

Abstract
Purpose
The strong dose-dependent association between anthracyclines and cardiomyopathy is further exacerbated by the co-occurrence of cardiovascular risk factors (diabetes and hypertension). The high morbidity associated with cardiomyopathy necessitates an understanding of the underlying pathogenesis so that targeted interventions can be developed.
Patients and Methods
By using a two-stage design, we investigated host susceptibility to anthracycline-related cardiomyopathy by using the ITMAT/Broad CARe cardiovascular single nucleotide polymorphism (SNP) array to profile common SNPs in 2,100 genes considered relevant to de novo cardiovascular disease.
Results
By using a matched case-control design (93 cases, 194 controls), we identified a common SNP, rs2232228, in the hyaluronan synthase 3 (HAS3) gene that exerts a modifying effect on anthracycline dose-dependent cardiomyopathy risk (P = 5.3 × 10−7). Among individuals with rs2232228 GG genotype, cardiomyopathy was infrequent and not dose related. However, in individuals exposed to high-dose (> 250 mg/m2) anthracyclines, the rs2232228 AA genotype conferred an 8.9-fold (95% CI, 2.1- to 37.5-fold; P = .003) increased cardiomyopathy risk compared with the GG genotype. This gene-environment interaction was successfully replicated in an independent set of 76 patients with anthracycline-related cardiomyopathy. Relative HAS3 mRNA levels measured in healthy hearts tended to be lower among individuals with AA compared with GA genotypes (P = .09).
Conclusion
Hyaluronan (HA) produced by HAS3 is a ubiquitous component of the extracellular matrix and plays an active role in tissue remodeling. In addition, HA is known to reduce reactive oxygen species (ROS) –induced cardiac injury. The high cardiomyopathy risk associated with AA genotype could be due to inadequate remodeling and/or inadequate protection of the heart from ROS-mediated injury on high anthracycline exposure.
INTRODUCTION
Anthracyclines are one of the most effective classes of chemotherapeutic agents currently available for cancer diagnosed across the age spectrum. The therapeutic potential of anthracyclines, however, is limited because of their strong dose-dependent relation with progressive and irreversible cardiomyopathy leading to congestive heart failure.1–3 This dose-dependent risk is modified by the coexistence of cardiovascular disease risk factors such as hypertension and diabetes.4,5 An interindividual variability in cardiomyopathy risk has been observed, such that cumulative anthracycline exposure as low as 150 mg/m2 results in cardiomyopathy in some patients, although exposure as high as 1,000 mg/m2 is tolerated without cardiomyopathy by others.6
This study aimed to investigate reason(s) for observed interindividual variability by identifying single nucleotide polymorphisms (SNPs) that might modify the association between anthracycline exposure and risk of cardiotoxicity. Specifically, we used the ITMAT/Broad CARe (IBC) cardiovascular SNP array, which profiles SNPs in 2,100 genes considered relevant for cardiovascular disease in the general population,7 to identify interaction of SNPs known to increase the risk of cardiovascular disease in the general population with anthracycline exposure. We used a two-stage design; SNPs surpassing a prespecified threshold for statistical significance in the discovery stage were validated in the replication stage by using an independent set of cases with anthracycline-related cardiomyopathy. Functional significance of validated SNPs was evaluated by measuring gene expression levels in heart samples procured from the National Disease Research Interchange (Philadelphia, PA).
PATIENTS AND METHODS
Study Participants
Discovery set.
The discovery set was drawn from a Children's Oncology Group study (COG-ALTE03N1) that aims to understand the pathogenesis of cardiomyopathy in childhood cancer survivors. COG member institutions (Appendix Table A1, online only) enrolled patients after obtaining approval from local institutional review boards. Written informed consent/assent was obtained from patients/parents/legal guardians. Cases and controls were identified from individuals diagnosed with cancer at age 21 years or younger. Cases consisted of individuals who developed cardiomyopathy and were alive at study participation. For each case, one to four controls were randomly selected from the same COG childhood cancer survivor cohort by using the following matching criteria: cancer diagnosis, year of diagnosis (± 5 years), race/ethnicity, and duration of cardiomyopathy-free follow-up for controls to exceed time from cancer diagnosis to cardiomyopathy for the corresponding case. In all, 401 individuals (130 cases; 271 controls) participated in this study. All participants provided a biologic specimen (blood, 89%; buccal cells/saliva, 11%) for DNA.
Anthracycline-exposed participants had normal cardiac function before anthracycline exposure. Cases fulfilled American Heart Association criteria for cardiac compromise by presenting with symptoms (dyspnea, orthopnea, and/or fatigue) and/or signs (edema, hepatomegaly, and/or rales) of cardiac compromise, and echocardiographic evidence of left ventricular dysfunction, or, in the absence of symptoms/signs, had echocardiographic features of left ventricular dysfunction (ejection fraction [EF] ≤ 40% and/or fractional shortening [FS] ≤ 28%; Data Supplement).
Replication set.
An independent set of 76 patients (all ages and all racial/ethnic backgrounds) diagnosed with cardiomyopathy after anthracycline exposure were drawn from a single institution (Data Supplement).5 The study was approved by the institutional review board, and written informed consent/assent was obtained. Eligible cases met American Heart Association criteria and echocardiographic cutoffs for cardiac compromise.
Therapeutic Exposures
Lifetime anthracycline exposure was calculated by multiplying the cumulative dose (in milligrams per meter squared) of individual anthracyclines by a factor that reflects the drug's cardiotoxic potential (Data Supplement)8 and then summing the results. Total dose of radiation with heart in field (“chest radiation”) was computed in Gy.
Genotyping and Quality Control
Discovery set.
