

Abstract
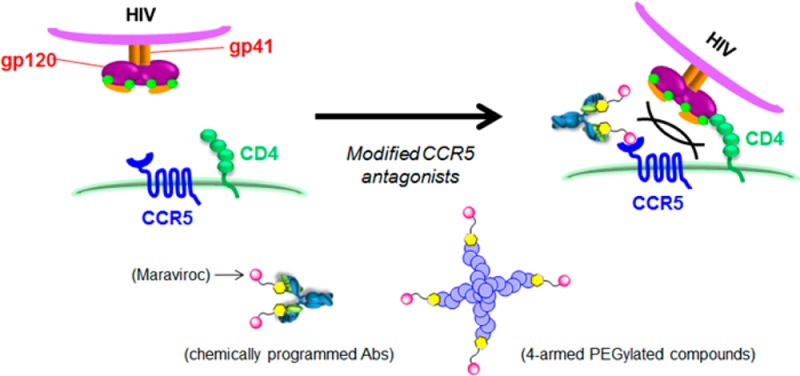
CCR5 antagonists are among the most advanced approaches in HIV therapy and may also be relevant to treatment of graft-versus-host disease and Staphylococcus aureus infection. To expand the potential of the only approved CCR5 antagonist, Maraviroc, we studied derivatives that would enable functional linkage of Maraviroc to long-lived carriers. Through targeted synthesis, we discovered an effective linkage site on Maraviroc and demonstrate the potential of these derivatives to prepare potent chemically programmed antibodies and PEGylated derivatives. The resulting compounds effectively neutralized a variety of HIV-1 isolates. Both chemically programmed antibody and PEGylation approaches extend the neutralization activity of serum circulating Maraviroc. Derivation of a successful conjugation strategy for Maraviroc should further enable its use in chemically programmed vaccines, novel bispecific antibodies, and topical microbicides.
Keywords: CCR5 antagonist, Maraviroc, chemically programmed antibody, PEGylation
HIV-1 infection is typically managed by a treatment regimen known as highly active antiretroviral therapy or HAART, which commonly involves the administration of combinations of reverse transcriptase and protease inhibitors. Viral escape, drug side effects, and compliance issues, however, continue to drive the development of novel approaches, which most recently have seen the approval of entry, fusion, and integrase inhibitors.1 Of these more recent innovations, entry inhibitors that target conserved host proteins are particularly intriguing. The accepted mechanism of HIV infection involves initial attachment of the virus to the host cell receptor CD4 via interaction with the viral gp120 envelope protein. This binding event then triggers a conformational change in the envelope protein that provides for binding to chemokine coreceptors CCR5 or CXCR4 and finally membrane fusion after viral gp41 insertion into the target cell (Figure 1).2−7 Blockade of chemokine receptor engagement by the virus therefore blocks infection. To date the only approved chemokine receptor targeted inhibitor is Maraviroc (1, Figure 2), a potent CCR5 antagonist that received FDA approval in 2007.8,9 Maraviroc treatment regimens, however, require twice daily dosing with as much as 1.2 g of drug per day, making patient compliance an issue. Significantly, the biology of CCR5 is not limited to HIV-1, and recent studies have demonstrated beneficial activity of Maraviroc therapy in graft-versus-host disease10 and Staphylococcus aureus pathogenesis.11 Thus, the development of Maraviroc derivatives with extended pharmacokinetic profiles could be a valuable contribution to therapy.
Figure 1.
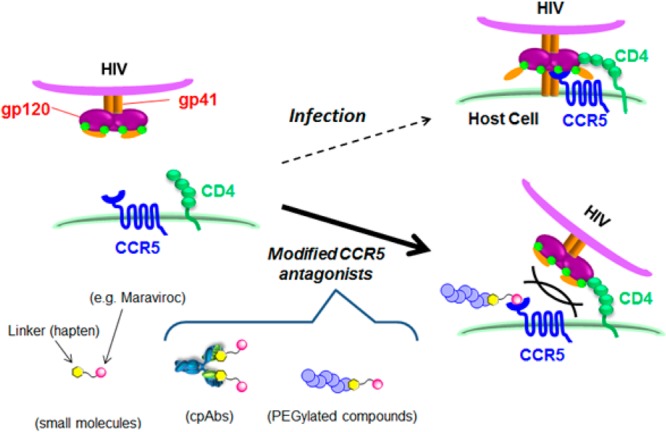
Schematic representations of mechanism of HIV-1 infection of a host cell and inhibition of the viral entry by CCR5 antagonists.
