

Abstract
AMP-activated protein kinase (AMPK), an established metabolic stress sensor, has gained popularity in cancer biology due to its ability to control cellular growth and mediate cell cycle checkpoints in cancer cells in response to low energy levels. AMPK is a key effector of the tumor suppressor liver kinase B 1 (LKB1) which inhibits the cellular growth mediator mammalian target of rapamycin (mTOR) and activates checkpoint mediators such as p53 and the cyclin dependent kinase inhibitors p21cip1 and p27kip1. However, recent work describes a novel function for AMPK as a sensor of genomic stress and a participant of the DNA damage response (DDR) pathway. Ionizing radiation and chemotherapy activate AMPK in cancer cells to mediate signal transduction downstream of ataxia telangiectasia mutated (ATM) to activate p53- p21cip1/p27kip1 and inhibit mTOR. We discuss evidence on the transcriptional and post-translational regulation of AMPK by ionizing radiation and the role of the enzyme as a mediator of chemo- and radiation sensitivity in epithelial cancer cells. Furthermore, we review data on the participation of AMPK in cytokinesis and observations suggesting a physical association of this enzyme with the mitotic apparatus. The evidence available to date suggests that AMPK is a point of convergence of metabolic and genomic stress signals, which (1) control the activity of growth mediators, (2) propagate DDR, and (3) mediate the anti-proliferative effects of common cytotoxic cancer therapy such as radiation and chemotherapy. This highlights the importance of targeting AMPK with novel cancer therapeutics.
Keywords: AMPK, ionizing radiation, cell cycle, ATM, mitosis
Introduction
The ability of living organisms to maintain normal function and longevity is intricately linked with their ability to adapt to the physiological challenges that arise in a continuously changing environment. Genomic stress in the form of ionizing radiation, chemical carcinogens, infections, or metabolism generated reactive oxygen species (ROS),1 leads to accumulation of DNA aberrations, and this is opposed by molecular stress response pathways. To evade metabolic stress cells rapidly suppress anabolic processes, stall cell cycle progression, and promote efficient energy production.
AMP-activated protein kinase (AMPK) protects cells against physiological and pathological stress, including nutrient withdrawal, hypoxia, exercise, and heat shock by responding to reductions in the ATP:AMP/ADP ratio. Upon activation AMPK switches on metabolism to generate ATP, while systematically blocking energy expenditure.2 Consistent with this role, AMPK mediates cell cycle checkpoints, inhibits pro-survival growth pathways, and modulates mitotic progression, all of which allows damage repair or cellular death if cells sustain irreparable damage.3 Overall, these events are essential to maintain genomic stability and defend against carcinogenesis.4
AMPK: Structure and Regulation
AMPK is a serine/threonine protein kinase that is highly conserved across most eukaryotic organisms. It is a heterotrimeric complex and in mammals there are seven subunit isoforms of AMPK encoded by separate genes (PRKAA1–2, PRKAB1–2, and PRKAG1–3), including two α-subunits (α1–2), two β (β1–2), and three γ subunits (γ1–3) respectively. Theoretically this allows up to 12 heterotrimeric AMPKαβγ combinations.5
The α-subunit of AMPK has catalytic activity and is comprised of a kinase domain at the N-terminus, preceded by a regulatory domain that contains an auto-inhibitory sequence, and a subunit interacting domain that binds to the β-subunit.6 Phosphorylation of AMPK on its conserved αThr172 residue in the activation loop is necessary for full enzyme activity. Under conditions of metabolic stress the tumor suppressor liver kinase B 1 (LKB1) (also known as serine/threonine kinase 11: STK11) is the main AMPKα Thr-172 kinase, which enhances AMPK activity by >100-fold.6 LKB1 phosphorylates a family of 12 other AMPK-related kinases, but AMPKα1/2 are the only substrates of this family that respond to low energy conditions.7 However, in the hypothalamus, neurons, and T lymphocytes, AMPK is also regulated by calcium (Ca2+) signals and calmodulin-dependent protein kinase (CaMKK).8 Under these conditions, CaMKKβ appears to be the principal kinase that phosphorylates AMPKα on Thr172.8,9
The β subunits of AMPK act as a scaffold to bind α and γ-subunits to form a functional AMPK heterotrimeric complex.10,11 In addition, there is growing appreciation for their importance in regulating enzyme activity effects, that are mediated through a carbohydrate binding module (CBM) and myristolation sites. The latter aids in membrane localization,12 believed to facilitate activation of the enzyme through Thr172 phosphorylation. Ser108, within the CBM of the β1 subunit, aids in the allosteric activation and inhibition of Thr172 dephosphorylation in response to the direct activators A-76966213 and salicylate.14 Interestingly, this residue is not conserved in the β2 subunit, which is the predominant isoform expressed in skeletal muscle.15
The γ-subunit of AMPK contains four tandem cystathionine β synthase (CBS) repeats, which were initially defined by Bateman (1997) who observed that these repeats occur as two pairs of domains, now acknowledged as Bateman domains.16 They are congregated in a pseudosymmetrical fashion, exposing four clefts for ATP, ADP, and AMP binding. These clefts are characterized as sites 1–4 based on the number of the cleft harboring an aspartate residue associated with adenine nucleotide binding.17 Interestingly, three of these sites on AMPKγ bind adenine nucleotides, while one site (site 2) is always unoccupied. Furthermore, site 4 of AMPKγ associates with AMP very tightly, and does not exchange with ATP or ADP.17 Nucleotide binding to AMPKγ regulates the enzymes activity. During energy stress, increased ADP or AMP levels (compared with ATP) acts as a cellular read-out of energy depletion. ADP or AMP bindings increases AMPK activity through a conformational change that promotes α-subunit Thr172 phosphorylation, as well as inhibition of Thr172 de-phosphorylation by phosphatases.18 AMP, but not ADP, is shown to support allosteric activation of AMPK that has already been phosphorylated on its α-Thr172 residue by upstream kinases.19
Role of AMPK in the metabolic stress response and growth
Once active, AMPK directly phosphorylates a number of downstream substrates that acutely affect energy metabolism and growth, or induces gene expression that will lead to prolonged alterations in metabolic programming (Fig. 1).3 AMPK was initially described as a kinase that directly phosphorylated and inhibited acetyl CoA carboxylase (ACC) and 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMG-CoA), which are the rate limiting enzymes for fatty-acid and sterol synthesis.20 The suppression of this pathway allows for enhanced activation of carnitine palmitoyltransferase I (CPT-1), a mitochondrial enzyme that permits long-chain fatty acids to enter the mitochondrial matrix for β oxidization and generation of ATP.21

Figure 1. Control of growth and metabolic pathways by AMPK. Phosphorylation and activation of AMPK on α-Thr172 is regulated by the upstream kinases LKB1 and CaMKK. In turn, AMPK directly phosphorylates multiple downstream targets to balance cell growth with energy supply. Substrates that regulate proliferation and metabolism and are well-established to be phosphorylated directly by AMPK are shown. Abbreviations: LKB1, liver-kinase B1; CaMKK, calmodulin-dependent protein kinase kinase; AMPK, AMP-activated protein kinase; TSC2, tuberous sclerosis protein 2; HMGCoA, 3-hydroxy-3-methyl-glutaryl-CoA reductase; ACC1/2, acetyl CoA carboxylase 1/2; FOXO3, forkhead box O3; PGC1α, proliferator-activated receptor gamma coactivator 1-α; SREBP-1c, sterol regulatory element-binding protein-1c; and H2B, histone H2B.
Protein synthesis and cell proliferation are significant contributors to cellular energy expenditure. In an effort to conserve energy under metabolic stress AMPK inhibits protein translation and cellular growth through inhibition of the mammalian target of rapamycin (mTOR) which stimulates cell growth, proliferation, protein synthesis, and has been implicated in malignant transformation and cancer.4 mTOR is member of the phosphatidylinositol 3-kinase related kinase (PIKK) superfamily, a family of enzymes that respond to metabolic and genotoxic stresses and execute adaptive mechanisms to sustain cell survival.22 mTOR is a catalytic subunit of two distinctly different protein complexes, known as mTORC1 and mTORC2.23 mTORC1 is comprised of mTOR, the regulatory associated protein of mTOR (raptor), and mLST8, and is sensitive to inhibition by the immunosuppressor drug rapamycin. mTORC1 phosphorylates p70-S6 kinase (p70S6K) and eukaryotic initiation factor 4E (eIF4E) binding protein 1 (4EBP1) to initiate protein translation.23 Conversely, mTORC2 exists as a protein complex consisting of mTOR, rapamycin-insensitive companion of mTOR (rictor), as well as mSIN1, and can phosphorylate protein kinases involved in cellular growth including Akt and protein kinase C (PKC).24 Jointly, mTORC1 and mTORC2 control cell size, proliferation, and cell cycle progression in mammals.22
AMPK inhibits mTORC1 activation to conserve energy through multiple mechanisms. AMPK inhibits mTOR through phosphorylation of the tuberous sclerosis complex (TSC) (TSC1:TSC2). TSC1:TSC2 have GTPase activity toward the small G-protein Rheb, which activates mTORC1 when at its GTP-bound state. AMPK triggers TSC2 activity via direct phosphorylation on its Thr1227 and Ser1345 residues, which in turn inactivates Rheb by converting it to a GDP-bound confirmation.23 An alternative approach for AMPK to suppress mTORC1 is through phosphorylation and inhibition of the mTOR binding partner raptor, which prevents mTOR from phosphorylating downstream targets.25
AMPK also regulates a wide range of transcription factors, their co-activators, and histones to regulate gene expression and nuclear events leading to metabolic reprogramming and cell survival. It phosphorylates forkhead box 03a (FOXO3a), a transcription factor that plays a role in stress resistance, glucose metabolism, and apoptosis.26 To modulate lipid metabolism, AMPK inhibits the activity of sterol regulatory element binding protein-1c (SREBP-1c) transcription factor via Ser374 phosphorylation.27 Further, AMPK directly phosphorylates peroxisome proliferator-activated receptor gamma (PPAR-γ) coactivator-1-α (PGC-1α), a transcriptional co-activator that regulates numerous metabolic genes and mitochondrial biogenesis.28
AMPK is suggested to directly phosphorylate p53 on Ser15, a step that stabilizes the molecule,29 but alternative mechanisms of p53 stabilization by AMPK have also been described. Lee et al.,30 has suggested that AMPK mediates p53 stability through inhibition of its deacetylation by SIRT1, a NAD-dependent protein deacetylase that is involved in gene silencing and the homolog to the yeast Sir2 protein. Further, AMPK was also shown to phosphorylate the cyclin-dependent kinase inhibitor (CDKI) p27kip1 on Thr198 to sequester it in the cytoplasm and promote survival in response to nutrient or growth-factor withdrawal.31 Additionally, Bungard et al.32 (2010) showed that AMPK provokes transcriptional regulation of genes in response to bioenergetic strain through direct phosphorylation of histone H2B on Ser36. Taken together, AMPK, in an attempt to attenuate stress and modulate cell survival, targets multiple signaling pathways by acute phosphorylation or transcriptional control (as depicted in Fig. 1).