Genomic DNA was isolated from peripheral blood (QIAamp/Qiagen kits; Valencia, CA) and buccal cells/saliva (Puregene/Oragene kits; Minneapolis, MN). Genotyping was performed on the Illumina IBC cardiovascular SNP array (San Diego, CA) by the Center for Applied Genomics at The Children's Hospital of Philadelphia. The IBC cardiovascular SNP array7 uses a “cosmopolitan” approach to determine tagging SNPs for loci of interest to cover genetic diversity in populations of different ancestry.9
Quality control for genotype data was performed with PLINK.10 Of the 401 study participants, 399 (99.5%; 129 cases, 270 controls) met call rates of more than 95%. No duplicated samples or sample contamination was identified. The multidimensional scaling method10 was used to cluster individuals in the discovery set into non-Hispanic whites and “others.” To control for potential population stratification, 112 individuals in the other category were filtered out, retaining 287 non-Hispanic whites (93 cases, 194 controls) in the discovery stage analysis. After adjusting for the overall genomic control inflation factor (λ = 1.23), type I error appeared to be under control (Data Supplement). The total genotyping rate exceeded 98.9%. Of the 43,293 autosomal SNPs in the IBC SNP array, 988 that failed a missingness threshold for missing fraction more than 0.05 and 7,267 that failed a frequency threshold for a minor allele frequency less than 0.01 were removed. A check for Hardy-Weinberg equilibrium resulted in exclusion of 108 SNPs with P value less than .000001. The final data set retained 34,912 (81%) autosomal SNPs.
Replication set.
Genomic DNA was extracted from peripheral blood and buccal cells/saliva by using Qiagen kits (Valencia, CA). Genomic DNA from formalin-fixed paraffin-embedded bone marrow biopsies or unstained bone marrow slides was extracted by using QuickExtract formalin-fixed paraffin-embedded DNA Extraction Solution (Epicentre Biotechnologies, Madison, WI). Significant SNP(s) identified in the discovery stage were genotyped by using Sequenom iPLEX SNP chemistry on a MassARRAY system (San Diego, CA; call rate, 93.4%).
Gene Expression Analysis
Gene expression analysis was performed on heart samples procured from the National Disease Research Interchange (Philadelphia, PA). Tissue procurement, processing, genotyping, and gene expression analyses are detailed in the Data Supplement. Significant SNPs validated in the replication stage were genotyped with TaqMan genotyping assays (Applied Biosystems, Carlsbad, CA). Total RNA from heart tissue samples was reverse transcribed and amplified by using one-step QuantiTect SYBR Green reverse transcriptase polymerase chain reaction kits (Qiagen, Valencia, CA). Relative hyaluronan synthase 3 (HAS3) mRNA levels were obtained after normalization to reference gene (ACTB).11
Statistical Analyses
Discovery stage.
Discovery stage was designed as a genome-wide association study to examine the main effects of SNPs and gene-environment (anthracycline) interactions. Conditional logistic regression techniques (model 1; equation 1) were used in R (http://www.r-project.org/) for each SNP that passed quality control, minor allele frequency, and Hardy-Weinberg equilibrium filters.
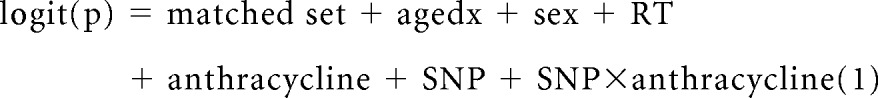 |
where p is the probability of cardiomyopathy in a patient conditional on matched set, agedx is the age at diagnosis of primary cancer (continuous variable), sex (male/female) of participants, RT is the chest radiation dose (continuous variable), anthracycline is the cumulative anthracycline dose in milligrams per meter squared (continuous variable), and SNP is the genotype for each SNP in additive coding.
The repeated sliding-window procedure of Purcell et al10 estimated 10,000 independent tests, taking into consideration linkage disequilibrium (LD) among SNPs. This allowed a P value less than 5 × 10−6 to serve as the threshold for the whole genome significance test, after accounting for multiple testing.12 Odds ratio (OR) estimates were interpreted as approximate relative risk estimates because of the relative rarity of cardiomyopathy.
Replication stage.
Replication stage used a case-only design to verify significant gene-environment interactions identified in the discovery stage. Cumulative anthracycline exposure was dichotomized as low to moderate dose (≤ 250 mg/m2) and high dose (> 250 mg/m2) on the basis of previous observations by us and others, of a significantly increased risk of cardiomyopathy associated with anthracycline dose exceeding 250 mg/m2.1,13 We treated the binary variable of anthracycline exposure as a dependent variable and used logistic regression techniques to conduct a gene-environment interaction analysis (model 2; equation 2).
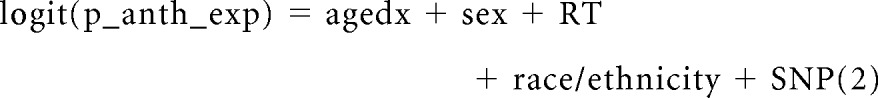 |
where p_anth_exp is the probability of being in the high-dose anthracycline group (anth_exp 0 is the low-to-moderate dose [≤ 250 mg/m2] and anth_exp 1 is the high dose [> 250 mg/m2]).
On the basis of the significantly increased risk of cardiomyopathy in anthracycline-exposed adult patients with coexisting diabetes or hypertension,5 we repeated the analysis after including these comorbidities.
Final model.
By using the combined data from the discovery and replication sets, we used model 2 to test for gene-environment interactions for significant SNP(s) identified in the discovery stage.
Gene expression.
Log-transformed mRNA levels were compared across genetic variants by using the t test.
RESULTS
Discovery Stage
Demographic and clinical characteristics.
Cases received higher cumulative anthracycline exposure (median dose: 300 v 152 mg/m2; P < .001) compared with the controls. Among the cases, median EF was 40% (range, 10% to 56%), and median FS was 23% (range, 5% to 33%; Table 1).
Table 1.