Figure 2.

Structure of Maraviroc (1) and linker-attached Maraviroc (2a, 2b, and 3).
Chemically programmed antibodies (cpAbs),12−19 which link a catalytic antibody to a small molecule drug, peptide, or aptamer dramatically extend the pharmacokinetic profile of the attached molecule. Chemical programming of the monoclonal antibody (mAb) 38C2 is facilitated by a low pKa lysine residue located in the 38C2-binding site. This lysine is key to 38C2 aldolase activity and can be site-selectively labeled with N-acyl-β-lactams to produce a chemically programmed antibody.20−22 The cpAb approach has demonstrated efficacy in a number of disease models including anti-infectives, and the relative merits of cpAbs over conventional mAbs have been well documented.12 For example, with a derivative of Zanamivir, a neuraminidase inhibitor, the cpAb approach provided long-term systemic exposure without loss of neuraminidase inhibitory activity.18 Another approach to extending the pharmacokinetic profiles of drugs involves their conjugation to polyethylene glycol (PEG), a process known as PEGylation. PEGylation often imparts other significant pharmacological advantages, such as improved solubility, minimized proteolytic cleavage, reduced dosage frequency, increased serum half-life, and reduced immunogenicity and antigenicity.23−25 PEGINTRON, an α-interferon derivative, is the first FDA-approved, PEG-modified drug. The plasma circulating half-life of PEGINTRON, which is used for treatment of hepatitis C, is about 10 times that of native IFN α-2b and allows weekly subcutaneous dosing.26 PEGylation also imparts desired properties on small molecule drugs.27,28 Pepinsky et al. reported that PEGylation dramatically improves pharmacodynamics and pharmacokinetic properties of an integrin α4β1 inhibitor.28 Key to the development of effective chemically programmed antibodies and PEGylated small molecules is the discovery of a linkage chemistry that minimally impacts activity of the parental drug. Herein, we describe the structure activity relationships of linked Maraviroc derivatives and macromolecular conjugates with mAb 38C2 and polyethylene glycol variants designed to create potent long-lived CCR5 antagonists.
In our previous study of the CCR5 antagonist Aplaviroc, we showed that linkage of the small molecule to mAb 38C2 through a benzoic acid moiety resulted in a cpAb with potent activity.16 Aplaviroc was dropped from clinical development due to toxicity. To avoid concerns regarding the toxicity of Aplaviroc, we were compelled to study the more challenging antagonist Maraviroc. We decided to explore two routes toward linked variants of Maraviroc. The first westerly linkage point was chosen based on ease of synthesis and known structural tolerance at the cyclohexyl position. To explore this linkage directionality, we synthesized compounds 2a and 2b. A docking study of CCR5 antagonists performed by Kondru et al. suggests that the benzoic acid moiety of Aplaviroc and the triazole moiety of Maraviroc overlap in the putative binding pocket.29 Since we had successfully introduced a linker on Aplaviroc at the benzoic acid position, the modeling encouraged us to explore introduction of a linker onto the triazole ring of Maraviroc (easterly connection), and we synthesized compound 3 to explore this point of connectivity.
The routes used for the syntheses of the mAb derivatives of Maraviroc are shown in Scheme 1. Azide compounds 2a and 3, synthesized as illustrated in Supporting Information (SI) Scheme S2 and S3, were converted to N-acyl-β-lactam compounds 4 and 5 by Click reaction with the corresponding alkyne compounds (SIS21, Scheme S4). N-Acyl-β-lactam compounds were conjugated to mAb 38C2 after incubation at room temperature in PBS. The time dependency of the conjugation reaction of 5 with mAb 38C2 was evaluated by measuring the catalytic activity of retro-aldol reaction of methodol.30 The reaction was complete in 2 h (SI Figure S1). The lack of catalytic activity of the product was confirmed (SI Figure S2), and cpAbs 6 and 7 were purified by size-exclusion column chromatography to remove excess small molecules.
Scheme 1. Syntheses of the cpAbs.
