

Abstract
Aims
The mechanisms of heart failure remain largely elusive. The present study determined a causative role of DNA methylation in norepinephrine-induced heart hypertrophy and reduced cardiac contractility.
Methods and results
Male adult rats were subjected to norepinephrine infusion for 28 days, some of which were treated with 5-aza-2′-deoxycytidine for the last 6 days of norepinephrine treatment. At the end of the treatment, hearts were isolated and left ventricular morphology and function as well as molecular assessments was determined. Animals receiving chronic norepinephrine infusion showed a sustained increase in blood pressure, heightened global genomic DNA methylation and changes in the expression of subsets of proteins in the left ventricle, left ventricular hypertrophy, and impaired contractility with an increase in the susceptibility to ischaemic injury. Treatment of animals with 5-aza-2′-deoxycytidine for the last 6 days of norepinephrine infusion reversed norepinephrine-induced hypermethylation, corrected protein expression patterns, and rescued the phenotype of heart hypertrophy and failure.
Conclusions
The findings provide novel evidence of a causative role of increased DNA methylation in programming of heart hypertrophy and reduced cardiac contractility, and suggest potential therapeutic targets of demethylation in the treatment of failing heart and ischaemic heart disease.
Keywords: Methylation, Heart, Hypertrophy, Failure, Proteomic
1. Introduction
Prolonged sympathetic nervous system activation with elevated circulating catecholamine levels have implications in human heart failure for both disease progression and survival.1–4 Catecholamine-induced heart injury may play an important role in prolonging myocardial damage following an infarction. Yet, the molecular mechanisms underlying the changes in the myocardium that occur following catecholamine infusion and subsequent acute ischaemia reperfusion injury remain largely elusive. Elevated reactive oxygen species (ROS) production and reprogramming of gene expression patterns in cardiomyocytes have been implicated in the pathophysiology of left ventricular hypertrophy and failure. This is characterized by the up-regulation of foetal genes and decreased expression of some subsets of adult ones.
Although previous studies with the approach of transgenic knockout (KO) or knockin of a single protein revealed important information, it is unlikely that heart hypertrophy and failure are caused by changes in a single protein or pathway. Thus, networks of proteins and pathways in the heart must be altered, leading to heart hypertrophy and failure. Recent clinical and experimental studies suggest a vital role of epigenetic mechanisms in the regulation of cardiac gene expression patterns in the progress of heart hypertrophy and failure.5–7 DNA methylation at CpG dinucleotides and histone modifications are the most common mechanisms of epigenetic modulation of gene expression. Whereas it has been suggested that histone modification may contribute to reprogramming of cardiac genes expression in heart failure,7,8 the role of DNA methylation is far less defined. Some recent studies showed that distinct epigenomic DNA methylation patterns existed in important DNA elements of the cardiac genome in human end-stage cardiomyopathy,6 and differential DNA methylation correlated with differential expression of angiogenic factors in human heart failure.5 Yet, whether aberrant DNA methylation plays a causative role in the progression of heart hypertrophy and failure remains unknown.
The present study tested the hypothesis that DNA methylation and altered cardiac gene expression patterns play a causative role in norepinephrine-induced heart hypertrophy and heightened ischaemic injury in a rodent model. Here, we present novel evidence in a rodent model that epigenetic modification via increased DNA methylation is a causative mechanism in norepinephrine-induced programming of heart hypertrophy and reduced cardiac contractility. Given that protein kinase C epsilon (PKCɛ) plays an important role in regulating heart hypertrophy and ischaemic injury, we also examined the specific effect of norepinephrine treatment on PKCɛ promoter methylation and protein expression in the heart. The findings suggest a potentially novel therapeutic approach of demethylation in the treatment of failing heart and ischaemic heart disease.
2. Methods
An expanded Methods section is available in Supplementary material online.
2.1. Experimental animals
Six-month-old Sprague–Dawley male rats were randomly divided into five groups: (i) saline control, (ii) norepinephrine 100 μg/kg/h, (iii) norepinephrine 200 μg/kg/h, (iv) saline plus 5-aza-2′-deoxycytidine 1 mg/kg/day, and (v) norepinephrine 100 μg/kg/h plus 5-aza-2′-deoxycytidine 1 mg/kg/day. Norepinephrine was continuously administered for 28 days via osmotic minipumps (2ML4, Alzet, Durect Corp., Cupertino, CA), as described previously.9 To implant the osmotic minipumps, animals were anaesthetized with 75 mg/kg ketamine and 5 mg/kg xylazine injected intramuscularly, and adequate anaesthesia was determined by loss of pedal withdrawal reflex. 5-Aza-2′-deoxycytidine was administered intraperitoneally for the last 6 days of norepinephrine infusion. All procedures and protocols were approved by the Institutional Animal Care and Use Committee guidelines, and followed the guidelines by the US National Institutes of Health Guide for the Care and Use of Laboratory Animals.
2.2. Measurement of arterial blood pressure
Animals were implanted with catheters in femoral arteries at the same time when the osmotic minipumps were implanted. The anaesthetic agent used, the dose used, and route of administration were stated as above. Blood pressure was recorded daily during the norepinephrine treatment.
2.3. Measurement of plasma norepinephrine concentrations
Blood samples were collected from animals, and plasma norepinephrine concentrations were determined by an high performance liquid chromatography (HPLC) with electrochemical detection.9
2.4. Measurement of cardiac function and ischaemia and reperfusion injury
Rats were anaesthetized with 75 mg/kg ketamine and 5 mg/kg xylazine injected intramuscularly and hearts were removed. The adequacy of anaesthesia was determined by the loss of a pedal withdrawal reflex and any other reaction from the animal in response to pinching the toe, tail, or ear of the animal. Additionally, even respiration rate of the animal under anaesthesia was closely monitored, and an increased respiration rate was used as a sign that anaesthesia was too light. Isolated hearts were retrogradely perfused via the aorta in a modified Langendorff apparatus, as described previously.10 Left ventricle end-diastolic pressure (LVEDP) was set at ∼5 mmHg. After baseline recording for 60 min, hearts were subjected to 20 min of global ischaemia followed by 60 min of reperfusion. Left ventricle developed pressure (LVDP), heart rate, dP/dtmax, dP/dtmin, and LVEDP were continuously recorded. At the end of reperfusion, left ventricles were collected and myocardial infarct size was measured with 1% triphenyltetrazolium chloride, as described previously.10 Lactate dehydrogenase (LDH) activity was measured in coronary effluent collected during reperfusion, using the TOX 7 assay kit.
