17 extra-cardiac tissue. CYP1A4 expression was not significantly altered relative to total RNA (GAPDH) in either HH17 hearts or extra-cardiac tissue at PBS, 8 or 800 ppb TCE exposure. C. CYP2C45 expression in HH17 hearts. D. CYP2C45 expression in HH17 extra-cardiac tissue. CYP2C45 expression was not significantly altered relative to total RNA (GAPDH) in either HH17 hearts or extra-cardiac tissue after PBS, 8 or 800 ppb TCE exposure. E. CYP2H1 expression in HH17 hearts. F. CYP2H1 expression in HH17 extra-cardiac tissue. CYP2H1 expression was significantly increased relative to total RNA (GAPDH) in HH17 hearts after 8 ppb TCE exposure (* = p-value < 0.0001, One-way ANOVA), while PBS and 800 ppb TCE exposure showed no significant change in expression. Real-time PCR results for CYP2H1 showed no significant change in expression at PBS, 8 or 800 ppb TCE exposure.
Lash et al. (2000) identified an additional minor cytochrome involved in TCE metabolism in humans and mice, CYP2C9. The avian homologue of this enzyme is CYP2C45 as identified in NCBI’s homologene database (Table 2a). CYP2C45 expression was also examined by real-time PCR in HH17 extra-cardiac and heart tissue (Fig 1C&D). Results show that although detected by PCR, CYP2C45 expression is not significantly altered after TCE exposure in either cardiac or extra-cardiac tissue at either the 8 ppb or 800 ppb TCE doses.
In the course of our study, we identified an additional CYP2C subfamily member, CYP2C18, whose expression after TCE exposure was not addressed in Lash et al. (2000). CYP2C18 expression was previously identified in human heart tissue (Thum and Borlak, 2000). The avian homologue of this enzyme is CYP2H1 as identified in NCBI’s homologene database (Table 2b). In HH17 heart tissue CYP2H1 demonstrated significantly increased expression at 8 ppb TCE exposure (One-way ANOVA: p < 0.0001), while after 800 ppb TCE exposure there was no significant change in expression. These data demonstrate that 8 ppb TCE is sufficient to produce a significant increase in the expression of CYP2H1 in the developing chick heart. Although the liver bud can be identified by marker expression as early as stage HH17, cellular organization begins around stage HH25 and albumin expression (as a measure of function) does not begin until stage HH30 (Yanai et al., 2005). Thus, the liver is insufficiently developed at the stage of collection (HH17) to function as a site of detoxification. This is confirmed by the lack of response in the extra-cardiac tissue sample. Significant upregulation of expression in the heart suggest this organ to be a primary site of phase I enzyme expression in the early embryo.
CYP2C Subfamily Protein Localization
The results above show that a cytochrome P450 enzyme transcript associated with TCE metabolism is expressed in the embryonic heart but not in the rest of the embryo (extra-cardiac tissue). CYP2H1 is significantly induced by 8ppb TCE exposure in the developing heart. To confirm protein expression in heart tissue and to identify the cells where expression takes place, we undertook an immunostaining procedure on embryonic chick tissue. As we were unable to obtain cytochrome-specific antibodies for the chick, we utilized a cytochrome 2C subfamily specific antibody and verified its ability to work in embryonic chick heart cell lysate by western blot (data not shown). CYP2H1 is an avian member of the CYP2C subfamily (Table 2a) (Mattschoss et al., 1986). Embryos were injected with TCE at stage 13 as in the previous experiments and were collected and fixed at stage 17. Sections were stained with anti-CYP2C antibody and photographed. More than 3 exposed and control thoraxes were examined. Cytochrome 2C staining in control sections was seen primarily in the myocardium without regard to regional specificity (Fig. 2A & C). TCE exposure (8ppb) showed an enhanced signal in the myocardium and endothelium compared to controls (compare 2A & B and 2C & D. Expression in the endothelium was somewhat variable. However, a sagittal section through a TCE-exposed cardiac cushion (valve forming tissue) in the developing AV canal shows a dramatic expression of CYP2C protein in the endothelial cells most proximal to the atrium (Fig. 2E). This is an area of constriction in the heart where blood flows from the atrium, through the AV canal to the single ventricle. This constriction that produces high rates of blood flow and shows endothelial cell proximity to the blood (Makwana et al., 2010). Endothelial expression appears to vary in the endothelium based upon regional or, perhaps, physiological parameters.
