

Abstract
Dermatomyositis (DM), polymyositis (PM), necrotizing myopathy (NM) and inclusion body myositis (IBM) are four distinct subtypes of idiopathic inflammatory myopathies – in short myositis. Recent studies have shed some light on the unique pathogenesis of each entity. Some of the clinical features are distinct, but muscle biopsy is indispensable for making a reliable diagnosis. The use of magnetic resonance imaging of skeletal muscles and detection of myositis-specific autoantibodies have become useful additions to our diagnostic repertoire. Only few controlled trials are available to substantiate current treatment approaches for myositis and hopes are high that novel modalities will become available within the next few years. In this review we provide an up-to-date overview of the pathogenesis and diagnostic approach of myositis. We aim to present a guide towards therapeutic and general management.
Keywords: muscle immunology/disease, myositis, neuroimmunology
Other Articles published in this series.
Paraneoplastic neurological syndromes. Clinical and Experimental Immunology 2014, 175: 336–48.
Disease-modifying therapy in multiple sclerosis and chronic inflammatory demyelinating polyradiculoneuropathy: common and divergent current and future strategies. Clinical and Experimental Immunology 2014, 175: 359–72.
Monoclonal antibodies in treatment of multiple sclerosis. Clinical and Experimental Immunology 2014, 175: 373–84.
CLIPPERS: chronic lymphocytic inflammation with pontine perivascular enhancement responsive to steroids. Review of an increasingly recognized entity within the spectrum of inflammatory central nervous system disorders. Clinical and Experimental Immunology 2014, 175: 385–96.
Requirement for safety monitoring for approved multiple sclerosis therapies: an overview. Clinical and Experimental Immunology 2014, 175: 397–407.
Myasthenia gravis: an update for the clinician. Clinical and Experimental Immunology 2014, 175: 408–18.
Cerebral vasculitis in adults: what are the steps in order to establish the diagnosis? Red flags and pitfalls. Clinical and Experimental Immunology 2014, 175: 419–24.
Multiple sclerosis treatment and infectious issues: update 2013. Clinical and Experimental Immunology 2014, 175: 425–38.
Introduction
Idiopathic inflammatory myopathies – in short myositis – include dermatomyositis (DM), polymyositis (PM), necrotizing myopathy (NM) and inclusion body myositis (IBM). They all present with muscle weakness. Diagnosis is based on the clinical examination (distribution of paresis) in combination with laboratory values, including creatine kinase (CK) and autoantibodies, electromyography (EMG) and the histopathology of the skeletal muscle. The use of magnetic resonance imaging (MRI) of the skeletal muscle is not only helpful to identify an adequate muscle for biopsy, but also to demonstrate the pattern of affected muscles beyond clinical appearance, which helps to exclude, for example, muscular dystrophies. While DM, PM and NM mainly respond well to treatment with immunosuppressants, IBM is usually resistant to these drugs, and only in few patients may immunoglobulins display a temporary beneficial effect.
Dermatomyositis (DM)
The incidence and prevalance of DM are 1·4 and 5·8 cases among 100 000 people in the United States [1]. It shows a female preponderance and a higher prevalence among older people. As juvenile DM (JDM), it can occur in children with a prevalence of 3·2 among 1 million children in the United Kingdom and is more common among girls [2].
Patients present with a symmetric proximal muscle weakness that develops within weeks or months, together with typical erythematous changes [3]. The skin changes can also precede or follow the myopathy. Typical signs are a heliotrophic rash, oedema of the eyelids, mechanic's hands, Gottron papules at extensor surfaces and subcutaneous calcification. Myalgia is not typical, but can occur. Patients with a severe course of DM can develop dysphagia and dysarthria. Other important complications are the detection of interstitial lung disease (ILD) [4] or tumour [5].
Clinically amyopathic DM (CADM) is a subtype in which patients present with typical skin changes and without or only minimal signs of a myopathy [6]. It makes up to 20% of all patients with DM and can also be associated with ILD [7]. For the anti-CADM-140 antibody, a correlation between DM/CADM and the prediction of outcome of a rapid progressive ILD has been described [4].
The pathology of DM includes binding of immune complexes to endothelium cells with subsequent activation of the complement system and cell lysis, mediated by the membrane-attack complex (MAC) [8]. This leads to necrosis of these cells, and a reduced number of capillaries in the muscle can be seen [9]. The blood supply becomes insufficient, which is believed to cause perifascicular atrophy.
This classical concept has been challenged recently, in that Greenberg's group [10] reported a type I interferon (IFN)-mediated cascade and suggest that this is a predominant element of the pathology. The type I IFN-(α/β)-induced genes are overexpressed in muscle, skin and blood and correlate significantly with the disease activity [11]. Dendritic cells are suggested as antigen-presenting cells and are a potential source of IFNs [10]. It is so far unclear as to which of these cascades precedes or is predominant.
Within the inflammatory tissue, there is an over-expression of proinflammatory mediators, including transforming growth factor (TGF)-β, major histocompatibility complex (MHC)-I, IL-1β, CCL-3, CCL-4, etc. [12–14]. The extravasation of immune cells to the muscle tissue is enhanced by up-regulation of the vascular cell adhesion molecule (VCAM)-1 and intercellular adhesion molecule (ICAM)-1 on endothelial cells and binding to their receptors very late antigen (VLA)-4 and lymphocyte function-associated antigen (LFA)-1 on immune cells [15]. As cellular components of the immune system, T cells, B cells, macrophages and plasmacytoid dendritic cells are found in perimysial and perivascular areas.
In the skin, typical findings are a vacuolar degeneration of epidermal basal cells with epidermal atrophy and dermatitis with lymphocytes and macrophages [16,17]. Depositions of MAC are found along the dermo–epidermal junction and along the vessels [18].
Polymyositis (PM)
The clinical symptoms of PM include a muscular weakness of proximal muscles, mainly with a subacute onset and marked elevation of the CK [9]. The weakness is usually most severe in the pelvic girdle and shoulders. The neck flexors are commonly affected, and in some patients also the neck extensors [19].
The age-and gender-adjusted incidence is 3·8 and the prevalence in the United States is 9·7 per 100 000 people [1]. In the opinion of the authors and other experts, PM is overdiagnosed, and not all studies have been based upon a diagnostic muscle biopsy.
A biopsy is indispensable to distinguish PM from other myopathies. Cellular infiltrates are composed of macrophages and cytotoxic CD8+ T cells [9]. These cells can surround or even invade non-necrotic fibres, a feature which is not usually observed in DM.
The pathogenesis is characterized by local activation of immune cells in skeletal muscle. The proinflammatory milieu includes expression of cytokines such as IFN-γ, IL-6, IL-1β, tumour necrosis factor (TNF)-α and TGF-β [12,20], as well as chemokines such as IL-8, CCL-2, CCL-3, CCL-4, CCL-5, CXCL-9 and CXCL-10, contributing to the local inflammation, and are the attracting stimulus for immune cells [14]. An efficient extravasation of immune cells is mediated by anchoring receptors on immune cells (e.g. VLA-4, LFA-1) and their binding to adhesion molecules, which are up-regulated on endothelial cells (e.g. ICAM-1 and VCAM-1). Extracellular matrix proteins can be degraded by metalloproteinases 2 and 9, which were identified in PM and can contribute to the migration of T cells [21]. These enzymes are induced by cytokines and secreted by inflammatory cells. MHC-I, which is up-regulated ubiquitously by muscle fibres under proinflammatory conditions, seems to be pivotal for the interaction of muscle fibres and immune cells [22,23].
Necrotizing myopathy (NM)
The clinical presentation of NM, recently also termed immune-mediated necrotizing myopathy or necrotizing autoimmune myopathy, includes a progressive, symmetrical weakness of the proximal muscles of arms and legs. Clinically, NM is indistinguishable from PM. Myalgia can occur in up to 80% [24]. In severe cases, dysphagia and dysarthria can develop [25,26].