2.5. Western blot analysis
Protein abundance of PKCε, DNA methyltransferase (Dnmt) 1, Dnmt 3a, and Dnmt 3b in left ventricles was determined with western blot analysis.10
2.6. Real-time RT-PCR
RNA was extracted from left ventricles, and mRNA abundance of PKCε, atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), and β-myosin heavy chain (β-MHC) were determined by real-time reverse transcription polymerase chain reaction (RT-PCR) using an Icycler Thermal cycler.10
2.7. Quantitative methylation-specific PCR
Genomic DNA was isolated from left ventricles and subjected to bisulphite modification.10,11 Bisulphite-treated DNA was used as a template for real-time methylation-specific PCR (MSP) using primers designed to amplify the Egr-1-binding site at rat PKCɛ promoter, as described previously.10,11 Real-time MSP was performed using the iQ SYBR Green Supermix with iCycler real-time PCR system.
2.8. Measurement of global DNA methylation
Genomic DNA was isolated from left ventricles and digested with a mixture of benzonase nuclease, shrimp alkaline phosphatase, and phosphodiesterse I from Crotalus adamanteus venom.12 Quantification of methyl-cytosine (Cm) and cytosine (C) was carried out by HPLC coupled with tandem mass spectrometry in multiple reaction monitoring mode (HPLC–MS/MS-MRM).13 Results were expressed as the Cm/C ratio.
2.9. Measurement of Dnmt activity
Dnmt activity assay was performed using an ELISA EpiQuik DNMT activity/inhibitor assay kit (Epigentek, Farmingdale, NY, USA). To measure tissue Dnmt activity, nuclear extract (NE) isolated from the left ventricle was incubated with S-adenosylmethionine (AdoMet) and a universal proprietary Dnmt substrate in the Dnmt assay buffer at 37°C for 2 h. The blank contained only AdoMet and the substrate without NE, while the positive control contained AdoMet/substrate with the purified Dnmt enzyme preparation containing both maintenance and de novo Dnmts, supplied in the kit. After the incubation, the capture antibody and detection antibody were added in sequence, followed by incubation with developer solution for 10 min at room temperature. Signal was measured by a dual wavelength microplate reader at 450/655 nm. To determine the effect of 5-aza-2′-deoxycytidine on individual Dnmt activity, purified human Dnmt 1, Dnmt 3a, or murine Dnmt 3b (Epigentek), instead of NE, were incubated with AdoMet and the Dnmt substrate in the absence or presence of 5-aza-2′-deoxycytidine.
2.10. Measurement of ROS
ROS in left ventricles were measured with an Oxiselect In Vitro ROS assay kit, as well as by imaging of dihydroethidium fluorescence in tissue slides with confocal microscopy.11
2.11. Proteomic analysis
Protein expression analysis was performed in the left ventricle by tandem mass tags (TMTs)–LC–MS/MS analysis with an LTQ-Orbitrap-Pro instrument as previously described.14,15 Proteins were isolated from the left ventricle of each animal (n = 5) in the five treatment groups, as listed in Section 2.1, and were TMT-labelled with one of the TMT reporters at m/z = 126.1 for saline control group, 127.1 for norepinephrine 100 µg/kg/h group, 128.1 for norepinephrine 200 µg/kg/h group, 129.1 for saline plus 5-aza-2′-deoxycytidine group, and 130.1 for norepinephrine 100 µg/kg/h plus 5-aza-2′-deoxycytidine group. Mass spectrometry data were processed and searched against the rat protein database (ipi.RAT.v3.73.fasta) through the Thermo Scientific Proteome Discoverer 1.3 platform using MASCOT™ search engine with parameters as previously described by Xiong et al.14 Cardiac protein expression levels obtained from five different animals in each group were expressed as norepinephrine-induced fold changes relative to the respective controls in the absence or presence of 5-aza-2′-deoxycytidine as the ratios of the intensities of the reporter ions, 127/126, 128/126, and 130/129, respectively. A fold-change cut-off value of 1.2 (>20% up- or down-regulation) was considered significant as published previously.16,17 The results were presented in Table 1 as mean ± SEM (n = 5).
Table 1.
5-Aza-2′-deoxycytidine caused a reversal of norepinephrine-mediated changes in the cardiac proteome
| Accession# | Entrez gene name | Location | 127/126 | 128/126 | 130/129 | Protein# |
|---|---|---|---|---|---|---|
| 55 391 508 | Albumin (ALB) | Extracellular space | 1.301 ± 0.05 | 4.047 ± 0.09 | 0.731 ± 0.07 | 1 |
| 25 990 263 | Aldehyde dehydrogenase 2 family (mitochondrial) (ALDH2) | Cytoplasm | 1.761 ± 0.03 | 1.422 ± 0.01 | No change | 2 |
| 6 978 545 | ATPase, Na+/K+ transporting, α 2 polypeptide (ATP1A2) | Plasma membrane | 0.712 ± 0.05 | 0.561 ± 0.03 | No change | 3 |
| 19 855 078 | ATPase, Na+/K+ transporting, α 3 polypeptide (ATP1A3) | Plasma membrane | 1.353 ± 0.01 | 1.272 ± 0.05 | No change | 4 |
| 8 392 983 | Biglycan (BGN) | Extracellular space | 1.742 ± 0.14 | 2.536 ± 0.07 | 1.340 ± 0.13 | 5 |
| 6 978 589 | Caldesmon 1 (Cald1) | Plasma membrane | 1.311 ± 0.06 | 1.641 ± 0.12 | No change | 6 |
| 38 505 168 | CAP-GLY domain-containing linker protein 1 (Clip1) | Cytoplasm | 1.409 ± 0.05 | 1.365 ± 0.04 | No change | 7 |