AMPK and cancer metabolism
A key hallmark of cancer is altered carbohydrate metabolism that involves aerobic glycolysis (enhanced glycolysis and reduced oxidative phosphorylation (OXPHOS) despite oxygen availability), a phenomenon first described in the 1920s by Otto Warburg (the Warburg effect).33 Warburg believed that this shift in glucose metabolism from OXPHOS to glycolysis was attributed to dysfunctional mitochondria within the cancer cells. While now it is widely accepted that tumor cells have normal functioning mitochondria,34,35 a potential explanation for the preferential use of glycolysis to obtain energy in cancer cells is that the rate of energy turn-over operates significantly faster than OXPHOS, although the energetic yield per molecule of glucose is less.36 In addition, glycolysis provides cancer cells with the glycolytic substrates (nucleotides, amino acids, and fatty acids) required for continued macromolecule biosynthesis that are necessary for cell division and growth.37
AMPK has been shown to oppose metabolic reprogramming that contributes to the Warburg phenotype observed in cancer cells.38 Aside from suppression of anabolic signals, AMPK also engages signaling pathways to re-establish the metabolism of glucose through OXPHOS.39 Importantly, AMPK inhibition promotes a metabolic shift toward the Warburg effect in both non-transformed and malignant cells,40 indicating that this enzyme is a negative regulator of glycolysis and suppressor of tumor development through regulation of metabolic pathways that support uncontrolled proliferation.
Role of AMPK in Genomic Stress Responses
Recently, AMPK was shown to participate in signaling events that respond to genomic stress and modulate cell survival. In response to UV radiation, AMPK was shown to associate with the promoter sequence of p21cip1 to increase cell survival.32 In addition, AMPK-dependent repression of p73α transcription was suggested to promote cell survival in response to DNA damage caused by cisplatin exposure.41 In this context AMPK would be suggested to facilitate cell survival after genotoxic stress. However, AMPK has also been found to induce mitochondrial p53 accumulation, which subsequently sensitized cells to Bak-induced apoptosis in Myc-driven tumors.42 Our work with epithelial cancer models, discussed below, suggests that AMPK is an effector of ataxia telangiectasia mutated (ATM), a key DNA damage sensor that regulates cell cycle and cellular sensitivity to genotoxic agents.
ATM as a mediator of the DNA damage response (DDR)
The DDR pathway senses a variety of lesions in the physical structure of DNA, including single stand breaks or potentially lethal double stand breaks.43 In eukaryotes, these DNA aberrations are repaired through non-homologous end-joining or homologous recombination repair.44 Members of the highly conserved DNA damage sensor MRN complex, including meiotic recombination 11 (MRE11), RAD50, and Nijmegen breakage syndrome 1 (NBS1), help recruit ATM to DSB's and activate this enzyme which in turn phosphorylate NBS1 and enhances DDR.45 ATM is a key transducer of DSB-induced DDR. It is a 370kDa protein, member of the PIKK family, which also includes DNA-dependent protein kinase (DNA-PK) that can also sense DSB, ATM and Rad3-related protein (ATR), which detects SSB, as well as mTOR.44 Mutation of the ATM gene gives rise to ataxia telangiectasia, an autosomal recessive disorder that appears in early childhood.43 Ataxia telangiectasia patients lack ATM protein expression by inheriting a missense or nonsense mutation in ATM, and show signs of ataxia associated with progressive loss of motor function. These patients also exhibit increased accumulation of cellular ROS, hypersensitivity to ionizing radiation, as well as an increased risk of developing cancer and type 2 diabetes.44,46
Ionizing radiation-induced double stand breaks alter chromatin structure and lead to activation of the p53 binding protein 53BP1,47 which facilitates activation of ATM via autophosphorylation on Ser1981; leading to dissociation of inactive ATM dimmers (this event is depicted in Fig. 2). ATM then initiates a network of signaling events leading to DNA repair, cell cycle arrest, or survival.43 Activated ATM phosphorylates histones, such as H2AX (on serine 139, known as γH2Ax), which orchestrates recruitment of repair complexes at the location of DNA damage (Fig. 3).48 ATM mediates ionizing radiation—DNA damage-induced cell cycle checkpoints through phosphorylation of downstream substrates. ATM phosphorylates p53 on Ser 15, leading to the transcriptional regulation of target genes including p21cip1, which mediate G1/S or G2/M phase cell cycle arrest.49 ATM also phosphorylates check point kinase 2 (Chk2) on Thr68, which in turn inhibits the CDC25A or CDC25C family of phosphatases that are required to promote S and G2/M phase cell cycle progression via activation of cyclic-dependent kinase 2 (CDK2) or cell division control protein 2 (CDC2) respectively.50 At times of extensive DNA damage ATM activation leads to apoptosis or cellular senescence through p53 regulation,44 as shown in Figure 2. A recent large-scale proteomic analysis of ATM identified several hundred presumed ATM target-proteins involved in cellular functions ranging from nucleotide regulation to cell proliferation in response to ionizing radiation.51

Figure 2. Role of ATM in the DNA damage response. ATM is rapidly phosphorylated (Ser1981) and activated by MRE11 or 53BP1 in response to radiation-induced double stranded DNA breaks. Active ATM in its monomeric state then initiates pathways that regulate cell cycle arrest, DNA repair, or if required apoptosis. Phosphorylation of H2AX on Ser139 by ATM promotes DNA repair signals which are required for survival following radiation, while the substrates Chk2 and p53 can arrest cell cycle progression or the latter can initiate cell death. Abbreviations: ATM, ataxia telangiectasia mutated; MRE11, meiotic recombination 11; NBS1, Nijmegen breakage syndrome 1; 53BP1, p53 binding protein 1; CDC25A, cell division cycle 25A; CDC25C, cell division cycle 25C; CDK2, cyclic-dependent kinase 2; CDC2, cell division control protein 2.

Figure 3. A model of acute regulation of AMPK by radiation. Ionizing radiation causes DNA damage leading to increased ATM phosphorylation, which leads to enhanced expression of the γH2AX marker. ATM leads to phosphorylation and activation of AMPK and its translocation from the nucleus to the cytoplasm. Ionizing radiation leads to AMPK-dependent checkpoint, associated with expression of the cycling-depended kinase inhibitor p21cip1 and a G2-M cell cycle arrest. Immunofluorescence microscopy images were obtained from untreated or radiated (1 h post irradiation) A549 cells, which were fixed and stained with Hoechst (blue), an antibody that detects phosphorylated AMPKα on its Thr172 residue (green, 1:00 dilution, Cell Signaling), as well as phosphorylated H2AX (γH2Ax) (red, 1:200 dilution, Cell Signaling). Abbreviations: ATM, ataxia telangiectasia mutated; AMPK, AMP-activated protein kinase.
While ATM is considered primarily a nuclear protein that responds to DNA damage, recent studies suggested that distinct pools of ATM are also localized in the cytoplasm and acts as an important regulator of oxidative stress, cell metabolism, and mediator of AMPK activation by genotoxic stress and pharmaceutical agents (see Fig. 4).46,52-54 Cytoplasmic ATM also exists as an active dimmer but, in contrast to nuclear ATM activation by DNA damage, it does not require Ser1981 phosphorylation for its activation.44,55 Cytoplasmic ATM is sensitive to redox signals and treatment with hydrogen peroxide (H2O2) activates ATM leading to ATM-mediated LKB1-Thr366 phosphorylation, an effect associated with increased AMPK activation,52 inhibition of mTOR, and signaling and stimulation of autophagy (discussed below).52 Interestingly, DNA-PK was recently found to interact with the γ1-subunit of AMPK under conditions of glucose withdrawal,56 suggesting that other members of the PIKK family may also regulate AMPK independently of genomic damage.

Figure 4. Participation of AMPK in chronic molecular responses of cancer cells to chemo- and radiotherapy. Chemo- and radiation therapy elicit signal transduction pathways at the level of DNA damage, starting with rapid activation of ATM in the nucleus at the site of the DNA lesion. To mediate cell cycle arrest ATM phosphorylates AMPK, which coordinates its activity with numerous cell cycle regulators including p53, p27kip1, and p21cip1 to arrest cell cycle progression. Additionally, AMPK can signal to p53 and its downstream effector SESN2 to generate a positive-feedback loop of sustained activity under times of genotoxic stress. Following DNA damage AMPK activity becomes redistributed to the cytoplasm whereby can modulate the radio-/chemo-sensitivity of cancer cells by inhibiting pathways of survival, including the Akt-mTOR signaling cascade. Abbreviations: ATM, ataxia telangiectasia mutated; AMPK, AMP-activated protein kinase; SESN2, sestrin 2; LKB1, liver-kinase B1; TSC, tuberous sclerosis proteins 1/2; PI3K, phosphatidylinositide 3-kinases; Rheb, Ras homolog enriched in brain; mTOR, mammalian target of rapamycin.
