

Abstract
Prostate cancer is a heterogeneous disease where the previous concept of “hormone resistance” has been changed by a new generation of hormonal therapies that have proven efficacy in the castration-resistant setting. The fact is that androgens play a crucial role in the whole clinical course of prostate cancer, even when a patient meets castration-resistance criteria. The development of abiraterone showed how important and clinically meaningful can be to achieve the lowest possible levels of testosterone, and androgen receptor overexpression, mutation, or enhanced crosstalk with other pathways, which can also be targeted with new agents tested in the last few years. New androgen biosynthesis inhibitors have been developed, such as orteronel (TAK-700), but also new antiandrogens (enzalutamide, ARN-509, ODM-201) or even agents with a dual mechanism of action (galeterone). In this review the development of new hormonal therapies following the arrival of abiraterone for the treatment of prostate cancer will be summarized.
Keywords: castration-resistant prostate cancer, abiraterone, enzalutamide, ARN-509, orteronel, TAK-700, galeterone, ODM-201
Introduction
Prostate cancer is the most common malignancy in men, accounting for 241 740 estimated new cases in the United States in 2012.1 Its mortality is relatively low, with 28 170 estimated deaths due to prostate carcinoma in the same time; even so, it is the second cause of cancer death among men, above other tumors such as colorectal or pancreatic cancer. Almost all of these deaths occur in patients with castration-resistant prostate carcinoma (CRPC), in whom disease has progressed despite castrate levels of testosterone, and after several hormonal therapies.
Estimated prostate cancer incidence rates are higher in developed countries,2 with 72% of the cases and 53% of the deaths (all regions of Europe plus North America, Australia/New Zealand, and Japan). Lowest incidence rates are seen in south central Asia, northern Africa and eastern Asia, with an age standardized ratio per 100 000 habitants (ASR) between 4 and 8. On the other hand, Australia/New Zealand area shows an ASR of 105, and North America also has one the highest ASR, of 85. In contrast, mortality is higher in the Caribbean (ASR 26.3), southern Africa (19.3) and western Africa (18,3), while it is lower in North America (ASR 9.9) and some areas of eastern and central Asia (ASR between 2 and 3). In most of these areas, prostate cancer incidence rates are rising over the past 10 years of observation, but mainly in less resourced countries. These countries also tend to show stable or rising mortality trends, compared with the highest income regions, where mortality is getting lower.3
CRPC, according to the European consensus and the Prostate Cancer Working Group-2 (PCWG2), is defined by the following criteria: castrate serum levels of testosterone (<50 ng/ml); three consecutive rises of prostatespecific antigen (PSA), 1 week apart, resulting in two 50% increases over the nadir; antiandrogen withdrawal for at least 4 weeks for flutamide and for at least 6 weeks for bicalutamide; PSA progression despite consecutive hormonal manipulations; and progression or appearance of 2 or more bone lesions in bone scintigraphy, or in soft tissue following Response Evaluation Criteria in Solid Tumors (RECIST) criteria, or nodes >2 cm in diameter.4
In this setting of metastatic CRPC (mCRPC), chemotherapy is a treatment option. This is mainly based in the TAX 327 study,5,6 a phase III trial that demonstrated an improvement in overall survival (OS) and quality of life (QoL) in patients with CRPC when they were treated with every-3-week docetaxel and daily prednisone compared with weekly docetaxel or mitoxantrone (OS 19.2 vs. 17.8 vs. 16.3 mo, P < 0.004). A similar trial, comparing docetaxel and estramustine vs. mitoxantrone and prednisone,7 also favored the docetaxel arm, with superior PFS (6.3 vs 3.2 mo, P < 0.001) and OS (17.5 vs 15.6 mo, P = 0.02). These results supported the use of docetaxel-based chemotherapy as first-line therapy for patients with mCRPC.
After progression to docetaxel, therapeutic options were scarce until 2010, when cabazitaxel and abiraterone showed improvements in OS in the post-docetaxel setting.
Cabazitaxel8 is a novel tubulin-binding taxane that proved superiority over mitoxantrone, both in combination with prednisone, with better OS (15.1 vs 12.7 mo, P < 0.0001), better progression-free survival (PFS) (2.8 vs 1.4 mo, P < 0.0001) and better PSA response rate (39.2 vs 17.8%, P = 0.0002). Main toxicities associated with cabazitaxel were neutropenia (82% grade ≥ 3), febrile neutropenia (8%), and anemia (11% grade ≥ 3). Thus, cabazitaxel was approved by the Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for the treatment of mCPRC that had progressed after docetaxel therapy.
On the other hand, abiraterone is a novel inhibitor of androgen biosynthesis that has also been tested in this setting. In the phase III trial COU-AA-301, published in 2011,9 abiraterone plus prednisone compared with placebo plus prednisone demonstrated a superior OS (14.8 vs 10.9 mo, P < 0.001), PFS (5.6 vs 3.6 mo, P < 0.001) and PSA response rate (29% vs 6%, P < 0.001). Time to first skeletal related event (SRE) was significantly longer with abiraterone, and the rate of pain palliation was also higher. Side effects of abiraterone were comparable to the placebo arm, being more common in the abiraterone group fluid retention and edema (31% all grades, < 3% grade ≥ 3) and hypokalemia (17% all grades, < 4% grade ≥ 3). Updated results published in 2012 have confirmed this data.10
The clinical development of abiraterone has already moved to the pre-chemotherapy setting: results of the COU-AA-302 trial were published at the beginning of 2013.11 In this trial, patients with mCRPC that were asymptomatic or mildly symptomatic, and chemotherapy-naïve, were randomized to abiraterone or placebo. There was a significant improvement in radiologic PFS (NR vs 8.3 mo, P < 0.0001), and a clear trend toward OS improvement (NR vs 27.2 mo, P = 0.0097). Secondary endpoints such as time to chemotherapy initiation, time to opioid use for cancer-related pain, time to ECOG-PS deterioration and time to PSA progression, also favored the abiraterone arm. Updated results from the 2013 ASCO meeting were consistent with this,12 with superiority of the abiraterone arm for radiologic PFS (16.5 vs 8.2 mo, P < 0.0001) and OS (35.3 vs 30.1 mo, P = 0.0151)
The emergence of abiraterone has somehow changed the paradigm of the treatment of prostate cancer, as it was assumed that the initiation of chemotherapy was in part because the disease had become “hormone-resistant”. But it has been demonstrated that the androgen receptor (AR) pathway still drives tumor progression in a wide majority of patients,13-15 even after progression to chemotherapy. Tumors can grow despite castration levels of testosterone through a wide variety of mechanisms: upregulation of androgen synthesis, mostly in a paracrine–autocrine manner,16,17 mutations affecting the androgen receptor and its downstream pathway,18-20 and enhanced crosstalk with other pathways,21,22 among others. These pathways are described in Figure 1.
