

Abstract
Multiple sclerosis (MS) and chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) represent chronic, autoimmune demyelinating disorders of the central and peripheral nervous system. Although both disorders share some fundamental pathogenic elements, treatments do not provide uniform effects across both disorders. We aim at providing an overview of current and future disease-modifying strategies in these disorders to demonstrate communalities and distinctions. Intravenous immunoglobulins (IVIG) have demonstrated short-and long-term beneficial effects in CIDP but are not effective in MS. Dimethyl fumarate (BG-12), teriflunomide and laquinimod are orally administered immunomodulatory drugs that are already approved or likely to be approved in the near future for the basic therapy of patients with relapsing–remitting MS (RRMS) due to positive results in Phase III clinical trials. However, clinical trials with these drugs in CIDP have not (yet) been initiated. Natalizumab and fingolimod are approved for the treatment of RRMS, and trials to evaluate their safety and efficacy in CIDP are now planned. Alemtuzumab, ocrelizumab and daclizumab respresent monoclonal antibodies in advanced stages of clinical development for their use in RRMS patients. Attempts to study the safety and efficacy of alemtuzumab and B cell-depleting anti-CD20 antibodies, i.e. rituximab, ocrelizumab or ofatumumab, in CIDP patients are currently under way. We provide an overview of the mechanism of action and clinical data available on disease-modifying immunotherapy options for MS and CIDP. Enhanced understanding of the relative effects of therapies in these two disorders may aid rational treatment selection and the development of innovative treatment approaches in the future.
Keywords: CIDP, immunosuppression, immunotherapy, multiple sclerosis
Other Articles published in this series.
Paraneoplastic neurological syndromes. Clinical and Experimental Immunology 2014, 175: 336–48.
Diagnosis, pathogenesis and treatment of myositis: recent advances. Clinical and Experimental Immunology 2014, 175: 349–58.
Monoclonal antibodies in treatment of multiple sclerosis. Clinical and Experimental Immunology 2014, 175: 373–84.
CLIPPERS: chronic lymphocytic inflammation with pontine perivascular enhancement responsive to steroids. Review of an increasingly recognized entity within the spectrum of inflammatory central nervous system disorders. Clinical and Experimental Immunology 2014, 175: 385–96.
Requirement for safety monitoring for approved multiple sclerosis therapies: an overview. Clinical and Experimental Immunology 2014, 175: 397–407.
Myasthenia gravis: an update for the clinician. Clinical and Experimental Immunology 2014, 175: 408–18.
Cerebral vasculitis in adults: what are the steps in order to establish the diagnosis? Red flags and pitfalls. Clinical and Experimental Immunology 2014, 175: 419–24.
Multiple sclerosis treatment and infectious issues: update 2013. Clinical and Experimental Immunology 2014, 175: 425–38.
Introduction
Multiple sclerosis (MS) and chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) share some fundamental immunological principles, with each representing a classic chronic, autoimmune demyelinating disorder of the central and peripheral nervous system [1,2].
MS is a chronic, autoimmune, inflammatory and degenerative disorder of the central nervous system (CNS). The majority of MS patients (80–90%) intially experience a relapsing−remitting disease course (RRMS), with alternating phases of clinical worsening, remission and stability. Over time, approximately half of MS patients convert from a relapsing−remitting to a secondary progressive disease course (SPMS), with continuous clinical worsening independent from relapses. In 10–20% of patients, the disorder presents with a primary progressive course (PPMS) with continuous clinical worsening, with and without additional relapses from the disease onset [2].
CIDP and its variants are chronic autoimmune inflammatory and degenerative disorders of the peripheral nervous system (PNS) that affect, to a varying extent, the spinal roots, plexus and nerve trunks in a multi-focal manner. CIDP evolves either in a chronic, progressive or relapsing manner, with partial or complete recovery between recurrences. Typically, a relapsing disease course presents in younger patients and a progressive disease course presents in older adults [1].
In both MS and CIDP, a dysfunction or failure of immune tolerance mechanisms is postulated to cause humoral and cellular autoimmunity to the complex of the myelin sheath and axon. Following their activation in secondary lymphatic organs (spleen, lymph node), myelin antigen-reactive CD4+ and CD8+ T cells are thought to transmigrate the blood−brain or blood−nerve barriers, leading to an influx of macrophages, B cells and antibodies into the CNS or PNS parenchyma. Damage to the myelin sheath and axon ensue due to several distinct molecular mechanisms (Fig. 1) [1,2]: first, a primary autoimmune response may result in damage to the complex of the myelin sheath and axon by (i) autoantibody and complement-mediated damage by macrophages and microglia, (ii) cytokine-mediated damage and (iii) cytotoxic damage by CD4+ and CD8+ T cells. Second, given an altered sensitivity of the immune system, primary damage to the myelin sheath or axons may trigger a secondary immune response. In addition to the proinflammatory, pathogenic effects of T and B cells, distinct subsets of these immune cells exert protective anti-inflammatory effects such as the release of neurotrophic factors and immunosuppressive cytokines.
Figure 1.
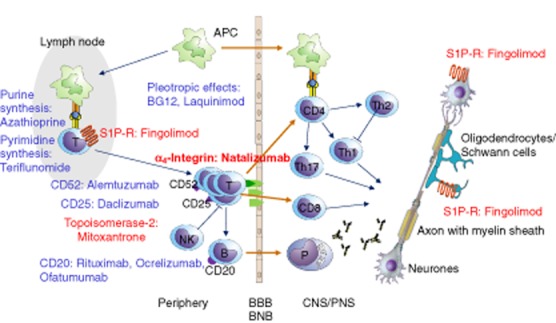
Common immunopathogenetic elements in multiple sclerosis (MS) and chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) and target molecules of approved (red) and potential future (blue) disease-modifying drugs (modified from [81]).
Disease-modifying immunotherapy approaches have provided great advances in the management of disorders such as MS or CIDP. Within the context of common pathogenic mechanisms, this review aims to summarize common or divergent clinical effects of disease-modifying treatment options across both disorders. This may deepen our understanding of the disease mechanism of each, and may assist with selecting the best treatment for each disorder.
As corticosteroids and plasma exchange are used predominantly to treat relapses and are not assumed to exert disease-modifying effects in both disorders, they are not the subject of this review. A detailed discussion of these treatment modalities can be found elsewhere [3–7].
Disease-modifying therapy in MS and CIDP
Immunomodulation with recombinant interferon-β (IFN-β) and glatiramer acetate (GA)
Immunmodulation with IFN-β
Preparations and applications: in clinically isolated syndrome (CIS) and RRMS, immunomodulation with recombinant IFN-β-1a [8–14], 1b [12–18] or GA [12,19–21] serves as basic therapy, which should be initiated as soon as possible after the diagnosis has been properly established. In addition, recombinant IFN-β may also be used in SPMS with residual inflammatory activity.
Four preparations are available in Europe and the United States for the treatment of MS patients with recombinant IFN-β (IFN-β-1a: Avonex®, Rebif®; IFN-β-1b: Betaferon®/Betaseron®, Extavia®). IFN-β-1b (Betaferon®/Betaseron®, Extavia®) is injected subcutaneously (s.c.) at a dose of 8 million IU every other day. IFN-β-1a is available in two different preparations: IFN-β-1a (Avonex®) is injected intramuscularly (i.m.) at a dose of 6 million IU (30 μg) once per week. IFN-β-1a (Rebif®) is injected subcutaneously at a dose of 22 μg or 44 μg thrice weekly.
Clinical trials: very recent data have emerged from a Phase III clinical trial that evaluated the 1-year efficacy and safety of peginterferon beta-1a in patients with RRMS. In this global, multi-centre, randomized, double-blind, parallel-group, placebo-controlled study (ADVANCE), more than 1500 patients with RRMS received either pegylated IFN-β-1a (125 μg) administered by s.c. injection every 2 or 4 weeks or placebo [22]. Pegylated IFN-β-1a provided a statistically significant reduction in the annualized relapse rate (ARR) by 35·6% (P < 0·001, 2-week dosing) and 27·5% (P < 0·02m 4-week dosing) compared to placebo. Moreover, pegylated IFN-β-1a reduced the risk of 12-week confirmed disability progression by 38% in both dosing arms (P < 0·04) and was superior to placebo across a range of MRI parameters.
Both dosing regimens showed favourable safety and tolerability profiles. The overall incidence of severe adverse events and adverse events was similar between the IFN-β-1a and placebo groups. The most common severe adverse events were infections (≤1% per group). The most commonly reported adverse events associated with pegylated IFN-β-1a treatment were redness at the injection site and influenza-like illness. Based on these data, Biogen is aiming for fast-track approval of pegylated IFN-β-1a for patients with RRMS in the United States and Europe in 2013.
In contrast, treatment with IFN-β-1a has failed to provide beneficial effects in patients with CIDP [23–25].
Adverse effects, frequent: flu-like symptoms, inflammation, redness and indurations at the side of puncture, induction or aggravation of depression and suicidality, aggravation of spasticity, elevation of liver enzymes; infrequent: aseptic skin necrosis, toxic hepatitis, leukopenia.
Immunmodulation with GA
Preparation and administration: in CIS and RRMS, immunomodulation with GA [12,19–21] serves as basic therapy, which should be initiated as soon as possible after the diagnosis has been properly established. GA (Copaxone®) is injected subcutaneously at a dose of 20 mg daily.