AMPK in Mediation Radiation Responses
Acute regulation of AMPK by IR
In line with the notion that AMPK participates in the DDR, we reported that in lung, prostate, and breast cancer cells, AMPK is rapidly phosphorylated on Thr172 and activated in response to clinical doses of radiation therapy.57 Phosphorylation of nuclear AMPKα was observed within minutes following treatment with radiation but over the course of an hour activated AMPK progressively translocated to the cytoplasm. While the significance of this subcellular migration is currently not known, we speculate that this event facilitates the cytoplasmic effects of AMPK on protein synthesis, metabolism, and mitochondrial function. Figure 3 illustrates a model of the acute regulation of AMPK by radiation in cancer cells.
AMPK as a Biphasic Gauge of Ionizing Radiation Signals
AMPK functions downstream of ATM
We have suggested that AMPK is a key effector of ATM signals (Figs. 3 and 4). Using chemical inhibition of ATM prior to ionizing radiation, with KU-55933, we observed attenuation of radiation-induced ATM activity and blockade of AMPK phosphorylation.57 We verified those observations with the newer and more specific ATM inhibitor KU60019,58 as well as molecular knockdown of ATM.58 Inhibition of ATM with either approach abolished radiation-induced phosphorylation of AMPK without affecting total AMPK levels.58 The exact mechanism that mediates the radiation-induced phosphorylation of AMPK downstream of ATM is not understood. ATM does not appear to phosphorylate directly AMPKα-Thr172.59 While earlier studies suggested that LKB1 is phosphorylated by radiation-activated ATM and this could provide an avenue for AMPK activation,60 our studies57,61 and those of other laboratories,53,62 demonstrate that LKB1 is not required for AMPK activation by radiation. LKB1-null A549 and H23 lung cancer cells showed a robust radiation activation of AMPK. Future studies need to investigate in depth the molecular interaction between ATM and AMPK phosphorylation of Thr712.
AMPK enables and stabilizes ATM signaling
In studies with mouse embryonic fibroblasts lacking AMPKα1/2 expression (AMPKα1/2−/−MEFs), a lack of AMPK was associated with enhanced total ATM levels and activity in untreated cells, indicated by increased nuclear γH2Ax foci.63 However, AMPKα1/2−/−MEFs lacked a clear ATM response to ionizing radiation in terms of both H2Ax and Chk2 phosphorylation. This indicates that AMPK has a role in modulating basal nuclear ATM activity potentially through a molecular feedback loop.
On the other hand, untreated AMPKα1/2−/−MEFs demonstrated upregulation of the Akt-mTOR pathway. Those cells exhibited enhanced, total Akt levels, Akt-Thr308 and -Ser473 phosphorylation, total mTOR levels, as well as enhanced phosphorylation of p70S6k and 4-EBP1. Further, increased p53 and p21cip1 levels were detected. However, AMPKα1/2−/−MEFs lacked response of both Akt-mTOR and p53/p21cip1 to radiation.63 Recently, we observed a similar robust activation of the Akt-mTOR-p70S6k/4-EBP1 pathway in A549 lung cancer cells in which AMPKα1/2 was knocked down with siRNAs.58 Those cells lacked the normal radiation-induced p21cip1 expression. In agreement with these findings, Bungard et al.32 showed that wild type MEFs treated with ionizing radiation or UV radiation promote transcription of the p53 and p21cip1 genes, but AMPKα1/2−/−MEFs showed a marked reduction in p53/p21cip1 following UV irradiation. In our experiments, both AMPKα1/2−/−MEFs and A549 lung cancer cells treated with AMPKα1/2 siRNAs demonstrated severe resistance to radiation induced cytoxicity (discussed below). Survival of those cells after radiation may be enhanced by the enhanced Akt-mTOR signals and their proliferation may be supported by the lack of radiation-induced expression of p53 and p21cip1. The mechanism of activation of Akt-mTOR pathway, in cells lacking AMPK activity is not presently understood, but ATM has been suggested to function as an Akt-S473 kinase (PDK2),64 which suggests an avenue for ATM to regulate Akt activity. Alternatively, lack of AMPK can enhance mTORC1 and mTORC2 activity, the latter of which has also been described to act as PDK2.65
Modulation of cell cycle control pathways
Cell cycle control pathways are promptly activated following ionizing radiation to facilitate DNA repair and regulate cell survival.66 ATM is known to mediate (1) Ser15 phosphorylation of p53 leading to stabilization of this tumor suppressor and (2) cell cycle check points through induction of p21cip1 and p27kip1.67 The mechanism by which ATM mediates these effects is not fully understood. We observed that inhibition of AMPK through the use of compound C or siRNA-mediated AMPKα1/2, attenuated the ability of ionizing radiation to enhance the expression of p53 and p21cip1 in lung cancer cells57,58 (Figs. 3 and 4). Of note, although p53 is known to regulate p21cip1 expression, this event was not required for radiation-mediated induction of p21cip1, which took place also in p53-null cells and was dependent on AMPK.57 Inhibition of AMPK, either in cancer cells57 or AMPKα1/2−/−MEFs,63 was sufficient to attenuate the ionizing radiation-induced G2/M cell cycle arrest. Inhibition of the radiation-induced G2/M checkpoint in the absence of AMPK may be explained by the blockade of typical ATM effectors such as p21cip1 and Chk2. However, their exact role in AMPK regulation of cell cycle should be investigated in detail.
Overall, our work to this point suggested that AMPK is a key effector of ATM which (1) mediates the acute regulation of p53 and p21cip1 and the radiation-induced cell cycle checkpoints and (2) regulates gene expression, activity and radiation responsiveness of the Akt-mTOR, Chk2, and p53/p21cip1 pathways (see Figs. 3 and 4). To this extent AMPK may play the role of a biphasic gauge in cellular radiation responses mediated by the ATM pathway.
Chronic regulation of AMPK signaling by ionizing radiation: potential mechanisms
Ionizing radiation does not only elicit acute signals that modulate DDR, but it also maintains such signals through the long-term regulation of gene expression, transcription, and translation.61 We observed that aside from acute AMPK phosphorylation, ionizing radiation modulated the long-term activity and expression of this enzyme in vitro and in vivo.61,63 In lung cancer cells AMPK α1, α2, β1, β2, γ1, and γ2 gene mRNA levels were all elevated 48 h after a single dose to 8Gy radiation, and this was associated with elevated levels of protein of each subunit.63 A549 and H1299 xenograft tumors treated with a single fraction of 10Gy radiation showed sustained expression of AMPK subunits, detected 8 weeks after irradiation, indicating long-term modulation of gene expression by radiation in tumors and a role of AMPK in the tumor response to radiotherapy.61 Long-term regulation of AMPK levels was not a phenomenon unique to lung cancer cells and tumors, and it was also observed in prostate cancer cells and xenografts.61 Importantly, the enhanced expression of AMPK by ionizing radiation was also associated with augmented expression of the ATM-p53 pathway and chronic suppression of the Akt-mTOR pro-survival pathway in these tumors.61
The above findings impose a key underlying question: What is the mechanism by which radiotherapy enhances chronically ATM-AMPK-p53 pathway expression and inhibits Akt/mTOR? A positive feedback loop exists between p53 and AMPK in response to genotoxic stress68 which may contribute to this effect. Radiation-mediated AMPK phosphorylation of Ser15 on p53 stabilizes this tumor suppressor.69 On the other hand, p53 regulates AMPK levels by at least two distinct mechanisms: (1) increased expression of sestrins that bind to AMPK and increase its activity70 and (2) via p53-dependent transcriptional upregulation of the AMPKβ1/2 genes.71
In regards to the first mechanism, Sestrin (SESN) members (SESN1 and SESN2) act as p53 target genes that accumulate in cells under physiologic stress. However, SESN2 expression can also be regulated by p53-independent stressors such as oxidative stress or hypoxia.72 Nevertheless, both SESN1 and SENS2 were shown to directly interact with AMPK and direct its activity toward inhibition of mTOR.70 We implicated SESN2 in the regulation AMPK expression following ionizing radiation in breast cancer cells,73 as seen in Figure 4. Overexpression of SESN2 mimicked the ability of ionizing radiation to increase AMPK levels, while siRNA against SESN2 prior to irradiation blocked radiation-induced AMPK activity and expression.73 We have detected enhanced SESN2 expression up to 8 weeks following a single fraction of ionizing radiation in A549 tumor xenografts, which closely correlates with increase AMPK phosphorylation and expression (Sanli and Tsakiridis, unpublished observations).
The second scenario was brought to light by Feng et al.,71 who identified putative p53 consensus binding sites on the AMPKβ1 and β2 promoters in colon cancer cells that could be triggered in response to ionizing radiation. While AMPKβ expression was found enhanced 24h following 10Gy ionizing radiation, it should be noted that no effect on AMPKα or AMPKγ subunit expression was observed at that early point in that study71 and p53-null cancer cells did not exhibit increases in AMPKβ expression. However, we observed increases in both SESN2 and AMPK subunit expression in a few epithelial tumor cells independent of p53 status.61 Thus, a reciprocal interplay between AMPK, p53, and SESN2 may account for the sustained activity and expression of the AMPK pathway, as well as inhibition of mTOR signaling in response to radiation in cancer cells.