Characteristics of the Study Population in the Discovery and Replication Sets
| Characteristic | Discovery Set |
Replication Set Cases (n = 76) |
|||||
|---|---|---|---|---|---|---|---|
| Cases (n = 93) |
Controls (n = 194) |
P* | |||||
| No. | % | No. | % | No. | % | ||
| Race/ethnicity† | Matched | ||||||
| Non-Hispanic whites | 93 | 100 | 194 | 100 | 45 | 59.2 | |
| Hispanics | 0 | 0 | 0 | 0 | 21 | 27.6 | |
| Other | 0 | 0 | 0 | 0 | 10 | 13.2 | |
| Age, years | |||||||
| At primary cancer diagnosis | 0.43 | ||||||
| Median | 6.9 | 6.3 | 48 | ||||
| Range | 0-20.2 | 0-20.6 | 13-68 | ||||
| At study participation | 0.35 | ||||||
| Median | 19.4 | 18.5 | 55 | ||||
| Range | 0.4-41.7 | 3.5-49.2 | 16-71 | ||||
| Females | 53 | 57.0 | 100 | 51.5 | .74 | 44 | 58 |
| Primary diagnosis† | Matched | ||||||
| Hodgkin lymphoma | 11 | 11.8 | 17 | 8.8 | 14 | 18.4 | |
| Non-Hodgkin lymphoma | 11 | 11.8 | 15 | 7.7 | 34 | 47.4 | |
| Bone tumors | 22 | 23.7 | 32 | 16.5 | 0 | 0 | |
| Soft tissue sarcoma | 9 | 9.7 | 10 | 5.2 | 0 | 0 | |
| ALL | 12 | 12.9 | 62 | 32.0 | 5 | 6.6 | |
| AML | 8 | 8.6 | 20 | 10.3 | 10 | 13.2 | |
| Other | 20 | 21.5 | 38 | 19.6 | 11 | 14.5 | |
| Year of primary cancer diagnosis† | Matched | ||||||
| 1990 or before | 45 | 48.4 | 55 | 28.3 | 9 | 11.8 | |
| 1991-2000 | 33 | 35.5 | 97 | 50.0 | 51 | 67.1 | |
| 2001-2008 | 15 | 16.1 | 42 | 21.7 | 16 | 21.1 | |
| Length of follow-up, years† | .10 | ||||||
| Median | 10.0 | 11.3 | 4.0 | ||||
| Range | 0.1-35.1 | 0.9-41.0 | 0.5-22.5 | ||||
| Cumulative anthracycline exposure, mg/m2 | <.001 | ||||||
| Median | 300 | 152 | 300 | ||||
| Range | 0-630 | 0-825 | 60-649 | ||||
| 0‡ | 7 | 7.5 | 43 | 22.2 | 0 | 0 | |
| 1-100 | 2 | 2.2 | 31 | 16.0 | 1 | 1.3 | |
| 101-150 | 6 | 6.5 | 22 | 11.3 | 8 | 10.5 | |
| 151-200 | 4 | 4.3 | 13 | 6.7 | 6 | 7.9 | |
| 201-250 | 9 | 9.7 | 21 | 10.8 | 12 | 15.8 | |
| 251-300 | 20 | 21.5 | 15 | 7.7 | 22 | 29.0 | |
| > 300 | 45 | 48.4 | 49 | 25.3 | 27 | 35.5 | |
| 1-250 | 21 | 22.6 | 87 | 44.9 | 27 | 35.5 | |
| > 250 | 65 | 69.9 | 64 | 33.0 | 49 | 64.5 | |
| Exposed to radiation to chest | 23 | 24.7 | 22 | 11.3 | .01 | 47 | 61.8 |
| Dose, Gy§ | .3 | ||||||
| Median | 36 | 35 | 12 | ||||
| Range | 12-54 | 7.5-55.8 | 12-50 | ||||
| Age at cardiomyopathy diagnosis, years | |||||||
| Median | 19.4 | N/A | 55 | ||||
| Range | 0.4-41.7 | 16-71 | |||||
| Ejection fraction (%) | |||||||
| Median | 40 | 66‖ | 39 | ||||
| Range | 10-56 | 55-81 | 13-50 | ||||
| HAS3 genotype¶ | .42 | ||||||
| GG | 13 | 14.0 | 38 | 19.6 | 14 | 18.4 | |
| GA | 50 | 53.8 | 91 | 47.2 | 32 | 42.1 | |
| AA | 30 | 32.2 | 64 | 33.2 | 30 | 39.5 | |
Abbreviations: ALL, acute lymphatic leukemia; AML, acute myelogenous leukemia; N/A, not applicable.
P values were obtained from conditional logistic regression or generalized linear model taking into consideration the matched set.
Because of variation in the number of controls per case, the percentage of controls and cases in each category of a specific matching variable may not be identical.
Fifteen patients with no exposure to anthracyclines received radiation to chest (five cases, 10 controls).
Among those who received radiation to chest.
Ejection fraction available for the 35 controls with anthracycline exposure.
One individual missed genotype call at SNP rs2232228 in the discovery data set.
Controls had no signs or symptoms of cardiac compromise at study participation. Of the 194 controls, 155 had normal echocardiographic features (median EF, 66; median FS, 37). Six of the 39 controls without echocardiograms did not receive any cardiotoxic exposure, and exclusion of the 33 anthracycline-exposed controls without echocardiograms did not alter results (Data Supplement); we opted to include them in the analysis.
Risk of cardiomyopathy.
In a multivariable conditional logistic regression analysis that included age at diagnosis of primary cancer, sex, and chest radiation dose in the model, among low-to-moderate-dose (≤ 250 mg/m2) and high-dose (> 250 mg/m2) anthracycline-exposed individuals cardiomyopathy risk was 7.3 (95% CI, 1.5 to 35.8; P = .01) and 49.5 (95% CI, 9.2 to 268.1; P < .001) times higher, respectively (P for trend < .001), than among unexposed patients.
Genome-wide association and gene-environment interaction analysis.
No main-effect association was observed between any of the SNPs examined and cardiomyopathy. However, one SNP (rs2232228) in the HAS3 gene on chromosome 16 exceeded the multiple-comparison-corrected threshold for significant SNP*anthracycline interaction (P = 5.3 × 10−7; Fig 1; Data Supplement).
Fig 1.

Results for test for a trend in the gene-environment (anthracycline) interaction between cardiomyopathy and each single nucleotide polymorphism (SNP) measured in the genome-wide association study. P values are shown for each SNP measured among the 93 cases with cardiomyopathy and 194 controls. Analyses are based on 34,912 SNPs (80.64%) on the ITMAT/Broad CARe SNP array. A result above the horizontal red line indicates strong evidence of association at P < 5 × 10−6.