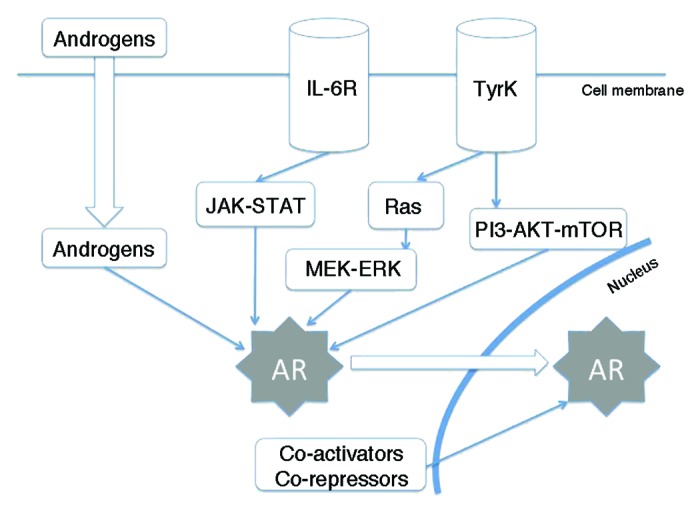
Figure 1. Summarized view of androgen receptor cross-talk with other pathways. AR, androgen receptor; TyrK, tyrosine-kinase receptor; IL-6R, interleukin-6 receptor.
In this review, I will briefly summarize the basis of prostate cancer molecular biology, as well as the new hormonal therapies targeting the androgen-related pathways that have been developed in the last few years, apart from abiraterone.
Basis of Prostate Cancer Molecular Biology
There are many mechanisms involved in prostate cancer initiation and progression. Throughout prostate development and maturation, genes such as NKX3.1, FOXA1, and AR function mediate gland formation and cellular differentiation.23 Aberrant activation of these and other pathways can promote hyperplasic proliferation and/or progression of prostate carcinoma.
NKX3.1 is one of the earliest and more maintained prostate-sp.ecific proteins at prostate differentiation. Although its mutations are relevant for disease occurrence and progression, NKX3.1 is not usually considered as a tumor suppressor gene since loss is not sufficient for tumorigenesis and it is still significantly expressed in aggressive prostate carcinoma.24 NKX3.1 loss, together with MYC overexpression, also promotes tumorigenesis in murine models;25 therefore, it is considered to have a crucial role in the initial stages of prostate cancer.
AR receptor activation plays a crucial role in the progression of prostate cancer, even in the CRPC stage. There are many different mechanisms to reactivate the AR, which will not be analyzed deeply as it is not the purpose of this review. The most common one is AR amplification/overexpression, being seen almost exclusively in tumors that recur after androgen deprivation therapy (ADT); this leads to more sensitivity to lower levels of androgens, and overrides the action of AR antagonists, such as bicalutamide.26,27 AR somatic mutations are not usually present at initial stages of the disease, as they represent a mechanism of resistance to ADT. Most of them are gain-of-function mutations, and recent studies with whole genome sequencing have shown that are almost universal in CRPC samples.28 Constitutively active splice variants of the AR have also been correlated with progression to the stage of CRPC.29 Also, several post-translational modifications (PTMs) of the AR influence its activity, structure and stability; the majority of them result in AR activation and are selected for during progression to CRPC.30 Nevertheless, most PTMs have not been studied in a clinical context, and it is unclear whether they have clinical utility. An alternative mechanism to reactivate AR in the absence of circulating androgens is the intratumoral synthesis of androgens.31
FOXA1 is a transcription factor that facilitates AR binding to chromatine. It appears to both inhibit and promote AR binding to regulate transcriptional programs.32 FOXA1 elevation in primary prostate cancer independently predicts for shorter time to death33 and is highly expressed in CRPC metastases.34
As with many other tumoral types, several oncogenes and tumor suppressor genes have been linked to prostate cancer initiation and progression. One of the most studied ones in prostate cancer is Myc, a proto-oncogene that acts as a transcription factor, regulating a wide range of cellular processes. Myc amplification is detected in about 30% of prostate carcinomas, and by itself can promote tumor initation;35 moreover, Myc amplification is more common in metastatic cancer compared with primary tumors, and its levels are correlated with tumor progression.36 Despite these data, Myc is considered nowadays “undruggable”, that is, cannot be targeted pharmacologically.
Recently, a fusion between the 5′-untranslated region of transmembrane protease serine-2 (TMPRSS2), and androgen regulated gene, and the 3′-exon of ERG, an erythroblast transformation-specific (ETS) transcription factor family member, was discovered;37 this fusion gene was initially considered to be involved in the initial stages of prostate carcinoma. However, several in vitro experiments seem to link this gene to the development of invasiveness and metastasis.38 Overall, approximately 50% of primary or metastatic tumors have a variant of this fusion; its presence has been correlated with a more aggressive phenotype in prostate cancer patients.39 Some other alterations, such as loss of phosphatase and tensin homolog (PTEN), mutations in phosphatidil-inosytol-3-kinase (PI3K) pathway, or malfunction of the retinobalstoma (Rb) gene, among others, have also been analyzed in prostate cancer, with very promising results.