This group of NM is heterogeneous and includes autoimmune inflammatory mechanisms, paraneoplastic conditions, exposure to toxins or drugs as well as combinations of these mechanisms [27]. Myositis-specific autoantibodies against single recognition particle (SRP) [28] or 3-hydroxy-3-methylglutaryl co-enzyme A reductase (HMGCR) [29] can be detected in a subset of 4–6% of patients with myositis and ˜60% of patients with NM [29,30]. Our knowledge about the epidemiology is scarce. In studies with small cohorts, a male preponderance of 61% was found [24]. The mean age at onset for SRP-associated NM is in the 5th decade [25,26] and for HMGCR-associated NM in the 6th decade [26,29].
A biopsy is required to make the diagnosis. The major findings are scattered necrotic muscle fibres [31,32]. Sparse inflammatory cells may surround the necrosis. Macrophages are predominant and few lymphocytes are present, which were identified as CD4+ and CD8+ T cells [27]. The interaction of these cells is hypothesized to play an important role in the progression and pathogenesis of NM, but the major part of the pathogenesis is still unknown. The expression of MHC-I in necrotic or regenerating fibres appears to be non-specific.
Most patients with SRP autoantibodies fulfil the criteria for NM; the diagnosis of PM is made only in few patients [30]. It is well known that statins and fibrates can lead to toxic myopathy and the risk increases with the daily dosage of the statin [33]. Two-thirds of myositis patients with HMGCR autoantibodies have previously been exposed to statins [29].
Inclusion body myositis (IBM)
IBM is considered to be the most frequently acquired myopathy after the 50th year of life. The prevalence in Australia per million people was found to be 9·3 in the general population and 51·3 in people aged over 50 years [34]. In contrast to PM and DM, there is a male preponderance [35,36].
IBM develops slowly, progressively and painlessly over years, leading to a mainly asymmetric paresis [35,36]. The flexion of hand and fingers and knee extension are typically affected. The development of dysphagia is typical for IBM, and at least minor swallowing difficulties are observed in 65–80% of the patients [37]. Dysphagia can precede the weakness in arms and legs. Ambulation of the patient is often impaired and assisting devices are commonly required during the course of the disease. The time from disease onset until the first use of a wheelchair ranges from 14 to 16 years [35,36]. After a median time of 24 years, patients are completely wheelchair-dependent [35]. Although the disease progresses slowly and leads to major disabilities, the patient's life expectancy is normal [35].
The diagnostic criteria proposed by Griggs et al. [38] rely very much on the histopathology. The criteria of the European Neuromuscular Centre (ENMC) define a definite or possible IBM by histological as well as several detailed clinical features [39,40].
Muscle biopsy reveals inflammatory and degenerative mechanisms. The inflammatory process is similar to PM, with an invasion of non-necrotic fibres by macrophages and cytotoxic CD8+ T cells [15]. There is an over-expression of metalloproteinaeses 2 and 9 on non-necrotic muscle fibres, as in PM [21]. The pattern of expression of chemokines and cytokines is also comparable or even higher than in PM [12]. Degenerative components include rimmed vacuoles and intracellular deposits of β-amyloid which are visualized, for example, by histochemical staining with Congo red [41] or thioflavine-S [12]. Other neurodegeneration-related proteins accumulate similarly, including p-tau, presenilin 1, apolipoprotein, γ-tubulin, clusterin, α-synuclein and gelsolin [41]. Cell stress appears to be a crucial component of the complex pathogenesis of IBM, as evidenced by co-localization of αB-crystallin and APP/β-amyloid [42]. It has been suggested that, under proinflammatory conditions, inducible nitric oxide synthase is up-regulated and causes fibre death [43]. Macroautophagic processing has been attributed to contribute to the accumulation of aberrant proteins [44], particularly under proinflammatory conditions [45,46].
Focal myositis and overlap syndromes
Focal forms of myositis involve only one or few distinct muscles of one arm, leg or eye [47]. The causes of focal myositis include sarcoidosis, systemic lupus erythematosus, Crohn's disease or anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis. Oral therapy with corticosteroids can be effective, but chronic forms may require immunosuppression or even local radiotherapy for ocular myositis.
Overlap syndromes are defined as a combination of DM/PM, rheumatoid arthritis, scleroderma, systemic lupus erythematosus or Sjögren's syndrome [48]. It is also hypothesized that overlap syndromes are defined clinical entities, and more than simply a combination of two diseases. This hypothesis is based on the finding of specific autoantibodies in some cases, e.g. in anti-synthetase syndromes. Extramuscular symptoms such as arthritis and Raynaud's phenomenon are most common [49]. Laboratory, electromyographic and muscle biopsy findings are similar to other forms of myositis. Overlap syndromes are associated with autoantibodies such as Jo-1, PM/SCL, U1RNP and others [50]. Patients are treated with corticosteroids, immunosuppressants or monoclonal antibodies, such as anti-TNF-α or anti-CD20 [48].
Myositis-specific autoantibodies (MSA) and associated diseases
MSA can be found in patients with myositis and support the correct diagnosis. Most common among these is the Jo-1 autoantibody, which is found in approximately 20% of adult patients with myositis [51,52]. It belongs to the group of aminoacyl-tRNA-synthetase autoantibodies and targets histidyl-tRNA-synthetase. Other autoantibodies, including PL-7, PL-12, OJ, EJ, KS, Ha and Zo, are directed against different synthetases and their frequency is below 5%. They lead to a similar phenotype, called anti-synthetase syndrome. Clinical manifestations are ILD, myositis, arthritis, fever, etc. With a frequency of 79–95%, ILD is the most common extramuscular manifestation [53]. The above-mentioned anti-CADM-140 also shows a strong association to ILD [4].
It has been well established that the Mi-2 autoantibody is a marker for DM, associated with the classical cutaneous phenotype without ILD or malignancy [54]. Some newly described autoantibodies are under further investigation, and evidence shows that MDA5, NXP2 and transcription intermediary factor (TIF)-1γ are also associated preferentially with DM [55,56]. The anti-155/140 autoantibody targets the TIF-1γ [55] and is associated strongly with malignancy [54], which should prompt thorough tumour screening in these patients.
Possible IBM-specific autoantibodies were detected in a recent study [57]: in sera of 25 IBM patients, 52% displayed an antibody against a 43 kDa protein while other samples from healthy controls or other diseases did not recognize this protein. Another study revealed the reactivity of recombinant immunoglobulins produced by plasma cells derived from IBM muscle tissue. For one of these recombinant immunoglobulins, desmin was identified as the target [58]. Two recent studies have identified an autoantibody to a muscle protein of 44 kDa (‘Mup44’), which was identified as the cytosolic 5'-nucleotidase 1A (cN1A) [59,60]. This antibody is present in ˜60% of patients with IBM, but it is not specific in view of other patients with myositis that are also positive. For all antibodies it is important to be aware of the fact that it is still unknown whether or not they are pathogenic or simply an epiphenomenon as the result of promiscuous activation of the immune system.
One recent analysis of epidemiological studies with the total number of 2439 patients revealed an underlying malignoma in up to 24% of DM and 10% of PM patients [5]. This association was found for several tumours, most of all lung tumours in western countries and nasopharyngeal cancer in Asia and northern Africa [5]. Most tumours are detected within the first years after the onset of myositis. This supports the hypothesis that the development of myositis in individual cases might be paraneoplastic [61]. NM has been shown to be associated with gastrointestinal tumours, small cell lung cancer or breast cancer [62]. For IBM, only few cases exist of the co-existence of a malignoma.