Figure 2.
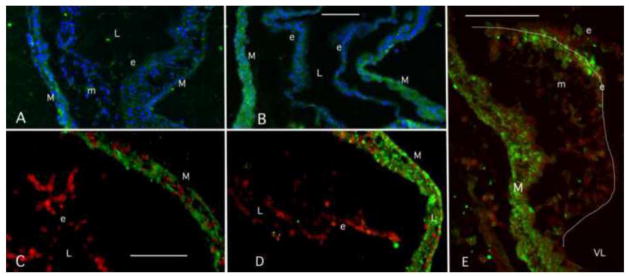
CYP2C Localization in Embryonic hearts. Representative staining is shown in two control and treatment pairs (A, B & C, D) from separate experiments. Each pair was treated together and examined with the same microscope settings. CYP2C antibody expression is observed primarily myocardium of the developing chick heart in control and 8 ppb TCE-treated samples. Some endothelial staining was seen in one control (A) while little was seen in the other (C). The extracellular matrix lies between the two cell layers. TCE-exposed embryos showed an increase in myocardial staining that corresponds with the PCR data. Increases in endothelial staining were variable and reflect region differences. Small amounts of staining are seen in mesenchymal cells. A sagittal section of a cardiac cushion shows strong staining on the atrial (inflow) side of the cushion where flow becomes constricted (E). Green= CYP2C, Blue is DAPI nuclear stain, Red is Topo3 nuclear stain. Outline of the cardiac cushion is indicated by the white line in E. M=myocardium, e = endothelium, m= mesenchyme (formed from endothelium), L= lumen, VL= ventricular lumen. Scale marker indicates 75 micrometers and the scale is the same in each matched pair.
Discussion
TCE exposure, during development, demonstrates cardiac specific effects for poorly understood reasons. Primary defects associated with human exposures include muscular ventricular septal defects, atrial septal defects, membranous ventricular septal defects and pulmonary stenosis (Yauck et al., 2004). Similarly, animal studies show almost identical cardiac defects after TCE exposure during development. In the avian model system, Loeber et al. (1988) observed atrial and ventricular septal defects and Drake et al. (2006a) observed altered valvuloseptal formation. Rufer et al. (2010) were able to observe the formation of cardiac defects in avian embryos post-hatch. In the murine system, Johnson et al. (2003) observed both valvuloseptal and myocardial defects after delivery of high doses (1100 ppm) of TCE in maternal drinking water. Their data show that TCE exposure during the organogenesis stage of development was critical.
A molecular survey in rat embryos exposed to TCE at 100 ppm in maternal drinking water identified Serca2a as transcriptionally downregulated (Collier et al., 2003). Further studies in mice and in a rat cardiomyocyte cell line highlighted transcriptional regulation of Serca2a and additional mediators of Ca2+ homeostasis by TCE exposure by doses as low as 10 ppb (Caldwell et al., 2010; Selmin et al., 2008). These results were confirmed in the H2C9 murine cardiomyocyte cell line by Caldwell et al. (2008) which show alterations in expression of the Ca2+ pumps, Ryr2 and Serca2a, after low dose TCE exposure with concomitant altered Ca2+ handling. Impaired cardiac output is known to be a mechanism for defective heart development (Groenendijk et al., 2005; Hogers et al., 1997). Reduced calcium fluxes result in reduced cardiac output and thus TCE can produce cardiac defects through altered cardiac function. This was confirmed in recent observations that markers of flow within the avian heart (KLF2 and NOS-3) were reduced after TCE exposure in ovo. Analysis of isolated myocytes from these embryos show TCE to have a persistent effect on myocardial contraction (Makwana et al., 2010).