These mechanisms of tumor initiation and progression are summarized in Figure 2.
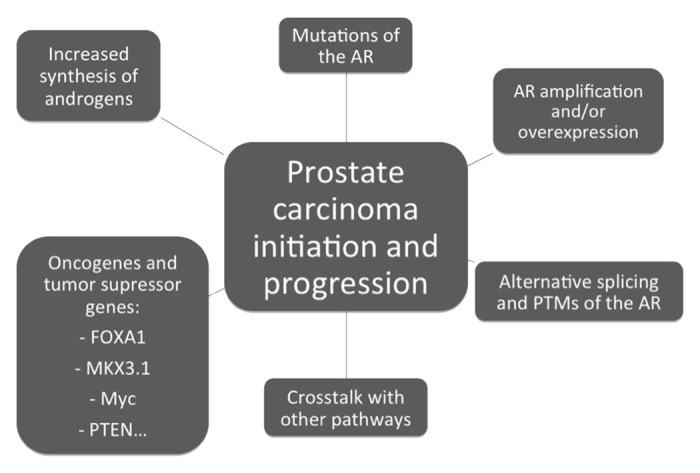
Figure 2. Mechanisms involved on prostate carcinoma initation and progression. AR, androgen receptor; PTMs, posttranslational modifications.
Enzalutamide (MDV3100)
Enzalutamide is a novel AR antagonist that has a triple mechanism of action. First, it has a much stronger affinity for the AR than other AR antagonists; second, inhibits AR nuclear translocation; and third, impairs AR binding to DNA and coactivator recruitment. In contrast to bicalutamide, MDV3100 is an AR pure antagonist, with no detectable agonist activity in cell lines that overexpress AR.40,41
A phase I–II study with MDV3100 conducted in 140 patients was published in 2010.42 Patients with progressive, metastatic, castration-resistant prostate cancer were enrolled in dose-escalation cohorts, from 30 mg to 600 mg. The primary objective was to identify the safety and tolerability profile of MDV3100 and to establish the maximum tolerated dose. Of the 140 patients, only 7 patients had a rising PSA as the only evidence of progression, with no detectable metastases; 78% of patients had bone metastases, and 9% of patients had visceral disease. Forty-six percent of patients were chemotherapy-naïve, 21% of patients had received four or more lines of hormonal treatment, and 45% of patients had been treated with ketoconazole.
After administration of one dose, the drug was absorbed rapidly, and time to maximum concentration was between 30 min and 4 h; the half-life was about one week. The maximum tolerated dose for sustained treatment (more than 28 d) was 240 mg. Median time on study drug was 21 weeks, with 70% of patients having discontinued treatment, mainly because of radiological progression (34%), PSA progression (12%), and clinical disease progression (11%). Toxicities with MDV3100 were generally mild, with fatigue being the only grade 3 or higher toxicity reported (11% of patients), and only at doses of 240 mg or higher.
Antitumor activity was seen at all dosage groups, with declines of PSA ≥50% in 56% of patients. The extent and proportion of PSA decrease was dose-dependent up to the dosage cohort of 150 mg, with no obvious additional benefit for increased dose. There were also confirmed responses in soft tissue (22%) and stabilizations in bone disease (56%). Median time to progression was 46 weeks for radiological progression (not reached for chemotherapy-naïve patients, and 29 weeks for patients with previous chemotherapy exposure). These encouraging data led to the design of a phase III trial.
The phase III randomized double-blind trial, named AFFIRM (A Study Evaluating the Efficacy and Safety of the Investigational Drug MDV3100), was conducted in patients with mCRPC that had been previously treated with one or two chemotherapy regimens, at least one of which contained docetaxel, and has been published in 2012.43 A total of 1199 patients were included from September 2009 to November 2010, being randomized in a 2:1 ratio to enzalutamide 160 mg (orally once daily as four 40 mg capsules) or placebo. The use of prednisone or other glucocorticoids was permitted but not required, and the study drug was given without regard to food intake. Primary endpoint was OS, and secondary endpoints included radiographic PFS, measures of response (in the PSA level, in soft tissue, and in QoL scores), time to PSA progression and time to first SRE.
The study was stopped after a planned interim analysis at the time of 520 deaths, showing a median overall survival of 18.4 mo for the enzalutamide group, compared with 13.6 mo in the placebo group (HR 0.63, P < 0.001). The overall survival benefit was maintained across all subgroups (regardless of age, pain intensity, geographic region, and type of progression at entry), and was consistent after adjustment for stratification factors and prognostic factors. On the basis of these results, an independent data and safety monitoring committee recommended that the study be halted and unblinded, with eligible patients in the placebo group offered treatment with enzalutamide. The superiority of enzalutamide over placebo was shown for all secondary endpoints: PSA level response rate (54% vs 2%, P < 0.001), soft-tissue response rate (29% vs 4%, P < 0.001), FACT-P QoL response (43% vs 18%, P < 0.001), time to PSA progression (8.3 vs 3 mo, P < 0.001), radiographic PFS (8.3 vs 2.9 mo, P < 0.001), and time to first SRE (16.7 vs 13.3 mo, P < 0.001). Some recently published data have also shown a clear benefit in health-related quality of life terms for patients taking enzalutamide vs. placebo.44
Safety data showed a similar rate of adverse events for the two groups, with the enzalutamide group having a lower incidence of adverse events of grade 3 or above (45.3% vs 53.1% in the placebo group). There was a higher incidence of all grades of fatigue, diarrhea, hot flashes, musculoskeletal pain and headache in the enzalutamide group. Seizures were reported in 5 of 800 patients (0.6%) receiving enzalutamide, several of whom had predisposing conditions or concomitant treatments; seizures were already seen in the phase I–II trial at doses of 360 mg or higher, and caution should be used in administering enzalutamide to patients with previous history of seizures or other predisposing conditions. The results of this trial have granted enzalutamide FDA approval on 31 August 2012 for the treatment of mCRPC patients previously treated with docetaxel.