Cardiac involvement is observed regularly in patients with DM or PM [63]. Typical findings are electrocardiographic changes, valve disease, coronary vasculitis, ischaemia, heart failure and myocarditis. In patients with myositis, extramuscular manifestations such as rapid-progressive ILD, severe cardiac involvement or tumour can lead to a poor prognosis.
Diagnostics
A precise medical history and medical examination are prerequisites for the accurate diagnosis of myositis. The paresis distribution may reveal a pattern typical for IBM, as opposed to a proximal weakness, which is similar in DM, PM and NM. Laboratory examination usually reveals an increased CK, and sometimes other enzymes such as LDH, AST or ALT and myositis-specific autoantibodies may be detected.
Needle EMG of affected muscles usually displays a myopathic pattern, and signs of acute damage may be noted. A retrospective Dutch study with 98 patients with myositis revealed that none of the patients with the diagnoses DM, PM or IBM had a normal needle EMG [64]. Alterations in signal intensity within the muscle can be detected using MRI [65]. In the early stages of myositis there is oedema; in later stages fatty transformation or muscle atrophy can be observed. Even though these changes are not specific to myositis and can also be seen, e.g. in injuries, muscle infarction, subacute denervation or rhabdomyolysis, they are valuable to identify affected muscle groups and make an adequate diagnosis. Most importantly, MRI may help to identify subclinical involvement of muscles, which may point to another disease such as a muscular dystrophy.
A biopsy is the most important and most invasive step to make the correct diagnosis; it is the only way to distinguish between the different subtypes of myositis. It is crucial to rule out muscular dystrophy or other forms of a hereditary myopathy. To achieve this, it is important to choose a representative muscle for the biopsy: usually, this muscle should demonstrate moderate paresis. MRI can help to identify end-stage changes of tissue destruction.
A chest X-ray should be performed for the detection of an ILD. High-resolution CT may be performed, particularly in patients with anti-synthetase autoantibodies and anti-CADM. This technique allows the detection of pulmonary changes prior to the appearance of clinical symptoms [66]. In patients with high risk of a cardiac involvement, regular cardiological examinations should take place. Assessment of the bone density should be considered during prednisone therapy at a daily dosage of more than 5 mg [67].
While dysphagia is reported to be highly frequent in IBM, patients with DM and PM also often display signs of swallowing difficulties [37,68]. A high incidence of self-reported dysphagia in patients with myositis was found, suggesting that every examination should include questions regarding this topic [69]. When dysphagia is reported, additional diagnostics may be conducted, including videofluoroscopy or flexible endoscopic evaluation of swallowing (FEES) [70].
Treatment of DM, PM and NM
Treatment is usually initiated with pulsed intravenous glucocorticosteroids, e.g. 250–1000 mg prednisolone per day for 3–5 days (Fig. 1). The following standard oral treatment usually consists of prednisone 1 mg/kg/day. This dosage is usually administered for at least 4 weeks. After initial stabilization, which may take 4–12 weeks, the dose can be tapered every 1 or 2 weeks by 10 mg until 20 mg/day is reached. Subsequently, the taper is slowed to a reduction by 5 mg until 10 mg daily and to 2·5 mg thereafter. The therapy is based on early reports suggesting a positive effect of corticosteroids on muscle strength [71], even though this effect has never been proved formally in a prospective double-blind study; nor has a scheme for the tapering ever been proved. The rate of reduction is dependent upon the patient's response, and in the case of renewed deterioration the taper has to be stopped or slowed or the dosage even increased. An alternate day regimen is a useful alternative and may help to reduce side-effects. Monthly 4-day courses of 40 mg dexamethasone as an ‘oral pulse therapy’ displayed comparable efficacy to daily prednisone, but significantly fewer side effects [72]. The risk of a fracture is increased under therapy with prednisone in a daily dosage of more than 5 mg or by duration of more than 3 months. Concomitant treatment with 1000 mg calcium carbonate and 500 IU vitamin D per day is advisable [67]. Steroid-induced myopathy is an important side effect of glucocorticosteroids, with slowly progressive proximal muscle weakness that can mimic a relapse [73]. Acute changes in EMG are typically expected in a relapse and may help to distinguish one from the other.
Figure 1.
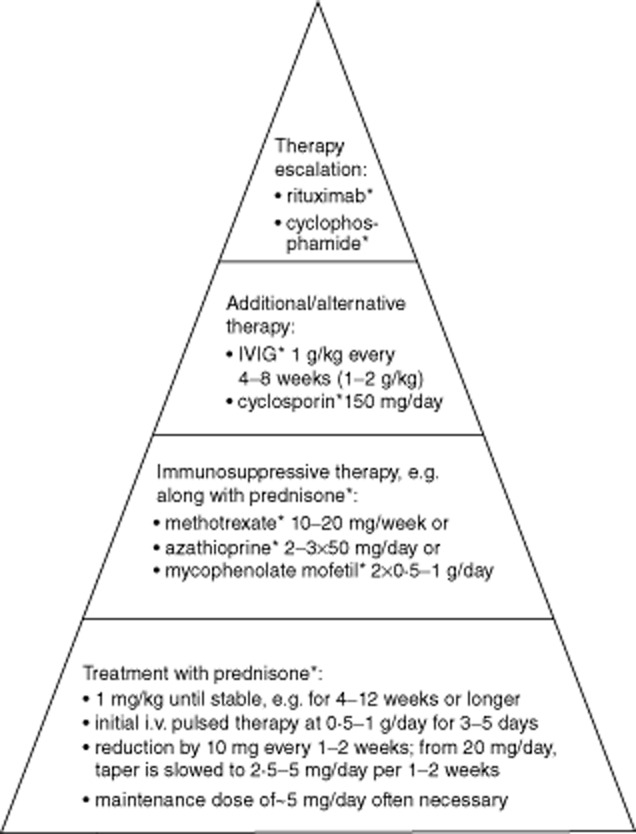
Therapy of dermatomyositis, polymyositis and necrotizing myopathy. The dose is to be adapted in each individual case; further information within the text. Adapted from [109] with the kind permission of Springer Science and Business Media.
Additional immunosuppressive therapy can be started simultaneously, unless only very mild symptoms are present. This treatment usually helps to spare prednisone. A recent Cochrane Review reveals the lack of randomized controlled trials (RCT) concerning immunosuppressive therapy in myositis [74]. Only 10 studies with a total of 258 patients were considered relevant and included. Six studies compared the therapy with immunoglobulin (IVIG), etanercept, eculizumab, infliximab, azathioprine or leucapheresis with placebo, and significant improvement in muscle strength was detected only for IVIG. The other four studies compared the effect of two immunosuppressive therapies and no statistically significant difference was found. It can be summarized that there is no convincing evidence for the efficacy of the commonly used immunosuppressive agents in myositis such as methotrexate (MTX), azathioprine and mycophenolate mofetil (MMF). However, in view of the immunopathogenesis, an international consensus of experts has recommended that immunosuppressants should be used. More prospective and controlled studies with an adequate cohort size are necessary.
MTX is administered in a weekly single dose of 5–20 mg, usually 10–15 mg, followed by leucoverin rescue on the subsequent day. The application of high-dose folic acid can reduce the toxicity and side effects of MTX as the drug interferes in the folate metabolism [75]. Common side effects include increase of the liver enzymes. Treatment with azathioprine is started with a daily dosage of 50 mg over 1 week. The dosage is increased weekly and monitored by the number of lymphocytes, which should be 600–1000/μl. A major potential side effect is bone marrow suppression, especially in patients with thiopurine methyltransferase deficiency. Thus, the activity of this enzyme should be measured before initiation of the treatment. A further frequent side effect is an increase of the liver enzymes. Frequent controls of the blood count and liver enzymes are essential during treatment with an immunosuppressant. The interval can be extended once the maintenance dosage is reached. MMF is the third common and orally administered immunopressant in myositis. Side effects are less frequent compared to MTX or azathioprine and include toxicity on kidney and liver. The therapy is started with a dosage of 500 mg twice daily and can be increased to 2 g or even 3 g per day. The use of cyclosporin as another alternative is limited by its common toxicity on liver and kidney.