Clinical trials: a Phase III clinical trial (a study in subjects with RRMS to assess the efficacy, safety and tolerability of GA injection 40 mg administered three times a week compared to placebo – GALA) compared efficacy, safety and tolerability of GA injected s.c. at a dose of 40 mg thrice weekly to placebo in 1404 RRMS patients. The annualized relapse rate was reduced by 34·4% in the GA group versus placebo (P < 0·0001). At 12 months, the cumulative number of new/enlarging T2 lesions (34·7% reduction, P < 0·0001) and gadolinium enhanced (GdE) lesions (44·8% reduction, P < 0·0001) were significantly lower in GA-treated patients. Hence, GA at 40 mg thrice weekly may provide a potential alternative therapeutic option of using a higher dose of GA at a reduced injection frequency, but direct comparison to the standard dosing regimen of 20 mg daily has not been performed [26].
GA has not (yet) been tested in patients with CIDP.
Adverse effects, frequent: local side effects at the site of puncture (itching, redness, swelling, inflammation), lymph node swelling; infrequent: systemic post-injection reaction (SPIR), anaphylactic reactions.
Immunomodulation with intravenous immunoglobulins (IVIG)
IVIG consist of pooled polyclonal immunoglobulins derived from healthy donors. The precise mechanism by which IVIG suppress autoimmune inflammation has not been established definitively, but is likely to involve a plethora of molecular effects via their Fab-or Fc-fragments [27]:
Preparation and administration: for induction therapy of neurological autoimmune diseases, IVIG are administered intravenously at a dose of 2 g/kg (corresponding to 5 × 0·4 g/kg/day) of body weight once monthly for 3–6 months. Subsequently, maintenance therapy dose range is 0·1–0·4 g/kg of body weight, approximately every 4 weeks (depending on the individual patient's clinical course). IVIG effects usually last between 2 weeks and 3 months.
Clinical trials: in MS, IVIG have been tested for their efficacy in (i) relapse treatment, their impact on the (ii) relapse rate and disease progression in RRMS and on (iii) disease progression in SPMS.
Two studies compared IVIG versus placebo as add-on treatment to methylprednisolone in acute MS relapse. There was no statistically significant difference between the treatment groups [28,29]. Thus, IVIG are currently not recommended for the treatment of acute relapses in MS.
A well-designed trial with 127 patients with RRMS did not show a statistically significant reduction of the relapse rate and disease progression between two IVIG treatments (0·2 g/kg and 0·4 g/kg monthly) and placebo [30]. Thus, despite earlier trials suggesting some efficacy of IVIG in RRMS they are currently not recommended as first-line therapy for the disease-modifying treatment of patients with RRMS.
A randomized, placebo-controlled clinical trial (ESIMS) including 318 patients with SPMS compared IVIG (1 g/kg monthly) to placebo and did not show a difference in clinical parameters [31]. With regard to MRI outcomes, there was a delayed reduction of brain volume in the IVIG group [32], but the relevance of this finding is not clear. These data do not allow for a recommendation of IVIG in progressive MS and it can be concluded that IVIG is not effective in delaying disease progression in SPMS. Thus, IVIG are currently not recommended for the disease-modifying treatment of patients with SPMS.
In CIDP, several short-term clinical trials showed beneficial effects of IVIG compared with placebo, plasma-exchange or steroids [33–35]. However, long-term data on the efficacy of IVIG in CIDP have emerged only recently.
A recent randomized, double-blind, placebo-controlled, response-conditional cross-over trial included 117 patients with CIDP (ICE trail). The long-term efficacy of IVIG (baseline loading dose of 2 g/kg over 2–4 days and then a maintenance dose of 1 g/kg over 1–2 days every 3 weeks for up to 24 weeks) was compared with placebo [36]. IVIG or placebo was administered for up to 24 weeks in an initial treatment period; patients who did not show an improvement in INCAT disability score of ≥1 point received the alternate treatment in a cross-over treatment period. Patients who showed an improvement and completed 24 weeks of treatment were eligible to be reassigned randomly in a blinded 24-week extension phase.
The primary outcome was the percentage of patients who had maintained an improvement from baseline in adjusted INCAT disability score of 1 point or more to week 24. Secondary efficacy outcomes were (i) mean change from baseline in maximum grip strength at end-point during the initial treatment period; (ii) mean change from baseline in the compound muscle action potential amplitude after stimulation of the most severely affected motor nerve at the proximal site at end-point during the first period; and (iii) time to relapse for patients who were first-period adjusted-INCAT responders or cross-over-period adjusted-INCAT responders to IVIG and entered the extension phase. Relapse during the extension phase was defined as worsening of adjusted INCAT disability score by 1 point or more from the extension baseline value.
During the initial treatment period, 54% of patients treated with IVIG and 21% of patients who received placebo had an improvement in adjusted INCAT disability score that was maintained to week 24 (treatment difference 33·5%; P = 0·0002). Improvements from baseline to end-point were also recorded for grip strength in the dominant hand (treatment difference 10·9 kPa; P = 0·0008) and the non-dominant hand (8·6 kPa; P = 0·005). Results were similar during the second cross-over period. During the extension phase, participants who continued to receive IVIG had a longer time to relapse than did patients treated with placebo (P = 0·011).
This is the first study that demonstrates clearly the long-term efficacy and tolerability of IVIG in CIDP.
Another recent, multi-centre, randomized, double-blind, placebo controlled, parallel-group study in 45 patients with CIDP compared the efficacy and tolerability of IVIG (0·5 g/kg/day for 4 consecutive days) to intravenous methylprednisolone (0·5 g/day for 4 consecutive days) given every month for 6 months [37]. After therapy discontinuation, patients were followed-up for 6 months to assess relapses. The primary outcome was the number of patients discontinuing either therapy owing to inefficacy or intolerance. Secondary end-points included the proportion of patients experiencing adverse events or worsening after therapy discontinuation.
More patients stopped methylprednisolone (52%) than IVIG (13%) (P = 0·0085). The reasons for discontinuation were lack of efficacy, adverse events or voluntary withdrawal. After therapy discontinuation, more patients on IVIG worsened and required further therapy (38%) than did those on methylprednisolone (none) (P = 0·0317). Thus, treatment of CIDP with IVIG for 6 months was discontinued less frequently because of inefficacy, adverse events or intolerance than treatment with intravenous methylprednisolone.
Another recent prospective, multi-centre, single-arm, open-label Phase III study [Privigen® Impact on Mobility and Autonomy (PRIMA) trial] evaluated the efficacy and safety of IVIG in 28 patients with CIDP [38]. Patients received one induction dose of IVIG (2 g/kg body weight) and up to seven maintenance doses (1 g/kg body weight) at 3-week intervals. The overall responder rate defined as an improvement of ≥1 point on the INCAT disability scale at completion was 60·7%. IVIG-pretreated patients demonstrated a higher responder rate than IVIG-naive patients (76·9 versus 46·7%). The INCAT score, the maximum grip strength and the Medical Research Council sum score all improved significantly at completion compared to baseline.
Thus, these recent trials provide evidence for the long-term efficacy of IVIG in patients with CIDP.
Adverse effects, frequent: headache, hypertension, allergic/anaphylactic reactions [especially in immunoglobulin (Ig)A-deficient patients], dermatitis; infrequent: infection (HIV or viral hepatitis) by contaminated blood product, pulmonary oedema from fluid overload, due to the high colloid oncotic pressure of IVIG, venous thrombosis, aseptic meningitis and haemolysis.
‘Classic’ oral immunosuppressive drugs
In MS, ‘classic’ non-selective oral immunosuppressive drugs showed some clinical and paraclinical efficacy in various randomized controlled clinical trials. In CIDP, such drugs either showed no significant benefit or there were no efficacy data available from randomized controlled clinical trials.
Azathioprine
Azathioprine is a purine analogue that is metabolized rapidly to the cytotoxic and immunosuppressant derivatives 6-mercaptopurine and thioinosinic acid. The latter inhibits purine synthesis, impairs activation and proliferation and causes apoptosis of T cells and B cells due to their lack of metabolic pathways for nucleotide salvage (‘recycling’). Azathioprine is used widely in organ transplantation and in autoimmune disorders. Azathioprine has been the most widely used immunosuppressive treatment in MS prior to approval of immunomodulatory therapies.
Preparations and administration: azathioprine is usually administered orally at a dose of 2−3 mg/kg/day in two to three single doses.
Clinical trials: in a recent meta-analysis of five controlled, randomized clinical trials involving 698 patients with RRMS, azathioprine at a dose of 2−3 mg/kg/day reduced the relapse rate compared with placebo during the first year of treatment [relative risk reduction (RRR) = 20%], at 2 years' (RRR = 23%) and at 3 years' (RRR = 18%) follow-up [39]. Moreover, in three small trials with a total of 87 patients, azathioprine reduced the number of patients with disability progression (RRR = 42%) at 3 years' follow-up compared to placebo [39]. Unfortunately, data on MRI paramenters of inflammation or degeneration were not available [39].
In CIDP, azathioprine showed no significant benefit on primary (clinical disability) or secondary (electrophysiological parameters, demand for corticosteroids and/or IVIG) outcomes measures in a recent meta-analysis that included only one controlled, randomized clinical trial with 27 patients [25]. Due to the limited size of the study, uncertainty remains about the effects of azathioprine and its use in patients with CIDP, in whom disease activity cannot otherwise be controlled.