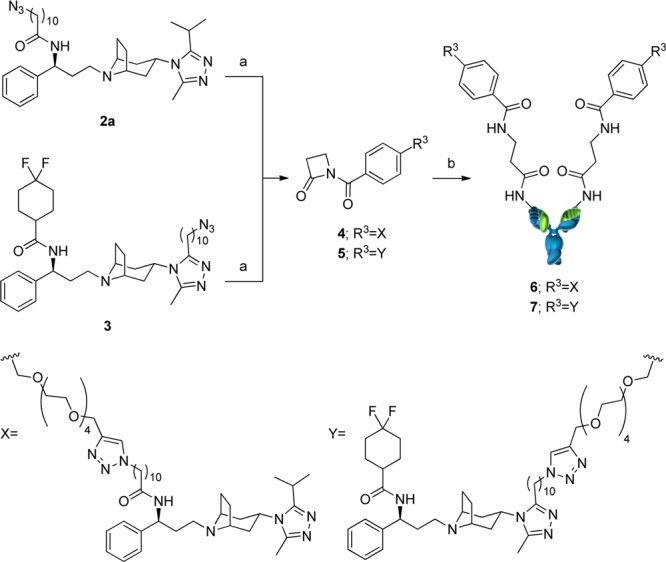
Reagents and conditions: (a) S21, THPTA, CuSO4, sodium ascorbate, H2O, t-BuOH, rt, 2 h, 4 100%, 5 67%; (b) mouse mAb 38C2, 100 mM PBS solution (pH 7.4), rt, 2 h.
Linear and branched PEGylated compounds were prepared by simple amidation of N-hydroxysuccinimide (NHS) esters (Scheme 2). PEGylation of n-propylamine or Maraviroc-attached propylamine (9), which was obtained by deprotection of the Boc group in compound 8, with the commercially available PEG-NHS derivatives, gave the desired PEGylated compounds (10, 11, 12, and 14). All PEGylated compounds were purified by precipitation from Et2O. None of the small molecule remained in the obtained solid, as confirmed by HPLC (SI Figure S6). MALDI-TOF and/or ESI mass analysis of cpAbs and PEGylated compounds was in agreement with expected values (SI Figure S3–5).
Scheme 2. Synthesis of the PEGylated Compounds.

Reagents and conditions: (a) S22, THPTA, CuSO4, sodium ascorbate, H2O, tert-BuOH, rt, 2 h, 82%; (b) TFA, CH2Cl2, rt, 1 h, quant; (c) SUNBRIGHT ME-050AS, ME-400AS, or PTE-400HS, DMF, rt, 3 h, 10, 46%, 11 82%, 12 92%, 14, 92%.
We initially studied the azide linker derivatives 2a and 2b and evaluated their activity for neutralization of HIV-1 JR-FL reporter virus. The activity of 2a (westerly connection replacing the cylcohexyl group) was 2 orders of magnitude less than that of parental Maraviroc 1 (Table 1). The activity of compound 2b with a PEG linker was further reduced relative to 2a. Previous studies of Maraviroc derivatives showed that the amide site we used for connectivity in these compounds should interact with a predominantly lipophilic binding site on the CCR5 receptor, and this result is consistent with the lower activity of 2b as compared with 2a.31 By contrast, compound 3, modified on the triazole ring, had potent activity (easterly connection).
Table 1. IC50 Values of Maraviroc Derivatives in Neutralization Assay Using HIV-1 Pseudotype JR-FL.
| compd | compd-type | IC50 (nM)a |
|---|---|---|
| 1 | Maraviroc | 1.6 ± 0.25b |
| 2a | small molecule | 360 ± 34a |
| 2b | small molecule | >1000a |
| 3 | small molecule | 0.96 ± 0.26a |
| 4 | small molecule | 27 ± 2.3a |
| 5 | small molecule | 2.6 ± 0.62a |
| 6 | cpAbs | 19 ± 2.6a |
| 7 | cpAbs | 7.7 ± 0.50a |
| 38C2 | mAb carrier control | >1000a |
| 8 | small molecule | 3.0 ± 0.58a |
| 10 | PEGylated compound | 43 ± 9.6a |
| 11 | PEGylated compound | 49 ± 13a |
| 12 | PEGylated compound | 5.6 ± 2.0c |
| 14 | PEGylated control | >1000a |
Mean ± SE (N = 3).