AMPK activity modulates cellular radiosensitivity
In response to ionizing radiation cancer cells rapidly stimulate pro-survival signals leading to gene transcription and translation. These events are regulated by ATM and AMPK also.66 We observed that inhibition of AMPK activity reduces cellular radio-sensitivity. AMPKα1/2−/−MEFs and lung cancer cells treated with AMPKα1/2 siRNA are resistance to radiotherapy.58,63 We and others provided strong evidence that AMPK works to suppress mTOR activation following irradiation,61,63,74 and loss of AMPK activity correlates with enhanced mTOR signaling and cell survival.58,63,74
Pre-clinical studies using AMPK activators in combination with radiation therapy have indicated that potentiation of this pathway may be a promising approach to bypass intrinsic or acquired radiation resistance in tumors. Over the past few years we have studied 3 activators of AMPK all of which enhanced AMPK activation by ionizing radiation and enhanced the cytotoxicity of radiation. The HMG-CoA reductase inhibitor lovastatin, a member of the statin family of anti-cholesterol agents, when used alone phosphorylated and activated AMPK, potentiated ionizing radiation-induced apoptosis and sensitized lung cancer cells to radiotherapy.75 A study by Fritz et al.76 also found that lovastatin could enhance the therapeutic response of cervical cancer cells to ionizing radiation by abrogation of G2-cell cycle arrest, leading to enhanced apoptosis.
In prostate cancer cells, we investigated the radio-modifying properties of the polyphenolic phytoalexin resveratrol, a compound with widely reported anti-aging and anti-cancer properties.77 Low dose (2.5–10 µM) resveratrol inhibited the Akt-mTOR pathway, enhanced activity of the AMPK-p53-p21cip1 axis, and sensitized radio-resistant prostate cancer cells to radiation.77 Similarly, work by Zoberi et al.78 also demonstrated that resveratrol promoted a dose-dependent enhancement of tumor cell killing in irradiated cervical cancer cell lines.
Finally, we explored the therapeutic potential of metformin to sensitize lung cancer cells to radiation therapy.57,58 In earlier work,57 we suggested that clinically achievable doses of metformin (µM) sensitize lung cancer cells to ionizing radiation, while recently we supported this notion further in work with both tissue culture and xenograft models of lung cancer.58 Skinner et al.79 made similar observations in p53-null head and neck cancer cells and Song et al.80 showed that metformin can suppress mTOR activity, be cytotoxic to cancer stem cells specifically, and effectively enhance ionizing radiation-induced cell death in breast cancer and sarcoma cells in vitro and in vivo. Inhibition of AMPK with compound C or specific siRNA largely attenuated metformin's ability to reduce clonogenic survival alone, or in combination with radiotherapy, underscoring the importance of AMPK signaling in modulating cellular radiosensitivity (Fig. 4).57,80 Further, potentiation of AMPK activity with metformin was associated with enhancement of the apoptotic effects of radiation in cancer cell and tumor and increased inhibition of angiogenesis, indicating that AMPK activity is associated with a global anti-tumor action in epithelial cancers.58
ATM-AMPK Activation by Chemotherapeutic Agents
The cytotoxicity of chemotherapeutic agents is mediated by induction of permanent DNA lesions that trigger ATM and DDR signaling. These events have also been linked to AMPK activation.81-83 The topoisomerase II inhibitor etoposide, which mediates DNA breaks by preventing re-ligation of DNA, was shown to induce ATM-dependent activation of AMPK which enhances apoptosis in prostate cancer cells compared with cells lacking functional LKB1-AMPK.81 In addition, cisplatin, which causes DNA damage by forming intra-strand crosslinks, has been reported to activate the ATM-AMPK network in multiple tumor types.54,82 Importantly, potentiation of this pathway via metabolic stress (nutrient deprivation) has been shown to further sensitize cancer cells to cisplatin in vitro and in vivo.83 Conversely, deregulation of ATM-mediated DNA damage signaling in oral cancers has been correlated with resistance to cisplatin.84
Doxorubicin, an anthracycline antibiotic that intercalates between the base pairs of DNA, was also recently demonstrated to activate AMPK through a mechanisms that involves increased ROS production.85 AMPK contributes to doxorubicin-induced apoptosis in multiple cancer cell lines, since (1) chemotherapy-induced AMPK activation inhibits mTORC1 and (2) molecular inhibition of AMPKα was shown to largely attenuate this drug’s effect on cancer cell survival.85 Combining known AMPK-activators, such as the AMP mimetic 5-aminoimidazole-4-carboxamide-1-b-4-ribofuranoside (AICAR), was found to sensitize cancer cells to doxorubicin-induced apoptosis.85 Interestingly, AICAR alone has been reported to signal through ATM in order to regulate AMPK activity.86 Overall, an underlying mechanism of chemotherapy-induced DNA damage appears to also involve the activation of AMPK by ATM, in order to regulate pathways that mediate cell cycle checkpoints and survival (as seen in Fig. 4).
The Role of AMPK in Mitosis and Genomic Stability
Relevant to its ability to match cell cycle progression to energy availability, AMPK is now proposed to play a role in regulating cell polarity, mitotic progression, and cytokinesis.87-89 An early glimpse into the importance of AMPK in cytokinesis and cell proliferation was observed in Drosophila melanogaster, where mutation of the α-catalytic subunit of AMPK led to disruptions in cell polarity and enhanced cell proliferation under metabolic stress.88 Another Drosophila study suggested that the enzyme supports the fidelity of cell division in early stages of development since AMPK-null germ-line clones exhibited gross mitotic defects, such as lagging chromosomes in anaphase, failure to complete cytokinesis, polyploidy, and embryonic lethality.90 In flies, AMPK was found to phosphorylate non-muscle myosin regulatory light chain (MRLC; also known as MRLC2) on Ser22, a protein that interacts with actin to facilitate changes in the actin cytoskeleton that are required for the maintenance of cell polarity. Consistently, a constitutively active form of MRLC was sufficient to rescue the mitotic defects observed in AMPK-null cells, suggesting MRLC is a vital downstream substrate of AMPK in the execution of cell replication.90 The notion arising from these observations suggests involvement of AMPK in the organized steps of cytokinesis as an upstream regulator of mechanical cytoskeletal events supporting mitosis (see Fig. 5).
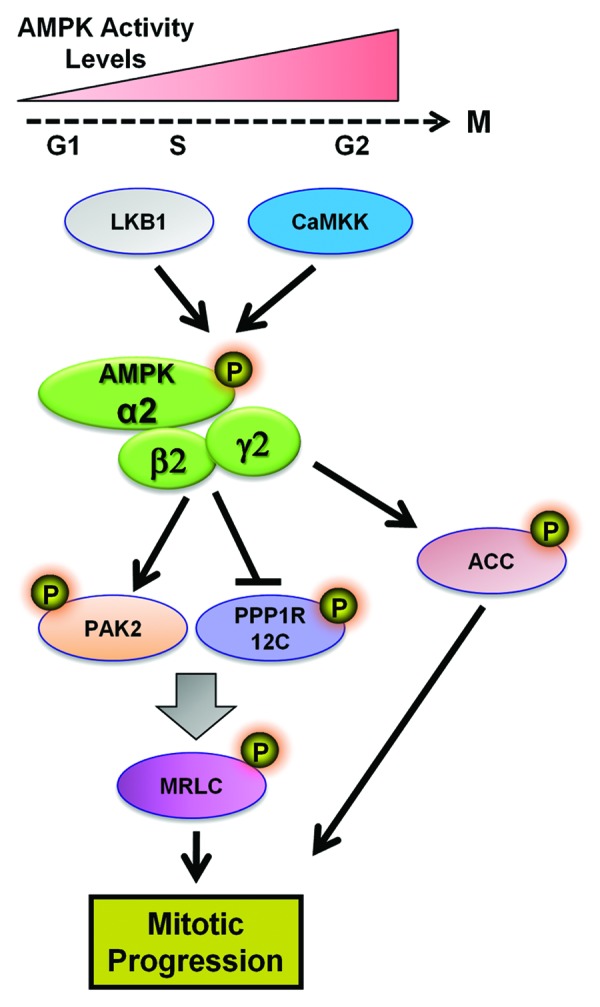
Figure 5. Involvement of AMPK in mitotic progression. The activity of AMPK progressively increases as cells move from G1-phase toward the G2/M stage of cell cycle. During M-phase AMPK is described to phosphorylate key substrates that regulate mitotic progression, including PAK2, PPP1R12C, and ACC. The AMPK α2β2γ2 enzymatic complex is believed to be responsive for this action. Phosphorylation of MLRC leads to regulation of the mechanical cytoskeletal events participating in mitosis. Abbreviations: PAK2, p21-activated protein kinase; PPP1R12C, protein phosphatase 1 regulatory subunit 12C; MRLC, myosin regulatory light chain; ACC, acetyl-CoA-carboxylase.
In mammalian cells, fluorescent microscopy studies showed that the active form of the catalytic AMPKα subunit is directly associated with multiple mitotic structures through each stage of mitosis in human epidermoid carcinoma cells.89,91 We have made similar observations in lung cancer cells (A549) where phosphorylated AMPK α1 and α2 Thr172 was associated with centrosomes in prophase, spindle poles in metaphase and cleavage furrow in anaphase and telophase (Fig. 6A). Consistent with our observations57 on the role of AMPK in mediating G2/M checkpoint, we detected enhanced AMPKα Thr172 phosphorylation in dividing lung cancer cells treated with 8 Gy radiation compared with surrounding non-dividing cells treated with the same dose (Fig. 6B). This suggests that AMPK may have a role in regulation of mitosis in cells under genomic stress. This notion is in agreement with the observations of Vazquez-Martin et al.92 They suggested that AMPK activity varies across different stages of the cell cycle, peaking at the G2/M transition prior to cell division and then decreasing as cells re-entered the G1/S phase (see model Fig. 5); which parallels the expression kinetics of other mitotic passenger proteins such as Aurora B and INCENP.