Adverse effects, frequent: gastrointestinal disturbances, bone marrow suppression and hepatic toxicity are the most frequent side effects. Infrequent: data from clinical trials and from cohort and case–control studies did not show an increase in risk of malignancy from azathioprine. However, a possible long-term risk of cancer from azathioprine may occur with treatment duration longer than 10 years or cumulative doses above 600 g [39].
Other ‘classic’ oral immunosuppressive drugs
In RRMS and CIDP, other ‘classic’ non-selective oral immunosuppressive drugs such as methotrexate, mycophenolate mofetil, tacrolimus/sirolimus and cyclosporin A (as monotherapies) either showed no significant benefit or there are no data available from randomized, controlled clinical trials to support a clinical benefit [25,40]. Due to the loss of patent protection of these drugs, it is unlikely that new studies will be performed to support their use as monotherapies in MS and CIDP. However, in a very recent retrospective multi-centre study with 344 RRMS patients either or not previously treated with other immunosuppressants, mycophenolate mofetil significantly reduced the annualized relapse rate from 1·11 ± 0·08 during a 1-year control period to 0·35 ± 0·05 (P < 0·0001) during a 1-year treatment period. Moreover, in the subgroup of patients without previous immunosuppressant treatment, there was no disability progression during the treatment period. Hence, mycophenolate mofetil might serve as an alternative therapy for RRMS [41]. Moreover, recent studies examined the safety and efficacy of combinations of ‘classic’ immunosuppressive drugs with recombinant IFN-β and showed equivocal results [42]. Moreover, some novel oral immunomodulatory drugs have recently been tested alone or in combination with IFN-β or GA in Phase III trials in patients with CIS or RRMS (see below). A parallel approach, however, is lacking in CIDP.
Established intravenous immunosuppressive drugs: mitoxantrone and cyclophosphamide
Mitoxantrone
Mitoxantrone is an anthracenedione derivative related to the anthracyclines doxorubicin and daunorubicin. It interacts with topoisomerase-2, stabilizes its cleavable complex with DNA, and thus prevents the ligation of DNA strands and consecutively delays cell-cycle progression.
Preparations and administration: mitoxantrone is approved in Europe for the disease-modifying monotherapy of patients with highly active RRMS and SPMS (‘escalation therapy’) [43]. Its use, however, is limited by cardiotoxicity (the standard cumulative lifetime dose of mitoxantrone is 96 mg/m2, which can be extended up to a maximum lifetime dose of 140 mg/m2 under careful risk–benefit weighting and monitoring) and the risk of therapy-associated leukaemia (especially acute myelogenous leukaemia, AML). Given these limitations and the broadening spectrum of drugs available for patients with highly active RRMS, the use of mitoxantrone is limited in clinical practice to patients with SPMS. Mitoxantrone is administered intravenously at a dosage of 12 mg/m2 every 3 months for a total of 2 years, according to the mitoxantrone in MS study (MIMS) [44]. To extend the total administration period, the dosage can be reduced to 5 mg/m2 upon clinical stabilization.
Clinical trials: there are no recent clinical trials with mitoxantrone in MS. Moreover, due to a lack of evidence from randomized, controlled clinical trials the use of mitoxantrone in CIDP is not established.
Adverse effects, frequent: secondary amenorrhoea/azoospermia, nausea and vomiting, myelosuppression; infrequent: alopecia, cardiotoxicity, secondary leukaemia (especially AML) [45,46].
Contraindications: severe active infections, chronic or relapsing infections, cardiomyopathy, treatment with other cardiotoxic drugs, severe liver or kidney dysfunction, pregnancy and lactation.
Cyclophosphamide
Due to a lack of evidence from randomized, controlled clinical trials, the use of cyclophosphamide in MS and CIDP is not properly established [25,47].
Potential novel oral immunosuppressive and immunmodulatory drugs: teriflunomide, dimethyl fumarate, laquinimod and cladribine
Teriflunomide
Teriflunomide is the biologically active metabolite of leflunomide, which is approved for the treatment of rheumatoid arthritis. Teriflunomide inhibits mitochondrial dihydroorotate dehydrogenase (DHODH), a key enzyme in the biosynthesis of pyrimidine. The resulting inhibition of de-novo synthesis of pyrimidine nucleotides reduces the proliferation and function of activated lymphocytes.
Preparations and administration: teriflunomide (Aubagio®) is approved in the United States and Europe for the basic therapy of patients with RRMS. It is administered orally at a dose of 7 or 14 mg once daily.
Clinical trials: a Phase III trial (teriflunomide MS oral – TEMSO) involving more than 1000 patients with RRMS compared teriflunomide (1 × 7 mg/day or 1 × 14 mg/day for 108 weeks) to placebo [48]. Teriflunomide reduced the annualized relapse rate at both doses by approximately 31% from 0·54 to 0·37 (P < 0·001). Moreover, the proportion of patients with confirmed disability progression was significantly lower with teriflunomide 7 mg (21·7%, P = 0·08) and 14 mg (20·2%, P = 0·03) than with placebo (27·3%). Teriflunomide at both doses was also superior to placebo with regard to various MRI parameters.
Positive results from another Phase III trial confirmed the safety and efficacy of teriflunomide in RRMS [49]. Both studies were criticized for their short observation periods and high attrition bias (26·8% and 36·4% attrition, respectively) [50]. Currently, ongoing clinical trials evaluate teriflunomide as monotherapy in patients with CIS (Phase III study with teriflunomide versus placebo in patients with first clinical symptom of MS – TOPIC) and as add-on therapy in combination with IFN-β (Phase II study of teriflunomide as adjunctive therapy to IFN-β in subjects with MS) and GA (Phase II study of teriflunomide as adjunctive therapy to GA in subjects with MS) in RRMS.
Clinical trials with teriflunomide – to the best of our knowledge – have not yet been performed in patients with CIDP or its variants.
Adverse effects: in both Phase III clinical trials, side effects such as diarrhoea, nausea and vomiting, hair thinning and (reversible) hair loss were more frequent with teriflunomide than placebo. Moreover, mildly elevated liver enzymes (>1 × UNL) and lymphopenia were more frequent with teriflunomide than placebo, whereas pronounced liver enzyme elevations (>3 × UNL) were observed with equal frequency in all three study groups. Severe infections occurred with similar frequency among teriflunomide-and placebo-treated patients.
Dimethyl fumarate
Dimethyl fumarate (BG-12) is an orally administered derivative of fumarate. Fumarate itself is used traditionally in the therapy of psoriasis. BG-12 and its main metabolite, monomethyl fumarate, exhibit pleiotrophic effects: they modulate – among others – the nuclear factor E2-related factor-2 (Nrf2) transcription pathway, which is important in the regulation of oxidative stress and the immune response. Activation of the Nrf2 pathway is known to protect oligodendrocytes and neurones from inflammatory and metabolic damage [51]. In addition, BG-12 shows immunomodulatory effects by inhibiting the expression of proinflammatory cytokines and certain adhesion molecules, as well as by functional alteration of antigen-presenting cells (APCs) [51].
Preparations and administration: BG-12 (Tecfidera®) was approved in March 2013 for the treatment of patients with RRMS by the US regulatory Food and Drug Administration (FDA) and received a positive CHMP opinion from the European Medicines Agency (EMA). BG-12 is administered orally at a dose of 240 mg twice daily.
Clinical trials: a Phase III trial (determination of the efficacy and safety of oral fumarate in RRMS − DEFINE) with more than 1200 patients with RRMS compared BG-12 (2 × 240 mg/day or 3 × 240 mg/day for 96 weeks) to placebo [52]. BG-12 reduced the annualized relapse rate by about 53% from 0·36 to 0·17 (twice daily, P < 0·0001) and 48% from 0·36 to 0·19 (thrice daily, P < 0·0001). The proportion of patients with confirmed disability progression was lowered from 27% (placebo) to 16% (twice daily, P = 0·005) and 18% (thrice daily, P = 0·013). BG-12 at both dosages was also superior to placebo with regard to various MRI parameters.
Another Phase III trial (comparator and an oral fumarate in RRMS – CONFIRM) with more than 1200 patients with RRMS compared BG-12 (2 × 240 mg/day or 3 × 240 mg/day for 96 weeks) to GA (20 mg/day s.c.) and placebo [53]. Importantly, the study was not powered to detect a difference between BG-12 and GA. BG-12 reduced the annualized relapse rate by 44% (0·22, twice daily, P < 0·001) and 51% (0·20, thrice daily, P < 0·001), whereas GA caused a reduction of 29% (0·29, P = 0·01) compared to placebo (0·40). BG-12 reduced the proportion of patients with confirmed disability progression by 21% (twice daily) and 24% (thrice daily), whereas GA caused a reduction of 7% compared to placebo. However, the latter results did not reach statistical significance in a preliminary analysis, due possibly to a very low disability progression within the control group. BG-12 was also superior to placebo with regard to various MRI parameters.
Participants from these two Phase III clinical trials may have continued into the ongoing extension phase (long-term safety and efficacy study of oral BG00012 monotherapy in relapsing−remitting MS – ENDORSE).
To the best of our knowledge, clinical trials with BG-12 have not yet been performed in patients with CIDP or its variants.
Adverse effects: in both Phase III clinical trials flush, diarrhoea, nausea, vomiting and abdominal pain as well as lymphopenia occurred more frequently with BG-12 compared with placebo; severe infections or deaths were not more common with BG-12 treatment compared to placebo. However, during the extension phase of both clinical trials, there were 14 malignancies in 13 patients – six in patients who continued on BG-12 and eight in patients who switched from placebo to BG-12. There were three deaths, none of which were considered related to the study drug [54].