| 51 854 229 | Carnitine O-acetyltransferase (CRAT) | Cytoplasm | 0.794 ± 0.02 | 0.659 ± 0.04 | No change | 8 |
| 16 924 004 | Cysteine and glycine-rich protein3 (cardiac LIM protein) (CSRP3) | Nucleus | 1.262 ± 0.07 | 1.547 ± 0.11 | No change | 9 |
| 18 543 351 | Cytoglobin (CYGB) | Cytoplasm | 1.569 ± 0.14 | 1.439 ± 0.15 | No change | 10 |
| 56 057 | Decorin (DCN) | Extracellular space | 1.347 ± 0.05 | 1.578 ± 0.10 | No change | 11 |
| 8 393 296 | Eukaryotic translation elongation factor 2 (EEF2) | Cytoplasm | No change | 1.268 ± 0.06 | No change | 12 |
| 19 924 061 | ERO1-like (Saccharomyces cerevisiae) (ERO1L) | Cytoplasm | 1.281 ± 0.07 | 1.663 ± 0.17 | No change | 13 |
| 13 928 940 | Four and a half LIM domains 2 (FHL2) | Nucleus | 0.557 ± 0.03 | 0.591 ± 0.05 | No change | 14 |
| 46 485 440 | Glucose-6-phosphate isomerase (GPI) | Extracellular space | 1.230 ± 0.06 | 1.282 ± 0.05 | No change | 15 |
| 21 955 178 | Hepatoma-derived growth factor/related protein 3 (HDGFRP3) | Nucleus | 1.376 ± 0.06 | 1.587 ± 0.09 | No change | 16 |
| 28 467 005 | Heat-shock protein 90 kDa α, Class A Member 1 (HSP90AA1) | Cytoplasm | No change | 1.579 ± 0.09 | No change | 17 |
| 47 059 179 | Heat-shock 70-kDa protein 1A (HSPA1A/HSPA1B) | Cytoplasm | 1.324 ± 0.09 | 3.628 ± 0.15 | 1.251 ± 0.01 | 18 |
| 94 400 790 | Heat-shock 27-kDa protein 1 (HSPB1) | Cytoplasm | No change | 2.103 ± 0.05 | No change | 19 |
| 58 865 372 | Heat-shock 105-kDa/110-kDa protein 1 (HSPH1) | Cytoplasm | 1.420 ± 0.09 | 2.034 ± 0.05 | No change | 20 |
| 203 734 | Keratin 8 (KRT8) | Cytoplasm | 0.724 ± 0.04 | 0.639 ± 0.08 | 1.401 ± 0.07 | 21 |
| 13 591 983 | Lumican (LUM) | Extracellular space | 1.776 ± 0.03 | 2.367 ± 0.09 | No change | 22 |
| 6 679 961 | Myotrophin (MTPN) | Nucleus | 1.396 ± 0.04 | 1.547 ± 0.14 | No change | 23 |
| 33 086 454 | Polymerase (RNA) II (DNA directed) polypeptide L (Polr2 l) | Nucleus | 0.079 ± 0.01 | 0.144 ± 0.03 | 1.270 ± 0.05 | 24 |
| 5 031 981 | Proteasome 26S subunit, non-ATPase, 14 (PSMD14) | Cytoplasm | No change | 1.337 ± 0.09 | No change | 25 |
| 13 592 079 | S100 calcium-binding protein A10 (S100A10) | Cytoplasm | 1.324 ± 0.08 | 1.438 ± 0.09 | No change | 26 |
| 25 742 657 | Sarcosine dehydrogenase (SARDH) | Cytoplasm | 0.677 ± 0.07 | 0.725 ± 0.06 | No change | 27 |
| 56 090 265 | Seryl-tRNA synthetase (SARS) | Cytoplasm | 0.776 ± 0.02 | 0.763 ± 0.01 | No change | 28 |
| 20 302 113 | Stress-induced-phosphoprotein 1 (STIP1) | Cytoplasm | No change | 1.512 ± 0.07 | No change | 29 |
| 6 678 369 | Troponin C Type 1 (slow) (TNNC1) | Cytoplasm | No change | 1.291 ± 0.04 | No change | 30 |
| 62 286 645 | Ts translation elongation factor, mitochondrial (Tsfm) | Unknown | 0.556 ± 0.09 | 0.607 ± 0.08 | No change | 31 |
| 32 401 233 | Titin (cardiac titin N2B isoform) (Ttn) | Cytoplasm | 0.740 ± 0.03 | 0.557 ± 0.06 | No change | 32 |
| 4 389 299 | Vimentin (VIM) | Cytoplasm | 1.391 ± 0.01 | 1.630 ± 0.09 | No change | 33 |
127/126: Norepinephrine 100 µg/kg/h vs. control.
128/126: Norepinephrine 200 µg/kg/h vs. control.
130/129: Norepinephrine 100 µg/kg/h vs. control in the presence of 5-aza-2′-deoxycytidine.
Data are mean ± SEM, n = 5.
2.12. Statistical analysis
Data were expressed as means ± SEM obtained from the number (n) of experimental animals given. Statistical significance (P < 0.05) was determined by the analysis of variance followed by Newman–Keuls post hoc testing or Student's t-test, where appropriate.
3. Results
3.1. Norepinephrine treatment increased blood pressure
Chronic infusion of norepinephrine to rats for 28 days increased plasma norepinephrine concentrations from the control level of 28.4 ± 7.7 to 112.2 ± 20.9 (100 µg/kg/h) and 175.6 ± 55.9 (200 µg/kg/h) pg/μL, respectively, and produced a sustained increase in arterial blood pressure during the norepinephrine treatment (Figure 1).
Figure 1.

Norepinephrine induced an increase in arterial blood pressure. Rats were treated with saline control or norepinephrine (100 µg/kg/h, NE 100) for 28 days. SBP, systolic blood pressure; DBP, diastolic blood pressure; MAP, mean arterial blood pressure. Data are mean ± SEM of five animals. *P < 0.05, NE vs. control.
3.2. 5-Aza-2′-deoxycytidine inhibited Dnmt activity
The norepinephrine treatment resulted in a significant increase in Dnmt activity in the left ventricle, which was blocked by 5-aza-2′-deoxycytidine (Figure 2A). Whereas protein abundance of Dnmt 1, 3a, and 3b was barely detectable in the control heart, the norepinephrine treatment induced an increase in the expression of Dnmt 1, 3a, and 3b in the left ventricle (Figure 2B). To assess the direct inhibitory effect of 5-aza-2′-deoxycytidine on the individual Dnmt, an EpiQuik DNMT Activity/Inhibitor Screening Assay Kit was used to determine the direct effect of 5-aza-2′-deoxycytidine on the activity of purified human Dnmt 1, Dnmt 3a, and murine Dnmt 3b. As shown in Figure 2C, 5-aza-2′-deoxycytidine dose-dependently inhibited all three Dnmt activities.
Figure 2.