Figure 6. Active AMPK associates with the mitotic apparatus. (A) Untreated A549 cells were fixed and stained with Hoechst (blue) and an antibody that detects phosphorylated AMPKα on its Thr172 residue (green, 1:00 dilution, Cell Signaling). The cells were imaged at 40× progressing through each phase of mitosis. A representative image for each phase is shown. (B) A549 cells were left untreated or exposed to a single dose of 8Gy IR and imaged one hour later. IR enhanced nuclear and cytoplasmic AMPK phosphorylation (green) in mitotic (arrow) and interphase cells. (C) AMPK activity in A549 cells was inhibited using either the chemical agent compound C (Comp C, 1 μM, Calbiochem) or molecular knockdown of AMPKα with siRNA (Qiagen). AMPK phoshporylation (green) was present in untreated dividing cells, but blocked in cells treated with Comp C or AMPKα siRNA. Phosphorylated histone H3 (Ser10, in red 1:100 dilution, Cell Signaling), which occurs during chromosome condensation during mitosis, was used here are a marker of mitotic progression. Phospho-Ser10-H3 signal and mitosis were detected despite inhibition of AMPK. (D) Mouse embryonic fibroblasts (MEF) that are wild-type or lack AMPKα expression (AMPKα1/2−/−) were fixed and stained with Hoechst (blue) and an antibody that detects phosphorylated AMPKα on its Thr172 residue (red). Dividing and non-dividing cells were imaged at 40× and a representative section of cells are shown.
It is believed that phosphorylation of mitosis-associated AMPK is dependent on the activity of upstream kinases (LKB1 or CaMKK). Inhibition of CaMKK in LKB1-deficient cells was shown to induce spindle mis-orientation, similar to direct inhibition of AMPK, suggesting that at least one of these upstream regulators of AMPK is necessary to facilitate AMPK-mediated mitotic progression.93 Some reports suggested that disruption of AMPK activity with the chemical inhibitor compound C, or molecular knock-down of the AMPKα-catalytic subunit resulted in misshaped spindle poles, prolonged the duration of cells in mitosis, and increased the number of multinucleated cells.93,94
From a mechanistic standpoint, Pinter and colleagues showed that the specific heterotrimeric complex of AMPK that associates with the mitotic apparatus is comprised of the isoforms α2β2γ2.95 In line with this notion, Banko et al.94 conducted a chemical genetic screen of novel AMPK α2-subunit substrates, which uncovered numerous new signaling molecules that are enriched from proteins involved in cytoskeletal dynamics, mitosis, and cytokinesis. Of particular interest were the substrates; protein phosphatase 1 regulatory subunit 12C (PPP1R12C) and p21-activated protein kinase (PAK2), because they both influence the activity of mammalian MRLC.94 AMPK-mediated phosphorylation of both PP1R12C (Ser452) and PAK2 (Ser20) was proposed to activate the mammalian MRLC by phosphorylation on its Ser19 residue, which similar to its Drosophila ortholog, regulates mitotic progression and cell polarity.90,94
Interestingly, previous reports have indicated that disruption of ACC, a major downstream substrate of AMPK, resulted in abnormal mitotic progression and G2/M cell cycle arrest in yeast independent of its metabolic function.96 Recently, phosphorylated ACC (Ser79) was found to associate with mitotic structures in lung carcinoma cells.97 Ser-79 ACC was observed at the spindle poles and cytokinesis furrow during mitosis, but this effect was suppressed in the presence of the AMPK inhibitor compound C,97 indicating that AMPK may target ACC when cells enter mitosis. The functional relevance of this association between mitotic AMPK–ACC in not yet known, but it points to the possibility that metabolic enzymes may also have alternate roles in mitosis. These results support a model of AMPK action illustrated in Figure 5.
However, other studies using dominant negative expression,29 tissue-specific ablation,11 or knockout models98 of AMPK suggested that this enzyme is not vital for normal cell division, since in most scenarios mammalian cells can still continue to replicate in the absence of AMPK. Our own observations are consistent with the latter notion where in lung cancer cells inhibition of AMPK with compound C or knockdown of AMPKα subunits with siRNAs did not inhibit progression of the cell cycle in untreated cells (Fig. 6C). Of note, these observations were made in A549 lung cancer cells which lack LKB1 activity. In addition, AMPKα1/2−/−MEFs do not show defects in mitosis (Fig. 6D). Furthermore, we observed increased proliferation and a survival advantage for AMPKα1/2−/−MEFs and A549 lung cancer cells with inhibited AMPK when they were treated with therapeutic doses of ionizing radiation compared with wild-type MEFs or control cells.57,58,63
The above studies suggest that AMPK may be a passenger of the mitotic apparatus that is not required for normal mitotic progression, but may function as a “genomic fidelity checkpoint regulator” that can trigger cell cycle arrest and alter mitotic progression in the face of genotoxic stress. The evidence supporting this conclusion comes from findings that AMPK mediates a radiation-induced G2/M checkpoint through induction of cell cycle regulators (including p53 and p21cip1), and that this event coincides with the temporal accumulation of activated AMPK and other chromosomal passenger proteins during this phase of cell cycle.92 By this virtue, AMPK may be “primed” to regulate cell cycle signals near the cellular transition between interphase and mitosis if challenged by genotoxic agents (see the model in Fig. 7).
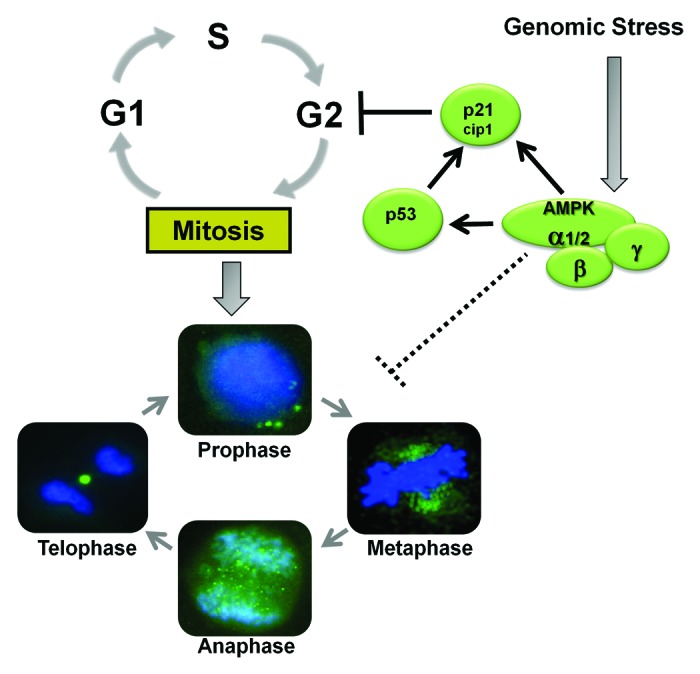
Figure 7. In mammalian cells AMPK mediates a G2/M checkpoint in response to genomic stress. During cell division, activated AMPK associates with the mitotic apparatus in spindle fibers in prophase, the centrosomes in metaphase, mitotic spindle in anaphase, and the cleavage furrow in telophase. However, this association is not necessary for the progression of the cell cycle and mitosis in untreated cells, and AMPK may simply be a passenger molecule. AMPK modulates mitotic progression in cells undergoing genomic stress.
Conceptually, it is perplexing to understand how AMPK can operate to suppress proliferation and enhance energy levels under times of cellular stress on one hand, but also actively support mitosis on the other (Figs. 5 and 7). However, the two functions appear to be independent. Glucose deprivation was not found to affect AMPK’s role in modulating execution of mitosis,94 suggesting that AMPK may have mutually exclusive functions under different types of cellular stress. Distinct AMPK heterotrimers may have distinct roles in cellular function. A unifying hypothesis would suggest that (1) the AMPK α2β2γ2 complex may indeed support molecular events leading to mitotic progression but this role of this enzyme in mammalian cells may be redundant, and (2) AMPK is a passenger of mitotic apparatus that is not required for cellular division but can actively mediate the G2/M checkpoint and inhibit mitotic progression at times of genomic stress.
Genomic Stress-Mediated Activation of Autophagy by AMPK
Finally, consideration should be given to potential survival mechanisms that are regulated by AMPK. Much of the discussion here has supported a role of AMPK as a mediator of tumor suppression that can augment cell cytotoxicity in response to chemo- or radiation therapy. However, induction of the AMPK pathway by various stress agents can also stimulate autophagy, a reversible state of cellular metabolism whereby cells under energy stress consume cellular organelles and substrates for energy production and survival.52,99 Stress-induced initiation of autophagy requires activation of the Unc-51-like kinase (ULK) complex, which consists of ULK1, ULK2, Atg13, and FIP200, and is tightly regulated by AMPK and mTOR.100 Starvation and stress-activated AMPK phosphorylates ULK1 on Ser317 and Ser777 to initiate autophagy. Conversely, under nutrient-depleted conditions, mTORC1 phosphorylates ULK1 on Ser757, which displaces the interaction of AMPK with ULK1 and suppresses autophagy.101
In early stages of carcinogenesis the physiological process of autophagy has been viewed as protective against tumorigenesis.102 Breast tumors are found to be mutated in the autophagy induction gene beclin 1 (BECN1). This may provide them with a growth advantage.103 In fact breast cancer cells carrying deletion in the BECN1 allele will exhibit reduced tumorigenicity if the BECN1 gene is re-expressed by undergoing the signal transduction pathways leading to AMPK activation and autophagy.103
Conversely, in advanced stages of tumor progression, autophagy is suggested to provide a survival advantage against cancer therapies by suppressing the apoptosis pathway (protective autophagy).102 Autophagy is linked to radiation resistance since it is conceived to mediate metabolic recycling of damaged organelles to help cells survive conditions of increased metabolic demand.104 Cancer cells may undergo protective autophagy in response chemotherapy treatment. Autophagy was implicated in chemo-resistance to etoposide and this effect was reversed by inhibition of AMPK, which led to apoptosis in HepG2 cancer cells.105 While it has been reported that AMPK-activators such as metformin may work well in combination with chemotherapy in cancer cells,106 it has been suggested that this may only offer benefit in tumors that lack p53 which cannot induce protective autophagy triggered by metformin-mediated metabolic stress.107
Although the role of autophagy in malignant cells may be multifaceted, manipulation of this process with activators or inhibitors of AMPK may still provide an additional therapeutic option for cancer treatment. Our work suggests that combination treatment with AMPK activators and ionizing radiation leads preferentially to inhibition of pro-survival pathways and increased cytotoxicity. Although AMPK is suggested to stimulate autophagy, which could function as a survival mechanism in cells under genotoxic stress, there is adequate evidence that potentiation of AMPK activity is a valuable tool to sensitize tumor cells to radiotherapy and certain chemotherapeutic agents.