Laquinimod
The chinoline derivative, laquinimod, is an orally administered immunodulator, which diminishes infiltration of T cells and macrophages into the CNS and causes a shift from a T helper type 1 (Th1) to a Th2 immune response. In addition, direct neuroprotective effects of laquinimod have been proposed.
Preparations and administration: TEVA applied for approval of laquinimod for the treatment of RRMS in the United States and Europe. However, due to the unexpected benefit of laquinimod on reducing disability progression, which is much more pronounced than its impact on inflammatory activity, additional efficacy data have been requested in the United States; approval is under consideration in Europe. Laquinimod is administered orally at a dose of 0·6 mg once daily.
Clinical trials: a Phase III trial (assessment of oral laquinimod in preventing progression in MS – ALLEGRO) with more than 1100 patients with RRMS compared laquinimod (1 × 0·6 mg/day for 24 months) to placebo [55]. Laquinimod reduced the annualized relapse rate by 23% from 0·39 to 0·30 (P < 0·002). The proportion of patients with confirmed disability progression was lowered from 15·7 to 11·1% (P = 0·01). Laquinimod was also superior to placebo with regard to various MRI parameters.
Another Phase III trial [laquinimod double-blind placebo-controlled study in RRMS patients with a rater-blinded reference arm of IFN-β-1a (Avonex) – BRAVO] with more than 1300 patients with RRMS compared laquinimod (1 × 0·6 mg/day for 24 months) to IFN-β-1a (30 μg/week i.m.) and placebo [56]. Laquinimod reduced (after correction for differences between study groups) the annualized relapse rate by 21% (P = 0·026) and the proportion of patients with confirmed disability progression by 33·5% (P = 0·044). In this trial, IFN-β-1a lowered the annualized relapse rate but had no significant impact on disability progression compared to placebo. Laquinimod was also superior to placebo with regard to various MRI parameters.
Due to the request for additional efficacy data in the United States, a third Phase III trial (efficacy and safety and tolerability of laquinimod in subjects with RRMS – CONCERTO) has recently been initiated to evaluate two doses of laquinimod (0·6 mg and 1·2 mg) in approximately 1800 patients for up to 24 months. The primary outcome measure will be confirmed disability progression [57].
To the best of our knowledge, clinical trials with laquinimod have not yet been performed in patients with CIDP or its variants.
Adverse effects: in both Phase III clinical trials, elevated liver enzymes (>3 × UNL) were more frequent with laquinimod than with placebo. However, severe infections, tumours or deaths did not occur more frequently with laquinimod treatment compared to placebo.
Immunotherapy by blocking α4-integrin and sphingosin-1 phosphate-receptor-mediated lymphocyte trafficking
Natalizumab and firategrast
Natalizumab is a humanized monoclonal antibody against α4-integrin that recognizes very late antigen-4 (VLA-4) on the surface of various immune cell types. Binding of natalizumab to VLA-4 inhibits the interaction with its ligand vascular cell adhesion molecule 1 (VCAM-1) on the surface of endothelial cells of the blood−brain barrier and impairs trafficking of lymphocytes and monocytes into the inflamed CNS parenchyma.
Preparations and administration: natalizumab (Tysabri®) [58,59] is approved for disease-modifying monotherapy of patients with highly active RRMS in Europe and the United States (escalation therapy) in two subgroups of patients:
Patients with high disease activity despite treatment with either IFN-β or GA. These patients should have had at least one relapse in the past 12 months and at least nine T2-hyperintense lesions or at least one gadolinum-enriching lesion on cerebral MRI.
Patients with high disease activity showing at least two relapses with confirmed disability progression in the past 12 months and at least one gadolinum-enriching lesion or a significant increase in the number of T2-hyperintense lesions on cerebral MRI within the past 6–12 months.
Natalizumab is administered intravenously at a dose of 300 mg every 4 weeks.
Clinical trials: a recent Phase II clinical trial (study of SB-683699 compared to placebo in subjects with RRMS) assessed the safety and efficacy of firategrast, a small oral anti-α4β-integrin molecule, in 343 patients with RRMS [60]. Patients received one of four treatments twice daily: firategrast 150 mg, firategrast 600 mg or firategrast 900 mg (women) or 1200 mg (men) or placebo. A 49% reduction (P = 0·0026) in the cumulative number of new gadolinium-enhancing MRI lesions was seen with 900 mg or 1200 mg of firategrast. In the 600 mg group, a non-significant 22% reduction (P = 0·2657) occurred in the mean number of new gadolinium-enhanced lesions relative to placebo. Interestingly, in the 150 mg group, a significant 79% increase (P = 0·0353) occurred relative to placebo.
In one case of CIDP, clinical and paraclinical effects of natalizumab treatment were studied [61]. T cells expressing the α4-integrin were found in the inflamed peripheral nerve, and natalizumab bound with high affinity to the α4-integrin on T lymphocytes. However, the patient's clinical condition and paraclinical measures of disease activity deteriorated despite natalizumab treatment. Hence, natalizumab cannot be recommended in CIDP at present but warrants further exploration in future controlled clinical trials.
Adverse effects, frequent: hypersensitivity reactions, elevations of liver enzymes; infrequent: treatment with natalizumab is associated with the risk of developing progressive multi-focal leukoencephalopathy (PML), i.e. an opportunistic infection of the CNS with the JC-virus that leads eventually to death (approximately 20%) or severe neurological sequelae [45,46]. Risk of PML increases with long treatment duration (>2 years), preceding immunosuppressive treatment (independent from its duration and strength as well as the time interval to the natalizumab treatment), or a positive serological status for JC-virus [62]. Therefore, careful clinical surveillance including cognitive and neuropsychological assessments is mandatory.
Contraindications: active bacterial infections (urinary tract, lung, hepatitis), systemic mycosis in the past 6 months; viral infections: herpes zoster or herpes simplex infections with acute reactivations in the past 3 months; HIV-infection and subsequent opportunistic infections in the past 3 months; other chronic or recurrent viral or bacterial infections, malignant tumours, organ transplantation with ongoing immunosuppression, pregnancy and lactation.
Fingolimod
Fingolimod (FTY 720) has a unique immunoregulatory mechanism of action. Following its in-vivo phosphorylation, FTY720 becomes FTY720-phosphate(p), a non-selective, high-affinity antagonist of sphingosine 1-phosphate receptors (S1P-R). FTY720-p binds directly to S1P-Rs on lymphocytes, precipitating internalization and degradation of the receptor. This functional antagonism impairs the egress of autoreactive lymphocytes from lymph nodes along an endogenous chemotactic S1P-gradient. FTY720-p also binds to S1P-Rs on endothelial cells of the lymph node, which impairs the transmigration of lymphocytes from the medullary parenchyma to draining regions of lymph nodes. Hence, fingolimod retains T cells and B cells in secondary lymphatic organs, causes a pronounced lymphopenia in the blood and thus impairs invasion of lymphocytes into the inflamed CNS parenchyma. Fingolimod may also exert direct protective effects on parenchymal cells (neurones, oligodendrocytes) in the CNS.
Preparations and administration: in the United States, fingolimod [63,64] is approved for basic therapy, whereas in Europe fingolimod is approved for the escalation therapy of patients with RRMS. Fingolimod is administered orally at a dose of 0·5 mg once daily.
Clinical trials: a Phase III clinical trial is currently being initiated to compare oral fingolimod (0·5 mg/day) to placebo in patients with CIDP (‘Evaluate efficacy and safety of fingolimod 0·5 mg orally once daily versus placebo in chronic inflammatory demyelinating polyradiculoneuropathy patients’).
Adverse effects, frequent: infections, headache, gastrointestinal disturbances, bradycardia, elevation of liver enzymes; infrequent: sinuatrial block and/or atrioventricular block I–II°, increased arterial blood pressure, macula oedema.
Contraindications: immunodeficency, severe active infections, chronic active infections (hepatitis, tuberculosis), active malignancies, severe liver dysfunction, pregnancy and lactation.
Potential novel monoclonal antibodies: alemtuzumab, rituximab, ocrelizumab, ofatumumab, daclizumab
Alemtuzumab
Alemtuzumab is a humanized monoclonal antibody binding specifically to the CD52 antigen on the surface of B, T and natural killer (NK) cells, as well as monocytes and macrophages. It depletes these immune cell types by inducing complement-mediated cell lysis. Currently, alemtuzumab is approved for the treatment of patients with chronic lymphatic leukaemia of the B cell type (B-CLL).
Preparations and administration: Sanofi/Genzyme applied for approval of alemtuzumab (Lemtrada™) for RRMS in the United States and Europe, and received a positive CHMP opinion for active MS patients in 2013. Alemtuzumab is administered intravenously at a dosage of 12 mg/day on days 1–5 of the first year and days 1–3 of the second year.
Clinical trials: a first Phase III trial (comparison of alemtuzumab and Rebif® efficacy in MS – CARE-MS I) with 581 patients with RRMS without preceding disease-modifying therapy compared alemtuzumab (at a dosage of 12 mg/day on days 1–5 of the first year and days 1–3 of the second year) to IFN-β 1a (3 × 44 μg/week) for 2 years [65]. Alemtuzumab reduced the relapse rate by 55% compared to IFN-β 1a (P < 0·0001). The proportion of patients with confirmed disability progression was reduced from 11% (IFN-β-1a) to 8% (alemtuzumab, P = 0·22) [65].