The development of enzalutamide has already moved to the pre-chemotherapy setting. A phase III randomized, double-blind, placebo controlled trial, named PREVAIL (A Safety and Efficacy Study of Oral MDV3100 in Chemotherapy-Naïve Patients with Progressive Metastatic Prostate Cancer, NCT01212991), is being conducted in patients with mCRPC before chemotherapy, and recruitment was completed in June 2012 with 1680 patients, with results still pending. First interim analysis is expected for late 2013.
One of the main potential advantages of enzalutamide is that concomitant administration of corticosteroids is not mandatory. In the AFFIRM trial, patients were allowed but not required to take corticosteroids. Nevertheless, approximately 30% of the patients received corticosteroids at baseline and 48% were initiated on steroid therapy during the trial. In a recently presented post-hoc analysis of the AFFIRM trial, on-study corticosteroid use was associated with reduced OS and higher rates of grade 3–4 adverse events.45 This may be explained by the fact that patients with more advanced disease have more disease related-symptoms that may require steroid therapy; but we should also consider that corticosteroids may induce aberrant androgen receptors through promiscuous binding to the AR, inducing a more rapid disease progression.46 Importantly, the positive effect of enzalutamide was confirmed regardless of whether or not steroids were also co-administered.
There are several clinical trials with enzalutamide. Some of them are testing it in earlier stages of the disease; for example, one is testing enzalutamide monotherapy in hormone-naïve prostate cancer patients (NCT01302041). First results were seen at the ASCO 2013 Annual Meeting,47 showing a PSA response at week 25 of 93%, and a median PSA decrease of −99.6%, with similar efficacy to castration. Some other trials are testing the combination of enzalutamide with abiraterone in mCRPC patients (NCT01650194), or with immunotherapy in non-metastatic CRPC (NCT01875250), but definitive results are still unpublished.
ARN-509
ARN-509 is a new antiandrogen, a competitive inhibitor of AR that is fully antagonistic to AR overexpression, which is a very common feature in CRPC. This new agent binds AR and inhibits growth and androgen-mediated gene transcription in AR overexpressing prostate cancer cells, but also impairs nuclear localization of AR and DNA binding, and therefore it has shown a potent inhibition of tumor growth in murine xenograft models of CRPC. A maximal therapeutic effect was observed at 30 mg/kg/day, compared with 100 mg/kg/day with enzalutamide.48 Besides that, it seems to penetrate less effectively the blood-brain barrier than enzalutamide in this mouse model, suggesting that the chance of developing seizures may be less than with enzalutamide.49
Data from a phase I–II trial with ARN-509 (NCT01171898) were presented at the ASCO Genitourinary Cancer Symposium 2012.50 In the phase I part of the study, 24 patients were enrolled, receiving an orally dose of ARN-509 on a continuous daily dosing schedule, and testing seven different doses. The most common treatment-related grade 1–2 adverse events were fatigue (38%), nausea (29%), and pain (24%), with only one grade 3 event (abdominal pain) at the dose of 300 mg, possibly related to a higher pill burden, that was not seen in the other three patients with this dosing schedule. Pharmacokinetic was shown to be linear and dose-dependent, and 240 mg was the selected dose for the phase II part of the study. Fifty-five percent of patients had ≥50% PSA declines, showing promising activity.
The phase II trial included three different cohorts:51 non-metastatic treatment-naïve CRPC patients, mCRPC treatment-naïve patients, and mCRPC patients pre-treated with abiraterone. Primary endpoint was PSA response rate at 12 weeks. A total of 46 patients with mCRPC were enrolled: 25 treatment-naïve and 21 post-abiraterone. At 12 weeks, the PSA response was 88% (treatment-naïve) and 29% (post-abiraterone). The most common treatment related adverse events were fatigue (30%), abdominal pain (24%), nausea (22%), and diarrhea (17%). In the non-metastatic CRPC population,52 47 patients were enrolled between November 2011 and May 2012, with a 12-week response of 91%. Time to PSA progression has not been reached. Adverse events were similar to previous studies.
Orteronel (TAK-700)
Orteronel (TAK-700) is a novel oral, selective, reversible, non-steroidal androgen synthesis inhibitor of the 17.20 lyase activity, one of the two enzymatic reactions catalyzed by CYP17.53 This agent, as it does not inhibit the 17-α-hydroxylase activity of CYP17, is less likely to require corticosteroid replacement, an important advantage due to the drawbacks associated with long-term corticosteroid use.54
Preliminary phase I–II data were presented at the ASCO Genitourinary Cancer Symposium 2012.55 A total of 97 patients with mCRPC received orteronel at different doses (300 mg twice a day, 400 mg twice a day, 600 mg twice a day, or 600 mg daily), and with or without prednisone. Most common adverse events were fatigue (76%), nausea (47%), and constipation (38%), with grade ≥ 3 fatigue in 12% of the patients, and hypokalemia grade ≥ 3 in 8% of the patients. PSA response rates (≥50% decrease) were seen in approximately 50% of patients, with no clear relationship with dose ≥ 300 mg or with the addition of prednisone. Of 51 RECIST-evaluable patients, 10 had partial responses, 22 stable disease, and 15 progressive disease.
Another phase II trial (NCT01046916) was presented at the ASCO 2012 Annual Meeting.56 It enrolled 39 patients with non-metastatic CRPC, with only PSA progression, but without evidence of metastatic disease. Orteronel was given at a dose of 300 mg twice a day, without concomitant corticoids. PSA responses were seen in 78% of the patients, with a PFS of 14.8 mo. Main grade 3–4 toxicities were hypertension (14%), dyspnea (8%), and pneumonitis (7%).
Due to these results, phase III trials with orteronel have been designed, one in docetaxel-resistant mCRPC patients (NCT01193257), and another one in chemotherapy-naïve mCRPC patients (NCT01193244). Both have completed accrual, but the trial in the post-chemotherapy setting has been recently unblinded because of an interim analysis that indicated the study would likely not meet the primary endpoint of overall survival.