The main effect association between SNP rs2232228 genotype and cardiomyopathy risk is provided in Table 2. In addition, the modifying effect of rs2232228 genotype on the dose-dependent association between anthracycline and cardiomyopathy risk is shown graphically (Fig 2) and in a tabular format (Table 2). At low-to-moderate-dose anthracycline exposure, the risk of cardiomyopathy did not differ significantly by rs2232228 genotype (GG, GA, or AA; Fig 2). However, among individuals with AA genotype exposed to higher doses of anthracyclines, cardiomyopathy risk increased substantially with anthracycline exposure (anthracycline exposure > 450 mg/m2: OR, 56.5). Although in individuals with GG genotype, cardiomyopathy risk was not increased at any anthracycline dose, the odds of developing cardiomyopathy were approximately 1 at all doses, and at cumulative anthracycline doses exceeding 450 mg/m2, the risk was not increased (OR, 0.6; Fig 2). Furthermore, as delineated in Table 2, among individuals exposed to high-dose (> 250 mg/m2) anthracyclines, the presence of AA genotype conferred an 8.9-fold (95% CI, 2.1- to 37.5-fold; P = .003) increased cardiomyopathy risk when compared with individuals with GG genotype.
Table 2.
Main and Modifying Effects of HAS3 rs2232228 Genotypes on Dose-Dependent Risk of Anthracycline-Related Cardiomyopathy in Discovery Set
| Cumulative Anthracycline Exposure (mg/m2) | HAS3 rs2232228 Genotype Status | Risk of Cardiomyopathy |
||
|---|---|---|---|---|
| OR | 95% CI | P | ||
| Main Effect of HAS3 Genotype | ||||
| All exposures* | ||||
| GG | 1.0 | — | ||
| GA | 2.0 | 0.8 to 4.6 | .12 | |
| AA | 1.8 | 0.7 to 4.7 | .20 | |
| Modifying Effect of HAS3 Genotype† | ||||
| 0-250‡ | GG | 1.0 | — | |
| GA | 0.5 | 0.2 to 1.8 | .3 | |
| AA | 0.2 | 0.1 to 0.8 | .03 | |
| > 250‡ | GG | 1.1 | 0.3 to 4.8 | .9 |
| GA | 5.2 | 1.6 to 17.4 | .007 | |
| AA | 9.9 | 2.4 to 40.9 | .002 | |
| Patients Exposed to High-Dose Anthracyclines§ | ||||
| > 250 | GG | 1.0 | ||
| GA | 4.7 | 1.4 to 16.2 | .02 | |
| AA | 8.9 | 2.1 to 37.5 | .003 | |
Abbreviation: OR, odds ratio.
ORs were obtained from conditional logistic regression adjusting for age at diagnosis, sex, chest radiation dose, and anthracycline exposure (continuous).
Reference group: 0-250 mg/m2 anthracycline exposure and rs2232228 GG genotype.
ORs were obtained from conditional logistic regression adjusting for age at diagnosis, sex, and chest radiation doses.
Reference group: rs2232228 GG genotype for the anthracycline exposure level of > 250 mg/m2.
Fig 2.

Risk of cardiomyopathy by anthracycline dose and genotype status (AA, GA, GG). Odds ratios (ORs) were calculated based on model 1, treating anthracycline dose as a continuous variable (reference group: GG genotype with no anthracycline exposure).
Replication Stage
Demographic and clinical characteristics.
Median cumulative anthracycline exposure was 300 mg/m2, and median EF was 39% (Table 1).
Gene-environment interaction.
Compared with cases with GG genotype, the odds for cases with GA and AA genotype of being in the high-dose anthracycline group were 3.6 (95% CI, 0.9 to 15.0; P = .07) and 4.5 (95% CI, 1.1 to 18.7; P = .04) times higher, respectively (Table 3). After inclusion of the presence or absence of hypertension and diabetes in the model, the odds of cases with AA genotype for being in the high-dose anthracycline group were 4.9 times higher (95% CI, 1.1 to 22.8; P = .04).
Table 3.
Analysis of Gene-Environment Interaction for Among Cases Only
| HAS3 rs2232228 Genotype Status | Cumulative Anthracycline Exposure (mg/m2) |
OR* | 95% CI | P | |||
|---|---|---|---|---|---|---|---|
| ≤ 250 |
> 250 |
||||||
| No. | % | No. | % | ||||
| Replication set | |||||||
| GG | 8 | 29.6 | 6 | 12.2 | 1.0 | ||
| GA | 11 | 40.8 | 21 | 42.9 | 3.6 | 0.9 to 15.0 | .07 |
| AA | 8 | 29.6 | 22 | 44.9 | 4.5 | 1.1 to 18.7 | .04 |
| Combined discovery† and replication set | |||||||
| GG | 12 | 25.0 | 12 | 10.5 | 1.0 | ||
| GA | 23 | 47.9 | 56 | 49.1 | 2.6 | 1.0 to 6.9 | .05 |
| AA | 13 | 27.1 | 46 | 40.4 | 3.7 | 1.3 to 10.2 | .01 |
Abbreviation: OR, odds ratio.
ORs were calculated based on multivariable logistic regression adjusting for age at diagnosis, sex, chest radiation dose, and race/ethnicity (white v other).
Cases of cardiomyopathy in the discovery set with no anthracycline exposure (n = 7) were excluded from this analysis.
Final Model
We combined cases from the discovery and replication sets to test gene-environment interaction at SNP rs2232228. Cases with AA genotype had 3.7 (95% CI, 1.3 to 10.2; P = .01) times higher odds of being in the high-dose anthracycline group than those with GG genotype (Table 3).
Gene Expression
The relative HAS3 mRNA levels in heart samples with homozygous A genotype (n = 16; geometric mean, 1.9) tended to be lower than the relative HAS3 mRNA levels for heart samples carrying the GA genotype (n = 9; geometric mean, 7.8; P = .09; Data Supplement). Because of the small number (n = 3), hearts with GG genotype were not included in the analysis.