Thus, we and others have shown that TCE is a specific teratogen in the embryonic heart but there is an unexplained aspect to the dose response. Epithelial mesenchymal transition in vitro is inhibited at 50–250 ppb (Boyer et al., 2000). Cushion (valve progenitor) proliferation is stimulated at 80 ppb (Mishima et al., 2006) and Caldwell et al. (2008) noted a biphasic response curve within a cardiomyocyte cell line. Our published work (Makwana et al., 2010) and that of Drake et al. (2006b) show that blood flow is more sensitive to 8 ppb than 800 ppb. However, Rufer et al. (2010) found that 400ppb TCE produced more embryo loss than 8 ppb exposure. This suggests that TCE may affect different developmental processes at different exposures or that issues of compartmentalization or metabolism may play a role in the non-monotonic dose response curve.
Cytochrome P450 expression in the early embryo is not well studied. Previous studies of these enzymes in the embryo focus almost entirely on the role of cytochromes in endogenous retinoic acid metabolism or in response to dioxin exposure. CYP26 family members are expressed in the head region of the chick embryo as early as stage 4 and are postulated to be involved in creating morphogenetic gradients involved in patterning (Reijntjes et al., 2004). Mouse embryos null for cytochrome P450 reductase, an electron donor to cytochromes die at E9.5 and show defects in vasculogenesis. The observed abnormalities appear to coincide strongly with known defects from disruption of retinoic acid homeostasis (Otto et al., 2003). Dioxin (TCDD) is a teratogen that produces a number of defects including malformation of the heart. TCDD exposure induces CYP1A expression in the endothelium of the zebrafish heart (Mizell and Romig, 1997). CYP1A4 is upregulated in isolated embryonic chick cardiomyocytes after dioxin exposure (Jones and Kennedy, 2009). While these data show that Cytochrome P450s are present in the embryo they do not provide a context for more widespread xenobiotic metabolism.
The data described here demonstrate that CYP2H1 is significantly upregulated in the heart after TCE exposure and that CYP2C45 is present but un-induced. CYP2H1 upregulation precedes the development of the liver and is consistent with suggestions that the heart is uniquely sensitive to TCE exposure. The early expression of these CYPs as well as CYP1A enzymes is consistent with general observations that environmental xenobiotic exposure can lead to heart defects. Localization of expression by antibody suggests that there is both myocardial expression and endothelial expression at a stage when these are the two major cell types in the heart. While both cell types appear to upregulate expression, the strong expression seen in the endothelial cells of the atrioventricular canal is interesting as these are precursors of the heart valves. Additionally, these endothelial cells are arranged in the most constricted part of the developing heart where they may serve a surveillance function. Of course, the avian embryo develops in ovo and the expression of phase I enzymes in the heart could be specific to non-placental animals as maternal protection is not available. Though we have not formally explored this expression in mammals, we note that microarray data from the hearts of TCE-exposed murine embryos show upregulation of numerous cytochromes (Caldwell et al., 2010).