Conclusions
AMPK is a critical regulator of cellular energy homeostasis. This is why it comes as no surprise that it is also tightly linked with the regulation of biochemical pathways that influence cell survival and proliferation. Work with cancer models from a number of laboratories implicates AMPK as sensor of genomic stress, suppressor of survival signals, and regulator of mitotic progression. Recent studies demonstrate a role for the enzyme as a transducer of signals in the ATM pathway and bring AMPK to the center stage of tumor radiation biology. AMPK complexes associate with most stages of mitotic progression but in mammalian cells the enzyme appears to function as a passenger protein that modulates cell cycle progression and mitosis only at times of cellular stress. The knowledge gained from the analysis of the non-metabolic actions of AMPK made apparent the value of targeting this enzyme in cancer therapy. Ongoing studies with pharmacological activators of AMPK and availability of animal models of tissue-specific or whole-body AMPK subunit deletions are expected to help us understand better the role of AMPK in regulating normal and cancer cell fate and genomic stability.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Glossary
Abbreviations:
- AMPK
AMP-activated protein kinase
- ATM
ataxia telangiectasia mutated
- ACC
acetyl CoA carboxylase
- LKB1
liver-kinase B1
- mTOR
mammalian target of rapamycin
- CaMKK
calmodulin-dependent protein kinase
- HMG-CoA
hydroxy-3-methyl-glutaryl-CoA reductase
- CPT-1
carnitine palmitoyltransferase I
- TSC1/2
tuberous sclerosis complex 1/2
- FOXO3a
forkhead box 03a
- SREBP-1c
sterol regulatory element binding protein-1c
- PGC-1α
peroxisome proliferator-activated receptor gamma coactivator-1-α
- OXPHOS
oxidative phosphorylation
- MRE11
meiotic recombination 11
- NBS1
Nijmegen breakage syndrome 1
- DNA-PK
DNA-dependent protein kinase
- ATR
ATM and Rad3-related protein
- MEF
mouse embryonic fibroblasts
- SESN1/2
sestrins 1/2
- AICAR
5-aminoimidazole-4-carboxamide-1-b-4-ribofuranoside
- MRLC
myosin regulatory light chain
- PPP1R12C
protein phosphatase 1 regulatory subunit 12C
- PAK2
p21-activated protein kinase
- ULK
Unc-51-like kinase
- BECN1
beclin 1
- P70S6K
p70-S6 kinase
- 4EBP1
eukaryotic initiation factor 4E binding protein 1
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/26726
References
- 1.Kourtis N, Tavernarakis N. Cellular stress response pathways and ageing: intricate molecular relationships. EMBO J. 2011;30:2520–31. doi: 10.1038/emboj.2011.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oakhill JS, Scott JW, Kemp BE. Structure and function of AMP-activated protein kinase. Acta Physiol (Oxf) 2009;196:3–14. doi: 10.1111/j.1748-1716.2009.01977.x. [DOI] [PubMed] [Google Scholar]
- 3.Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016–23. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shaw RJ. LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol (Oxf) 2009;196:65–80. doi: 10.1111/j.1748-1716.2009.01972.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hardie DG, Carling D, Gamblin SJ. AMP-activated protein kinase: also regulated by ADP? Trends Biochem Sci. 2011;36:470–7. doi: 10.1016/j.tibs.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 6.Hardie DG, Hawley SA. AMP-activated protein kinase: the energy charge hypothesis revisited. Bioessays. 2001;23:1112–9. doi: 10.1002/bies.10009. [DOI] [PubMed] [Google Scholar]
- 7.Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9:563–75. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Woods A, Dickerson K, Heath R, Hong SP, Momcilovic M, Johnstone SR, Carlson M, Carling D. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005;2:21–33. doi: 10.1016/j.cmet.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 9.Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 10.Dzamko N, van Denderen BJ, Hevener AL, Jørgensen SB, Honeyman J, Galic S, Chen ZP, Watt MJ, Campbell DJ, Steinberg GR, et al. AMPK beta1 deletion reduces appetite, preventing obesity and hepatic insulin resistance. J Biol Chem. 2010;285:115–22. doi: 10.1074/jbc.M109.056762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Neill HM, Maarbjerg SJ, Crane JD, Jeppesen J, Jørgensen SB, Schertzer JD, Shyroka O, Kiens B, van Denderen BJ, Tarnopolsky MA, et al. AMP-activated protein kinase (AMPK) beta1beta2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc Natl Acad Sci U S A. 2011;108:16092–7. doi: 10.1073/pnas.1105062108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oakhill JS, Chen ZP, Scott JW, Steel R, Castelli LA, Ling N, Macaulay SL, Kemp BE. β-Subunit myristoylation is the gatekeeper for initiating metabolic stress sensing by AMP-activated protein kinase (AMPK) Proc Natl Acad Sci U S A. 2010;107:19237–41. doi: 10.1073/pnas.1009705107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scott JW, van Denderen BJ, Jorgensen SB, Honeyman JE, Steinberg GR, Oakhill JS, Iseli TJ, Koay A, Gooley PR, Stapleton D, et al. Thienopyridone drugs are selective activators of AMP-activated protein kinase beta1-containing complexes. Chem Biol. 2008;15:1220–30. doi: 10.1016/j.chembiol.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 14.Hawley SA, Fullerton MD, Ross FA, Schertzer JD, Chevtzoff C, Walker KJ, Peggie MW, Zibrova D, Green KA, Mustard KJ, et al. The ancient drug salicylate directly activates AMP-activated protein kinase. Science. 2012;336:918–22. doi: 10.1126/science.1215327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Steinberg GR, O’Neill HM, Dzamko NL, Galic S, Naim T, Koopman R, Jørgensen SB, Honeyman J, Hewitt K, Chen ZP, et al. Whole body deletion of AMP-activated protein kinase beta2 reduces muscle AMPK activity and exercise capacity. J Biol Chem. 2010;285:37198–209. doi: 10.1074/jbc.M110.102434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bateman A. The structure of a domain common to archaebacteria and the homocystinuria disease protein. Trends Biochem Sci. 1997;22:12–3. doi: 10.1016/S0968-0004(96)30046-7. [DOI] [PubMed] [Google Scholar]
- 17.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–62. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xiao B, Sanders MJ, Underwood E, Heath R, Mayer FV, Carmena D, Jing C, Walker PA, Eccleston JF, Haire LF, et al. Structure of mammalian AMPK and its regulation by ADP. Nature. 2011;472:230–3. doi: 10.1038/nature09932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Corton JM, Gillespie JG, Hawley SA, Hardie DG. 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur J Biochem. 1995;229:558–65. doi: 10.1111/j.1432-1033.1995.tb20498.x. [DOI] [PubMed] [Google Scholar]
- 20.Carlson CA, Kim KH. Regulation of hepatic acetyl coenzyme A carboxylase by phosphorylation and dephosphorylation. J Biol Chem. 1973;248:378–80. [PubMed] [Google Scholar]
- 21.Linher-Melville K, Zantinge S, Sanli T, Gerstein H, Tsakiridis T, Singh G. Establishing a relationship between prolactin and altered fatty acid β-oxidation via carnitine palmitoyl transferase 1 in breast cancer cells. BMC Cancer. 2011;11:56. doi: 10.1186/1471-2407-11-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Veelen W, Korsse SE, van de Laar L, Peppelenbosch MP. The long and winding road to rational treatment of cancer associated with LKB1/AMPK/TSC/mTORC1 signaling. Oncogene. 2011;30:2289–303. doi: 10.1038/onc.2010.630. [DOI] [PubMed] [Google Scholar]
- 24.Ikenoue T, Inoki K, Yang Q, Zhou X, Guan KL. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J. 2008;27:1919–31. doi: 10.1038/emboj.2008.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–26. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Greer EL, Oskoui PR, Banko MR, Maniar JM, Gygi MP, Gygi SP, Brunet A. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J Biol Chem. 2007;282:30107–19. doi: 10.1074/jbc.M705325200. [DOI] [PubMed] [Google Scholar]
- 27.Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B, Park O, Luo Z, Lefai E, Shyy JY, et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011;13:376–88. doi: 10.1016/j.cmet.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jäger S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A. 2007;104:12017–22. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18:283–93. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 30.Lee CW, Wong LL, Tse EY, Liu HF, Leong VY, Lee JM, Hardie DG, Ng IO, Ching YP. AMPK promotes p53 acetylation via phosphorylation and inactivation of SIRT1 in liver cancer cells. Cancer Res. 2012;72:4394–404. doi: 10.1158/0008-5472.CAN-12-0429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M, Kondo S, Dumont DJ, Gutterman JU, Walker CL, et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol. 2007;9:218–24. doi: 10.1038/ncb1537. [DOI] [PubMed] [Google Scholar]