5-Aza-2′-deoxycytidine inhibited Dnmt activity. Rats were treated with saline control or norepinephrine 100 µg/kg/h (NE 100) for 28 days. 5-Aza-2′-deoxycytidine (Aza, i.p. 1 mg/kg/day) was administered for the last 6 days of norepinephrine infusion. Dnmt activity (A) and protein abundance (B) were determined in the left ventricle. Data are mean ± SEM of five animals. *P < 0.05, NE 100 vs. control. (C) 5-Aza-2′-deoxycytidine inhibited the activity of purified human Dnmt 1, Dnmt 3a, or murine Dnmt 3b. Data are mean ± SEM. *P < 0.05, Aza vs. control, n = 5.
3.3. 5-Aza-2′-deoxycytidine reversed norepinephrine-induced heart hypertrophy and reduced cardiac contractility
Norepinephrine treatment for 28 days produced a concentration-dependent increase in the left ventricle to body weight ratio (Figure 3A). This was associated with a significant increase in mRNA expression of foetal genes ANP, BNP, and βMHC in the left ventricle (Figure 3B–D). Additionally, the norepinephrine treatment caused significant decreases in LVDP and dP/dtmax, determined in a Langendorff preparation (Figure 3E and F). Strikingly, treatment of rats with a DNA-demethylating agent 5-aza-2′-deoxycytidine for the last 6 days of norepinephrine infusion caused a complete reversal of norepinephrine-induced left ventricular hypertrophy and reduced contractility (Figure 3A–F).
Figure 3.

5-Aza-2′-deoxycytidine reversed norepinephrine-induced heart hypertrophy and failure. Rats were treated with saline control or norepinephrine 100 µg/kg/h (NE 100) or 200 µg/kg/h (NE 200) for 28 days. 5-Aza-2′-deoxycytidine (Aza, i.p. 1 mg/kg/day) was administered for the last 6 days of norepinephrine infusion. Cardiac function was determined in a Langendorff preparation. LV, left ventricle; BW, body weight; LVDP, left ventricle developed pressure. Data are mean ± SEM of five animals. aP < 0.05, NE vs. control; bP < 0.05, NE 200 vs. NE 100.
3.4. 5-Aza-2′-deoxycytidine abrogated norepinephrine-mediated increase in heart susceptibility to ischaemic injury
The norepinephrine treatment resulted in a concentration-dependent increase in LVEDP (Figure 4A), myocardial infarct size (see Supplementary material online, Figure S1 and Figure 4B), and LDH release (Figure 4C), resulted from 20 min of global ischaemia and 60 min of reperfusion in a Langendorff preparation. This was associated with significant decreases in post-ischaemic recovery of LVDP (Figure 4E) and dP/dtmax (Figure 4F). Of importance, treatment of rats with 5-aza-2′-deoxycytidine for the last 6 days of norepinephrine infusion abrogated the norepinephrine-mediated increase in heart susceptibility to ischaemia and reperfusion injury (Figure 4B, C, D, G, and H).
Figure 4.

5-Aza-2′-deoxycytidine abrogated a norepinephrine-mediated increase in heart susceptibility to ischaemic injury. Rats were treated with saline control or norepinephrine 100 µg/kg/h (NE 100) or 200 µg/kg/h (NE 200) for 28 days. 5-Aza-2′-deoxycytidine (Aza, i.p. 1 mg/kg/day) was administered for the last 6 days of norepinephrine infusion. Hearts were subjected to 20 min of ischaemia and 60 min of reperfusion in a Langendorff preparation. LVEDP, left ventricle end-diastolic pressure; LDH, lactate dehydrogenase; LVDP, left ventricle developed pressure. Data are mean ± SEM of five animals. aP < 0.05, NE vs. control; bP < 0.05, NE 200 vs. NE 100.
3.5. 5-Aza-2′-deoxycytidine abolished norepinephrine-induced ROS production
Elevated ROS production is implicated in the pathophysiology of left ventricular hypertrophy and failure. The norepinephrine treatment caused a significant increase in ROS production in the left ventricle, which was abolished by 5-aza-2′-deoxycytidine (see Supplementary material online, Figure S2).
3.6. 5-Aza-2′-deoxycytidine reversed norepinephrine-induced hypermethylation
The norepinephrine treatment produced a concentration-dependent increase in global genomic DNA methylation (Figure 5A), as well as an increase in a gene-specific CpG methylation of the Egr-1-binding site at PKCɛ promoter region (Figure 5B) in the left ventricle. The treatment of rats with 5-aza-2′-deoxycytidine for the last 6 days of norepinephrine infusion reversed hypermethylation induced by norepinephrine (Figure 5A and B). The functional significance of 5-aza-2′-deoxycytidine-mediated demethylation in rescuing gene expression was demonstrated by the finding that 5-aza-2′-deoxycytidine abrogated norepinephrine-induced PKCɛ gene repression in the left ventricle (Figure 5C–E).
Figure 5.

5-Aza-2′-deoxycytidine reversed norepinephrine-mediated hypermethylation and restored PKCε gene expression. Rats were treated with saline control or norepinephrine 100 µg/kg/h (NE 100) or 200 µg/kg/h (NE 200) for 28 days. 5-Aza-2′-deoxycytidine (Aza, i.p. 1 mg/kg/day) was administered for the last 6 days of norepinephrine infusion. Global genomic DNA methylation (Cm/C) in left ventricles was determined by HPLC–MS/MS-MRM. CpG methylation of the Egr-1-binding site at PKCε promoter was determined by MSP. PKCε mRNA and protein abundance in left ventricles were determined by qRT-PCR and western immunoblotting. Data are mean ± SEM of five animals. aP < 0.05, NE vs. control; bP < 0.05, NE 200 vs. NE 100.
3.7. 5-Aza-2′-deoxycytidine caused a reversal of norepinephrine-induced changes in cardiac proteome
To elucidate protein pathways and networks that mediated norepinephrine-induced left ventricular hypertrophy and failure, protein expression patterns were determined in the left ventricle by TMT–LC–MS/MS analysis with an LTQ-Orbitrap-Pro instrument. A total of 1054 proteins were identified in the left ventricle of all hearts in all the treatment groups by one-dimensional LC–MS/MS approach. Of these proteins, 33 demonstrated a consistent modulation pattern by norepinephrine, most of which showed concentration-dependent changes induced by norepinephrine (Table 1). Hierarchical clustering analysis of Table 1 is presented in Supplementary material online, Figure S3. These subsets of proteins included heat-shock chaperon proteins, oxidative stress-related proteins, contractile proteins, and fibrotic response proteins (Figure 6). Of importance, the proteomic analysis revealed that norepinephrine induced an up-regulation of myotrophin and a down-regulation of four and one-half LIM protein 2 (FHL2; Table 1 and Figure 6). Ingenuity pathway analysis (IPA) using the stringent filter (rodent) revealed five separate sub-networks of proteins modulated by norepinephrine (see Supplementary material online, Figure S4). The merged protein networks that were induced by norepinephrine, along with respective sub-cellular localization, are shown in Supplementary material online, Figure S4F, G, and H. Strikingly, treatment of rats with 5-aza-2′-deoxycytidine for the last 6 days of norepinephrine infusion caused a correction in the expression of most of these proteins to baseline in the left ventricle (Table 1) and also a near complete reversal of norepinephrine-activated protein pathways and networks in the left ventricle (see Supplementary material online, Figure S4E and H).