Mean ± SE (N = 12).
Mean ± SE (N = 5).
N-Acyl-β-lactam derivatives of these compounds were prepared through a standard Click reaction, and the lactams were used for conjugation to mAb 38C2. Surprisingly, the activity of N-acyl-β-lactam-linked Maraviroc derivative 4 was significantly higher than that of 2a. In contrast, the activity of N-acyl-β-lactam 5 was 2.6 nM, less active than 3 and comparable to that of Maraviroc 1. Unmodified antibody, mAb 38C2, did not neutralize the JR-FL HIV-1 reporter virus. The activity of the antibody conjugate of 5, cpAb 7, was higher than that of the conjugate of 4, cpAb 6. As shown in Figure 3 and Table S1, we evaluated the binding affinity of both cpAbs by FACS using TZM-bl cells, which derive from a HeLa cell line and are transfected to express CCR5, and CCR5 negative control cells (HeLa). cpAbs 6 and 7 bound to CCR5-expressing TZM-bl cells as did positive control mAb 2D7. None bound CCR5-negative HeLa cells (Figure 3). The IC50 of cpAbs 6 was 19 nM, and that of 7 was 7.7 nM. The neutralization activity for these cpAbs with the JR-FL HIV-1 strain is summarized in Table 1.
Figure 3.
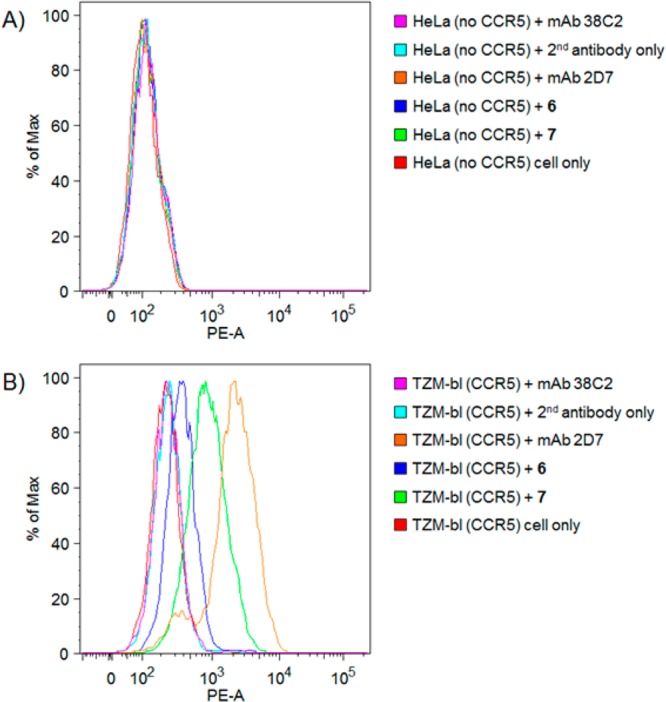
Binding of mAb 38C2, mAb conjugates 6 and 7 and positive control mAb 2D7 to (A) HeLa cells, which do not express CCR5, and to (B) TZM-bl cells, which are CCR5-positive, by FACS: mAb 38C2 (pink), secondary antibody (light blue), mAb 2D7 (orange), compound 6 (blue), compound 7 (light green), cell only (red).
Past studies have indicated that short PEG chains may not provide enough protection from metabolism and may not alter clearance rates significantly; however, long PEG chains may interfere with binding to target proteins.32 The constitution (linear vs branched) of the polymer also affects half-life.33 Boc-protected compound 8, the precursor for the PEGylated compound, had activity similar to that of N-acyl-β-lactam compound 5. Derivatives of Maraviroc with 5 kDa and 40 kDa linear PEG chains were synthesized by amidation of 9 with the NHS-PEG derivatives. In this case, the length of a PEG chain did not significantly influence the activity, but PEGylated compounds 10 (5 kDa) and 11 (40 kDa) were approximately 15 times less active than the parent small molecule 8. The antibody conjugate 7 carries two molar equivalents of Maraviroc and was approximately 6-fold more active than the monovalent PEGylated Maraviroc derivatives. This difference was likely due to enhanced avidity by the multiple interactions of the cpAb with target.34 To improve the activity of monovalent PEGylated compound, a branched derivative was synthesized. Attachment of four Maraviroc moieties to a single PEG (compound 12; >98% purity) dramatically enhanced neutralization activity relative to the linear PEGylated compounds.