- 32.Bungard D, Fuerth BJ, Zeng PY, Faubert B, Maas NL, Viollet B, Carling D, Thompson CB, Jones RG, Berger SL. Signaling kinase AMPK activates stress-promoted transcription via histone H2B phosphorylation. Science. 2010;329:1201–5. doi: 10.1126/science.1191241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Warburg O, Wind F, Negelein E. The Metabolism of Tumors in the Body. J Gen Physiol. 1927;8:519–30. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;9:425–34. doi: 10.1016/j.ccr.2006.04.023. [DOI] [PubMed] [Google Scholar]
- 35.Moreno-Sánchez R, Rodríguez-Enríquez S, Marín-Hernández A, Saavedra E. Energy metabolism in tumor cells. FEBS J. 2007;274:1393–418. doi: 10.1111/j.1742-4658.2007.05686.x. [DOI] [PubMed] [Google Scholar]
- 36.Bartrons R, Caro J. Hypoxia, glucose metabolism and the Warburg’s effect. J Bioenerg Biomembr. 2007;39:223–9. doi: 10.1007/s10863-007-9080-3. [DOI] [PubMed] [Google Scholar]
- 37.Shaw RJ. Glucose metabolism and cancer. Curr Opin Cell Biol. 2006;18:598–608. doi: 10.1016/j.ceb.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 38.Smolková K, Plecitá-Hlavatá L, Bellance N, Benard G, Rossignol R, Ježek P. Waves of gene regulation suppress and then restore oxidative phosphorylation in cancer cells. Int J Biochem Cell Biol. 2011;43:950–68. doi: 10.1016/j.biocel.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 39.Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev. 2009;23:537–48. doi: 10.1101/gad.1756509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z, Dupuy F, Chambers C, Fuerth BJ, Viollet B, et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab. 2013;17:113–24. doi: 10.1016/j.cmet.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee YG, Lee SW, Sin HS, Kim EJ, Um SJ. Kinase activity-independent suppression of p73alpha by AMP-activated kinase alpha (AMPKalpha) Oncogene. 2009;28:1040–52. doi: 10.1038/onc.2008.452. [DOI] [PubMed] [Google Scholar]
- 42.Nieminen AI, Eskelinen VM, Haikala HM, Tervonen TA, Yan Y, Partanen JI, Klefström J. Myc-induced AMPK-phospho p53 pathway activates Bak to sensitize mitochondrial apoptosis. Proc Natl Acad Sci U S A. 2013;110:E1839–48. doi: 10.1073/pnas.1208530110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bensimon A, Aebersold R, Shiloh Y. Beyond ATM: the protein kinase landscape of the DNA damage response. FEBS Lett. 2011;585:1625–39. doi: 10.1016/j.febslet.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 44.Ditch S, Paull TT. The ATM protein kinase and cellular redox signaling: beyond the DNA damage response. Trends Biochem Sci. 2012;37:15–22. doi: 10.1016/j.tibs.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zakikhani M, Bazile M, Hashemi S, Javeshghani S, Avizonis D, St Pierre J, Pollak MN. Alterations in cellular energy metabolism associated with the antiproliferative effects of the ATM inhibitor KU-55933 and with metformin. PLoS One. 2012;7:e49513. doi: 10.1371/journal.pone.0049513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Armata HL, Golebiowski D, Jung DY, Ko HJ, Kim JK, Sluss HK. Requirement of the ATM/p53 tumor suppressor pathway for glucose homeostasis. Mol Cell Biol. 2010;30:5787–94. doi: 10.1128/MCB.00347-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schultz LB, Chehab NH, Malikzay A, Halazonetis TD. p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J Cell Biol. 2000;151:1381–90. doi: 10.1083/jcb.151.7.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fernandez-Capetillo O, Lee A, Nussenzweig M, Nussenzweig A. H2AX: the histone guardian of the genome. DNA Repair (Amst) 2004;3:959–67. doi: 10.1016/j.dnarep.2004.03.024. [DOI] [PubMed] [Google Scholar]
- 49.Pawlik TM, Keyomarsi K. Role of cell cycle in mediating sensitivity to radiotherapy. Int J Radiat Oncol Biol Phys. 2004;59:928–42. doi: 10.1016/j.ijrobp.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 50.Stracker TH, Roig I, Knobel PA, Marjanović M. The ATM signaling network in development and disease. Front Genet. 2013;4:37. doi: 10.3389/fgene.2013.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, 3rd, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–6. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 52.Alexander A, Kim J, Walker CL. ATM engages the TSC2/mTORC1 signaling node to regulate autophagy. Autophagy. 2010;6:672–3. doi: 10.4161/auto.6.5.12509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sun Y, Connors KE, Yang DQ. AICAR induces phosphorylation of AMPK in an ATM-dependent, LKB1-independent manner. Mol Cell Biochem. 2007;306:239–45. doi: 10.1007/s11010-007-9575-6. [DOI] [PubMed] [Google Scholar]
- 54.Alexander A, Walker CL. The role of LKB1 and AMPK in cellular responses to stress and damage. FEBS Lett. 2011;585:952–7. doi: 10.1016/j.febslet.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 55.Alexander A, Cai SL, Kim J, Nanez A, Sahin M, MacLean KH, Inoki K, Guan KL, Shen J, Person MD, et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc Natl Acad Sci U S A. 2010;107:4153–8. doi: 10.1073/pnas.0913860107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Amatya PN, Kim HB, Park SJ, Youn CK, Hyun JW, Chang IY, Lee JH, You HJ. A role of DNA-dependent protein kinase for the activation of AMP-activated protein kinase in response to glucose deprivation. Biochim Biophys Acta. 2012;1823:2099–108. doi: 10.1016/j.bbamcr.2012.08.022. [DOI] [PubMed] [Google Scholar]
- 57.Sanli T, Rashid A, Liu C, Harding S, Bristow RG, Cutz JC, Singh G, Wright J, Tsakiridis T. Ionizing radiation activates AMP-activated kinase (AMPK): a target for radiosensitization of human cancer cells. Int J Radiat Oncol Biol Phys. 2010;78:221–9. doi: 10.1016/j.ijrobp.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 58.Storozhuk YH, Hopmans SN, Sanli T, Barron C, Tsiani E, Cutz JC, Pond G, Wright J, Singh G, Tsakiridis T. Metformin inhibits growth and enhances radiation response of non-small cell lung cancer (NSCLC) through ATM and AMPK. Br J Cancer. 2013;108:2021–32. doi: 10.1038/bjc.2013.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Woods A, Leiper JM, Carling D. The role of ATM in response to metformin treatment and activation of AMPK. Nat Genet. 2012;44:360–1. doi: 10.1038/ng.2235. [DOI] [PubMed] [Google Scholar]
- 60.Sapkota GP, Deak M, Kieloch A, Morrice N, Goodarzi AA, Smythe C, Shiloh Y, Lees-Miller SP, Alessi DR. Ionizing radiation induces ataxia telangiectasia mutated kinase (ATM)-mediated phosphorylation of LKB1/STK11 at Thr-366. Biochem J. 2002;368:507–16. doi: 10.1042/BJ20021284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Storozhuk Y, Sanli T, Hopmans SN, Schultz C, Farrell T, Cutz JC, Steinberg GR, Wright J, Singh G, Tsakiridis T. Chronic modulation of AMP-Kinase, Akt and mTOR pathways by ionizing radiation in human lung cancer xenografts. Radiat Oncol. 2012;7:71. doi: 10.1186/1748-717X-7-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fu X, Wan S, Lyu YL, Liu LF, Qi H. Etoposide induces ATM-dependent mitochondrial biogenesis through AMPK activation. PLoS One. 2008;3:e2009. doi: 10.1371/journal.pone.0002009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sanli T, Storozhuk Y, Linher-Melville K, Bristow RG, Laderout K, Viollet B, Wright J, Singh G, Tsakiridis T. Ionizing radiation regulates the expression of AMP-activated protein kinase (AMPK) in epithelial cancer cells: modulation of cellular signals regulating cell cycle and survival. Radiother Oncol. 2012;102:459–65. doi: 10.1016/j.radonc.2011.11.014. [DOI] [PubMed] [Google Scholar]
- 64.Viniegra JG, Martínez N, Modirassari P, Hernández Losa J, Parada Cobo C, Sánchez-Arévalo Lobo VJ, Aceves Luquero CI, Alvarez-Vallina L, Ramón y Cajal S, Rojas JM, et al. Full activation of PKB/Akt in response to insulin or ionizing radiation is mediated through ATM. J Biol Chem. 2005;280:4029–36. doi: 10.1074/jbc.M410344200. [DOI] [PubMed] [Google Scholar]
- 65.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 66.Braunstein S, Badura ML, Xi Q, Formenti SC, Schneider RJ. Regulation of protein synthesis by ionizing radiation. Mol Cell Biol. 2009;29:5645–56. doi: 10.1128/MCB.00711-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bristow RG, Hill R. Molecular and Cellular Basis of Radiotherapy. The Basic Science of Oncology. Toronto: McGraw-Hill, 1998:295-321. [Google Scholar]
- 68.Levine AJ, Feng Z, Mak TW, You H, Jin S. Coordination and communication between the p53 and IGF-1-AKT-TOR signal transduction pathways. Genes Dev. 2006;20:267–75. doi: 10.1101/gad.1363206. [DOI] [PubMed] [Google Scholar]
- 69.Maclaine NJ, Hupp TR. The regulation of p53 by phosphorylation: a model for how distinct signals integrate into the p53 pathway. Aging (Albany NY) 2009;1:490–502. doi: 10.18632/aging.100047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008;134:451–60. doi: 10.1016/j.cell.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Feng Z, Hu W, de Stanchina E, Teresky AK, Jin S, Lowe S, Levine AJ. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007;67:3043–53. doi: 10.1158/0008-5472.CAN-06-4149. [DOI] [PubMed] [Google Scholar]
- 72.Budanov AV, Shoshani T, Faerman A, Zelin E, Kamer I, Kalinski H, Gorodin S, Fishman A, Chajut A, Einat P, et al. Identification of a novel stress-responsive gene Hi95 involved in regulation of cell viability. Oncogene. 2002;21:6017–31. doi: 10.1038/sj.onc.1205877. [DOI] [PubMed] [Google Scholar]