Figure 6.
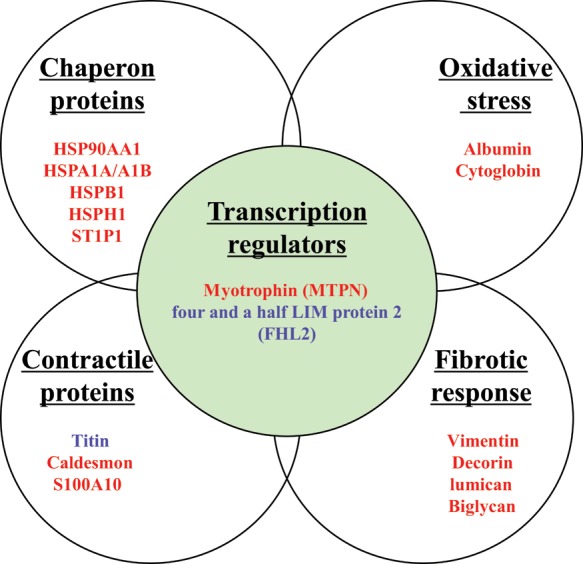
Norepinephrine changed expression of subsets of proteins. Rats were treated with saline control or norepinephrine (100 µg/kg/h or 200 µg/kg/h) for 28 days. Protein expression analysis was performed in left ventricles by TMT–LC–MS/MS analysis with an LTQ-Orbitrap-Velos instrument. Red coloured molecules indicate up-regulated proteins. Blue coloured molecules indicate down-regulated proteins. The list of modulated proteins is presented in Table 1.
4. Discussion
The novelty of the present study is two-fold. First, it provides clear evidence of a causative mechanism of heightened DNA methylation in the progression of heart hypertrophy in vivo in an animal model. Secondly, it demonstrates that increased DNA methylation and the phenotype of heart hypertrophy and reduced cardiac contractility can be reversed by a DNA-demethylating agent, providing a potentially novel therapeutic approach of demethylation in the treatment of failing heart and ischaemic heart disease.
Heart failure is often associated with increased sympathetic activity and elevated circulating catecholamine levels.1–4 Plasma norepinephrine levels are highly variable, and human studies showed a normal value of around 200 pg/mL to plasma norepinephrine values of >900 pg/mL in patients with congestive heart failure.4,18 Whereas rodent plasma norepinephrine levels may not be directly comparable with humans, the proximal 4- to 6-fold increases of plasma levels by the norepinephrine treatments in the present study are in agreement with clinical conditions in patients with heart failure. Consistent with the recent findings of distinct epigenomic DNA methylation patterns in the cardiac genome in heart failure patients,5,6 the present study demonstrated that norepinephrine-mediated left ventricle hypertrophy and reduced cardiac contractility were associated with an increase in global genomic DNA methylation and a gene-specific CpG methylation of PKCɛ gene promoter in the left ventricle. More importantly, the present study demonstrated that treatment of rats with a DNA-demethylating agent 5-aza-2′-deoxycytidine for the last 6 days of norepinephrine infusion caused a reversal of norepinephrine-induced hypermethylation and rescued the phenotype of heart hypertrophy and reduced contractility. 5-Aza-2′-deoxycytidine alone at the dose given had no significant effects on cardiac function and gene expression. These findings provide clear evidence of a causative mechanism of heightened DNA methylation in norepinephrine-induced progression of heart hypertrophy and reduced cardiac contractility in an animal model. 5-Aza-2′-deoxycytidine inhibited a norepinephrine-induced increase in Dnmt activity in the heart, and this inhibitory effect appeared a direct action on both maintenance (Dnmt 1) and de novo (Dnmt 3a and 3b) Dnmts.
The functional significance of 5-aza-2′-deoxycytidine-mediated demethylation in rescuing gene expression was demonstrated in the left ventricle, in which 5-aza-2′-deoxycytidine reversed norepinephrine-induced PKCɛ gene repression. This is in agreement with previous findings showing that 5-aza-2′-deoxycytidine abrogated promoter hypermethylation and rescued PKCɛ gene expression in the foetal heart.9–11 Consistent with the present finding of 5-aza-2′-deoxycytidine-mediated demethylation in the heart, previous studies in adult rats demonstrated that 5-aza-2′-deoxycytidine, given intraperitoneally (i.p. 1 mg/kg/day) for 3–7 days, caused demethylation of 11β-hydroxysteroid dehydrogenase type 2 (11βHSD2) gene promoter in the kidney, lung, and liver, resulting in higher expression of the 11βHSD2 gene in these organs.19 PKCɛ is the best-characterized PKC isoform in the contribution to cardiac hypertrophy and heart failure.20–23 In addition, PKCɛ plays a pivotal role of cardioprotection in heart ischaemia and reperfusion injury,22,24,25 and a causative mechanism of endogenous PKCɛ in protecting the heart against ischaemia and reperfusion injury has been demonstrated in rats.10 The present findings that 5-aza-2′-deoxycytidine abrogated promoter hypermethylation, rescued PKCɛ gene expression, and reversed heightened susceptibility of ischaemic injury in the heart provide additional evidence of a causative mechanism of increased DNA methylation in heightened vulnerability of ischaemic injury in the heart.