The activities of cpAb 7 and PEGylated derivative 12, which had the highest activity against HIV-1 JR-FL of those derivatives tested, were evaluated in neutralization assays against HIV-1 strains from clades A, B, and C. Representative inhibition curves are shown in Figure 4 and SI Figures S7–S10, and IC50 values are listed in Table 2. Both Maraviroc conjugates had broad spectra of activity similar to that of the parent compound.
Figure 4.
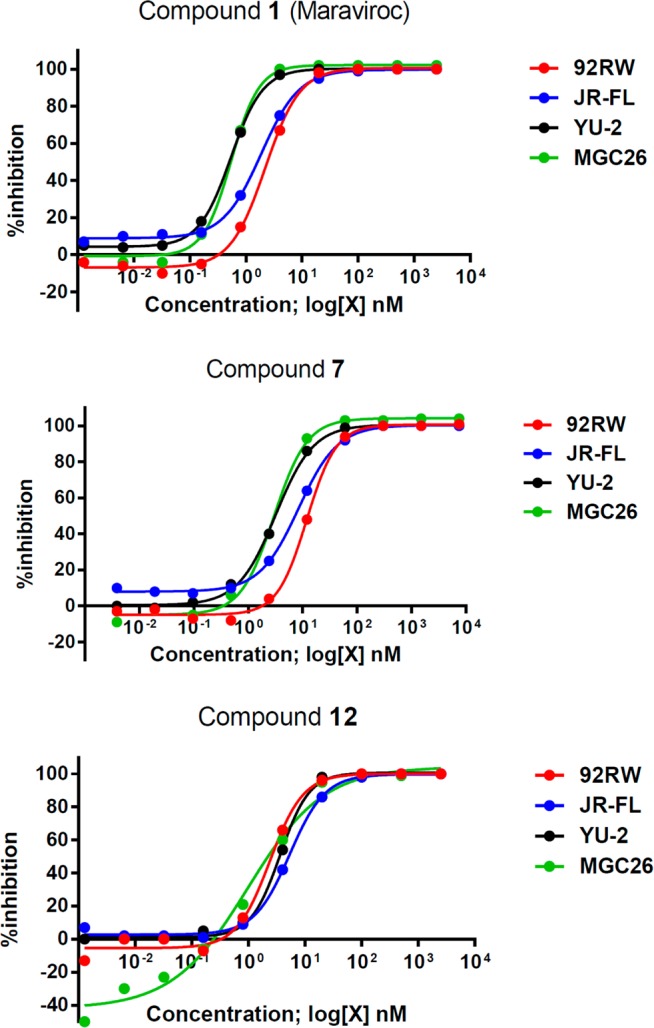
Evaluation of inhibition of HIV-1 replication in CCR5-positive cells by Maraviroc 1, mAb conjugate 7, and PEGylated compound 12: clade A 92RW020 (red line), clade B JR-FL (blue line), clade B YU-2 (black line), and clade C MGC26 (green line). All tests were performed in duplicate.
Table 2. IC50 Values of Maraviroc Derivatives in Neutralization Assays against Indicated Strains.
| IC50 (nM) of Maraviroc derivativesa |
|||
|---|---|---|---|
| HIV-1 strain | 1 | 7 | 12 |
| 92RW (clade A) | 1.3 ± 0.5b | 8.1a | 3.0a |
| JR-FL (clade B) | 1.6 ± 0.3c | 7.7 ± 0.5b | 5.6 ± 2.0d |
| YU-2 (clade B) | 0.43 ± 0.1b | 2.8a | 2.5a |
| MGC26 (clade C) | 0.37 ± 0.01b | 2.7a | 0.8a |
Average (N = 2).
Mean ± SE (N = 3).
Mean ± SE (N = 12),
Mean ± SE (N = 5).