The regional expression of CYP2H1 in the heart suggests the possibility that the basis for the TCE dose response curve lies directly in this tissue. It was found that an oxidative metabolite of TCE, trichloracetic acid (TCA) was more toxic than TCE itself in a study of heart defects (Johnson et al., 1998). Oxidation of TCE by cytochrome enzymes can lead to TCA production (Larson and Bull, 1992; Templin et al., 1995). Lumpkin et al. (2003) demonstrate that TCA was able to bind to plasma proteins in mice, rats and humans resulting in an increased half-life of TCA in the bloodstream. This regionally specific metabolism of TCE in the developing heart may imply that the heart is a site of bioaccumulation of the potential oxidative metabolites of TCE: TCA (trichloroacetic acid) and/or DCA (dichloroacetic acid). If these metabolites bind to proteins in the extracellular matrix (ECM) as well as the blood, they may be retained in the heart where they can act on myocardial function. Several investigators have noted that TCE metabolism can be destructive to the very cytochrome P450s catalyzing the metabolic reaction (Ensley, 1991; Miller and Guengerich, 1982; 1983). The unusual dose response curve may be due to a combination of production of toxic metabolites and the synthesis or destruction of CYPs. The heart may be susceptible at low doses because it contains competent CYPs to metabolize TCE into TCA. However, at higher doses, CYPs are degraded faster than they can be synthesized and less formation of TCA occurs. This suggests that cardio-specific toxicity is most potent prior to the onset of liver development as TCA in the heart would be reduced if TCE is metabolised elsewhere. This suggestion is consistent with the window of toxicity noted by Rufer et al. (2010).
In summary, our findings demonstrate that the early heart is a site of phase I enzyme expression in the early embryo. We show that TCE is sufficient to induce upregulation of CYP2H1 and the dose sensitivity of the response recapitulates the non-monotonic response to TCE seen by a number of laboratories. This suggests that CYP2H1 is capable of metabolizing TCE and that local metabolism can mediate the effects of TCE on cardiac teratogenesis. While the basis for the non-monotonic dose response curve remains unclear, there are a variety of molecular and physiological markers that confirm this response. Future studies will to examine transcriptional regulation of perturbed markers in order to resolve a global cell signaling effect of localized cardiac specific metabolism of TCE.
The data uniquely localizing cytochrome expression to the embryonic heart, as well as its induction by TCE exposure, suggest the idea that the developing heart is a unique site of xenobiotic metabolism in the early embryo. Such metabolism could be an important component of the particular sensitivity of the heart to developmental teratogens.
Acknowledgments
Funding
This work was supported by the American Heart Association [0810020Z to O.M.] and the National Institutes of Health [HL82851 to R.B.R. and ES00694 to the Southwest Environmental Health Sciences Center (SWEHSC)].
We would like to thank Dr. André L. Tavares for his guidance and support.
Bibliography
- ATSDR. Agency for Toxic Substances and Disease Registry (ATSDR) Toxicological Profile for Trichloroethylene (Update) Atlanta, GA: U.S. Department of Health and Human Services, U.S. Public Health Service; 2003. pp. 1–2. Jul 28, 2003 ed. [Google Scholar]