IVIG can be tried as an alternative when the side effects from immunosuppressants outweigh their clinical benefit. It can also be used as an add-on treatment during a relapse or when immunosuppressants are not sufficiently effective. In addition, IVIG is a treatment option when immunosuppression is not wanted, e.g. in child-bearing women or adolescent patients. Usually, a total dosage of 2 g/kg is administered initially. The therapy is repeated regularly every 4–8 weeks with a dose of 1 g/kg, and tapering depends upon the treatment effect. Relevant side effects are an increased risk for thrombosis, fever and allergic reactions/anaphylaxia [76]. The risk for the allergic reaction is increased in patients with an immunoglobulin (Ig)A deficiency, which should be excluded before starting the treatment.
Women of childbearing potential can be treated with IVIG as a safe option [77]. MTX, azathioprin and MMF are potentially teratogenic, which makes an effective contraception essential [78]. A pregnancy should not be planned before withdrawal from immunosuppressants for several months, as active metabolites can persist in the tissues for several weeks [79].
In recent studies, the effect of monoclonal antibodies in the treatment of myositis was investigated: rituximab leads to a depletion of B cells by targeting CD20. A prospective, double-blind trial with rituximab in myositis revealed a steroid-sparing effect and 83% of the patients who were refractory to prior treatments improved during a period of 44 weeks [80]. Similar results were found in smaller cohorts [81,82]. The TNF-α inhibitor etanercept was tested in a small, double-blind, placebo-controlled study including 16 patients with DM [83]. Even though the patients experienced no improvement, a significant steroid-sparing effect was detected.
Other substances, such as cyclosporin or cyclophosphamide, are used less often, due partly to a higher risk of side effects. However, these drugs can be very useful for escalation therapy in individual patients, particularly when all other treatments have failed.
Based on our clinical experience, we treat our patients with 1 mg/kg oral prednisone daily. Upon improvement, e.g. after 6–8 weeks, we slowly taper the prednisone and start with azathioprin at 50–150 mg/day. We consider MMF and MTX as second or third choices, e.g. in the case of side effects from azathioprin. If treatment with any of these drugs alone remains ineffective, we additionally administer intravenous immunoglobulins. Options for treatment escalation include a CD20 blockade with rituximab or maximal immunosuppression with cyclophosphamide. We recommend regular physiotherapy, e.g. twice a week, as an essential part of the treatment. There is growing evidence for the safety and beneficial effects of physiotherapy and home exercise programmes in myositis [84].
Treatment of IBM
Treatment of IBM is a challenge, despite increasing understanding of its pathology. It remains controversial whether or not the inflammatory mechanisms are cause or consequence of the degeneration or if both cascades occur independently. An underlying degenerative cascade could explain the resistance of IBM to immunosuppression. Prednisone, the standard therapy of the other subtypes of myositis, is usually not effective in IBM [85]; however, individual patients may experience at least a temporary improvement. Conversely, one recent study revealed that progression towards handicap for walking was more rapid among patients receiving immunosuppressive drugs [36]. This finding could be explained by either a side effect of the treatment or the suggestion that more severely affected patients receive treatment more often.
Other studies with MTX, compared to MTX and anti-T lymphocyte globulin [86], etanercept [87], oxandrolone [88] or normal or high-dose IFN-β [89,90], failed to identify clinical efficacy. MTX compared to placebo led to a significant decrease of the CK but the disease progression was unaltered [91]. More encouraging is a proof-of-principle study in which alemtuzumab seemed to reduce the disease progression for up to 6 months, and in some patients the muscle strength was improved [92]. However, these data should be interpreted with care, as this was an unblinded study and the rate of the yearly disease progression was higher compared to recent natural history studies [35,36]. Major side effects of alemtuzumab are the development of an autoimmune thyroiditis or idiopathic thrombocytopenic purpura [92]. In three controlled prospective studies with IVIG, no increase of the limb strength could be observed [93–96], whereas the dysphagia was improved significantly [94,97,98].
Most recently, pilot trials with other drugs have been performed. Simvastatin is supposed to have anti-inflammatory effects, but it failed to improve muscle weakness after 12 months of treatment [99]. Two other trials in IBM have been completed: lithium, an inhibitor of the tau-phosphorylating enzyme glycogen synthase kinase-3β, and arimoclomol, a drug that reduces the heat shock response and has been studied in amyotrophic lateral sclerosis [100] [NCT00917956 (http://www.clinicaltrials.gov/ct2/show/NCT00917956); NCT00769860 (http://www.clinicaltrials.gov/ct2/show/NCT00769860)]. Further treatment studies on IBM include BYM338/bimagrumab and follistatin gene transfer and are currently ongoing [NCT01925209 (http://www.clinicaltrials.gov/ct2/show/NCT01925209); NCT01519349 (http://www.clinicaltrials.gov/ct2/show/NCT01519349)].
In a small open study, a statistically significant improvement of the most affected muscle groups in IBM was shown by a 16-week twice-a-day home exercise programme [84]. In view of the resistance of IBM to immunosuppressive therapy, we consider physiotherapy and regular home exercise to be an essential element of the therapy.
Treatment of dysphagia
As noted previously, dysphagia can occur in all subtypes of myositis. A high percentage of patients with IBM is affected [35–37]. Dysphagia leads to a reduced quality of life and the risk for malnutrition and aspiration pneumonia is increased [101]. Treatment with IVIG improves swallowing in IBM (see above). IVIG is also beneficial for patients with prednisolone-resistant dysphagia and DM or PM. In one study, approximately 82% of 73 patients were able to return to oral feeding [102].
Other uncontrolled studies, each with a small number of patients, addressed the effect of the following interventions in IBM: cricopharyngeal myotomy, pharyngoesophageal dilatation, percutaneous endoscopic gastrostomy (PEG) and injection of botulinum toxin. A benefit was shown for both myotomy and dilatation, but the results were limited by the number of only 10 patients [98,103,104]. So far, treatment with botulinum toxin has shown different results, ranging between no effect and improvement in swallowing for several months [104,105]. Among the causes of death in patients with IBM, cachexia and (aspiration) pneumonia are increased significantly in comparison to the normal population [35,36]. PEG may be helpful as prevention, but further studies regarding this point are missing. In one retrospective study, five of six patients with PEG died because of aspiration and respiratory failure, while the cause of death of the sixth patient remains unknown. This finding could be explained by the fact that patients with severe dysphagia and PEG still exhibit an increased risk for aspiration pneumonia [106]. Similar to IBM, a balloon dilatation might be beneficial in PM and improve swallowing [107].
In summary, IVIG might be useful in patients with severe swallowing disturbances. The literature concerning the benefit of invasive interventions is controversial and, at present, no procedure can be recommended. A recent study concerning the underlying mechanisms of dysphagia in myositis revealed that an abnormal hyolaryngeal excursion is more likely to be the reason than the often-supposed failed relaxation of the upper oesophageal sphincter [108]. The authors would like to point out that the often-performed myotomy is not indicated in patients with normal relaxation. Controlled trials are much needed to address the treatment of dysphagia, particularly in IBM.
Conclusions
The four different forms of myositis (DM, PM, NM, IBM) can be distinguished by certain clinical clues on examination and additional diagnostics, particularly a muscle biopsy. Despite many recent studies and growing knowledge of the pathogenesis of myositis, our treatment of DM, PM and NM is still based more on experience than on prospective double-blind studies with an adequate number of patients. IBM remains a challenge due to its complex pathogenesis and lack of effective treatment. In view of the growing interest in rare disorders and the development of new therapeutic approaches, including novel biologicals, hope for better care of these patients appears to be justified.