A second Phase III trial (comparison of alemtuzumab and Rebif® efficacy in MS – CARE-MS II) with 667 patients with RRMS with sustained disease activity despite prior disease-modifying therapy compared alemtuzumab at a dosage of 12 mg/day on days 1 to 5 of the first year and days 1 to 3 of the second year to IFN-β-1a (3 × 44 μg/week) for 2 years [66]. Alemtuzumab reduced relapse rate by 49% (P < 0·0001) and the proportion of patients with confirmed diability progression by 42% (P = 0·008) compared to IFN-β-1a [66].
Based on the efficacy of alemtuzumab in the treatment of RRMS, this treatment is now being evaluated in patients with CIDP. In a small study, four of seven CIDP patients showed improvement following alemtuzumab; two of these achieved complete remission [67]. An open-label Phase IV clinical trial is currently being initiated to evaluate the impact of alemtuzumab in patients with CIDP (an open-label trial of alemtuzumab in CIDP).
Adverse effects: in both Phase III clinical trials, most frequent adverse events with alemtuzumab were infusion reactions and infections (infections of the upper respiratory tract, urinary tract, sinusitis and herpes simplex infections). There were no treatment-associated life-threatening or fatal infections with alemtuzumab treatment. Autoimmune thyroiditis occurred in 16% of patients treated with alemtuzumab and autoimmune thrombocytopenia in 1%, with one fatal outcome. Secondary B cell-mediated autoimmunity is an established phenomenon that occurs in patients with MS treated with alemtuzumab. These complications were detected by careful study-monitoring and treated accordingly.
Rituximab
Rituximab is a chimeric antibody specifically binding to the CD20 antigen on the surface of B cells. It depletes these cells by inducing complement-mediated cell lysis.
Preparations and administration: rituxmab (MabThera®, Rituxan®) is currently approved for the treatment of patients with non-Hodgkin lymphoma, rheumatoid arthritis and anti-neutrophil cytoplasmic antibody (ANCA)-associated systemic vasculitits. Rituximab is commonly administered i.v. either at a dose of 1000 mg on days 1 and 15, or 375 mg/m2 in four weekly doses.
Clinical trials: in a small Phase II trial with 69 patients with RRMS, rituximab at a dose of 1000 mg on days 1 and 15 exerted beneficial effects compared to placebo [68]. Compared with placebo, rituximab reduced the number of total and total new gadolinium-enhancing lesions at weeks 12, 16, 20 and 24 (P < 0·001); these results were sustained for 48 weeks (P < 0·001). Rituximab-treated patients showed a significantly lower relapse rate than placebo-treated patients at week 24 (14·5 versus 34·3%, P = 0·02) and week 48 (20·3 versus 40·0%, P = 0·04).
In CIDP, rituximab (1000 mg on days 1 and 15, or 375 mg/m2 in four weekly doses) provided clinical improvement after 2–12 months in up to 50% of patients [69–72]. High-quality evidence from randomized, controlled clinical trials, however, is still warranted [25].
Adverse effects, frequent: infusion-associated adverse events within 24 h after the first infusion, infections (nasopharyngitis, upper respiratory tract infections, urinary tract infections and sinusitis); infrequent: toxic epidermal necrolysis (Lyell syndrome) and Stevens–Johnson syndrome, progressive multi-focal leucoencephalopathy in patients with cancer and autoimmune diseases.
Ocrelizumab and ofatumumab
Ocrelizumab is a humanized, monoclonal, B cell-depleting anti-CD20 antibody, and ofatumumab represents a human monoclonal B cell-depleting anti-CD20 antibody. Both are expected to provide better tolerability than rituximab; therefore, more recent clinical trials to investigate B cell-depleting strategies are conducted preferentially using these newer agents.
Ocrelizumab
Ocrelizumab is a humanized monoclonal B cell-depleting anti-CD20 antibody.
Preparations and administration: ocrelizumab is administered intravenously on days 1 and 15.
Clinical trials: a Phase II trial (a study of the efficacy and safety of ocrelizumab in patients with RRMS) with 220 patients with RRMS compared ocrelizumab (300 mg/day or 1000 mg/day i.v. on days 1 and 15) to IFN-β 1a (30 μg/week i.m.) and placebo for 24 weeks. Ocrelizumab reduced the absolute number of gadolinium-enhancing lesions on MRI by 89% (600 mg, P < 0·0001) and 96% (2000 mg, P < 0·0001) compared to placebo. Moreover, annualized relapse rate was reduced by 80% (300 mg, P = 0·0005) and 73% (1000 mg, P = 0·0014), respectively, compared to placebo [73]. In an extension phase for a total of 96 weeks, there were no newly occurring gadolinium-enhancing lesions on MRI and a sustained reduction of the annualized relapse rate was observed in both ocrelizumab treatment groups [74].
Based on these results, two Phase III trials with 800 patients with RRMS have been initiated (a randomized, double-blind, double-dummy, parallel-group study to evaluate the efficacy and safety of ocrelizumab in comparison to IFN-β-1a (Rebif®) in patients with relapsing MS – OPERA I and II) to compare ocrelizumab (1 × 600 mg i.v. every 24 weeks) plus placebo (3×/week s.c.) to IFN-β-1a (3 × 44 μg/week s.c.) plus placebo (1 × i.v. every 24 weeks) on the annualized relapse rate, the confirmed disability progression and different MRI parameters for 96 weeks [74].
To the best of our knowledge, there is currently no clinical trial testing ocrelizumab in CIDP.
Adverse effects: in the Phase II clinical trial, severe adverse events occurred with similar frequency in both ocrelizumab treatment groups. Severe adverse events were systemic inflammatory response syndrome (SIRS), hypersensitivity reactions, oral herpes simplex, squamous cell carcinoma of the skin (based on a preexisting lesion) and fear. Moreover, one case of death occurred due to SIRS with high-dose ocrelizumab.
Ofatumumab
Ofatumumab is a human monoclonal B cell-depleting anti-CD20 antibody.
Preparations and administration: ofatumumab is currently approved for the treatment of chronic lymphatic leukaemia. It is administered intravenously on days 1 and 15.
Clinical trials: in a small Phase II trial (a double-blind, randomized, placebo-controlled, multi-centre, dose-finding trial of ofatumumab in RRMS patients) a total of 38 patients with RRMS received either ofatumumab (2 × 100 mg, 2 × 300 mg or 2 × 700 mg i.v.) or placebo for 24 weeks and were switched to either placebo or ofatumumab for another 24 weeks, respectively. Patients in both study groups exhibited a sustained reduction of inflammatory lesions on MRI at the end of the study [75].
Another Phase II trial (a randomized, double-blind, placebo-controlled, parallel-group, dose-ranging study to investigate the MRI efficacy and safety of 6 months' administration of ofatumumab in subjects with RRMS) is currently ongoing to compare ofatumumab (1 × 3 mg, 1 × 30 mg or 1 × 60 mg s.c. every 12 weeks or 1 × 60 mg s.c. every 4 weeks for a total of 24 weeks with subsequent observation for another 24 weeks) to placebo in approximately 200 patients with RRMS with regard to its impact on different MRI parameters as well as safety and tolerability [76].
To the best of our knowledge, there is currently no clinical trial that has evaluated ofatumumab in patients with CIDP.
Adverse effects: in the Phase II clinical trial there were no dose-limiting toxic effects or unexpected safety risks with ofatumumab [75].
Daclizumab
Daclizumab is a humanized, monoclonal antibody which binds and inactivates the alpha-chain of the IL-2-receptor (CD25 antigen) on T cells. IL-2 is crucial for the activation and proliferation of T cells. Daclizumab is also supposed to increase the number of natural killer cells which, in turn, attack (autoreactive) T cells.
Preparations and administration: daclizumab is administered subcutaneously every 2–4 weeks.
Clinical trials: a Phase II trial (daclizumab in patients with active, relapsing MS on concurrent interferon-beta therapy – CHOICE) with 230 patients with RRMS compared daclizumab (2 mg/kg every 2 weeks or 1 mg/kg every 4 weeks s.c.) plus IFN-β-1a (3 × 44 μg/week) to placebo plus IFN-β-1a for 24 weeks. High-but not low-dose daclizumab reduced the number of newly occurring or enlarging gadolinium-enhancing lesions on MRI by 72% (P = 0·004) [77].
Another Phase II trial (safety and efficacy study of daclizumab HYP to treat RRMS – SELECT) with 600 patients with RRMS compared daclizumab high-yield process (DAC HYP) (150 mg or 300 mg every 4 weeks s.c.) to placebo for 1 year. DAC HYP reduced the annualized relapse rate by 54% (150 mg, P < 0·0001) or 50% (300 mg, P = 0·0002), respectively, compared to placebo. DAC HYP also reduced the confirmed disability progression in a highly significant manner by 57% (150 mg) and 43% (300 mg). Further, DAC HYP caused a significant reduction of the cumulative number of new gadolinium-enhancing lesions between weeks 6 and 24 (150 mg: 69%; 300 mg: 78%) and the number of new or newly enlarging T2-hyperintense lesions after 1 year (150 mg: 70%; 300 mg: 79%) [78].
A Phase III trial (efficacy and safety of DAC-HYP versus IFN-β-1a in patients with RRMS – DECIDE) with about 1500 patients with RRMS is ongoing to compare daclizumab (150 mg every 4 weeks s.c.) to IFN-β-1a (3 × 44 μg/week) for 2 to 3 years with regard to its impact on the annualized relapse rate, the confirmed disability progression and different MRI parameters [74].