DISCUSSION
We used the high-density IBC cardiovascular SNP array to study the association between anthracycline-related cardiomyopathy and 34,912 SNPs in 2,100 carefully curated genes known to be associated with de novo cardiovascular disease.7 Among individuals with a homozygous G genotype on SNP rs2232228, cardiomyopathy risk did not demonstrate any dose-dependent increase. However, in individuals with AA genotype, cardiomyopathy risk increased substantially as anthracycline exposure increased, such that among individuals exposed to high-dose anthracyclines, the presence of AA genotype conferred an 8.9-fold increased cardiomyopathy risk when compared with the GG genotype. This significant gene-environment interaction at SNP rs2232228 was successfully replicated by using an independent set of cases with anthracycline-related cardiomyopathy.
The HAS3 gene, located on chromosome 16, encodes for an enzyme that produces low-molecular-weight hyaluronan (HA). HA is a ubiquitous component of extracellular matrix (ECM)14 and plays a dynamic role in ECM organization following injury by providing a matrix to support cell migration and adhesion.14–20 HA is especially enriched in matrices undergoing remodeling. Anthracyclines injure heart muscle through induction of cardiomyocyte apoptosis, which is then replaced by fibrosis.21 Anthracycline-related injury is directly linked to the amount of anthracyclines in the heart.22 The ECM provides a scaffold for alignment of cardiomyocytes, fibroblasts, endothelial cells, and vasculature after injury.23 In fact, cardiac fibroblasts play a pivotal role in the repair and remodeling of the heart that occur following myocardial infarction, serving as a central mediator of cardiac remodeling by using the ECM as a scaffold.24 An example of the role of HA in an injured myocardium is evidenced by the observation that HA accumulates after myocardial infarction in rats.25 These observations suggest that ECM is involved in cardiac remodeling after anthracycline-related injury. The extent of remodeling and repair is possibly modulated by variability in HA production in the ECM, consistent with the genotype-dependent cardiomyopathy risk observed in this study.
Anthracyclines are cardiotoxic per se but gain further toxicity after one-electron reduction with reactive oxygen species (ROS) overproduction. Variants of genes involved in ROS production (nicotinamide adenine dinucleotide phosphate oxidase)26 contribute to anthracycline-induced toxicity. HA has antioxidant activity27 and interacts specifically with the CD44 receptor on cardiomyocytes,28 maintaining integrity of the cardiomyocytes during ROS damage by stimulating cell proliferation29 as well as preventing activation of death receptors, thus maintaining survival and function.30 Recent in vitro data support the ability of HA to reduce ROS-mediated cardiac injury and activate the damage surveillance system.31 Our genotype-phenotype analysis suggests a trend toward higher HAS3 mRNA expression in heart samples with HAS3 rs2232228 GA genotype (minimal risk of anthracycline-related cardiomyopathy) as compared with hearts with AA genotype (high cardiomyopathy risk at high doses of anthracyclines). Taken together, these data suggest that lower cardiac HAS3 mRNA expression (AA genotype) may result in decreased synthesis of the antioxidant HA, supporting our findings of the high cardiomyopathy risk in individuals with AA genotype.
The SNP rs2232228 on exon 2 of HAS3 (67701078 bp) resides on chromosome 16 and appears to be in a region of low LD. Our data contain seven SNPs in HAS3, but the LD between rs2232228 and the other six SNPs was not strong (the largest r2 was 0.42 between rs2232228 and rs8047014); gene-environment interaction for rs8047014 was not significant. We also imputed the entire chromosome 16 by using 1,000 genome SNPs as reference. No gene-environment interaction was identified in the analysis based on SNPs with imputed r2 more than 0.5. Finally, we examined the HapMap CEU data (Utah residents with Northern and Western European ancestry) within 1 Mb of rs2232228 and identified only two SNPs, both with weak LD (rs8082856 r2 = 0.56; rs9332431 r2 = 0.579).
The case-only design used to replicate significant findings is well established as an efficient and valid method for evaluating gene-environment interactions.32 A positive (> two-fold) association implies a relevant gene-environment interaction.33 This study demonstrated that cases homozygous for the A allele of rs2232228 in HAS3 had a 3.7-fold increased odds of being in the high-dose anthracycline group, suggesting a valid gene-environment interaction in the replication stage.
For logistical reasons, we used a prevalent case-control study design. Prevalent case-control studies are vulnerable to the underestimation of effect size for genotypes associated with both increased disease risk and disease-associated lethality.34,35 Applying this hypothetical scenario to this study, the lack of association between the GG genotype and cardiomyopathy risk could possibly represent a false-negative finding (ie, the GG genotype could be associated with an increased risk of cardiomyopathy and cardiomyopathy-related death), making the GG-associated cardiomyopathy cases unavailable for enrollment onto our study. But because there is no published data to support high cardiomyopathy-related lethality associated with the G allele of rs2232228, we believe that the lack of association between the GG genotype and the risk of cardiomyopathy is not affected by the study design. Furthermore, the replication set included all consecutive patients with cardiomyopathy (alive and deceased) from a cohort of hematopoietic cell transplantation recipients at a single institution. The frequency of GG genotype among cases in the discovery set (14%) and replication set (18%) was comparable (P = .3). Thus, successful replication of significant findings identified in the discovery set indicates that survival bias is likely not a significant issue with the prevalent cases and controls in the discovery set.
Of note, although the discovery set was limited to non-Hispanic white survivors of childhood cancer, the replication set included cases drawn from survivors of childhood and adult-onset cancer from all racial/ethnic backgrounds. The clear dose-response relation between anthracycline exposure and cardiomyopathy in both pediatric1 and adult populations5 suggests shared mechanisms for anthracycline-induce toxicity. Moreover, the successful replication of the finding in a clinically and demographically diverse population speaks to the robustness of the association between the common variants in rs2232228 in HAS3 gene and anthracycline-related cardiomyopathy.
We used the IBC cardiovascular SNP array with a carefully curated yet comprehensive list of genes enriched for their association with de novo cardiovascular disorders. Our choice of the IBC array was based on our clinical observation that cardiovascular risk factors (diabetes, hypertension) interact with anthracycline exposure in cancer survivors to increase the risk of heart failure.36 Thus, the main focus of this study was to identify the interaction of SNPs on the IBC array with anthracycline exposure and not to examine the main effects of these SNPs.