Orteronel is also being tested in earlier stages of prostate cancer. There is a phase III trial recruiting mCRPC treatment-naïve patients, comparing luteinizing hormone-releasing hormone (LHRH) agonists with bicalutamide vs. LHRH agonists with orteronel (NCT01809691).
Galeterone (TOK-001)
Galeterone (TOK-001) is a selective inhibitor of 17-α-hydroxylase and 17.20-lyase activity, but also downregulates AR expression, competitively inhibits androgen binding, and impairs AR translocation to the nucleus.57 The preliminary results of an open-label, multicenter dose-finding phase I study were presented at the ASCO 2012 Annual Meeting.58 Chemotherapy-naïve CRPC patients were enrolled in dose cohorts from 650 to 2600 mg of galeterone daily for 12 weeks. Thirty-six of 49 patients completed 12 weeks of treatment, being toxicity (6 patients) and progression (5 patients) the most common reasons for discontinuation. Maximum tolerated dose was not reached, with no trend for increasing toxicity with dose escalation. There were 8% of grade 3 adverse events, and 1% of grade 4 adverse events. 22% of patients demonstrated >50% PSA decline, and 26% had 30–50% declines. A 2-part, phase II trial of galeterone is currently recruiting participants (ARMOR2 trial, NCT01709734).
Recent data have shown that, compared with other novel hormonal therapies, galeterone seems to be the most potent and selective CYP17 lyase inhibitor, while abiraterone most selectively inhibited CYP 17 hydroxylase activity.59 Galeterone produced minimal changes in cortisol levels and other intermediate precursors; in contrast, abiraterone increased significantly the levels of progresterone, and abiraterone and orteronel decreased cortisol levels by 70–90%.
ODM-201
ODM-201 is a new generation AR antagonist that does not cross the blood-brain barrier, unlike other antiandrogens, and inhibits AR nuclear translocation. ODM-201 has no agonist activity when the AR is overexpressed. The results of the first-in-man, multicenter, phase I–II dose escalation trial were presented at the European Society of Medical Oncology (ESMO) 2012 Meeting.60 This study planned to enroll 3 to 6 patients with metastatic CRPC, either treatment-naïve or pre-treated with docetaxel, in every dose cohort of 100, 200, 300, 500, 700, and 900 mg daily. Eighty-seven percent of the first 21 patients reached a PSA decline ≥50%, and all evaluable patients to date showed a partial response or stable disease at 12 weeks following RECIST criteria. There were no significant emergent adverse events with ODM-201, asthenia, nausea, and diarrhea being the most common ones. The phase II portion of this study started in June 2012, and plans to enroll 100 patients.
Future Research Directions in Prostate Cancer Hormonal Therapy
The field of hormonal therapies in CRPC has widened dramatically in the last few years. As some of them will probably move to the pre-chemotherapy scenario, we will be able to delay the need for initiating chemotherapy and its associated side effects. But nowadays there are still many questions that remain unanswered. The optimal timing and sequencing of these new agents with standard therapy is still unknown.
There is also interest about when to stop the CYP17 inhibition, and if it should be maintained throughout the whole course of the disease, as we usually do with LH-RH analogs. Potential synergistic mechanisms have been proposed for the combination of CYP17 inhibitors, such as abiraterone, and antiandrogens, mainly enzalutamide, and some early phase trials are exploring this issue. One of these trials is a phase II trial being conducted at the MD Anderson Cancer Center, exploring the combination of enzalutamide and abiraterone in mCRPC with bone metastases (NCT01650194). Some other trials also explore the potential role of androgen receptor pathway cross-talks with other pathways; for example, some trials are testing the combination of abiraterone with BEZ235, an inhibitor of PI3K and mTOR (NCT01717898), or with BKM-120, an inhibitor of PI3K (NCT01741753).
Another area in need of further development is biomarker discovery, as there will be a vast number of new agents that will hopefully get approval by regulatory agencies in the next years. There is evidence pointing that TMPRSS2–ERG fusion gene may be correlated with improved response to abiraterone;61 very recently, a work presented at ASCO 2013 Annual Meeting62 showed that patients with mCRPC chemotherapy-naïve treated with abiraterone derived a greater benefit if they had ERG fusions (mainly those with interstitial deletion and duplication of fusion sequences). Also, circulating tumor cells (CTCs), both baseline and after therapy, have been correlated with OS, and could be useful surrogate markers for future studies.63,64
The role of inflammation in prostate cancer initiation and progression may have also some interest for further research. There is epidemiological evidence that inflammatory stimuli play a role in prostate carcinogenesis,65 and targeting these features with anti-inflammatory drugs, such as nonsteroidal anti-inflammatory drugs (NSAIDS) or cyclooxygenase inhibitors (COX), could improve the results of hormonal therapies for prostate cancer.