Disclosures
J. S. has received payments for consultancies, talks, honoraria, reimbursements for travel and research grants from Bayer, Biotest, CSL Behring, Novartis and Octapharma.
References
- 1.Furst DE, Amato AA, Iorga SR, Gajria K, Fernandes AW. Epidemiology of adult idiopathic inflammatory myopathies in a U.S. managed care plan. Muscle Nerve. 2012;45:676–683. doi: 10.1002/mus.23302. [DOI] [PubMed] [Google Scholar]
- 2.Martin N, Li CK, Wedderburn LR. Juvenile dermatomyositis: new insights and new treatment strategies. Ther Adv Musculoskelet Dis. 2012;4:41–50. doi: 10.1177/1759720X11424460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Callen JP. Dermatomyositis. Lancet. 2000;355:53–57. doi: 10.1016/S0140-6736(99)05157-0. [DOI] [PubMed] [Google Scholar]
- 4.Sato S, Kuwana M, Fujita T, Suzuki Y. Anti-CADM-140/MDA5 autoantibody titer correlates with disease activity and predicts disease outcome in patients with dermatomyositis and rapidly progressive interstitial lung disease. Mod Rheumatol. 2013;23:496–502. doi: 10.1007/s10165-012-0663-4. [DOI] [PubMed] [Google Scholar]
- 5.Zahr ZA, Baer AN. Malignancy in myositis. Curr Rheumatol Rep. 2011;13:208–215. doi: 10.1007/s11926-011-0169-7. [DOI] [PubMed] [Google Scholar]
- 6.Ghazi E, Sontheimer RD, Werth VP. The importance of including amyopathic dermatomyositis in the idiopathic inflammatory myositis spectrum. Clin Exp Rheumatol. 2013;31:128–134. [PMC free article] [PubMed] [Google Scholar]
- 7.Sun Y, Liu Y, Yan B, Shi G. Interstitial lung disease in clinically amyopathic dermatomyositis (CADM) patients: a retrospective study of 41 Chinese Han patients. Rheumatol Int. 2013;33:1295–1302. doi: 10.1007/s00296-012-2545-7. [DOI] [PubMed] [Google Scholar]
- 8.Kissel JT, Mendell JR, Rammohan KW. Microvascular deposition of complement membrane attack complex in dermatomyositis. N Engl J Med. 1986;314:329–334. doi: 10.1056/NEJM198602063140601. [DOI] [PubMed] [Google Scholar]
- 9.Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet. 2003;362:971–982. doi: 10.1016/S0140-6736(03)14368-1. [DOI] [PubMed] [Google Scholar]
- 10.Greenberg SA. Dermatomyositis and type 1 interferons. Curr Rheumatol Rep. 2010;12:198–203. doi: 10.1007/s11926-010-0101-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baechler EC, Bilgic H, Reed AM. Type I interferon pathway in adult and juvenile dermatomyositis. Arthritis Res Ther. 2011;13:249. doi: 10.1186/ar3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schmidt J, Barthel K, Wrede A, Salajegheh M, Bahr M, Dalakas MC. Interrelation of inflammation and APP in sIBM: IL-1 beta induces accumulation of beta-amyloid in skeletal muscle. Brain. 2008;131:1228–1240. doi: 10.1093/brain/awn053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Civatte M, Schleinitz N, Krammer P, et al. Class I MHC detection as a diagnostic tool in noninformative muscle biopsies of patients suffering from dermatomyositis (DM) Neuropathol Appl Neurobiol. 2003;29:546–552. doi: 10.1046/j.1365-2990.2003.00471.x. [DOI] [PubMed] [Google Scholar]
- 14.De Paepe B, Creus KK, De Bleecker JL. Role of cytokines and chemokines in idiopathic inflammatory myopathies. Curr Opin Rheumatol. 2009;21:610–616. doi: 10.1097/BOR.0b013e3283317b31. [DOI] [PubMed] [Google Scholar]
- 15.Schmidt J, Dalakas MC. Pathomechanisms of inflammatory myopathies: recent advances and implications for diagnosis and therapies. Expert Opin Med Diagn. 2010;4:241–250. doi: 10.1517/17530051003713499. [DOI] [PubMed] [Google Scholar]
- 16.Magro CM, Dyrsen M, Kerns MJ. Cutaneous lesions of dermatomyositis with supervening fibrosis. J Cutan Pathol. 2008;35:31–39. doi: 10.1111/j.1600-0560.2007.00770.x. [DOI] [PubMed] [Google Scholar]
- 17.Hausmann G, Herrero C, Cid MC, Casademont J, Lecha M, Mascaro JM. Immunopathologic study of skin lesions in dermatomyositis. J Am Acad Dermatol. 1991;25:225–230. doi: 10.1016/0190-9622(91)70186-6. [DOI] [PubMed] [Google Scholar]
- 18.Mascaro JM, Jr, Hausmann G, Herrero C, et al. Membrane attack complex deposits in cutaneous lesions of dermatomyositis. Arch Dermatol. 1995;131:1386–1392. doi: 10.1001/archderm.1995.01690240040007. [DOI] [PubMed] [Google Scholar]
- 19.Mastaglia FL, Garlepp MJ, Phillips BA, Zilko PJ. Inflammatory myopathies: clinical, diagnostic and therapeutic aspects. Muscle Nerve. 2003;27:407–425. doi: 10.1002/mus.10313. [DOI] [PubMed] [Google Scholar]
- 20.De Paepe B, Creus KK, De Bleecker JL. The tumor necrosis factor superfamily of cytokines in the inflammatory myopathies: potential targets for therapy. Clin Dev Immunol. 2012;2012:369432. doi: 10.1155/2012/369432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choi YC, Dalakas MC. Expression of matrix metalloproteinases in the muscle of patients with inflammatory myopathies. Neurology. 2000;54:65–71. doi: 10.1212/wnl.54.1.65. [DOI] [PubMed] [Google Scholar]
- 22.Das L, Blumbergs PC, Manavis J, Limaye VS. Major histocompatibility complex class I and II expression in idiopathic inflammatory myopathy. Appl Immunohistochem Mol Morphol. 2013 doi: 10.1097/PAI.0b013e31827d7f16. doi: 10.1097/PAI.0b013e31827d7f16. [DOI] [PubMed] [Google Scholar]
- 23.Karpati G, Pouliot Y, Carpenter S. Expression of immunoreactive major histocompatibility complex products in human skeletal muscles. Ann Neurol. 1988;23:64–72. doi: 10.1002/ana.410230111. [DOI] [PubMed] [Google Scholar]
- 24.Ellis E, Ann TJ, Lester S, et al. Necrotizing myopathy: clinicoserologic associations. Muscle Nerve. 2012;45:189–194. doi: 10.1002/mus.22279. [DOI] [PubMed] [Google Scholar]
- 25.Miller T, Al-Lozi MT, Lopate G, Pestronk A. Myopathy with antibodies to the signal recognition particle: clinical and pathological features. J Neurol Neurosurg Psychiatry. 2002;73:420–428. doi: 10.1136/jnnp.73.4.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stenzel W, Goebel HH, Aronica E. Immune-mediated necrotizing myopathies – a heterogeneous group of diseases with specific myopathological features [Review] Neuropathol Appl Neurobiol. 2012;38:632–646. doi: 10.1111/j.1365-2990.2012.01302.x. [DOI] [PubMed] [Google Scholar]
- 27.Preusse C, Goebel HH, Held J, et al. Immune-mediated necrotizing myopathy is characterized by a specific Th1-M1 polarized immune profile. Am J Pathol. 2012;181:2161–2171. doi: 10.1016/j.ajpath.2012.08.033. [DOI] [PubMed] [Google Scholar]