In order to investigate the stability of cpAb 7 and the PEGylated Maraviroc derivative 12, we incubated each compound with human serum at 37 °C and evaluated their HIV neutralization activity at multiple time points. Unlike 1, both 7 and 12 retained their full activity for up to 10 days (SI Figure S11), indicating that cpAbs and PEGylation are potentially effective means for enhancing Maraviroc serum stability and extending neutralization activity. Lastly, while Maraviroc is known to cause only minor side effects, the toxicity of conjugates 7 and 12 is unknown.
In conclusion, we discovered that the triazole ring of the CCR5 antagonist Maraviroc could be derivatized for linkage to macromolecules without significant loss of activity. In contrast, modifications on the westerly side of the molecule, which presents a lipophilic amide in the parental Maraviroc, reduced activity relative to the parent. Antibody conjugate 7 and PEGylated compound 12 effectively neutralized HIV-1 strains from four clades with IC50 values similar to those of Maraviroc. As previously reported, cpAbs and PEGylation strategies dramatically extend the circulating serum half-life of conjugated molecules relative to small molecule,18,28 peptide, and protein parent drugs. Thus, we anticipate that 7 and 12 will have dramatically extended pharmacokinetic properties and warrant further study in anti-HIV models. While the discovery of a viable site of conjugation for this promising drug has allowed us to establish good antiviral activity in the case of a chemically programmed antibody and a PEGylated derivative, their application in a chemically programmed vaccine,35 chemical approaches to bispecific antibodies,36 and topical microbicides is also hereby facilitated.
Acknowledgments
We thank Angelica Cuevas and Khoa Le for performing HIV-1 neutralization assays.
Glossary
Abbreviations
- CCR5
C–C chemokine receptor type 5
- CXCR4
C-X-C chemokine receptor type 4
Supporting Information Available
Synthetic procedures, analytical data, and procedures for neutralization assay and FACS. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by NIH Grant AI095038.
The authors declare the following competing financial interest(s): Patents have been filed.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Mehellou Y.; De Clercq E. Twenty-six years of anti-HIV drug discovery: where do we stand and where do we go?. J. Med. Chem. 2010, 53, 521–38. [DOI] [PubMed] [Google Scholar]
- Deng H.; Liu R.; Ellmeier W.; Choe S.; Unutmaz D.; Burkhart M.; Di Marzio P.; Marmon S.; Sutton R. E.; Hill C. M.; Davis C. B.; Peiper S. C.; Schall T. J.; Littman D. R.; Landau N. R. Identification of a major co-receptor for primary isolates of HIV-1. Nature 1996, 381, 661–6. [DOI] [PubMed] [Google Scholar]
- Dragic T.; Litwin V.; Allaway G. P.; Martin S. R.; Huang Y.; Nagashima K. A.; Cayanan C.; Maddon P. J.; Koup R. A.; Moore J. P.; Paxton W. A. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature 1996, 381, 667–73. [DOI] [PubMed] [Google Scholar]
- Doranz B. J.; Berson J. F.; Rucker J.; Doms R. W. Chemokine receptors as fusion cofactors for human immunodeficiency virus type 1 (HIV-1). Immunol. Res. 1997, 16, 15–28. [DOI] [PubMed] [Google Scholar]
- Broder C. C.; Collman R. G. Chemokine receptors and HIV. J. Leukocyte Biol. 1997, 62, 20–9. [DOI] [PubMed] [Google Scholar]
- Bredeek U. F.; Harbour M. J. CCR5 antagonists in the treatment of treatment-naive patients infected with CCR5 tropic HIV-1. Eur. J. Med. Res. 2007, 12, 427–34. [PubMed] [Google Scholar]
- Dorr P.; Perros M. CCR5 inhibitors in HIV-1 therapy. Expert Opin. Drug Discovery 2008, 3, 1345–61. [DOI] [PubMed] [Google Scholar]