- Bove F, Shim Y, Zeitz P. Drinking water contaminants and adverse pregnancy outcomes: a review. Environmental health perspectives. 2002;110(Suppl 1):61–74. doi: 10.1289/ehp.02110s161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer AS, Finch WT, Runyan RB. Trichloroethylene inhibits development of embryonic heart valve precursors in vitro. Toxicological sciences : an official journal of the Society of Toxicology. 2000;53(1):109–117. doi: 10.1093/toxsci/53.1.109. [DOI] [PubMed] [Google Scholar]
- Brzezinski MR, Boutelet-Bochan H, Person RE, Fantel AG, Juchau MR. Catalytic activity and quantitation of cytochrome P-450 2E1 in prenatal human brain. The Journal of pharmacology and experimental therapeutics. 1999;289(3):1648–1653. [PubMed] [Google Scholar]
- Caldwell PT, Manziello A, Howard J, Palbykin B, Runyan RB, Selmin O. Gene expression profiling in the fetal cardiac tissue after folate and low-dose trichloroethylene exposure. Birth defects research Part A, Clinical and molecular teratology. 2010;88(2):111–127. doi: 10.1002/bdra.20631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell PT, Thorne PA, Johnson PD, Boitano S, Runyan RB, Selmin O. Trichloroethylene disrupts cardiac gene expression and calcium homeostasis in rat myocytes. Toxicological sciences : an official journal of the Society of Toxicology. 2008;104(1):135–143. doi: 10.1093/toxsci/kfn078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter SP, Lasker JM, Raucy JL. Expression, induction, and catalytic activity of the ethanol-inducible cytochrome P450 (CYP2E1) in human fetal liver and hepatocytes. Molecular pharmacology. 1996;49(2):260–268. [PubMed] [Google Scholar]
- Collier JM, Selmin O, Johnson PD, Runyan RB. Trichloroethylene effects on gene expression during cardiac development. Birth defects research Part A, Clinical and molecular teratology. 2003;67(7):488–495. doi: 10.1002/bdra.10073. [DOI] [PubMed] [Google Scholar]
- Drake VJ, Koprowski SL, Hu N, Smith SM, Lough J. Cardiogenic effects of trichloroethylene and trichloroacetic acid following exposure during heart specification of avian development. Toxicological sciences : an official journal of the Society of Toxicology. 2006a;94(1):153–162. doi: 10.1093/toxsci/kfl083. [DOI] [PubMed] [Google Scholar]
- Drake VJ, Koprowski SL, Lough J, Hu N, Smith SM. Trichloroethylene exposure during cardiac valvuloseptal morphogenesis alters cushion formation and cardiac hemodynamics in the avian embryo. Environmental health perspectives. 2006b;114(6):842–847. doi: 10.1289/ehp.8781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugard PH. Effects of trichloroethylene (TCE) on an in vitro chick atrioventricular canal culture. Toxicological sciences : an official journal of the Society of Toxicology. 2000;56(2):437–438. doi: 10.1093/toxsci/56.2.437. [DOI] [PubMed] [Google Scholar]
- Ensley BD. Biochemical diversity of trichloroethylene metabolism. Annual review of microbiology. 1991;45:283–299. doi: 10.1146/annurev.mi.45.100191.001435. [DOI] [PubMed] [Google Scholar]
- Gannon M, Gilday D, Rifkind AB. TCDD induces CYP1A4 and CYP1A5 in chick liver and kidney and only CYP1A4, an enzyme lacking arachidonic acid epoxygenase activity, in myocardium and vascular endothelium. Toxicology and applied pharmacology. 2000;164(1):24–37. doi: 10.1006/taap.1999.8864. [DOI] [PubMed] [Google Scholar]
- Goldberg SJ, Lebowitz MD, Graver EJ, Hicks S. An association of human congenital cardiac malformations and drinking water contaminants. Journal of the American College of Cardiology. 1990;16(1):155–164. doi: 10.1016/0735-1097(90)90473-3. [DOI] [PubMed] [Google Scholar]
- Groenendijk BC, Hierck BP, Vrolijk J, Baiker M, Pourquie MJ, Gittenberger-De Groot AC, Poelmann RE. Changes in shear stress-related gene expression after experimentally altered venous return in the chicken embryo. Circulation research. 2005;96(12):1291–1298. doi: 10.1161/01.RES.0000171901.40952.0d. [DOI] [PubMed] [Google Scholar]