The IBC cardiovascular SNP array does not include genes that regulate anthracycline metabolism or disposition.1,26,37 Thus, genes implicated in anthracycline-related cardiomyopathy such as CBR31 and SLC28A337 are not included on the IBC array. However, some SNPs from ABCC138 and ABCC226,39 genes are included, but the smallest P values did not meet the cutoff (5 × 10−6) for our discovery stage (Data Supplement). As opposed to 10,000 independent associations examined in this study, these previous studies used a limited candidate gene approach with a higher likelihood of identifying associations without the penalty of a lower α level due to multiple testing.
In this study, we provide evidence that SNP rs2232228 in the HAS3 gene alters the risk of anthracycline-related cardiomyopathy among patients exposed to high-dose anthracyclines. Our investigation establishes a foundation for understanding the functional significance of SNP rs2232228 and sets the stage for validation in a prospective cohort.
Supplementary Material
Acknowledgment
We thank our research staff, particularly Pamela McGill, Natalie Lowery, Sean Freeman, and Nancy Kornegay, as well as patients and families for their participation.
Appendix
Table A1.
Participating Children's Oncology Group Institutions
| A.B. Chandler Medical Center-University of Kentucky |
| A.I. duPont Hospital for Children |
| Advocate Hope Children's Hospital |
| All Children's Hospital |
| Allan Blair Cancer Centre |
| Baptist Children's Hospital |
| British Columbia's Children's Hospital |
| Brooklyn Hospital Center |
| C.S. Mott Children's Hospital |
| Cancer Research Center of Hawaii |
| CancerCare Manitoba |
| Cedars-Sinai Medical Center |
| Children's Healthcare of Atlanta, Emory University |
| Children's Hospital and Clinics, Minneapolis and St. Paul |
| Children's Hospital London Health Sciences |
| Children's Hospital Los Angeles |
| Children's Hospital Medical Center, Akron, Ohio |
| Children's Hospital Oakland |
| Children's Hospital of Eastern Ontario |
| Children's Hospital of Michigan |
| Children's Hospital of Philadelphia |
| Children's Hospital of the Greenville Hospital System |
| Children's Hospital of the King's Daughters |
| Children's Medical Center Dayton |
| Children's Memorial Medical Center at Chicago |
| Children's National Medical Center, Washington, DC |
| Children's of New Orleans/Louisiana State University Medical Center Community Clinical Oncology Program |
| Cincinnati Children's Hospital Medical Center |
| City of Hope National Medical Center |
| Connecticut Children's Medical Center |
| Cook Children's Medical Center |
| Dana-Farber Cancer Institute and Children's Hospital |
| Driscoll Children's Hospital |
| East Tennessee Children's Hospital |
| East Tennessee State University |
| Eastern Maine Medical Center |
| Emanuel Hospital-Health Center |
| Hackensack University Medical Center |
| Helen DeVos Children's Hospital |
| Hospital Sainte-Justine |
| Hospital for Sick Children |
| Hurley Medical Center |
| Indiana University-Riley Children's Hospital |
| Inova Fairfax Hospital |
| IWK (Izaak Walton Killam) Health Centre |
| Kaiser Permanente Medical Group, Inc., Northern California |
| Kalamazoo Center for Medical Studies |
| Kingston General Hospital/Kingston Regional Cancer |
| Kosair Children's Hospital |
| MD Anderson Cancer Center |
| Maimonides Medical Center |
| Mayo Clinic and Foundation |
| McGill University Health Center-Montreal Children's Hospital |
| McMaster University |
| Medical College of Georgia Children's Medical Center |
| Memorial Sloan-Kettering Cancer Center |
| Methodist Children's Hospital of South Texas |
| Miami Children's Hospital |
| Michigan State University |
| Midwest Children's Cancer Center |
| Nationwide Children's Hospital |
| Nemours Children's Clinic-Jacksonville |
| Nevada Cancer Research Foundation-Community Clinical Oncology Program |
| New York Medical College |
| Newark Beth Israel Medical Center |
| Primary Children's Medical Center |
| Princess Margaret Hospital for Children |
| Rady Children's Hospital San Diego |
| Rainbow Babies and Children's Hospital |
| Royal Children's Hospital, Brisbane |
| Royal Children's Hospital, University of Melbourne |
| Sacred Heart Children's Hospital |
| Sacred Heart Hospital |
| Saint Barnabas Medical Center |
| Saint Peter's University Hospital |
| Saskatoon Cancer Center |
| Scott and White Memorial Hospital |
| Seattle Children's |
| South Carolina Cancer Center |
| St. John Hospital and Medical Center |
| St. Joseph's Hospital and Medical Center |
| St. Jude Children's Research Hospital-Memphis |
| St. Vincent Children's Hospital-Indiana |
| St. Vincent Hospital-Wisconsin |
| Stanford University Medical Center |
| State University of New York at Stony Brook |
| Stollery Children's Hospital |
| State University of New York Upstate Medical University |
| Swiss Pediatric Oncology Group-Geneva |
| Tampa Children's Hospital |
| Texas Children's Cancer Center at Baylor College of Medicine |
| Texas Tech University Health Sciences Center-Amarillo |
| The Children's Hospital, Denver, CO |
| The Children's Hospital of Southwest Florida Lee Memorial Health System |
| The Children's Mercy Hospital |
| The University of Chicago Comer Children's Hospital |
| Tulane University Medical Center |
| University of California at Los Angeles David Geffen School of Medicine |
| University of Alabama |
| University of Florida |
| University of Iowa Hospitals and Clinics |
| University of Kansas Medical Center |
| University of Minnesota Cancer Center |
| University of Mississippi Medical Center Children's Hospital |
| University of Missouri-Columbia |
| University of New Mexico School of Medicine |
| University of North Carolina at Chapel Hill |
| University of Oklahoma Health Sciences Center |
| University of Pittsburgh |
| University of Texas Health Science Center at San Antonio |
| University of Vermont College of Medicine |
| University of Wisconsin-Children's Hospital Madison |
| University of Texas Southwestern Medical Center |
| Vanderbilt Children's Hospital |
| Virginia Commonwealth University Health System |
| Wake Forest University School of Medicine |
| Washington University Medical Center |
| West Virginia University Health Sciences Center-Charleston |
| Winthrop University Hospital |
| Women's and Children's Hospital, Adelaide |
| Yale University School of Medicine |
Footnotes
Supported by Grants No. U10 CA98543 from the National Cancer Institute (NCI), National Institutes of Health (NIH); No. 6093-08 from the Leukemia and Lymphoma Society; No. U01 GM92666 and GM73646 from the National Institute of General Medical Sciences Pharmacogenomics Research Network, NIH; and No. P30CA033572 from NCI, NIH, and American Lebanese Syrian Associated Charities.