Conclusions
Prostate cancer therapy has entered a new era. Several new agents have been approved in the past two years, and some more will prove their usefulness and join the therapeutic options available for the treatment of this disease. Recent evidence show that CRPC remains hormone-driven, even in advanced stages, and with deeper knowledge of the underlying biology of prostate cancer, we will be able to select patients more likely to respond to a specific drug, and therefore sequence these agents to derive the greatest benefit for the individual patient.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/26724
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 3.Center MM, Jemal A, Lortet-Tieulent J, Ward E, Ferlay J, Brawley O, Bray F. International variation in prostate cancer incidence and mortality rates. Eur Urol. 2012;61:1079–92. doi: 10.1016/j.eururo.2012.02.054. [DOI] [PubMed] [Google Scholar]
- 4.Mottet N, Bellmunt J, Bolla M, Joniau S, Mason M, Matveev V, Schmid HP, Van der Kwast T, Wiegel T, Zattoni F, et al. EAU guidelines on prostate cancer. Part II: Treatment of advanced, relapsing, and castration-resistant prostate cancer. Eur Urol. 2011;59:572–83. doi: 10.1016/j.eururo.2011.01.025. [DOI] [PubMed] [Google Scholar]
- 5.Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN, Oudard S, Théodore C, James ND, Turesson I, et al. TAX 327 Investigators Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004;351:1502–12. doi: 10.1056/NEJMoa040720. [DOI] [PubMed] [Google Scholar]
- 6.Berthold DR, Pond GR, Soban F, de Wit R, Eisenberger M, Tannock IF. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer: updated survival in the TAX 327 study. J Clin Oncol. 2008;26:242–5. doi: 10.1200/JCO.2007.12.4008. [DOI] [PubMed] [Google Scholar]
- 7.Petrylak DP, Tangen CM, Hussain MH, Lara PN, Jr., Jones JA, Taplin ME, Burch PA, Berry D, Moinpour C, Kohli M, et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med. 2004;351:1513–20. doi: 10.1056/NEJMoa041318. [DOI] [PubMed] [Google Scholar]
- 8.de Bono JS, Oudard S, Ozguroglu M, Hansen S, Machiels JP, Kocak I, Gravis G, Bodrogi I, Mackenzie MJ, Shen L, et al. TROPIC Investigators Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet. 2010;376:1147–54. doi: 10.1016/S0140-6736(10)61389-X. [DOI] [PubMed] [Google Scholar]
- 9.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, Chi KN, Jones RJ, Goodman OB, Jr., Saad F, et al. COU-AA-301 Investigators Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fizazi K, Scher HI, Molina A, Logothetis CJ, Chi KN, Jones RJ, Staffurth JN, North S, Vogelzang NJ, Saad F, et al. COU-AA-301 Investigators Abiraterone acetate for treatment of metastatic castration-resistant prostate cancer: final overall survival analysis of the COU-AA-301 randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2012;13:983–92. doi: 10.1016/S1470-2045(12)70379-0. [DOI] [PubMed] [Google Scholar]
- 11.Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, de Souza PL, Fizazi K, Mainwaring P, Piulats JM, Ng S, et al. COU-AA-302 Investigators Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med. 2013;368:138–48. doi: 10.1056/NEJMoa1209096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rathkopf DE, Smith MR, De Bono JS, Logothetis C, Shore N, De Souza PL, et al. Long-term safety and efficacy analysis of abiraterone acetate plus prednisone in metastatic castration-resistant prostate cancer without prior chemotherapy (COU-AA-302) J Clin Oncol. 2013;31(suppl; abstr 5009) [Google Scholar]
- 13.Chen Y, Clegg NJ, Scher HI. Anti-androgens and androgen-depleting therapies in prostate cancer: new agents for an established target. Lancet Oncol. 2009;10:981–91. doi: 10.1016/S1470-2045(09)70229-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Massard C, Fizazi K. Targeting continued androgen receptor signaling in prostate cancer. Clin Cancer Res. 2011;17:3876–83. doi: 10.1158/1078-0432.CCR-10-2815. [DOI] [PubMed] [Google Scholar]
- 15.Mohler JL. Castration-recurrent prostate cancer is not androgen-independent. Adv Exp Med Biol. 2008;617:223–34. doi: 10.1007/978-0-387-69080-3_21. [DOI] [PubMed] [Google Scholar]
- 16.Stanbrough M, Bubley GJ, Ross K, Golub TR, Rubin MA, Penning TM, Febbo PG, Balk SP. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006;66:2815–25. doi: 10.1158/0008-5472.CAN-05-4000. [DOI] [PubMed] [Google Scholar]
- 17.Mostaghel EA, Montgomery B, Nelson PS. Castration-resistant prostate cancer: targeting androgen metabolic pathways in recurrent disease. Urol Oncol. 2009;27:251–7. doi: 10.1016/j.urolonc.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nacusi LP, Tindall DJ. Androgen receptor abnormalities in castration-recurrent prostate cancer. Expert Rev Endocrinol Metab. 2009;4:417–22. doi: 10.1586/eem.09.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, Han M, Partin AW, Vessella RL, Isaacs WB, et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009;69:16–22. doi: 10.1158/0008-5472.CAN-08-2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–43. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu ML, Kyprianou N. Androgen receptor and growth factor signaling cross-talk in prostate cancer cells. Endocr Relat Cancer. 2008;15:841–9. doi: 10.1677/ERC-08-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lamont KR, Tindall DJ. Minireview: Alternative activation pathways for the androgen receptor in prostate cancer. Mol Endocrinol. 2011;25:897–907. doi: 10.1210/me.2010-0469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schrecengost R, Knudsen KE. Molecular pathogenesis and progression of prostate cancer. Semin Oncol. 2013;40:244–58. doi: 10.1053/j.seminoncol.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chuang AY, DeMarzo AM, Veltri RW, Sharma RB, Bieberich CJ, Epstein JI. Immunohistochemical differentiation of high-grade prostate carcinoma from urothelial carcinoma. Am J Surg Pathol. 2007;31:1246–55. doi: 10.1097/PAS.0b013e31802f5d33. [DOI] [PubMed] [Google Scholar]