- Latinovic O.; Kuruppu J.; Davis C.; Le N.; Heredia A. Pharmacotherapy of HIV-1 Infection: Focus on CCR5 Antagonist Maraviroc. Clin. Med. Ther. 2009, 1, 1497–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poveda E.; Paredes R.; Moreno S.; Alcami J.; Cordoba J.; Delgado R.; Gutierrez F.; Llibre J. M.; Garcia Deltoro M.; Hernandez-Quero J.; Pulido F.; Iribarren J. A.; Garcia F. Update on clinical and methodological recommendations for genotypic determination of HIV tropism to guide the usage of CCR5 antagonists. AIDS Rev. 2012, 14, 208–17. [PubMed] [Google Scholar]
- Reshef R.; Luger S. M.; Hexner E. O.; Loren A. W.; Frey N. V.; Nasta S. D.; Goldstein S. C.; Stadtmauer E. A.; Smith J.; Bailey S.; Mick R.; Heitjan D. F.; Emerson S. G.; Hoxie J. A.; Vonderheide R. H.; Porter D. L. Blockade of lymphocyte chemotaxis in visceral graft-versus-host disease. N. Engl. J. Med. 2012, 367, 135–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonzo F. 3rd; Kozhaya L.; Rawlings S. A.; Reyes-Robles T.; DuMont A. L.; Myszka D. G.; Landau N. R.; Unutmaz D.; Torres V. J. CCR5 is a receptor for Staphylococcus aureus leukotoxin ED. Nature 2013, 493, 51–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rader C.; Sinha S. C.; Popkov M.; Lerner R. A.; Barbas C. F. 3rd Chemically programmed monoclonal antibodies for cancer therapy: adaptor immunotherapy based on a covalent antibody catalyst. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 5396–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popkov M.; Rader C.; Gonzalez B.; Sinha S. C.; Barbas C. F. 3rd. Small molecule drug activity in melanoma models may be dramatically enhanced with an antibody effector. Int. J. Cancer 2006, 119, 1194–207. [DOI] [PubMed] [Google Scholar]
- Gavrilyuk J. I.; Wuellner U.; Barbas C. F. 3rd Beta-lactam-based approach for the chemical programming of aldolase antibody 38C2. Bioorg. Med. Chem. Lett. 2009, 19, 1421–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuellner U.; Gavrilyuk J. I.; Barbas C. F. 3rd Expanding the concept of chemically programmable antibodies to RNA aptamers: chemically programmed biotherapeutics. Angew. Chem., Int. Ed. Engl. 2010, 49, 5934–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavrilyuk J.; Uehara H.; Otsubo N.; Hessell A.; Burton D. R.; Barbas C. F. 3rd Potent inhibition of HIV-1 entry with a chemically programmed antibody aided by an efficient organocatalytic synthesis. ChemBioChem 2010, 11, 2113–8. [DOI] [PubMed] [Google Scholar]
- Huang H.; Lai J. Y.; Do J.; Liu D.; Li L.; Del Rosario J.; Doppalapudi V. R.; Pirie-Shepherd S.; Levin N.; Bradshaw C.; Woodnutt G.; Lappe R.; Bhat A. Specifically targeting angiopoietin-2 inhibits angiogenesis, Tie2-expressing monocyte infiltration, and tumor growth. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 2011, 17, 1001–11. [DOI] [PubMed] [Google Scholar]
- Hayakawa M.; Toda N.; Carrillo N.; Thornburg N. J.; Crowe J. E. Jr.; Barbas C. F. 3rd A chemically programmed antibody is a long-lasting and potent inhibitor of influenza neuraminidase. ChemBioChem 2012, 13, 2191–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S.; Inokuma T.; Otsubo N.; Burton D. R.; Barbas C. F. 3rd. Chemically Programmed Antibodies as HIV-1 Attachment Inhibitors. ACS Med. Chem. Lett. 2013, 4, 460–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner J.; Lerner R. A.; Barbas C. F. 3rd Efficient aldolase catalytic antibodies that use the enamine mechanism of natural enzymes. Science 1995, 270, 1797–800. [DOI] [PubMed] [Google Scholar]
- Zhong G.; Lerner R. A.; Barbas I. C. Broadening the Aldolase Catalytic Antibody Repertoire by Combining Reactive Immunization and Transition State Theory: New Enantio- and Diastereoselectivities. Angew. Chem., Int. Ed. Engl. 1999, 38, 3738–3741. [DOI] [PubMed] [Google Scholar]