Presented at the 13th International Conference on Long-Term Complications of Treatment of Children and Adolescents for Cancer, Memphis, TN, June 13-15, 2013, and at the 49th Annual Meeting of the American Society of Clinical Oncology, Chicago, IL, May 31-June 4, 2013.
S.B. has full access to all the data in the study and takes responsibility for the integrity of the data and accuracy of the data analysis.
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The author(s) indicated no potential conflicts of interest.
AUTHOR CONTRIBUTIONS
Conception and design: Leslie L. Robison, Mary V. Relling, Smita Bhatia
Financial support: Mary V. Relling, Smita Bhatia
Administrative support: Wendy Landier, Smita Bhatia
Provision of study materials or patients: Jill P. Ginsberg, Joseph P. Neglia, Sharon M. Castellino, Melissa M. Hudson, Smita Bhatia
Collection and assembly of data: Hakon Hakonarson, Lindsey Hageman, Yan Ding, Wendy Landier, Javier G. Blanco, Jill P. Ginsberg, Frank Keller, Joseph P. Neglia, Sharon M. Castellino, Irene Cherrick, Melissa M. Hudson, Mary V. Relling, Smita Bhatia
Data analysis and interpretation: Xuexia Wang, Wei Liu, Can-Lan Sun, Saro H. Armenian, Lu Chen, Adolfo Quiñones, Daniel Ferguson, Naomi Winick, Joseph P. Neglia, Sunil Desai, Charles A. Sklar, ZoAnn E. Dreyer, Yutaka Yasui, Mary V. Relling, Smita Bhatia
Manuscript writing: All authors
Final approval of manuscript: All authors
REFERENCES
- 1.Blanco JG, Sun CL, Landier W, et al. Anthracycline-related cardiomyopathy after childhood cancer: Role of polymorphisms in carbonyl reductase genes—A report from the Children's Oncology Group. J Clin Oncol. 2012;30:1415–1421. doi: 10.1200/JCO.2011.34.8987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grenier MA, Lipshultz SE. Epidemiology of anthracycline cardiotoxicity in children and adults. Semin Oncol. 1998;25:72–85. [PubMed] [Google Scholar]
- 3.Barry EV, Lipshultz SE, Sallan SE. Anthracycline-induced cardiotoxicity: Natural history, risk factors, and prevention. Presented at 44th Annual Meeting Am Soc Clin Oncol meeting; May 30-June 3, 2008; Chicago, IL. [Google Scholar]
- 4.Puma N, Ruggiero A, Ridola V, et al. Anthracycline-related cardiotoxicity: Risk factors and therapeutic options in childhood cancers. SIGNA VITAE. 2008;3:30–34. [Google Scholar]
- 5.Armenian SH, Sun CL, Shannon T, et al. Incidence and predictors of congestive heart failure following autologous hematopoietic cell transplantation. Blood. 2011;118:6023–6029. doi: 10.1182/blood-2011-06-358226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bryant J, Picot J, Levitt G, et al. Cardioprotection against the toxic effects of anthracyclines given to children with cancer: A systematic review. Health Technol Assess. 2007;11:iii, ix–x, 1–84. doi: 10.3310/hta11270. [DOI] [PubMed] [Google Scholar]
- 7.Keating BJ, Tischfield S, Murray SS, et al. Concept, design and implementation of a cardiovascular gene-centric 50 k SNP array for large-scale genomic association studies. PLoS ONE. 2008;3:e3583. doi: 10.1371/journal.pone.0003583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shankar SM, Marina N, Hudson MM, et al. Monitoring for cardiovascular disease in survivors of childhood cancer: Report from the Cardiovascular Disease Task Force of the Children's Oncology Group. Pediatrics. 2008;121:e387–e396. doi: 10.1542/peds.2007-0575. [DOI] [PubMed] [Google Scholar]
- 9.de Bakker PI, Burtt NP, Graham RR, et al. Transferability of tag SNPs in genetic association studies in multiple populations. Nat Genet. 2006;38:1298–1303. doi: 10.1038/ng1899. [DOI] [PubMed] [Google Scholar]
- 10.Purcell S, Neale B, Todd-Brown K, et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adamia S, Maxwell CA, Pilarski LM. Hyaluronan and hyaluronan synthases: Potential therapeutic targets in cancer. Curr Drug Targets Cardiovasc Haematol Disord. 2005;5:3–14. doi: 10.2174/1568006053005056. [DOI] [PubMed] [Google Scholar]
- 12.Cappola TP, Li M, He J, et al. Common variants in HSPB7 and FRMD4B associated with advanced heart failure. Circ Cardiovasc Genet. 2010;3:147–154. doi: 10.1161/CIRCGENETICS.109.898395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mulrooney DA, Yeazel MW, Kawashima T, et al. Cardiac outcomes in a cohort of adult survivors of childhood and adolescent cancer: Retrospective analysis of the Childhood Cancer Survivor Study cohort. BMJ. 2009;339:b4606. doi: 10.1136/bmj.b4606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spicer AP, Tien JY. Hyaluronan and morphogenesis. Birth Defects Res C Embryo Today. 2004;72:89–108. doi: 10.1002/bdrc.20006. [DOI] [PubMed] [Google Scholar]
- 15.Jiang D, Liang J, Fan J, et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med. 2005;11:1173–1179. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- 16.Laurent TC, Fraser JR. Hyaluronan. FASEB J. 1992;6:2397–2404. [PubMed] [Google Scholar]
- 17.Knudson CB, Knudson W. Hyaluronan-binding proteins in development, tissue homeostasis, and disease. FASEB J. 1993;7:1233–1241. [PubMed] [Google Scholar]