Whereas 5-aza-2′-deoxycytidine-induced DNA hypomethylation is likely to affect many genes in the heart, this may be, indeed, important in its effect in rescuing heart hypertrophy and failing heart because of the inevitable involvement of networks of proteins and pathways in reprogramming of heart hypertrophy and failure. Indeed, the proteomic approach revealed multiple protein pathways and networks that may be involved in norepinephrine-induced hypermethylation and left ventricle hypertrophy and reduced cardiac contractility. Whether these were the direct effects of norepinephrine on the heart or the indirect effects through its action on other organs and tissues remain to be determined. The finding of up-regulation of a group of heat-shock and oxidative stress-related proteins indicates a state of stress in the heart caused by norepinephrine treatment. Although the primary physiological function of heat-shock proteins in non-stressed conditions is to perform the chaperoning activity, in stress when the general protein synthesis equilibrium is lost, new heat-shock proteins appear in various cellular compartments and mediate protection to proteins.26 Additionally, both albumin and cytoglobin may function as ROS scavengers in the heart,27,28 and increases of these proteins are likely to be a compensatory mechanism in response to norepinephrine-induced increase in ROS production in the heart. Increasing evidence indicates heightened ROS production in the myocardium of patients and animal models with heart failure.29,30 5-Aza-2′-deoxycytidine abrogated norepinephrine-induced ROS production, resulting in a reversal of stress proteins to baseline levels in the left ventricle.
Consistent with the finding of a failing heart, several proteins related to the contractile machinery of cardiomyocytes were modulated by norepinephrine. Some of these proteins are implicated in heart failure. Thus, cardiac titin N2B isoform, a critical protein that supports cellular acto-myosin fabric and its down-regulation is implicated in cardiac dilation and contractile dysfunction,31,32 was indeed concentration-dependently down-regulated by norepinephrine treatment. Titin is highly susceptible to various forms of cellular damage including oxidation by free radicals.33 Another example is caldesmon that binds to actin and inhibits myosin binding to actin, resulting in inhibition of actin-activated myosin ATPase activity.34 A norepinephrine-induced increase in caldesmon expression in the left ventricle is thus expected to cause increased inhibition of contractility. In addition to these changes in contractile proteins leading to reduced contractility of a failing heart, norepinephrine treatment also induced increases of a group of proteins in association with cardiac tissue fibrotic response, or cardiac remodelling in heart failure. For example, vimentin is an intermediate filament protein and a mesenchymal marker of fibrosis. The increase of vimentin expression in norepinephrine-treated hearts is thus consistent and may indicate an onset of fibrosis in cardiac tissue. Additionally, the three different proteoglycans, decorin, lumican and biglycan, have been up-regulated by norepinephrine in the left ventricle. Although decorin may confer cardioprotection,35 increased expression of other two proteoglycans could either be a consequence of cardiac tissue fibrotic response (e.g. lumican), or heart failure associated cardiac remodelling (e.g. biglycan). Thus, up-regulated expression of lumican is necessary for collagen fibre assembly in fibrosis, and the expression level of lumican is greater in and around the fibrotic lesions of ischaemic and reperfused rat hearts than it is in normal hearts.36 In an analogous fashion, biglycan expression is up-regulated in failing hearts and is implicated in cardiac remodelling in particular for organization of collagen fibrils.37 Biglycan is a potent proteoglycan that promotes the supramolecular assembly of collagen VI.38 The finding that 5-aza-2′-deoxycytidine inhibited norepinephrine-induced changes in contractile proteins as well as proteins related to fibrotic response, and cardiac remodelling is consistent with its function in blocking hypertrophy and rescuing contractility of the left ventricle, and suggests a causative mechanism of heightened DNA methylation in regulating expression of subsets of cardiac proteins in the progression of heart hypertrophy and reduced cardiac contractility.
Of importance, proteomic analysis revealed that norepinephrine induced an up-regulation of myotrophin and a down-regulation of FHL2. Myotrophin is a transcription regulator. Overexpression of myotrophin in cardiac tissues triggers myocardial hypertrophy and heart failure in transgenic mice.39 This process of myotrophin-induced cardiac hypertrophy involves the activation of nuclear factor NF-κβ40 and other cardiac hypertrophy indicator early response mRNAs, namely, c-myc, c-jun, c-fos, ANF, α-actin, β-MHC, and connexin 43 mRNA.41 Similar to rodents, in human hearts, myotrophin is found at higher levels in dilated cardiomyopathy, and increased levels of cardiac myotrophin have been proposed to play a role in the initiation of cardiac hypertrophy.42 On the other hand, FHL2 was significantly down-regulated in norepinephrine-treated hearts. Studies with FHL2 KO transgenic mice revealed that FHL2 function was not necessary for normal cardiac development, but hearts in adult FHL2 KO mice became much more sensitive in cardiac mass increase in response to chronic infusion of β-adrenergic agonist isoproterenol, when compared with wild-type littermates.43 FHL2 acts both as a transcription co-activator and an adaptor protein binding with cardiac titin-N2B segment, which has potential stretch-sensing function. Several studies have revealed that a decrease in FHL2 binding with the titin-N2B segment is one of the pathways to dilated cardiomyopathy.44,45 Although the functional significance of myotrophin and FHL2 in cardiac hypertrophy and failure is well recognized, the regulatory mechanisms of their gene expression are much less clear. The present finding that 5-aza-2′-deoxycytidine reversed norepinephrine-induced changes in the expression of myotrophin and FHL2 provides a novel mechanism of DNA methylation in epigenetic regulation of myotrophin and FHL2 expression patterns in the heart in response to sympathetic overactivity and pressure overload.
The present study provides novel evidence of a causative mechanism of heightened DNA methylation in the progression of heart hypertrophy and reduced cardiac contractility in vivo in an animal model. Furthermore, our study demonstrates that increased DNA methylation and the phenotype of heart hypertrophy can be reversed by a DNA-demethylating agent. It should be noted that whether the gene expression and methylation profiles in the present study would be found in a chronically failing heart remains to be determined. Given the increasing evidence showing the existence of distinct epigenomic patterns of DNA methylation in end-stage failing human hearts, and highly dynamic changes of DNA methylation and demethylation in adult tissues,46 the present finding that a DNA-demethylating agent may rescue the phenotype of heart hypertrophy and failing heart provides exciting therapeutic targets for novel treatments.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported by the National Institutes of Health (grants HL082779 to L.Z., HL083966 to L.Z., and DA032510 D.X.).
Supplementary Material
Acknowledgements
The authors thank Charles Hewitt for his technical expertise in the measurement of plasma norepinephrine levels in this study. The authors also thank Prof. Kala Seal, Loyola Marymount University, College of Business Administration for his valuable suggestions with proteomic analysis.
Conflict of interest: none declared.