- 28.Hengstman GJ, terLaak HJ, Vree Egberts WT, et al. Anti-signal recognition particle autoantibodies: marker of a necrotising myopathy. Ann Rheum Dis. 2006;65:1635–1638. doi: 10.1136/ard.2006.052191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mammen AL, Chung T, Christopher-Stine L, et al. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase in patients with statin-associated autoimmune myopathy. Arthritis Rheum. 2011;63:713–721. doi: 10.1002/art.30156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Benveniste O, Drouot L, Jouen F, et al. Correlation of anti-signal recognition particle autoantibody levels with creatine kinase activity in patients with necrotizing myopathy. Arthritis Rheum. 2011;63:1961–1971. doi: 10.1002/art.30344. [DOI] [PubMed] [Google Scholar]
- 31.Liang C, Needham M. Necrotizing autoimmune myopathy. Curr Opin Rheumatol. 2011;23:612–619. doi: 10.1097/BOR.0b013e32834b324b. [DOI] [PubMed] [Google Scholar]
- 32.Bronner IM, Hoogendijk JE, Wintzen AR, et al. Necrotising myopathy, an unusual presentation of a steroid-responsive myopathy. J Neurol. 2003;250:480–485. doi: 10.1007/s00415-003-1027-y. [DOI] [PubMed] [Google Scholar]
- 33.Mastaglia FL, Needham M. Update on toxic myopathies. Curr Neurol Neurosci Rep. 2012;12:54–61. doi: 10.1007/s11910-011-0232-9. [DOI] [PubMed] [Google Scholar]
- 34.Needham M, Corbett A, Day T, Christiansen F, Fabian V, Mastaglia FL. Prevalence of sporadic inclusion body myositis and factors contributing to delayed diagnosis. J Clin Neurosci. 2008;15:1350–1353. doi: 10.1016/j.jocn.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 35.Cox FM, Titulaer MJ, Sont JK, Wintzen AR, Verschuuren JJ, Badrising UA. A 12-year follow-up in sporadic inclusion body myositis: an end stage with major disabilities. Brain. 2011;134:3167–3175. doi: 10.1093/brain/awr217. [DOI] [PubMed] [Google Scholar]
- 36.Benveniste O, Guiguet M, Freebody J, et al. Long-term observational study of sporadic inclusion body myositis. Brain. 2011;134:3176–3184. doi: 10.1093/brain/awr213. [DOI] [PubMed] [Google Scholar]
- 37.Cox FM, Verschuuren JJ, Verbist BM, Niks EH, Wintzen AR, Badrising UA. Detecting dysphagia in inclusion body myositis. J Neurol. 2009;256:2009–2013. doi: 10.1007/s00415-009-5229-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Griggs RC, Askanas V, DiMauro S, et al. Inclusion body myositis and myopathies. Ann Neurol. 1995;38:705–713. doi: 10.1002/ana.410380504. [DOI] [PubMed] [Google Scholar]
- 39.Badrising UA, Maat-Schieman M, vanDuinen SG, et al. Epidemiology of inclusion body myositis in the Netherlands: a nationwide study. Neurology. 2000;55:1385–1387. doi: 10.1212/wnl.55.9.1385. [DOI] [PubMed] [Google Scholar]
- 40.Hilton-Jones D, Miller A, Parton M, Holton J, Sewry C, Hanna MG. Inclusion body myositis: MRC Centre for Neuromuscular Diseases, IBM workshop, London, 13 June 2008. Neuromuscul Disord. 2010;20:142–147. doi: 10.1016/j.nmd.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 41.Askanas V, Engel WK, Nogalska A. Pathogenic considerations in sporadic inclusion-body myositis, a degenerative muscle disease associated with aging and abnormalities of myoproteostasis. J Neuropathol Exp Neurol. 2012;71:680–693. doi: 10.1097/NEN.0b013e31826183c8. [DOI] [PubMed] [Google Scholar]
- 42.Muth IE, Barthel K, Bahr M, Dalakas MC, Schmidt J. Proinflammatory cell stress in sporadic inclusion body myositis muscle: overexpression of alphaB-crystallin is associated with amyloid precursor protein and accumulation of beta-amyloid. J Neurol Neurosurg Psychiatry. 2009;80:1344–1349. doi: 10.1136/jnnp.2009.174276. [DOI] [PubMed] [Google Scholar]
- 43.Schmidt J, Barthel K, Zschuntzsch J, et al. Nitric oxide stress in sporadic inclusion body myositis muscle fibres: inhibition of inducible nitric oxide synthase prevents interleukin-1beta-induced accumulation of beta-amyloid and cell death. Brain. 2012;135:1102–1114. doi: 10.1093/brain/aws046. [DOI] [PubMed] [Google Scholar]
- 44.Lunemann JD, Schmidt J, Schmid D, et al. Beta-amyloid is a substrate of autophagy in sporadic inclusion body myositis. Ann Neurol. 2007;61:476–483. doi: 10.1002/ana.21115. [DOI] [PubMed] [Google Scholar]
- 45.Keller CW, Fokken C, Turville SG, et al. TNF-alpha induces macroautophagy and regulates MHC class II expression in human skeletal muscle cells. J Biol Chem. 2011;286:3970–3980. doi: 10.1074/jbc.M110.159392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Keller CW, Schmitz M, Munz C, Lunemann JD, Schmidt J. TNF-alpha upregulates macroautophagic processing of APP/beta-amyloid in a human rhabdomyosarcoma cell line. J Neurol Sci. 2013;325:103–107. doi: 10.1016/j.jns.2012.12.011. [DOI] [PubMed] [Google Scholar]
- 47.Fraser CL, Skalicky SE, Gurbaxani A, McCluskey P. Ocular myositis. Curr Allergy Asthma Rep. 2013;13:315–321. doi: 10.1007/s11882-012-0319-7. [DOI] [PubMed] [Google Scholar]
- 48.Laccarino L, Gatto M, Bettio S, et al. Overlap connective tissue disease syndromes. Autoimmun Rev. 2013;12:363–373. doi: 10.1016/j.autrev.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 49.Romans B, Cohen S. A rheumatologist's view of polymyositis/dermatomyositis: extracutaneous and extramuscular involvement and overlap syndromes. Clin Dermatol. 1988;6:15–22. doi: 10.1016/0738-081x(88)90046-6. [DOI] [PubMed] [Google Scholar]
- 50.Venables PJ. Polymyositis-associated overlap syndromes. Br J Rheumatol. 1996;35:305–306. doi: 10.1093/rheumatology/35.4.305. [DOI] [PubMed] [Google Scholar]
- 51.Mimori T, Nakashima R, Hosono Y. Interstitial lung disease in myositis: clinical subsets, biomarkers, and treatment. Curr Rheumatol Rep. 2012;14:264–274. doi: 10.1007/s11926-012-0246-6. [DOI] [PubMed] [Google Scholar]
- 52.Gunawardena H, Betteridge ZE, McHugh NJ. Myositis-specific autoantibodies: their clinical and pathogenic significance in disease expression. Rheumatology (Oxf) 2009;48:607–612. doi: 10.1093/rheumatology/kep078. [DOI] [PubMed] [Google Scholar]
- 53.Richards TJ, Eggebeen A, Gibson K, et al. Characterization and peripheral blood biomarker assessment of anti-Jo-1 antibody-positive interstitial lung disease. Arthritis Rheum. 2009;60:2183–2192. doi: 10.1002/art.24631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hamaguchi Y, Kuwana M, Hoshino K, et al. Clinical correlations with dermatomyositis-specific autoantibodies in adult Japanese patients with dermatomyositis: a multicenter cross-sectional study. Arch Dermatol. 2011;147:391–398. doi: 10.1001/archdermatol.2011.52. [DOI] [PubMed] [Google Scholar]