- Shabat D.; Rader C.; List B.; Lerner R. A.; Barbas C. F. 3rd. Multiple event activation of a generic prodrug trigger by antibody catalysis. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 6925–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinckerhoff L. H.; Kalashnikov V. V.; Thompson L. W.; Yamshchikov G. V.; Pierce R. A.; Galavotti H. S.; Engelhard V. H.; Slingluff C. L. Jr. Terminal modifications inhibit proteolytic degradation of an immunogenic MART-1(27–35) peptide: implications for peptide vaccines. Int. J. Cancer 1999, 83, 326–34. [DOI] [PubMed] [Google Scholar]
- Cheng T. L.; Chen B. M.; Chan L. Y.; Wu P. Y.; Chern J. W.; Roffler S. R. Poly(ethylene glycol) modification of beta-glucuronidase-antibody conjugates for solid-tumor therapy by targeted activation of glucuronide prodrugs. Cancer Immunol. Immunother. 1997, 44, 305–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffee S.; Mary A.; Stiehm E. R.; Girault D.; Fischer A.; Hershfield M. S. IgG antibody response to polyethylene glycol-modified adenosine deaminase in patients with adenosine deaminase deficiency. J. Clin. Invest. 1992, 89, 1643–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. S.; Youngster S.; Grace M.; Bausch J.; Bordens R.; Wyss D. F. Structural and biological characterization of pegylated recombinant interferon alpha-2b and its therapeutic implications. Adv. Drug Delivery Rev. 2002, 54, 547–70. [DOI] [PubMed] [Google Scholar]
- Greenwald R. B.; Pendri A.; Conover C.; Gilbert C.; Yang R.; Xia J. Drug delivery systems. 2. Camptothecin 20-O-poly(ethylene glycol) ester transport forms. J. Med. Chem. 1996, 39, 1938–40. [DOI] [PubMed] [Google Scholar]
- Pepinsky R. B.; Lee W. C.; Cornebise M.; Gill A.; Wortham K.; Chen L. L.; Leone D. R.; Giza K.; Dolinski B. M.; Perper S.; Nickerson-Nutter C.; Lepage D.; Chakraborty A.; Whalley E. T.; Petter R. C.; Adams S. P.; Lobb R. R.; Scott D. M. Design, synthesis, and analysis of a polyethelene glycol-modified (PEGylated) small molecule inhibitor of integrin {alpha}4{beta}1 with improved pharmaceutical properties. J. Pharmacol. Exp. Ther. 2005, 312, 742–50. [DOI] [PubMed] [Google Scholar]
- Kondru R.; Zhang J.; Ji C.; Mirzadegan T.; Rotstein D.; Sankuratri S.; Dioszegi M. Molecular interactions of CCR5 with major classes of small-molecule anti-HIV CCR5 antagonists. Mol. Pharmacol. 2008, 73, 789–800. [DOI] [PubMed] [Google Scholar]
- Sinha S. C.; Das S.; Li L. S.; Lerner R. A.; Barbas C. F. 3rd. Preparation of integrin alpha(v)beta3-targeting Ab 38C2 constructs. Nat. Protoc. 2007, 2, 449–56. [DOI] [PubMed] [Google Scholar]
- Wood A.; Armour D. The discovery of the CCR5 receptor antagonist, UK-427,857, a new agent for the treatment of HIV infection and AIDS. Prog. Med. Chem. 2005, 43, 239–71. [DOI] [PubMed] [Google Scholar]
- Yamaoka T.; Tabata Y.; Ikada Y. Distribution and tissue uptake of poly(ethylene glycol) with different molecular weights after intravenous administration to mice. J. Pharm. Sci. 1994, 83, 601–6. [DOI] [PubMed] [Google Scholar]
- Bailon P. B. W. Polyethylene glycol-conjugated pharmaceutical proteins. Pharm. Sci. Technol. Today 1998, 1, 352–356. [Google Scholar]
- Rudnick S. I.; Adams G. P. Affinity and avidity in antibody-based tumor targeting. Cancer Biother. Radiopharm. 2009, 24, 155–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popkov M.; Gonzalez B.; Sinha S. C.; Barbas C. F. 3rd. Instant immunity through chemically programmable vaccination and covalent self-assembly. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 4378–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavrilyuk J.; Ban H.; Uehara H.; Sirk S. J.; Saye-Francisco K.; Cuevas A.; Zablowsky E.; Oza A.; Seaman M. S.; Burton D. R.; Barbas C. F. 3rd. Antibody conjugation approach enhances breadth and potency of neutralization of anti-HIV-1 antibodies and CD4-IgG. J. Virol. 2013, 87, 4985–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.