References
- 1.Cohn JN, Levine TB, Olivari MT, Garberg V, Lura D, Francis GS, et al. Plasma norepinephrine as a guide to prognosis in patients with chronic congestive heart failure. N Engl J Med. 1984;311:819–824. doi: 10.1056/NEJM198409273111303. [DOI] [PubMed] [Google Scholar]
- 2.Kaye DM, Lefkovits J, Jennings GL, Bergin P, Broughton A, Esler MD. Adverse consequences of high sympathetic nervous activity in the failing human heart. J Am Coll Cardiol. 1995;26:1257–12563. doi: 10.1016/0735-1097(95)00332-0. [DOI] [PubMed] [Google Scholar]
- 3.Floras JS. Sympathetic nervous system activation in human heart failure: clinical implications of an updated model. J Am Coll Cardiol. 2009;54:375–385. doi: 10.1016/j.jacc.2009.03.061. [DOI] [PubMed] [Google Scholar]
- 4.Francis GS, Cohn JN, Johnson G, Rector TS, Goldman S, Simon A. Plasma norepinephrine, plasma renin activity, and congestive heart failure. Relations to survival and the effects of therapy in V-HeFT II. The V-HeFT VA Cooperative Studies Group. Circulation. 1993;87:V140–V148. [PubMed] [Google Scholar]
- 5.Movassagh M, Choy MK, Goddard M, Bennett MR, Down TA, Foo RS. Differential DNA methylation correlates with differential expression of angiogenic factors in human heart failure. PLoS ONE. 2010;5:e8564. doi: 10.1371/journal.pone.0008564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Movassagh M, Choy MK, Knowles DA, Cordeddu L, Haider S, Down T, et al. Distinct epigenomic features in end-stage failing human hearts. Circulation. 2011;124:2411–2422. doi: 10.1161/CIRCULATIONAHA.111.040071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trivedi CM, Luo Y, Yin Z, Zhang M, Zhu W, Wang T, et al. Hdac2 regulates the cardiac hypertrophic response by modulating Gsk3 beta activity. Nat Med. 2007;13:324–331. doi: 10.1038/nm1552. [DOI] [PubMed] [Google Scholar]
- 8.Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell. 2002;110:479–488. doi: 10.1016/s0092-8674(02)00861-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lawrence J, Chen M, Xiong F, Xiao D, Zhang H, Buchholz JN, et al. Foetal nicotine exposure causes PKCepsilon gene repression by promoter methylation in rat hearts. Cardiovasc Res. 2011;89:89–97. doi: 10.1093/cvr/cvq270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Patterson AJ, Chen M, Xue Q, Xiao D, Zhang L. Chronic prenatal hypoxia induces epigenetic programming of PKCε gene repression in rat hearts. Circ Res. 2011;107:365–673. doi: 10.1161/CIRCRESAHA.110.221259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiong F, Xiao D, Zhang L. Norepinephrine causes epigenetic repression of PKCε gene in rodent hearts by activating Nox1-dependent reactive oxygen species production. FASEB J. 2012;26:2753–2763. doi: 10.1096/fj.11-199422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quinlivan EP, Gregory JF., III DNA digestion to deoxyribonucleoside: a simplified one-step procedure. Anal Biochem. 2008;373:383–385. doi: 10.1016/j.ab.2007.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang K, Haynes TA, Filippova M, Filippov V, Duerksen-Hughes PJ. Quantification of ceramide levels in mammalian cells by high performance liquid chromatography coupled to tandem mass spectrometry with multiple-reaction-monitoring mode (HPLC-MS/MS-MRM) Anal Methods. 2011;3:1193–1197. [Google Scholar]
- 14.Xiong L, Darwanto A, Sharma S, Herring J, Hu S, Filippova M, et al. Mass spectrometric studies on epigenetic interaction networks in cell differentiation. J Biol Chem. 2011;286:13657–13668. doi: 10.1074/jbc.M110.204800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang K, Schrag M, Crofton A, Trivedi R, Vinters H, Kirsch W. Targeted proteomics for quantification of histone acetylation in Alzheimer's disease. Proteomics. 2012;12:1261–1268. doi: 10.1002/pmic.201200010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Unwin RD, Smith DL, Blinco D, Wilson CL, Miller CJ, Evans CA, et al. Quantitative proteomics reveals posttranslational control as a regulatory factor in primary hematopoietic stem cells. Blood. 2006;107:4687–4694. doi: 10.1182/blood-2005-12-4995. [DOI] [PubMed] [Google Scholar]
- 17.van Ulsen P, Kuhn K, Prinz T, Legner H, Schmid P, Baumann C, et al. Identification of proteins of Neisseria meningitidis induced under iron-limiting conditions using the isobaric tandem mass tag (TMT) labeling approach. Proteomics. 2009;9:1771–1781. [Google Scholar]
- 18.Masuo K, Katsuya T, Sugimoto K, Kawaguchi H, Rakugi H, Ogihara T, et al. High plasma norepinephrine levels associated with beta2-adrenoceptor polymorphisms predict future renal damage in nonobese normotensive individuals. Hypertens Res. 2007;30:503–511. doi: 10.1291/hypres.30.503. [DOI] [PubMed] [Google Scholar]
- 19.Alikhani-Koopaei R, Fouladkou F, Frey FJ, Frey BM. Epigenetic regulation of 11 beta-hydroxysteroid dehydrogenase type 2 expression. J Clin Invest. 2004;114:1146–1157. doi: 10.1172/JCI21647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dorn GW, II, Force T. Protein kinase cascades in the regulation of cardiac hypertrophy. J Clin Invest. 2005;115:527–537. doi: 10.1172/JCI24178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu G, Toyokawa T, Hahn H, Dorn GW., II Epsilon protein kinase C in pathological myocardial hypertrophy. Analysis by combined transgenic expression of translocation modifiers and Galphaq. J Biol Chem. 2000;275:29927–29930. doi: 10.1074/jbc.C000380200. [DOI] [PubMed] [Google Scholar]