To the best of our knowledge, there is currently no clinical trial testing daclizumab in CIDP.
Adverse effects: in the CHOICE study, the incidence of common adverse events was similar in all groups. The most frequent severe adverse events were infections. There were no opportunistic infections or deaths, and all infections resolved with standard therapies. Two patients, both of whom were treated with daclizumab, developed malignant diseases. One patient with a family history of breast cancer developed breast cancer (ductal carcinoma in situ) more than 1 year after her last daclizumab dose. Another patient had pseudomyxoma peritonei, a recurrence of a pre-existing condition [77].
In the SELECT study, adverse events and treatment discontinuations occurred in all study groups with similar frequency. However, severe infections, severe skin reactions and pronounced elevations of liver enzymes (>5 UNL) were more frequent in the DAC HYP group than in the placebo group. One case of death occurred due to a muscular abscess in a patients recovering form a severe skin reaction [78].
Conclusion
This review summarizes the immune mechanisms and common or divergent clinical effects of a range of treatment options for potential use in MS or CIDP (Table 1). IVIG have been shown to exert short-and long-term beneficial effects in CIDP, but are not recommended in MS. Recombinant IFN-β and GA are approved for basic therapy of CIS and RRMS, but there is no evidence of their efficacy in CIDP. Evidence from randomized, controlled trials exists for azathioprine in RRMS but not in CIDP. Dimethyl fumarate (BG-12), teriflunomide and laquinimod represent three orally administered immunomodulatory drugs, either already approved or likely to be approved in the near future for basic therapy of patients with RRMS due to positive results in Phase III clinical trials. However, clinical trials with these drugs in CIDP have not (yet) been initiated. Natalizumab and fingolimod are approved for therapy of RRMS and trials testing their safety and efficacy are under way in patients with CIDP. Moreover, alemtuzumab, ocrelizumab and daclizumab respresent three monoclonal antibodies in advanced stages of clinical development. Their future role in the therapeutic armentarium against RRMS cannot yet be definitely foreseen. However, due to their strong effects on the immune system, they are likely to be used in patients with highly active RRMS. Attempts to study the safety and efficacy of alemtuzumab and a B cell-depleting anti-CD20 antibody (rituximab, ocrelizumab or ofatumumab) in patients with CIDP are currently under way.
Table 1.
Summary overview of evidence to date for immunotherapy options in multiple sclerosis (MS) or chronic inflammatory demyelinating polyradiculoneuropathy (CIDP).
| MS |
CIDP |
|||||
|---|---|---|---|---|---|---|
| Positive data | Negative data | Ongoing studies | Positive data | Negative data | Ongoing studies | |
| IFN-β | ✓ | ✓ | ||||
| Glatiramer acetate | ✓ | |||||
| IVIG | ✓ | ✓ | ||||
| Azathioprine | ✓ | |||||
| BG-12 | ✓ | |||||
| Teriflunomide | ✓ | |||||
| Laquinimod | ✓ | |||||
| Natalizumab | ✓ | ✓ | ||||
| Fingolimod | ✓ | ✓ | ||||
| Mitoxantrone | ✓ | |||||
| Alemtuzumab | ✓ | ✓ | ✓ | ✓ | ||
| Rituximab | ✓ | ✓ | ||||
| Ocrelizumab | ✓ | |||||
| Ofatumumab | ✓ | |||||
| Daclizumab | ✓ | ✓ | ||||
BG-12: dimethyl fumarate; IFN: interferon; IVIG: intravenous immunoglobulins.
Consideration of the relative clinical effects of treatment options across MS and CIDP may provide deeper insights into the immunopathogenesis of these disorders and their relationship to one another: positive data on rituximab und alemtuzumab represent a very strong hint on the pathogenic role of both B cells and T cells in both disorders. However, as alemtuzumab targets both cell types and rituximab may also critically influence T cell responses due to the antigen-presenting function of B cells, it is currently difficult to discern the individual contribution of both cell types. However, in light of these facts, it is very reasonable to expect clinical benefits of B and T cell-trapping in lymphnotes by fingolimod in CIDP, as in MS.
The strong clinical efficacy of natalizumab in MS together with the lack of an effect (in one case of) CIDP may point towards a difference in the mechanism of lymphocyte trafficking across the blood–brain and blood–nerve barriers.
In contrast, due to the wealth of molecular effects of both IFN-β and IVIG, it is difficult to speculate on the underlying immunopathogenic differences between MS and CIDP that causes the opposing clinical effects in both diseases.
Clearly, many more treatments have been evaluated and demonstrated clinical benefits in MS, highlighting an urgent need to focus research efforts on other immune disorders such as CIDP. Nevertheless, it is important to consider that the clinical effects of all these treatments beyond 2 years are uncertain [80] due to the limited follow-up of trial cohorts which should be mandatory for future investigations. It is hoped that resulting enhanced understanding may enable the progression of more effective treatment regimens for these chronic, debilitating disorders.
Literature search
We compare clinical trial evidence for established treatment strategies in MS and CIDP and report major findings from recent phase II and III clinical trials from the past 5 years in MS and corresponding evidence in CIDP.
Acknowledgments
The scientific and clinical work of the authors is supported by the German research foundation (DFG), the BMBF, the IZKF Münster, the IMF Münster and industry. N. M. received honoria for lecturing and travel expenses for attending meetings from Teva, Biogen Idec, GlaxoSmith Kline and Fresenius Medical Care and has received financial research support from Fresenius Medical Care. S. G. M. received honoraria for lecturing and travel expenses for attending meetings and has received financial research support from Bayer, Biogen Idec, Sanofi-Aventis, Bayer Schering, Merck Serono, Novo Nordisk, Genzyme, MSD and Teva.
Disclosures
All authors declare no relevant conflicts of interest.
References
- 1.Koller H, Kieseier BC, Jander S, Hartung HP. Chronic inflammatory demyelinating polyneuropathy. N Engl J Med. 2005;352:1343–1356. doi: 10.1056/NEJMra041347. [DOI] [PubMed] [Google Scholar]
- 2.Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- 3.Hughes RA, Mehndiratta MM. Corticosteroids for chronic inflammatory demyelinating polyradiculoneuropathy. Cochrane Database Syst Rev. 2012;(8) doi: 10.1002/14651858.CD002062.pub2. CD002062. [DOI] [PubMed] [Google Scholar]
- 4.Mehndiratta MM, Hughes RA. Plasma exchange for chronic inflammatory demyelinating polyradiculoneuropathy. Cochrane Database Syst Rev. 2012;(9) doi: 10.1002/14651858.CD003906.pub3. CD003906. [DOI] [PubMed] [Google Scholar]
- 5.Ciccone A, Beretta S, Brusaferri F, Galea I, Protti A, Spreafico C. Corticosteroids for the long-term treatment in multiple sclerosis. Cochrane Database Syst Rev. 2008;(1) doi: 10.1002/14651858.CD006264.pub2. CD006264. [DOI] [PubMed] [Google Scholar]
- 6.Burton JM, O'Connor PW, Hohol M, Beyene J. Oral versus intravenous steroids for treatment of relapses in multiple sclerosis. Cochrane Database Syst Rev. 2012;(12) doi: 10.1002/14651858.CD006921.pub3. CD006921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tumani H. Corticosteroids and plasma exchange in multiple sclerosis. J Neurol. 2008;255(Suppl. 6):36–42. doi: 10.1007/s00415-008-6007-9. [DOI] [PubMed] [Google Scholar]
- 8.Randomised double-blind placebo-controlled study of interferon beta-1a in relapsing/remitting multiple sclerosis. PRISMS (Prevention of Relapses and Disability by Interferon beta-1a Subcutaneously in Multiple Sclerosis) Study Group. Lancet. 1998;352:1498–1504. [PubMed] [Google Scholar]
- 9.Jacobs LD, Beck RW, Simon JH, et al. Intramuscular interferon beta-1a therapy initiated during a first demyelinating event in multiple sclerosis. CHAMPS Study Group. N Engl J Med. 2000;343:898–904. doi: 10.1056/NEJM200009283431301. [DOI] [PubMed] [Google Scholar]
- 10.Randomized controlled trial of interferon-beta-1a in secondary progressive MS: clinical results. Neurology. 2001;56:1496–1504. doi: 10.1212/wnl.56.11.1496. [DOI] [PubMed] [Google Scholar]
- 11.Li DK, Zhao GJ, Paty DW. Randomized controlled trial of interferon-beta-1a in secondary progressive MS: MRI results. Neurology. 2001;56:1505–1513. doi: 10.1212/wnl.56.11.1505. [DOI] [PubMed] [Google Scholar]
- 12.Clerico M, Faggiano F, Palace J, Rice G, Tintore M, Durelli L. Recombinant interferon beta or glatiramer acetate for delaying conversion of the first demyelinating event to multiple sclerosis. Cochrane Database Syst Rev. 2008;(2) doi: 10.1002/14651858.CD005278.pub3. CD005278. [DOI] [PubMed] [Google Scholar]
- 13.La Mantia L, Vacchi L, Di Pietrantonj C, et al. Interferon beta for secondary progressive multiple sclerosis. Cochrane Database Syst Rev. 2012;(1) doi: 10.1002/14651858.CD005181.pub3. CD005181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rice GP, Incorvaia B, Munari L, et al. Interferon in relapsing–remitting multiple sclerosis. Cochrane Database Syst Rev. 2001;(4) doi: 10.1002/14651858.CD002002. CD002002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kappos L, Freedman MS, Polman CH, et al. Long-term effect of early treatment with interferon beta-1b after a first clinical event suggestive of multiple sclerosis: 5-year active treatment extension of the phase 3 BENEFIT trial. Lancet Neurol. 2009;8:987–997. doi: 10.1016/S1474-4422(09)70237-6. [DOI] [PubMed] [Google Scholar]
- 16.Kappos L, Freedman MS, Polman CH, et al. Effect of early versus delayed interferon beta-1b treatment on disability after a first clinical event suggestive of multiple sclerosis: a 3-year follow-up analysis of the BENEFIT study. Lancet. 2007;370:389–397. doi: 10.1016/S0140-6736(07)61194-5. [DOI] [PubMed] [Google Scholar]
- 17.Kappos L, Polman CH, Freedman MS, et al. Treatment with interferon beta-1b delays conversion to clinically definite and McDonald MS in patients with clinically isolated syndromes. Neurology. 2006;67:1242–1249. doi: 10.1212/01.wnl.0000237641.33768.8d. [DOI] [PubMed] [Google Scholar]
- 18.Placebo-controlled multicentre randomised trial of interferon beta-1b in treatment of secondary progressive multiple sclerosis. European Study Group on interferon beta-1b in secondary progressive MS. Lancet. 1998;352:1491–1497. [PubMed] [Google Scholar]
- 19.Comi G, Martinelli V, Rodegher M, et al. Effect of glatiramer acetate on conversion to clinically definite multiple sclerosis in patients with clinically isolated syndrome (PreCISe study): a randomised, double-blind, placebo-controlled trial. Lancet. 2009;374:1503–1511. doi: 10.1016/S0140-6736(09)61259-9. [DOI] [PubMed] [Google Scholar]
- 20.La Mantia L, Munari LM, Lovati R. Glatiramer acetate for multiple sclerosis. Cochrane Database Syst Rev. 2010;(5) doi: 10.1002/14651858.CD004678.pub2. CD004678. [DOI] [PubMed] [Google Scholar]
- 21.Johnson KP, Brooks BR, Cohen JA, et al. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind placebo-controlled trial. The Copolymer 1 Multiple Sclerosis Study Group. Neurology. 1995;45:1268–1276. doi: 10.1212/wnl.45.7.1268. [DOI] [PubMed] [Google Scholar]
- 22.Biogen Idec. 2013. Biogen Idec announces positive top-line results from Phase 3 study of peginterferon beta-1a in multiple sclerosis. Weston, MA: Biogen Idec.