- 18.Toole BP. Hyaluronan and its binding proteins, the hyaladherins. Curr Opin Cell Biol. 1990;2:839–844. doi: 10.1016/0955-0674(90)90081-o. [DOI] [PubMed] [Google Scholar]
- 19.West DC, Hampson IN, Arnold F, et al. Angiogenesis induced by degradation products of hyaluronic acid. Science. 1985;228:1324–1326. doi: 10.1126/science.2408340. [DOI] [PubMed] [Google Scholar]
- 20.Zhang W, Watson CE, Liu C, et al. Glucocorticoids induce a near-total suppression of hyaluronan synthase mRNA in dermal fibroblasts and in osteoblasts: A molecular mechanism contributing to organ atrophy. Biochem J. 2000;349:91–97. doi: 10.1042/0264-6021:3490091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perik PJ, de Vries EG, Gietema JA, et al. The dilemma of the strive for apoptosis in oncology: Mind the heart. Crit Rev Oncol Hematol. 2005;53:101–113. doi: 10.1016/j.critrevonc.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 22.Meissner K, Sperker B, Karsten C, et al. Expression and localization of P-glycoprotein in human heart: Effects of cardiomyopathy. J Histochem Cytochem. 2002;50:1351–1356. doi: 10.1177/002215540205001008. [DOI] [PubMed] [Google Scholar]
- 23.Corda S, Samuel JL, Rappaport L. Extracellular matrix and growth factors during heart growth. Heart Fail Rev. 2000;5:119–130. doi: 10.1023/A:1009806403194. [DOI] [PubMed] [Google Scholar]
- 24.Burlew BS, Weber KT. Connective tissue and the heart: Functional significance and regulatory mechanisms. Cardiol Clin. 2000;18:435–442. doi: 10.1016/s0733-8651(05)70154-5. [DOI] [PubMed] [Google Scholar]
- 25.Waldenström A, Martinussen HJ, Gerdin B, et al. Accumulation of hyaluronan and tissue edema in experimental myocardial infarction. J Clin Invest. 1991;88:1622–1628. doi: 10.1172/JCI115475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wojnowski L, Kulle B, Schirmer M, et al. NAD(P)H oxidase and multidrug resistance protein genetic polymorphisms are associated with doxorubicin-induced cardiotoxicity. Circulation. 2005;112:3754–3762. doi: 10.1161/CIRCULATIONAHA.105.576850. [DOI] [PubMed] [Google Scholar]
- 27.Young IS, Woodside JV. Antioxidants in health and disease. J Clin Pathol. 2001;54:176–186. doi: 10.1136/jcp.54.3.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hellström M, Johansson B, Engström-Laurent A. Hyaluronan and its receptor CD44 in the heart of newborn and adult rats. Anat Rec A Discov Mol Cell Evol Biol. 2006;288:587–592. doi: 10.1002/ar.a.20332. [DOI] [PubMed] [Google Scholar]
- 29.Wang YZ, Cao ML, Liu YW, et al. CD44 mediates oligosaccharides of hyaluronan-induced proliferation, tube formation and signal transduction in endothelial cells. Exp Biol Med (Maywood) 2011;236:84–90. doi: 10.1258/ebm.2010.010206. [DOI] [PubMed] [Google Scholar]
- 30.Pauloin T, Dutot M, Liang H, et al. Corneal protection with high-molecular-weight hyaluronan against in vitro and in vivo sodium lauryl sulfate-induced toxic effects. Cornea. 2009;28:1032–1041. doi: 10.1097/ICO.0b013e3181a0a3f8. [DOI] [PubMed] [Google Scholar]
- 31.Law CH, Li JM, Chou HC, et al. Hyaluronic acid-dependent protection in H9C2 cardiomyocytes: A cell model of heart ischemia-reperfusion injury and treatment. Toxicology. 2013;303:54–71. doi: 10.1016/j.tox.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 32.Begg CB, Zhang ZF. Statistical analysis of molecular epidemiology studies employing case-series. Cancer Epidemiol Biomarkers Prev. 1994;3:173–175. [PubMed] [Google Scholar]
- 33.VanderWeele TJ, Hernández-Díaz S, Hernán MA. Case-only gene-environment interaction studies: When does association imply mechanistic interaction? Genet Epidemiol. 2010;34:327–334. doi: 10.1002/gepi.20484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hernán MA, Hernández-Díaz S, Robins JM. A structural approach to selection bias. Epidemiology. 2004;15:615–625. doi: 10.1097/01.ede.0000135174.63482.43. [DOI] [PubMed] [Google Scholar]
- 35.Anderson CD, Nalls MA, Biffi A, et al. The effect of survival bias on case-control genetic association studies of highly lethal diseases. Circ Cardiovasc Genet. 2011;4:188–196. doi: 10.1161/CIRCGENETICS.110.957928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Armenian SH, Sun CL, Vase T, et al. Cardiovascular risk factors in hematopoietic cell transplantation survivors: Role in development of subsequent cardiovascular disease. Blood. 2012;120:4505–4512. doi: 10.1182/blood-2012-06-437178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Visscher H, Ross CJ, Rassekh SR, et al. Pharmacogenetic prediction of anthracycline-induced cardiotoxicity in children. J Clin Oncol. 2012;30:1422–1428. doi: 10.1200/JCO.2010.34.3467. [DOI] [PubMed] [Google Scholar]
- 38.Semsei AF, Erdelyi DJ, Ungvari I, et al. ABCC1 polymorphisms in anthracycline-induced cardiotoxicity in childhood acute lymphoblastic leukemia. Cell Biol Int. 2012;36:79–86. doi: 10.1042/CBI20110264. [DOI] [PubMed] [Google Scholar]
- 39.Armenian SH, Ding Y, Mills G, et al. Genetic susceptibility to anthracycline-related congestive heart failure in survivors of haematopoietic cell transplantation. Br J Haematol, 2013;163:205–213. doi: 10.1111/bjh.12516. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.