- Hardin BD, Kelman BJ, Brent RL. Trichloroethylene and dichloroethylene: a critical review of teratogenicity. Birth defects research Part A, Clinical and molecular teratology. 2005;73(12):931–955. doi: 10.1002/bdra.20192. [DOI] [PubMed] [Google Scholar]
- Hogers B, DeRuiter MC, Gittenberger-De Groot AC, Poelmann RE. Unilateral vitelline vein ligation alters intracardiac blood flow patterns and morphogenesis in the chick embryo. Circulation research. 1997;80(4):473–481. doi: 10.1161/01.res.80.4.473. [DOI] [PubMed] [Google Scholar]
- Jelinek D, Patrick SM, Kitt KN, Chan T, Francis GA, Garver WS. Physiological and coordinate downregulation of the NPC1 and NPC2 genes are associated with the sequestration of LDL-derived cholesterol within endocytic compartments. Journal of cellular biochemistry. 2009;108(5):1102–1116. doi: 10.1002/jcb.22339. [DOI] [PubMed] [Google Scholar]
- Johnson PD, Dawson BV, Goldberg SJ. Cardiac teratogenicity of trichloroethylene metabolites. Journal of the American College of Cardiology. 1998;32(2):540–545. doi: 10.1016/s0735-1097(98)00232-0. [DOI] [PubMed] [Google Scholar]
- Johnson PD, Goldberg SJ, Mays MZ, Dawson BV. Threshold of trichloroethylene contamination in maternal drinking waters affecting fetal heart development in the rat. Environmental health perspectives. 2003;111(3):289–292. doi: 10.1289/ehp.5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SP, Kennedy SW. Chicken embryo cardiomyocyte cultures--a new approach for studying effects of halogenated aromatic hydrocarbons in the avian heart. Toxicological sciences : an official journal of the Society of Toxicology. 2009;109(1):66–74. doi: 10.1093/toxsci/kfp039. [DOI] [PubMed] [Google Scholar]
- Larson JL, Bull RJ. Species differences in the metabolism of trichloroethylene to the carcinogenic metabolites trichloroacetate and dichloroacetate. Toxicology and applied pharmacology. 1992;115(2):278–285. doi: 10.1016/0041-008x(92)90333-n. [DOI] [PubMed] [Google Scholar]
- Lash LH, Fisher JW, Lipscomb JC, Parker JC. Metabolism of trichloroethylene. Environmental health perspectives. 2000;108(Suppl 2):177–200. doi: 10.1289/ehp.00108s2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeber CP, Hendrix MJ, Diez De Pinos S, Goldberg SJ. Trichloroethylene: a cardiac teratogen in developing chick embryos. Pediatric research. 1988;24(6):740–744. doi: 10.1203/00006450-198812000-00018. [DOI] [PubMed] [Google Scholar]
- Lumpkin MH, Bruckner JV, Campbell JL, Dallas CE, White CA, Fisher JW. Plasma binding of trichloroacetic acid in mice, rats, and humans under cancer bioassay and environmental exposure conditions. Drug metabolism and disposition: the biological fate of chemicals. 2003;31(10):1203–1207. doi: 10.1124/dmd.31.10.1203. [DOI] [PubMed] [Google Scholar]
- Makwana O, King NM, Ahles L, Selmin O, Granzier H, Runyan RB. Exposure to low-dose trichloroethylene alters shear stress gene expression and function in the developing chick heart. Cardiovascular Toxicology. 2010;10(2):100–107. doi: 10.1007/s12012-010-9066-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattschoss LA, Hobbs AA, Steggles AW, May BK, Elliott WH. Isolation and characterization of genomic clones for two chicken phenobarbital-inducible cytochrome P-450 genes. The Journal of biological chemistry. 1986;261(20):9438–9443. [PubMed] [Google Scholar]
- Miller RE, Guengerich FP. Oxidation of trichloroethylene by liver microsomal cytochrome P-450: evidence for chlorine migration in a transition state not involving trichloroethylene oxide. Biochemistry. 1982;21(5):1090–1097. doi: 10.1021/bi00534a041. [DOI] [PubMed] [Google Scholar]