- 23.Hadden RD, Sharrack B, Bensa S, Soudain SE, Hughes RA. Randomized trial of interferon beta-1a in chronic inflammatory demyelinating polyradiculoneuropathy. Neurology. 1999;53:57–61. doi: 10.1212/wnl.53.1.57. [DOI] [PubMed] [Google Scholar]
- 24.Hughes RA, Gorson KC, Cros D, et al. Intramuscular interferon beta-1a in chronic inflammatory demyelinating polyradiculoneuropathy. Neurology. 2010;74:651–657. doi: 10.1212/WNL.0b013e3181d1a862. [DOI] [PubMed] [Google Scholar]
- 25.Mahdi-Rogers M, vanDoorn PA, Hughes RA. Immunomodulatory treatment other than corticosteroids, immunoglobulin and plasma exchange for chronic inflammatory demyelinating polyradiculoneuropathy. Cochrane Database Syst Rev. 2013;(6) doi: 10.1002/14651858.CD003280.pub4. CD003280. [DOI] [PubMed] [Google Scholar]
- 26.Khan O, Rieckmann P, Boyko A, Selmaj K, Zivadinov R. 2013. A multinational, multicenter, randomized, placebo-controlled, double-blind study to assess the efficacy, safety, and tolerability of glatiramer acetate 40 mg injection three times a week in subjects with RRMS: efficacy and safety results of the GALA study. Abstract, 65th American Academy of Neurology Annual Meeting.
- 27.Gelfand EW. Intravenous immune globulin in autoimmune and inflammatory diseases. N Engl J Med. 2012;367:2015–2025. doi: 10.1056/NEJMra1009433. [DOI] [PubMed] [Google Scholar]
- 28.Sorensen PS, Haas J, Sellebjerg F, Olsson T, Ravnborg M. IV immunoglobulins as add-on treatment to methylprednisolone for acute relapses in MS. Neurology. 2004;63:2028–2033. doi: 10.1212/01.wnl.0000145798.61383.39. [DOI] [PubMed] [Google Scholar]
- 29.Visser LH, Beekman R, Tijssen CC, et al. A randomized, double-blind, placebo-controlled pilot study of i.v. immune globulins in combination with i.v. methylprednisolone in the treatment of relapses in patients with MS. Mult Scler. 2004;10:89–91. doi: 10.1191/1352458504ms978sr. [DOI] [PubMed] [Google Scholar]
- 30.Fazekas F, Lublin FD, Li D, et al. Intravenous immunoglobulin in relapsing–remitting multiple sclerosis: a dose-finding trial. Neurology. 2008;71:265–271. doi: 10.1212/01.wnl.0000318281.98220.6f. [DOI] [PubMed] [Google Scholar]
- 31.Hommes OR, Sorensen PS, Fazekas F, et al. Intravenous immunoglobulin in secondary progressive multiple sclerosis: randomised placebo-controlled trial. Lancet. 2004;364:1149–1156. doi: 10.1016/S0140-6736(04)17101-8. [DOI] [PubMed] [Google Scholar]
- 32.Filippi M, Rocca MA, Pagani E, et al. European study on intravenous immunoglobulin in multiple sclerosis: results of magnetization transfer magnetic resonance imaging analysis. Arch Neurol. 2004;61:1409–1412. doi: 10.1001/archneur.61.9.1409. [DOI] [PubMed] [Google Scholar]
- 33.Hughes R, Bensa S, Willison H, et al. Randomized controlled trial of intravenous immunoglobulin versus oral prednisolone in chronic inflammatory demyelinating polyradiculoneuropathy. Ann Neurol. 2001;50:195–201. doi: 10.1002/ana.1088. [DOI] [PubMed] [Google Scholar]
- 34.Mendell JR, Barohn RJ, Freimer ML, et al. Randomized controlled trial of IVIg in untreated chronic inflammatory demyelinating polyradiculoneuropathy. Neurology. 2001;56:445–449. doi: 10.1212/wnl.56.4.445. [DOI] [PubMed] [Google Scholar]
- 35.Hahn AF, Bolton CF, Zochodne D, Feasby TE. Intravenous immunoglobulin treatment in chronic inflammatory demyelinating polyneuropathy. A double-blind, placebo-controlled, cross-over study. Brain. 1996;119:1067–1077. doi: 10.1093/brain/119.4.1067. [DOI] [PubMed] [Google Scholar]
- 36.Hughes RA, Donofrio P, Bril V, et al. Intravenous immune globulin (10% caprylate-chromatography purified) for the treatment of chronic inflammatory demyelinating polyradiculoneuropathy (ICE study): a randomised placebo-controlled trial. Lancet Neurol. 2008;7:136–144. doi: 10.1016/S1474-4422(07)70329-0. [DOI] [PubMed] [Google Scholar]
- 37.Nobile-Orazio E, Cocito D, Jann S, et al. Intravenous immunoglobulin versus intravenous methylprednisolone for chronic inflammatory demyelinating polyradiculoneuropathy: a randomised controlled trial. Lancet Neurol. 2012;11:493–502. doi: 10.1016/S1474-4422(12)70093-5. [DOI] [PubMed] [Google Scholar]
- 38.Leger JM, De Bleecker JL, Sommer C, et al. Efficacy and safety of Privigen((R)) in patients with chronic inflammatory demyelinating polyneuropathy: results of a prospective, single-arm, open-label Phase III study (the PRIMA study) J Peripher Nerv Syst. 2013;18:130–140. doi: 10.1111/jns5.12017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Casetta I, Iuliano G, Filippini G. Azathioprine for multiple sclerosis. Cochrane Database Syst Rev. 2007;(4) doi: 10.1002/14651858.CD003982.pub2. CD003982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gray O, McDonnell GV, Forbes RB. Methotrexate for multiple sclerosis. Cochrane Database Syst Rev. 2004;(2) doi: 10.1002/14651858.CD003208.pub2. CD003208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Michel L, Vukusic S, De Seze J, et al. Mycophenolate mofetil in multiple sclerosis: a multicentre retrospective study on 344 patients. J Neurol Neurosurg Psychiatry. 2013 doi: 10.1136/jnnp-2013-305298. http://dx.doi.org/10.1136/jnnp-2013-305298. [DOI] [PubMed] [Google Scholar]
- 42.Conway D, Cohen JA. Combination therapy in multiple sclerosis. Lancet Neurol. 2010;9:299–308. doi: 10.1016/S1474-4422(10)70007-7. [DOI] [PubMed] [Google Scholar]
- 43.Martinelli Boneschi F, Vacchi L, Rovaris M, Capra R, Comi G. Mitoxantrone for multiple sclerosis. Cochrane Database Syst Rev. 2013;(5) doi: 10.1002/14651858.CD002127.pub3. CD002127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hartung HP, Gonsette R, Konig N, et al. Mitoxantrone in progressive multiple sclerosis: a placebo-controlled, double-blind, randomised, multicentre trial. Lancet. 2002;360:2018–2025. doi: 10.1016/S0140-6736(02)12023-X. [DOI] [PubMed] [Google Scholar]
- 45.Rommer PS, Zettl UK, Kieseier B, et al. Requirement for safety monitoring for approved multiple sclerosis therapies: an overview. Clin Exp Immunol. 2014;175:397–407. doi: 10.1111/cei.12206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Winkelmann A, Loebermann M, Reisinger EC, Zettl UK. Multiple sclerosis treatment and infectious issues: update 2013. Clin Exp Immunol. 2014;175:425–438. doi: 10.1111/cei.12226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.La Mantia L, Milanese C, Mascoli N, D'Amico R, Weinstock-Guttman B. Cyclophosphamide for multiple sclerosis. Cochrane Database Syst Rev. 2007;(1) doi: 10.1002/14651858.CD002819.pub2. CD002819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.O'Connor P, Wolinsky JS, Confavreux C, et al. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med. 2011;365:1293–1303. doi: 10.1056/NEJMoa1014656. [DOI] [PubMed] [Google Scholar]
- 49.Sanofi/Genzyme. 2012. Genzyme reports positive top-line results of TOWER, a pivotal Phase III trial for AUBAGIOTM* (teriflunomide) in relapsing multiple sclerosis. Sanofi/Genzyme.