- 55.Casciola-Rosen L, Mammen AL. Myositis autoantibodies. Curr Opin Rheumatol. 2012;24:602–608. doi: 10.1097/BOR.0b013e328358bd85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gunawardena H, Betteridge ZE, McHugh NJ. Newly identified autoantibodies: relationship to idiopathic inflammatory myopathy subsets and pathogenesis. Curr Opin Rheumatol. 2008;20:675–680. doi: 10.1097/BOR.0b013e328313bff4. [DOI] [PubMed] [Google Scholar]
- 57.Salajegheh M, Lam T, Greenberg SA. Autoantibodies against a 43 KDa muscle protein in inclusion body myositis. PloS One. 2011;6:e20266. doi: 10.1371/journal.pone.0020266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ray A, Amato AA, Bradshaw EM, et al. Autoantibodies produced at the site of tissue damage provide evidence of humoral autoimmunity in inclusion body myositis. PLOS One. 2012;7:e46709. doi: 10.1371/journal.pone.0046709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Larman HB, Salajegheh M, Nazareno R, et al. Cytosolic 5′-nucleotidase 1A autoimmunity in sporadic inclusion body myositis. Ann Neurol. 2013;73:408–418. doi: 10.1002/ana.23840. [DOI] [PubMed] [Google Scholar]
- 60.Pluk H, vanHoeve BJ, vanDooren SH, et al. Autoantibodies to cytosolic 5′-nucleotidase 1A in inclusion body myositis. Ann Neurol. 2013;73:397–407. doi: 10.1002/ana.23822. [DOI] [PubMed] [Google Scholar]
- 61.Danko K, Ponyi A, Molnar AP, Andras C, Constantin T. Paraneoplastic myopathy. Curr Opin Rheumatol. 2009;21:594–598. doi: 10.1097/BOR.0b013e3283317fa5. [DOI] [PubMed] [Google Scholar]
- 62.Wegener S, Bremer J, Komminoth P, Jung HH, Weller M. Paraneoplastic necrotizing myopathy with a mild inflammatory component: a case report and review of the literature. Case Rep Oncol. 2010;3:88–92. doi: 10.1159/000308714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bazzani C, Cavazzana I, Ceribelli A, Vizzardi E, Dei CL, Franceschini F. Cardiological features in idiopathic inflammatory myopathies. J Cardiovasc Med (Hagerstown) 2010;11:906–911. doi: 10.2459/JCM.0b013e32833cdca8. [DOI] [PubMed] [Google Scholar]
- 64.Blijham PJ, Hengstman GJ, Hama-Amin AD, vanEngelen BG, Zwarts MJ. Needle electromyographic findings in 98 patients with myositis. Eur Neurol. 2006;55:183–188. doi: 10.1159/000093866. [DOI] [PubMed] [Google Scholar]
- 65.Curiel RV, Jones R, Brindle K. Magnetic resonance imaging of the idiopathic inflammatory myopathies: structural and clinical aspects. Ann NY Acad Sci. 2009;1154:101–114. doi: 10.1111/j.1749-6632.2009.04386.x. [DOI] [PubMed] [Google Scholar]
- 66.Winklehner A, Berger N, Maurer B, Distler O, Alkadhi H, Frauenfelder T. Screening for interstitial lung disease in systemic sclerosis: the diagnostic accuracy of HRCT image series with high increment and reduced number of slices. Ann Rheum Dis. 2012;71:549–552. doi: 10.1136/annrheumdis-2011-200564. [DOI] [PubMed] [Google Scholar]
- 67.Pereira RM, Carvalho JF, Paula AP, et al. Guidelines for the prevention and treatment of glucocorticoid-induced osteoporosis. Rev Bras Reumatol. 2012;52:580–593. [PubMed] [Google Scholar]
- 68.Ertekin C, Secil Y, Yuceyar N, Aydogdu I. Oropharyngeal dysphagia in polymyositis/dermatomyositis. Clin Neurol Neurosurg. 2004;107:32–37. doi: 10.1016/j.clineuro.2004.02.024. [DOI] [PubMed] [Google Scholar]
- 69.Mulcahy KP, Langdon PC, Mastaglia F. Dysphagia in inflammatory myopathy: self-report, incidence, and prevalence. Dysphagia. 2012;27:64–69. doi: 10.1007/s00455-011-9338-0. [DOI] [PubMed] [Google Scholar]
- 70.Langmore SE. Evaluation of oropharyngeal dysphagia: which diagnostic tool is superior? Curr Opin Otolaryngol Head Neck Surg. 2003;11:485–489. doi: 10.1097/00020840-200312000-00014. [DOI] [PubMed] [Google Scholar]
- 71.Micks RH, Mullaney J. Dermatomyositis successfully treated by prednisone. Ir J Med Sci. 1958;33:333–334. doi: 10.1007/BF02950398. [DOI] [PubMed] [Google Scholar]
- 72.van deVlekkert J, Hoogendijk JE, deHaan RJ, et al. Oral dexamethasone pulse therapy versus daily prednisolone in sub-acute onset myositis, a randomised clinical trial. Neuromuscul Disord. 2010;20:382–389. doi: 10.1016/j.nmd.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 73.Batchelor TT, Taylor LP, Thaler HT, Posner JB, DeAngelis LM. Steroid myopathy in cancer patients. Neurology. 1997;48:1234–1238. doi: 10.1212/wnl.48.5.1234. [DOI] [PubMed] [Google Scholar]
- 74.Gordon PA, Winer JB, Hoogendijk JE, Choy EH. Immunosuppressant and immunomodulatory treatment for dermatomyositis and polymyositis. Cochrane Database Syst Rev. 2012;(8) doi: 10.1002/14651858.CD003643.pub4. CD003643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Levitt M, Mosher MB, DeConti RC, et al. Improved therapeutic index of methotrexate with ‘leucovorin rescue’. Cancer Res. 1973;33:1729–1734. [PubMed] [Google Scholar]
- 76.Nydegger UE. Safety and side effects of i.v. immunoglobulin therapy. Clin Exp Rheumatol. 1996;14(Suppl. 15):S53–S57. [PubMed] [Google Scholar]
- 77.Lidar M, Langevitz P. Pregnancy issues in scleroderma. Autoimmun Rev. 2012;11:A515–A519. doi: 10.1016/j.autrev.2011.11.021. [DOI] [PubMed] [Google Scholar]
- 78.Makol A, Wright K, Amin S. Rheumatoid arthritis and pregnancy: safety considerations in pharmacological management. Drugs. 2011;71:1973–1987. doi: 10.2165/11596240-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 79.Ostensen M, Forger F. Management of RA medications in pregnant patients. Nat Rev Rheumatol. 2009;5:382–390. doi: 10.1038/nrrheum.2009.103. [DOI] [PubMed] [Google Scholar]
- 80.Oddis CV, Reed AM, Aggarwal R, et al. Rituximab in the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis: a randomized, placebo-phase trial. Arthritis Rheum. 2013;65:314–324. doi: 10.1002/art.37754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Levine TD. Rituximab in the treatment of dermatomyositis: an open-label pilot study. Arthritis Rheum. 2005;52:601–607. doi: 10.1002/art.20849. [DOI] [PubMed] [Google Scholar]
- 82.Mahler EA, Blom M, Voermans NC, vanEngelen BG, vanRiel PL, Vonk MC. Rituximab treatment in patients with refractory inflammatory myopathies. Rheumatology (Oxf) 2011;50:2206–2213. doi: 10.1093/rheumatology/ker088. [DOI] [PubMed] [Google Scholar]