- 73.Sanli T, Linher-Melville K, Tsakiridis T, Singh G. Sestrin2 modulates AMPK subunit expression and its response to ionizing radiation in breast cancer cells. PLoS One. 2012;7:e32035. doi: 10.1371/journal.pone.0032035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zannella VE, Cojocari D, Hilgendorf S, Vellanki RN, Chung S, Wouters BG, Koritzinsky M. AMPK regulates metabolism and survival in response to ionizing radiation. Radiother Oncol. 2011;99:293–9. doi: 10.1016/j.radonc.2011.05.049. [DOI] [PubMed] [Google Scholar]
- 75.Sanli T, Liu C, Rashid A, Hopmans SN, Tsiani E, Schultz C, Farrell T, Singh G, Wright J, Tsakiridis T. Lovastatin sensitizes lung cancer cells to ionizing radiation: modulation of molecular pathways of radioresistance and tumor suppression. J Thorac Oncol. 2011;6:439–50. doi: 10.1097/JTO.0b013e3182049d8b. [DOI] [PubMed] [Google Scholar]
- 76.Fritz G, Brachetti C, Kaina B. Lovastatin causes sensitization of HeLa cells to ionizing radiation-induced apoptosis by the abrogation of G2 blockage. Int J Radiat Biol. 2003;79:601–10. doi: 10.1080/09553000310001609233. [DOI] [PubMed] [Google Scholar]
- 77.Rashid A, Liu C, Sanli T, Tsiani E, Singh G, Bristow RG, Dayes I, Lukka H, Wright J, Tsakiridis T. Resveratrol enhances prostate cancer cell response to ionizing radiation. Modulation of the AMPK, Akt and mTOR pathways. Radiat Oncol. 2011;6:144. doi: 10.1186/1748-717X-6-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zoberi I, Bradbury CM, Curry HA, Bisht KS, Goswami PC, Roti Roti JL, Gius D. Radiosensitizing and anti-proliferative effects of resveratrol in two human cervical tumor cell lines. Cancer Lett. 2002;175:165–73. doi: 10.1016/S0304-3835(01)00719-4. [DOI] [PubMed] [Google Scholar]
- 79.Skinner HD, Sandulache VC, Ow TJ, Meyn RE, Yordy JS, Beadle BM, Fitzgerald AL, Giri U, Ang KK, Myers JN. TP53 disruptive mutations lead to head and neck cancer treatment failure through inhibition of radiation-induced senescence. Clin Cancer Res. 2012;18:290–300. doi: 10.1158/1078-0432.CCR-11-2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Song CW, Lee H, Dings RP, Williams B, Powers J, Santos TD, Choi BH, Park HJ. Metformin kills and radiosensitizes cancer cells and preferentially kills cancer stem cells. Sci Rep. 2012;2:362. doi: 10.1038/srep00362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Luo L, Huang W, Tao R, Hu N, Xiao ZX, Luo Z. ATM and LKB1 dependent activation of AMPK sensitizes cancer cells to etoposide-induced apoptosis. Cancer Lett. 2013;328:114–9. doi: 10.1016/j.canlet.2012.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Basu A, Krishnamurthy S. Cellular responses to Cisplatin-induced DNA damage. J Nucleic Acids. 2010;2010:pii: 201367. doi: 10.4061/2010/201367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shi Y, Felley-Bosco E, Marti TM, Orlowski K, Pruschy M, Stahel RA. Starvation-induced activation of ATM/Chk2/p53 signaling sensitizes cancer cells to cisplatin. BMC Cancer. 2012;12:571. doi: 10.1186/1471-2407-12-571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang L, Mosel AJ, Oakley GG, Peng A. Deficient DNA damage signaling leads to chemoresistance to cisplatin in oral cancer. Mol Cancer Ther. 2012;11:2401–9. doi: 10.1158/1535-7163.MCT-12-0448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ji C, Yang B, Yang YL, He SH, Miao DS, He L, Bi ZG. Exogenous cell-permeable C6 ceramide sensitizes multiple cancer cell lines to Doxorubicin-induced apoptosis by promoting AMPK activation and mTORC1 inhibition. Oncogene. 2010;29:6557–68. doi: 10.1038/onc.2010.379. [DOI] [PubMed] [Google Scholar]
- 86.Zajkowicz A, Rusin M. The activation of the p53 pathway by the AMP mimetic AICAR is reduced by inhibitors of the ATM or mTOR kinases. Mech Ageing Dev. 2011;132:543–51. doi: 10.1016/j.mad.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 87.Bettencourt-Dias M, Giet R, Sinka R, Mazumdar A, Lock WG, Balloux F, Zafiropoulos PJ, Yamaguchi S, Winter S, Carthew RW, et al. Genome-wide survey of protein kinases required for cell cycle progression. Nature. 2004;432:980–7. doi: 10.1038/nature03160. [DOI] [PubMed] [Google Scholar]
- 88.Mirouse V, Swick LL, Kazgan N, St Johnston D, Brenman JE. LKB1 and AMPK maintain epithelial cell polarity under energetic stress. J Cell Biol. 2007;177:387–92. doi: 10.1083/jcb.200702053. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 89.Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. The active form of the metabolic sensor: AMP-activated protein kinase (AMPK) directly binds the mitotic apparatus and travels from centrosomes to the spindle midzone during mitosis and cytokinesis. Cell Cycle. 2009;8:2385–98. doi: 10.4161/cc.8.15.9082. [DOI] [PubMed] [Google Scholar]
- 90.Lee JH, Koh H, Kim M, Kim Y, Lee SY, Karess RE, Lee SH, Shong M, Kim JM, Kim J, et al. Energy-dependent regulation of cell structure by AMP-activated protein kinase. Nature. 2007;447:1017–20. doi: 10.1038/nature05828. [DOI] [PubMed] [Google Scholar]
- 91.Vazquez-Martin A, Oliveras-Ferraros C, Lopez-Bonet E, Menendez JA. AMPK: Evidence for an energy-sensing cytokinetic tumor suppressor. Cell Cycle. 2009;8:3679–83. doi: 10.4161/cc.8.22.9905. [DOI] [PubMed] [Google Scholar]
- 92.Vazquez-Martin A, López-Bonet E, Oliveras-Ferraros C, Pérez-Martínez MC, Bernadó L, Menendez JA. Mitotic kinase dynamics of the active form of AMPK (phospho-AMPKalphaThr172) in human cancer cells. Cell Cycle. 2009;8:788–91. doi: 10.4161/cc.8.5.7787. [DOI] [PubMed] [Google Scholar]
- 93.Thaiparambil JT, Eggers CM, Marcus AI. AMPK regulates mitotic spindle orientation through phosphorylation of myosin regulatory light chain. Mol Cell Biol. 2012;32:3203–17. doi: 10.1128/MCB.00418-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Banko MR, Allen JJ, Schaffer BE, Wilker EW, Tsou P, White JL, Villén J, Wang B, Kim SR, Sakamoto K, et al. Chemical genetic screen for AMPKα2 substrates uncovers a network of proteins involved in mitosis. Mol Cell. 2011;44:878–92. doi: 10.1016/j.molcel.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pinter K, Jefferson A, Czibik G, Watkins H, Redwood C. Subunit composition of AMPK trimers present in the cytokinetic apparatus: Implications for drug target identification. Cell Cycle. 2012;11:917–21. doi: 10.4161/cc.11.5.19412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Saitoh S, Takahashi K, Nabeshima K, Yamashita Y, Nakaseko Y, Hirata A, Yanagida M. Aberrant mitosis in fission yeast mutants defective in fatty acid synthetase and acetyl CoA carboxylase. J Cell Biol. 1996;134:949–61. doi: 10.1083/jcb.134.4.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Vazquez-Martin A, Corominas-Faja B, Oliveras-Ferraros C, Cufí S, Dalla Venezia N, Menendez JA. Serine79-phosphorylated acetyl-CoA carboxylase, a downstream target of AMPK, localizes to the mitotic spindle poles and the cytokinesis furrow. Cell Cycle. 2013;12:1639–41. doi: 10.4161/cc.24700. http:// [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Viollet B, Horman S, Leclerc J, Lantier L, Foretz M, Billaud M, Giri S, Andreelli F. AMPK inhibition in health and disease. Crit Rev Biochem Mol Biol. 2010;45:276–95. doi: 10.3109/10409238.2010.488215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Amaravadi RK, Thompson CB. The roles of therapy-induced autophagy and necrosis in cancer treatment. Clin Cancer Res. 2007;13:7271–9. doi: 10.1158/1078-0432.CCR-07-1595. [DOI] [PubMed] [Google Scholar]
- 100.Mathew R, White E. Autophagy, stress, and cancer metabolism: what doesn’t kill you makes you stronger. Cold Spring Harb Symp Quant Biol. 2011;76:389–96. doi: 10.1101/sqb.2012.76.011015. [DOI] [PubMed] [Google Scholar]
- 101.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–41. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer. 2005;5:726–34. doi: 10.1038/nrc1692. [DOI] [PubMed] [Google Scholar]
- 103.Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–6. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 104.Apel A, Herr I, Schwarz H, Rodemann HP, Mayer A. Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res. 2008;68:1485–94. doi: 10.1158/0008-5472.CAN-07-0562. [DOI] [PubMed] [Google Scholar]
- 105.Xie BS, Zhao HC, Yao SK, Zhuo DX, Jin B, Lv DC, Wu CL, Ma DL, Gao C, Shu XM, et al. Autophagy inhibition enhances etoposide-induced cell death in human hepatoma G2 cells. Int J Mol Med. 2011;27:599–606. doi: 10.3892/ijmm.2011.607. [DOI] [PubMed] [Google Scholar]
- 106.Rocha GZ, Dias MM, Ropelle ER, Osório-Costa F, Rossato FA, Vercesi AE, Saad MJ, Carvalheira JB. Metformin amplifies chemotherapy-induced AMPK activation and antitumoral growth. Clin Cancer Res. 2011;17:3993–4005. doi: 10.1158/1078-0432.CCR-10-2243. [DOI] [PubMed] [Google Scholar]
- 107.Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, Viollet B, Thompson CB. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007;67:6745–52. doi: 10.1158/0008-5472.CAN-06-4447. [DOI] [PubMed] [Google Scholar]