- 50.He D, Xu Z, Dong S, et al. Teriflunomide for multiple sclerosis. Cochrane Database Syst Rev. 2012;(12) doi: 10.1002/14651858.CD009882.pub2. CD009882. [DOI] [PubMed] [Google Scholar]
- 51.Joshi G, Johnson JA. The Nrf2-ARE pathway: a valuable therapeutic target for the treatment of neurodegenerative diseases. Recent Pat CNS Drug Discov. 2012;7:218–229. doi: 10.2174/157488912803252023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gold R, Kappos L, Arnold DL, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. 2012;367:1098–1107. doi: 10.1056/NEJMoa1114287. [DOI] [PubMed] [Google Scholar]
- 53.Fox RJ, Miller DH, Phillips JT, et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med. 2012;367:1087–1097. doi: 10.1056/NEJMoa1206328. [DOI] [PubMed] [Google Scholar]
- 54.Phillips JT, Fox EJ, Selmaj K, et al. 2012. Long-term safety and tolerability of oral BG-12 (dimethyl fumarate) in relapsing–remitting multiple sclerosis: interim results from ENDORSE. 28th Congress of the European Committee for the Treatment and Research in Multiple Sclerosis.
- 55.Comi G, Jeffery D, Kappos L, et al. Placebo-controlled trial of oral laquinimod for multiple sclerosis. N Engl J Med. 2012;366:1000–1009. doi: 10.1056/NEJMoa1104318. [DOI] [PubMed] [Google Scholar]
- 56.TEVA Pharmaceutical Industries, Inc. 2011. Results of Phase III BRAVO trial reinforce unique profile of laquinimod for multiple sclerosis treatment Tikva, Israel: TEVA;
- 57.TEVA Pharmaceutical Industries, Inc. 2013. First patient enrolled in third Phase III of oral laquinimod for relapsing–remitting multiple sclerosis. Tikva, Israel: TEVA;
- 58.Polman CH, O'Connor PW, Havrdova E, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;354:899–910. doi: 10.1056/NEJMoa044397. [DOI] [PubMed] [Google Scholar]
- 59.Rudick RA, Stuart WH, Calabresi PA, et al. Natalizumab plus interferon beta-1a for relapsing multiple sclerosis. N Engl J Med. 2006;354:911–923. doi: 10.1056/NEJMoa044396. [DOI] [PubMed] [Google Scholar]
- 60.Miller DH, Weber T, Grove R, et al. Firategrast for relapsing remitting multiple sclerosis: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2012;11:131–139. doi: 10.1016/S1474-4422(11)70299-X. [DOI] [PubMed] [Google Scholar]
- 61.Wolf C, Menge T, Stenner MP, et al. Natalizumab treatment in a patient with chronic inflammatory demyelinating polyneuropathy. Arch Neurol. 2010;67:881–883. doi: 10.1001/archneurol.2010.143. [DOI] [PubMed] [Google Scholar]
- 62.Bloomgren G, Richman S, Hotermans C, et al. Risk of natalizumab-associated progressive multifocal leukoencephalopathy. N Engl J Med. 2012;366:1870–1880. doi: 10.1056/NEJMoa1107829. [DOI] [PubMed] [Google Scholar]
- 63.Kappos L, Radue EW, O'Connor P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362:387–401. doi: 10.1056/NEJMoa0909494. [DOI] [PubMed] [Google Scholar]
- 64.Cohen JA, Barkhof F, Comi G, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362:402–415. doi: 10.1056/NEJMoa0907839. [DOI] [PubMed] [Google Scholar]
- 65.Cohen JA, Coles AJ, Arnold DL, et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing–remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet. 2012;380:1819–1828. doi: 10.1016/S0140-6736(12)61769-3. [DOI] [PubMed] [Google Scholar]
- 66.Coles AJ, Twyman CL, Arnold DL, et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. Lancet. 2012;380:1829–1839. doi: 10.1016/S0140-6736(12)61768-1. [DOI] [PubMed] [Google Scholar]
- 67.Marsh EA, Hirst CL, Llewelyn JG, et al. Alemtuzumab in the treatment of IVIG-dependent chronic inflammatory demyelinating polyneuropathy. J Neurol. 2010;257:913–919. doi: 10.1007/s00415-009-5437-3. [DOI] [PubMed] [Google Scholar]
- 68.Hauser SL, Waubant E, Arnold DL, et al. B-cell depletion with rituximab in relapsing–remitting multiple sclerosis. N Engl J Med. 2008;358:676–688. doi: 10.1056/NEJMoa0706383. [DOI] [PubMed] [Google Scholar]
- 69.Munch C, Anagnostou P, Meyer R, Haas J. Rituximab in chronic inflammatory demyelinating polyneuropathy associated with diabetes mellitus. J Neurol Sci. 2007;256:100–102. doi: 10.1016/j.jns.2007.02.027. [DOI] [PubMed] [Google Scholar]
- 70.Briani C, Zara G, Zambello R, Trentin L, Rana M, Zaja F. Rituximab-responsive CIDP. Eur J Neurol. 2004;11:788. doi: 10.1111/j.1468-1331.2004.00911.x. [DOI] [PubMed] [Google Scholar]
- 71.Benedetti L, Briani C, Franciotta D, et al. Rituximab in patients with chronic inflammatory demyelinating polyradiculoneuropathy: a report of 13 cases and review of the literature. J Neurol Neurosurg Psychiatry. 2011;82:306–308. doi: 10.1136/jnnp.2009.188912. [DOI] [PubMed] [Google Scholar]
- 72.Kosmidis ML, Dalakas MC. Practical considerations on the use of rituximab in autoimmune neurological disorders. Ther Adv Neurol Disord. 2010;3:93–105. doi: 10.1177/1756285609356135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kappos L, Li D, Calabresi PA, et al. Ocrelizumab in relapsing-remitting multiple sclerosis: a phase 2, randomised, placebo-controlled, multicentre trial. Lancet. 2011;378:1779–1787. doi: 10.1016/S0140-6736(11)61649-8. [DOI] [PubMed] [Google Scholar]
- 74.Hoffmann-LaRoche. 2011. Phase II study showed ocrelizumab maintained significant reduction in disease activity for multiple sclerosis patients for almost two years. Basel, Switzerland: Hoffmann-LaRoche.
- 75.GlaxoSmithKline. 2010. Genmab announces results of ofatumumab Phase II study in multiple sclerosis. Princeton, NJ: GlaxoSmithKline.
- 76.GlaxoSmithKline. 2010. GlaxoSmithKline and Genmab refocus development programme for ofatumumab in autoimmune indications. Princeton, NJ: GlaxoSmith Kline.
- 77.Wynn D, Kaufman M, Montalban X, et al. Daclizumab in active relapsing multiple sclerosis (CHOICE study): a phase 2, randomised, double-blind, placebo-controlled, add-on trial with interferon beta. Lancet Neurol. 2010;9:381–390. doi: 10.1016/S1474-4422(10)70033-8. [DOI] [PubMed] [Google Scholar]
- 78.Gold R, Giovannoni G, Selmaj K, et al. Daclizumab high-yield process in relapsing-remitting multiple sclerosis (SELECT): a randomised, double-blind, placebo-controlled trial. Lancet. 2013;381:2167–2175. doi: 10.1016/S0140-6736(12)62190-4. [DOI] [PubMed] [Google Scholar]
- 79.Biogen Idec. 2010. Biogen Idec and Abbott announce enrollment of first patient in global Phase III study of daclizumab for relapsing–remitting multiple sclerosis. Weston, MA: Biogen Idec.
- 80.Filippini G, Del Giovane C, Vacchi L, et al. Immunomodulators and immunosuppressants for multiple sclerosis: a network meta-analysis. Cochrane Database Syst Rev. 2013;(6) doi: 10.1002/14651858.CD008933.pub2. CD008933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Linker RA, Kieseier BC, Gold R. Identification and development of new therapeutics for multiple sclerosis. Trends Pharmacol Sci. 2008;29:558–565. doi: 10.1016/j.tips.2008.07.012. [DOI] [PubMed] [Google Scholar]