- 83.Muscle Study Group. A randomized, pilot trial of etanercept in dermatomyositis. Ann Neurol. 2011;70:427–436. doi: 10.1002/ana.22477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Alexanderson H, Lundberg IE. Exercise as a therapeutic modality in patients with idiopathic inflammatory myopathies. Curr Opin Rheumatol. 2012;24:201–207. doi: 10.1097/BOR.0b013e32834f19f5. [DOI] [PubMed] [Google Scholar]
- 85.Lotz BP, Engel AG, Nishino H, Stevens JC, Litchy WJ. Inclusion body myositis. Observations in 40 patients. Brain. 1989;112:727–747. doi: 10.1093/brain/112.3.727. [DOI] [PubMed] [Google Scholar]
- 86.Lindberg C, Trysberg E, Tarkowski A, Oldfors A. Anti-T-lymphocyte globulin treatment in inclusion body myositis: a randomized pilot study. Neurology. 2003;61:260–262. doi: 10.1212/01.wnl.0000071852.27182.c7. [DOI] [PubMed] [Google Scholar]
- 87.Barohn RJ, Herbelin L, Kissel JT, et al. Pilot trial of etanercept in the treatment of inclusion-body myositis. Neurology. 2006;66:S123–S124. doi: 10.1212/01.wnl.0000192258.32408.54. [DOI] [PubMed] [Google Scholar]
- 88.Rutkove SB, Parker RA, Nardin RA, Connolly CE, Felice KJ, Raynor EM. A pilot randomized trial of oxandrolone in inclusion body myositis. Neurology. 2002;58:1081–1087. doi: 10.1212/wnl.58.7.1081. [DOI] [PubMed] [Google Scholar]
- 89.Muscle Study Group. Randomized pilot trial of betaINF1a (Avonex) in patients with inclusion body myositis. Neurology. 2001;57:1566–1570. doi: 10.1212/wnl.57.9.1566. [DOI] [PubMed] [Google Scholar]
- 90.Muscle Study Group. Randomized pilot trial of high-dose betaINF-1a in patients with inclusion body myositis. Neurology. 2004;63:718–720. doi: 10.1212/01.wnl.0000134675.98525.79. [DOI] [PubMed] [Google Scholar]
- 91.Badrising UA, Maat-Schieman ML, Ferrari MD, et al. Comparison of weakness progression in inclusion body myositis during treatment with methotrexate or placebo. Ann Neurol. 2002;51:369–372. doi: 10.1002/ana.10121. [DOI] [PubMed] [Google Scholar]
- 92.Dalakas MC, Rakocevic G, Schmidt J, et al. Effect of alemtuzumab (CAMPATH 1-H) in patients with inclusion-body myositis. Brain. 2009;132:1536–1544. doi: 10.1093/brain/awp104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Amato AA, Barohn RJ, Jackson CE, Pappert EJ, Sahenk Z, Kissel JT. Inclusion body myositis: treatment with intravenous immunoglobulin. Neurology. 1994;44:1516–1518. doi: 10.1212/wnl.44.8.1516. [DOI] [PubMed] [Google Scholar]
- 94.Dalakas MC, Sonies B, Dambrosia J, Sekul E, Cupler E, Sivakumar K. Treatment of inclusion-body myositis with IVIg: a double-blind, placebo-controlled study. Neurology. 1997;48:712–716. doi: 10.1212/wnl.48.3.712. [DOI] [PubMed] [Google Scholar]
- 95.Dalakas MC, Koffman B, Fujii M, Spector S, Sivakumar K, Cupler E. A controlled study of intravenous immunoglobulin combined with prednisone in the treatment of IBM. Neurology. 2001;56:323–327. doi: 10.1212/wnl.56.3.323. [DOI] [PubMed] [Google Scholar]
- 96.Walter MC, Lochmuller H, Toepfer M, et al. High-dose immunoglobulin therapy in sporadic inclusion body myositis: a double-blind, placebo-controlled study. J Neurol. 2000;247:22–28. doi: 10.1007/s004150050005. [DOI] [PubMed] [Google Scholar]
- 97.Cherin P, Pelletier S, Teixeira A, et al. Intravenous immunoglobulin for dysphagia of inclusion body myositis. Neurology. 2002;58:326–327. doi: 10.1212/wnl.58.2.326. [DOI] [PubMed] [Google Scholar]
- 98.Murata KY, Kouda K, Tajima F, Kondo T. Balloon dilation in sporadic inclusion body myositis patients with dysphagia. Clin Med Insights Case Rep. 2013;6:1–7. doi: 10.4137/CCRep.S10200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sancricca C, Mora M, Ricci E, Tonali PA, Mantegazza R, Mirabella M. Pilot trial of simvastatin in the treatment of sporadic inclusion-body myositis. Neurol Sci. 2011;32:841–847. doi: 10.1007/s10072-011-0657-6. [DOI] [PubMed] [Google Scholar]
- 100.Phukan J. Arimoclomol, a coinducer of heat shock proteins for the potential treatment of amyotrophic lateral sclerosis. IDrugs. 2010;13:482–496. [PubMed] [Google Scholar]
- 101.Ney DM, Weiss JM, Kind AJ, Robbins J. Senescent swallowing: impact, strategies, and interventions. Nutr Clin Pract. 2009;24:395–413. doi: 10.1177/0884533609332005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Marie I, Menard JF, Hatron PY, et al. Intravenous immunoglobulins for steroid-refractory esophageal involvement related to polymyositis and dermatomyositis: a series of 73 patients. Arthritis Care Res (Hoboken) 2010;62:1748–1755. doi: 10.1002/acr.20325. [DOI] [PubMed] [Google Scholar]
- 103.Oh TH, Brumfield KA, Hoskin TL, Stolp KA, Murray JA, Bassford JR. Dysphagia in inflammatory myopathy: clinical characteristics, treatment strategies, and outcome in 62 patients. Mayo Clin Proc. 2007;82:441–447. doi: 10.4065/82.4.441. [DOI] [PubMed] [Google Scholar]
- 104.Oh TH, Brumfield KA, Hoskin TL, Kasperbauer JL, Basford JR. Dysphagia in inclusion body myositis: clinical features, management, and clinical outcome. Am J Phys Med Rehabil. 2008;87:883–889. doi: 10.1097/PHM.0b013e31818a50e2. [DOI] [PubMed] [Google Scholar]
- 105.Liu LW, Tarnopolsky M, Armstrong D. Injection of botulinum toxin A to the upper esophageal sphincter for oropharyngeal dysphagia in two patients with inclusion body myositis. Can J Gastroenterol. 2004;18:397–399. doi: 10.1155/2004/360537. [DOI] [PubMed] [Google Scholar]
- 106.Kitamura T, Nakase H, Iizuka H. Risk factors for aspiration pneumonia after percutaneous endoscopic gastrostomy. Gerontology. 2007;53:224–227. doi: 10.1159/000100898. [DOI] [PubMed] [Google Scholar]
- 107.Nagano H, Yoshifuku K, Kurono Y. Polymyositis with dysphagia treated with endoscopic balloon dilatation. Auris Nasus Larynx. 2009;36:705–708. doi: 10.1016/j.anl.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 108.Langdon PC, Mulcahy K, Shepherd KL, Low VH, Mastaglia FL. Pharyngeal dysphagia in inflammatory muscle diseases resulting from impaired suprahyoid musculature. Dysphagia. 2012;27:408–417. doi: 10.1007/s00455-011-9384-7. [DOI] [PubMed] [Google Scholar]
- 109.Schmidt J, Vorgerd M. Standard treatment for myositis and muscular dystrophies. Nervenarzt. 2011;82:723–732. doi: 10.1007/s00115-010-2970-3. [DOI] [PubMed] [Google Scholar]