- 25.Anderson PD, McKissic SA, Logan M, Roh M, Franco OE, Wang J, Doubinskaia I, van der Meer R, Hayward SW, Eischen CM, et al. Nkx3.1 and Myc crossregulate shared target genes in mouse and human prostate tumorigenesis. J Clin Invest. 2012;122:1907–19. doi: 10.1172/JCI58540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Waltering KK, Helenius MA, Sahu B, Manni V, Linja MJ, Jänne OA, Visakorpi T. Increased expression of androgen receptor sensitizes prostate cancer cells to low levels of androgens. Cancer Res. 2009;69:8141–9. doi: 10.1158/0008-5472.CAN-09-0919. [DOI] [PubMed] [Google Scholar]
- 27.Donovan MJ, Osman I, Khan FM, Vengrenyuk Y, Capodieci P, Koscuiszka M, Anand A, Cordon-Cardo C, Costa J, Scher HI. Androgen receptor expression is associated with prostate cancer-specific survival in castrate patients with metastatic disease. BJU Int. 2010;105:462–7. doi: 10.1111/j.1464-410X.2009.08747.x. [DOI] [PubMed] [Google Scholar]
- 28.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–43. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008;68:5469–77. doi: 10.1158/0008-5472.CAN-08-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen S, Gulla S, Cai C, Balk SP. Androgen receptor serine 81 phosphorylation mediates chromatin binding and transcriptional activation. J Biol Chem. 2012;287:8571–83. doi: 10.1074/jbc.M111.325290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Knudsen KE, Penning TM. Partners in crime: deregulation of AR activity and androgen synthesis in prostate cancer. Trends Endocrinol Metab. 2010;21:315–24. doi: 10.1016/j.tem.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Augello MA, Hickey TE, Knudsen KE. FOXA1: master of steroid receptor function in cancer. EMBO J. 2011;30:3885–94. doi: 10.1038/emboj.2011.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sahu B, Laakso M, Ovaska K, Mirtti T, Lundin J, Rannikko A, Sankila A, Turunen JP, Lundin M, Konsti J, et al. Dual role of FoxA1 in androgen receptor binding to chromatin, androgen signalling and prostate cancer. EMBO J. 2011;30:3962–76. doi: 10.1038/emboj.2011.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gerhardt J, Montani M, Wild P, Beer M, Huber F, Hermanns T, Müntener M, Kristiansen G. FOXA1 promotes tumor progression in prostate cancer and represents a novel hallmark of castration-resistant prostate cancer. Am J Pathol. 2012;180:848–61. doi: 10.1016/j.ajpath.2011.10.021. [DOI] [PubMed] [Google Scholar]
- 35.Gallucci M, Merola R, Leonardo C, De Carli P, Farsetti A, Sentinelli S, Sperduti I, Mottolese M, Carlini P, Vico E, et al. Genetic profile identification in clinically localized prostate carcinoma. Urol Oncol. 2009;27:502–8. doi: 10.1016/j.urolonc.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 36.Hawksworth D, Ravindranath L, Chen Y, Furusato B, Sesterhenn IA, McLeod DG, Srivastava S, Petrovics G. Overexpression of C-MYC oncogene in prostate cancer predicts biochemical recurrence. Prostate Cancer Prostatic Dis. 2010;13:311–5. doi: 10.1038/pcan.2010.31. [DOI] [PubMed] [Google Scholar]
- 37.Mehra R, Tomlins SA, Yu J, Cao X, Wang L, Menon A, Rubin MA, Pienta KJ, Shah RB, Chinnaiyan AM. Characterization of TMPRSS2-ETS gene aberrations in androgen-independent metastatic prostate cancer. Cancer Res. 2008;68:3584–90. doi: 10.1158/0008-5472.CAN-07-6154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zong Y, Xin L, Goldstein AS, Lawson DA, Teitell MA, Witte ON. ETS family transcription factors collaborate with alternative signaling pathways to induce carcinoma from adult murine prostate cells. Proc Natl Acad Sci U S A. 2009;106:12465–70. doi: 10.1073/pnas.0905931106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Demichelis F, Fall K, Perner S, Andrén O, Schmidt F, Setlur SR, Hoshida Y, Mosquera JM, Pawitan Y, Lee C, et al. TMPRSS2:ERG gene fusion associated with lethal prostate cancer in a watchful waiting cohort. Oncogene. 2007;26:4596–9. doi: 10.1038/sj.onc.1210237. [DOI] [PubMed] [Google Scholar]
- 40.Dumas L, Payne H, Chowdhury S. The evolution of antiandrogens: MDV3100 comes of age. Expert Rev Anticancer Ther. 2012;12:131–3. doi: 10.1586/era.11.210. [DOI] [PubMed] [Google Scholar]
- 41.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, Wongvipat J, Smith-Jones PM, Yoo D, Kwon A, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787–90. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scher HI, Beer TM, Higano CS, Anand A, Taplin ME, Efstathiou E, Rathkopf D, Shelkey J, Yu EY, Alumkal J, et al. Prostate Cancer Foundation/Department of Defense Prostate Cancer Clinical Trials Consortium Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1-2 study. Lancet. 2010;375:1437–46. doi: 10.1016/S0140-6736(10)60172-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, de Wit R, Mulders P, Chi KN, Shore ND, et al. AFFIRM Investigators Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367:1187–97. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 44.Miller K, Scher HI, Fizazi K, Basch EM, Sternberg CN, Hirmand M, et al. Effect of enzalutamide on health-related quality of life (HRQoL) in men with metastatic castration resistant prostate cancer (mCRPC) following docetaxel-based therapy: results from the AFFIRM study. J Clin Oncol. 2013;31(suppl 6; abstr 17) [Google Scholar]
- 45.Scher HI, Fizazi K, Saad Chi KN, Taplin ME, Sternberg CN, et al. Impact of on-study corticosteroid use on efficacy and safety in the phase III AFFIRM study of enzalutamide, an androgen receptor inhibitor. J Clin Oncol. 2013;31(suppl 6; abstr 6) [Google Scholar]