- 22.Chen L, Hahn H, Wu G, Chen CH, Liron T, Schechtman D, et al. Opposing cardioprotective actions and parallel hypertrophic effects of delta PKC and epsilon PKC. Proc Natl Acad Sci USA. 2001;98:11114–11119. doi: 10.1073/pnas.191369098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mochly-Rosen D, Wu G, Hahn H, Osinska H, Liron T, Lorenz JN, et al. Cardiotrophic effects of protein kinase C epsilon: analysis by in vivo modulation of PKCepsilon translocation. Circ Res. 2000;86:1173–1179. doi: 10.1161/01.res.86.11.1173. [DOI] [PubMed] [Google Scholar]
- 24.Gray MO, Zhou HZ, Schafhalter-Zoppoth I, Zhu P, Mochly-Rosen D, Messing RO. Preservation of base-line hemodynamic function and loss of inducible cardioprotection in adult mice lacking protein kinase C epsilon. J Biol Chem. 2004;279:3596–3604. doi: 10.1074/jbc.M311459200. [DOI] [PubMed] [Google Scholar]
- 25.Ping P, Zhang J, Pierce WM, Jr, Bolli R. Functional proteomic analysis of protein kinase C epsilon signaling complexes in the normal heart and during cardioprotection. Circ Res. 2001;88:59–62. doi: 10.1161/01.res.88.1.59. [DOI] [PubMed] [Google Scholar]
- 26.Snoeckx LH, Cornelussen RN, Van Nieuwenhoven FA, Reneman RS, Van Der Vusse GJ. Heat shock proteins and cardiovascular pathophysiology. Physiol Rev. 2001;81:1461–1497. doi: 10.1152/physrev.2001.81.4.1461. [DOI] [PubMed] [Google Scholar]
- 27.Roche M, Rondeau P, Singh NR, Tarnus E, Bourdon E. The antioxidant properties of serum albumin. FEBS Lett. 2008;582:1783–1787. doi: 10.1016/j.febslet.2008.04.057. [DOI] [PubMed] [Google Scholar]
- 28.Trent JT, III, Hargrove MS. A ubiquitously expressed human hexacoordinate hemoglobin. J Biol Chem. 2002;277:19538–19545. doi: 10.1074/jbc.M201934200. [DOI] [PubMed] [Google Scholar]
- 29.Dhalla NS, Temsah RM, Netticadan T. Role of oxidative stress in cardiovascular diseases. J Hypertens. 2000;18:655–673. doi: 10.1097/00004872-200018060-00002. [DOI] [PubMed] [Google Scholar]
- 30.Pimentel DR, Amin JK, Xiao L, Miller T, Viereck J, Oliver-Krasinski J, et al. Reactive oxygen species mediate amplitude-dependent hypertrophic and apoptotic responses to mechanical stretch in cardiac myocytes. Circ Res. 2001;89:453–460. doi: 10.1161/hh1701.096615. [DOI] [PubMed] [Google Scholar]
- 31.Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366:619–628. doi: 10.1056/NEJMoa1110186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McNally EM. Genetics: broken giant linked to heart failure. Nature. 2012;483:281–282. doi: 10.1038/483281a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.LeWinter MM, Wu Y, Labeit S, Granzier H. Cardiac titin: structure, functions and role in disease. Clin Chim Acta. 2007;375:1–9. doi: 10.1016/j.cca.2006.06.035. [DOI] [PubMed] [Google Scholar]
- 34.Kordowska J, Huang R, Wang CL. Phosphorylation of caldesmon during smooth muscle contraction and cell migration or proliferation. J Biomed Sci. 2006;13:159–172. doi: 10.1007/s11373-005-9060-8. [DOI] [PubMed] [Google Scholar]
- 35.Yan W, Wang P, Zhao CX, Tang J, Xiao X, Wang DW. Decorin gene delivery inhibits cardiac fibrosis in spontaneously hypertensive rats by modulation of transforming growth factor-beta/Smad and p38 mitogen-activated protein kinase signaling pathways. Hum Gene Ther. 2009;20:1190–1200. doi: 10.1089/hum.2008.204. [DOI] [PubMed] [Google Scholar]
- 36.Baba H, Ishiwata T, Takashi E, Xu G, Asano G. Expression and localization of lumican in the ischemic and reperfused rat heart. Jpn Circ J. 2001;65:445–450. doi: 10.1253/jcj.65.445. [DOI] [PubMed] [Google Scholar]
- 37.Ahmed MS, Oie E, Vinge LE, Yndestad A, Andersen GG, Andersson Y, et al. Induction of myocardial biglycan in heart failure in rats—an extracellular matrix component targeted by AT1 receptor antagonism. Cardiovasc Res. 2003;60:557–568. doi: 10.1016/j.cardiores.2003.08.017. [DOI] [PubMed] [Google Scholar]
- 38.Wiberg C, Heinegard D, Wenglen C, Timpl R, Morgelin M. Biglycan organizes collagen VI into hexagonal-like networks resembling tissue structures. J Biol Chem. 2002;277:49120–49126. doi: 10.1074/jbc.M206891200. [DOI] [PubMed] [Google Scholar]
- 39.Sarkar S, Leaman DW, Gupta S, Sil P, Young D, Morehead A, et al. Cardiac overexpression of myotrophin triggers myocardial hypertrophy and heart failure in transgenic mice. J Biol Chem. 2004;279:20422–20434. doi: 10.1074/jbc.M308488200. [DOI] [PubMed] [Google Scholar]
- 40.Gupta S, Purcell NH, Lin A, Sen S. Activation of nuclear factor-kappaB is necessary for myotrophin-induced cardiac hypertrophy. J Cell Biol. 2002;159:1019–1028. doi: 10.1083/jcb.200207149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mukherjee DP, McTiernan CF, Sen S. Myotrophin induces early response genes and enhances cardiac gene expression. Hypertension. 1993;21:142–148. doi: 10.1161/01.hyp.21.2.142. [DOI] [PubMed] [Google Scholar]
- 42.Sil P, Misono K, Sen S. Myotrophin in human cardiomyopathic heart. Circ Res. 1993;73:98–108. doi: 10.1161/01.res.73.1.98. [DOI] [PubMed] [Google Scholar]
- 43.Kong Y, Shelton JM, Rothermel B, Li X, Richardson JA, Bassel-Duby R, et al. Cardiac-specific LIM protein FHL2 modifies the hypertrophic response to beta-adrenergic stimulation. Circulation. 2001;103:2731–2738. doi: 10.1161/01.cir.103.22.2731. [DOI] [PubMed] [Google Scholar]
- 44.Granzier HL, Radke MH, Peng J, Westermann D, Nelson OL, Rost K, et al. Truncation of titin's elastic PEVK region leads to cardiomyopathy with diastolic dysfunction. Circ Res. 2009;105:557–564. doi: 10.1161/CIRCRESAHA.109.200964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Radke MH, Peng J, Wu Y, MeNabb M, Nelson OL, Granzier H, et al. Targeted deletion of titin N2B region leads to diastolic dysfunction and cardiac atrophy. Proc Natl Acad Sci USA. 2007;104:3444–3449. doi: 10.1073/pnas.0608543104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guo JU, Su Y, Zhong C, Ming GL, Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145:423–434. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.