- Miller RE, Guengerich FP. Metabolism of trichloroethylene in isolated hepatocytes, microsomes, and reconstituted enzyme systems containing cytochrome P-450. Cancer research. 1983;43(3):1145–1152. [PubMed] [Google Scholar]
- Mishima N, Hoffman S, Hill EG, Krug EL. Chick embryos exposed to trichloroethylene in an ex ovo culture model show selective defects in early endocardial cushion tissue formation. Birth defects research Part A, Clinical and molecular teratology. 2006;76(7):517–527. doi: 10.1002/bdra.20283. [DOI] [PubMed] [Google Scholar]
- Mizell M, Romig ES. The aquatic vertebrate embryo as a sentinel for toxins: zebrafish embryo dechorionation and perivitelline space microinjection. Int J Dev Biol. 1997;41(2):411–423. [PubMed] [Google Scholar]
- Otto DME, Henderson CJ, Carrie D, Davey M, Gundersen TE, Blomhoff R, Adams RH, Tickle C, Wolf CR. Identification of novel roles of the cytochrome p450 system in early embryogenesis: effects on vasculogenesis and retinoic Acid homeostasis. Mol Cell Biol. 2003;23(17):6103–6116. doi: 10.1128/MCB.23.17.6103-6116.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page C, Check I, Found W, Pathways E, Fate B, Effects P, Instructions P, Home C, Profiles I, Learning I. Agency for Toxic Substances and Disease Registry Case Studies in Environmental Medicine (CSEM) Trichloroethylene Toxicity. 2001. [Google Scholar]
- Reijntjes S, Gale E, Maden M. Generating gradients of retinoic acid in the chick embryo: Cyp26C1 expression and a comparative analysis of the Cyp26 enzymes. Dev Dyn. 2004;230(3):509–517. doi: 10.1002/dvdy.20025. [DOI] [PubMed] [Google Scholar]
- Research Council N. Assessing the Human Health Risks of Trichloroethylene: Key Scientific Issues. The National Academies Press; 2006. p. 425. [Google Scholar]
- Rufer ES, Hacker TA, Flentke GR, Drake VJ, Brody MJ, Lough J, Smith SM. Altered cardiac function and ventricular septal defect in avian embryos exposed to low-dose trichloroethylene. Toxicological sciences : an official journal of the Society of Toxicology. 2010;113(2):444–452. doi: 10.1093/toxsci/kfp269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmin O, Thorne PA, Caldwell PT, Johnson PD, Runyan RB. Effects of trichloroethylene and its metabolite trichloroacetic acid on the expression of vimentin in the rat H9c2 cell line. Cell biology and toxicology. 2005;21(2):83–95. doi: 10.1007/s10565-005-0124-3. [DOI] [PubMed] [Google Scholar]
- Selmin O, Thorne PA, Caldwell PT, Taylor M. Trichloroethylene and trichloroacetic acid regulate calcium signaling pathways in murine embryonal carcinoma cells p19. Cardiovascular Toxicology. 2008;8(2):47–56. doi: 10.1007/s12012-008-9014-2. [DOI] [PubMed] [Google Scholar]
- Templin MV, Stevens DK, Stenner RD, Bonate PL, Tuman D, Bull RJ. Factors affecting species differences in the kinetics of metabolites of trichloroethylene. Journal of toxicology and environmental health. 1995;44(4):435–447. doi: 10.1080/15287399509531972. [DOI] [PubMed] [Google Scholar]
- Thum T, Borlak J. Gene expression in distinct regions of the heart. Lancet. 2000;355(9208):979–983. doi: 10.1016/S0140-6736(00)99016-0. [DOI] [PubMed] [Google Scholar]
- Yanai M, Tatsumi N, Endo F, Yokouchi Y. Analysis of gene expression patterns in the developing chick liver. Developmental dynamics : an official publication of the American Association of Anatomists. 2005;233(3):1116–1122. doi: 10.1002/dvdy.20413. [DOI] [PubMed] [Google Scholar]
- Yauck JS, Malloy ME, Blair K, Simpson PM, McCarver DG. Proximity of residence to trichloroethylene-emitting sites and increased risk of offspring congenital heart defects among older women. Birth defects research Part A, Clinical and molecular teratology. 2004;70(10):808–814. doi: 10.1002/bdra.20060. [DOI] [PubMed] [Google Scholar]