- 46.Golshayan AR, Antonarakis ES. Enzalutamide: an evidence-based review of its use in the treatment of prostate cancer. Core Evid. 2013;8:27–35. doi: 10.2147/CE.S34747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smith MR, Borre M, Rathenborg P, Werbrouck P, Van Poppel H, Heidenreich A, et al. Efficacy and safety of enzalutamide monotherapy in hormone-naïve prostate cancer. J Clin Oncol. 2013;31(suppl; abstr 5001) [Google Scholar]
- 48.Clegg NJ, Wongvipat J, Joseph JD, Tran C, Ouk S, Dilhas A, Chen Y, Grillot K, Bischoff ED, Cai L, et al. ARN-509: a novel antiandrogen for prostate cancer treatment. Cancer Res. 2012;72:1494–503. doi: 10.1158/0008-5472.CAN-11-3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Foster WR, Car BD, Shi H, Levesque PC, Obermeier MT, Gan J, Arezzo JC, Powlin SS, Dinchuk JE, Balog A, et al. Drug safety is a barrier to the discovery and development of new androgen receptor antagonists. Prostate. 2011;71:480–8. doi: 10.1002/pros.21263. [DOI] [PubMed] [Google Scholar]
- 50.Rathkopf DE, Danila DC, Morris MJ, Slovin SF, Steinbrecher JE, Arauz G, et al. Phase I/II safety and pharmacokinetic study of ARN-509 in patients with metastatic castration-resistant prostate cancer patients: phase I results of a Prostate Cancer Clinical Trials Consortium study. J Clin Oncol. 2012;30(suppl 5; abstr 43) [Google Scholar]
- 51.Rathkopf DE, Antonarakis ES, Shore ND, Tutrone R, Alumkal JJ, Ryan CJ, et al. ARN-509 in men with metastatic castration-resistant prostate cancer (mCPRC) J Clin Oncol. 2013;31:suppl 6; abstr 48. [Google Scholar]
- 52.Smith MR, Antonarakis ES, Ryan CJ, Berry WR, Shore N, Liu G, et al. ARN-509 in men with high-risk nonmetastatic castration-resistant prostate cancer (CRPC) J Clin Oncol. 2013;31(suppl 6; abstr)(7) [Google Scholar]
- 53.Kaku T, Hitaka T, Ojida A, Matsunaga N, Adachi M, Tanaka T, Hara T, Yamaoka M, Kusaka M, Okuda T, et al. Discovery of orteronel (TAK-700), a naphthylmethylimidazole derivative, as a highly selective 17,20-lyase inhibitor with potential utility in the treatment of prostate cancer. Bioorg Med Chem. 2011;19:6383–99. doi: 10.1016/j.bmc.2011.08.066. [DOI] [PubMed] [Google Scholar]
- 54.Ryan CJ, Tindall DJ. Androgen receptor rediscovered: the new biology and targeting the androgen receptor therapeutically. J Clin Oncol. 2011;29:3651–8. doi: 10.1200/JCO.2011.35.2005. [DOI] [PubMed] [Google Scholar]
- 55.Agus DB, Stadler WM, Shevrin DH, Hart L, MacVicar GR, Hamid O, et al. Safety, efficacy and pharmacodynamics of the investigational agent orteronel (TAK-700) in metastatic castration-resistant prostate cancer: updated data form a phase I/II study. J Clin Oncol. 2012;30(suppl 5; abstr 98) [Google Scholar]
- 56.George DJ, Corn PG, Michaelson MD, Hammers HJ, Alumkal JJ, Ryan CJ, et al. Safety and activity of the investigational agent orteronel without prednisone in men with nonmetastatic castration resistant prostate cancer and rising prostate-specific antigen: updated results of a phase II study. J Clin Oncol. 2012;X(suppl; abstr 4549) [Google Scholar]
- 57.Vasaitis T, Belosay A, Schayowitz A, Khandelwal A, Chopra P, Gediya LK, Guo Z, Fang HB, Njar VC, Brodie AM. Androgen receptor inactivation contributes to antitumor efficacy of 17α-hydroxylase/17,20-lyase inhibitor 3beta-hydroxy-17-(1H-benzimidazole-1-yl)androsta-5,16-diene in prostate cancer. Mol Cancer Ther. 2008;7:2348–57. doi: 10.1158/1535-7163.MCT-08-0230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Montgomery RB, Eisenberger MA, Rettig M, Chu F, Pili R, Stephenson J, et al. Phase I clinical trial of galeterone (TOK-001), a multifunctional antiandrogen and CYP17 inhibitor in castration resistant prostate cancer. J Clin Oncol. 2012;30(suppl; abstr 4665) [Google Scholar]
- 59.Jacoby DB, Williams M. Differential effects of galeterone, abiraterone, orteronel and ketoconazole on CYP 17 and steroidogenesis. J Clin Oncol. 2013;3(suppl 6; abstr 184) [Google Scholar]
- 60.Massard C, James N, Culine S, Jones R, Vuorela A, Mustonen M, et al. ARADES trial: a first-in-man, open-label, phase I/II safety, pharmacokinetic, and proof-of-concept study of ODM-201 in patients with progressive metastatic castration resistant prostate cancer. Ann Oncol. 2012;23(suppl 9) [Google Scholar]
- 61.Danila DC, Anand A, Sung CC, Heller G, Leversha MA, Cao L, Lilja H, Molina A, Sawyers CL, Fleisher M, et al. TMPRSS2-ERG status in circulating tumor cells as a predictive biomarker of sensitivity in castration-resistant prostate cancer patients treated with abiraterone acetate. Eur Urol. 2011;60:897–904. doi: 10.1016/j.eururo.2011.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Attard G, De Bono JS, Li W, Molina A, Griffin TW, San Kheoh T, et al. ERG rearrangements and association with clinical outcome in patients receiving abiraterone acetate: Results from the COU-AA-302 study in chemotherapy-naïve metastatic castration-resistant prostate cancer. J Clin Oncol. 2013;31(suppl; abstr 5004) [Google Scholar]
- 63.de Bono JS, Scher HI, Montgomery RB, Parker C, Miller MC, Tissing H, Doyle GV, Terstappen LW, Pienta KJ, Raghavan D. Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clin Cancer Res. 2008;14:6302–9. doi: 10.1158/1078-0432.CCR-08-0872. [DOI] [PubMed] [Google Scholar]
- 64.Economos C, Morrissey C, Vessella RL. Circulating tumor cells as a marker of response: implications for determining treatment efficacy and evaluating new agents. Curr Opin Urol. 2012;22:190–6. doi: 10.1097/MOU.0b013e3283519b58. [DOI] [PubMed] [Google Scholar]
- 65.Nakai Y, Nonomura N. Inflammation and prostate carcinogenesis. Int J Urol. 2013;20:150–60. doi: 10.1111/j.1442-2042.2012.03101.x. [DOI] [PubMed] [Google Scholar]