

Summary
The objective of the present study was to evaluate the pharmacokinetic (PK) and pharmacodynamic (PD) profiles of the new recombinant FVIII compound turoctocog alfa and a Glyco-PEGylated FVIII derivative thereof (N8-GP) in Haemophilia A dogs. Six haemophilic dogs divided into two groups were included in the study. Each dog was administered a dose of 125 U kg−1, blood samples were collected at predetermined time points for both pharmacokinetic (FVIII measured by one-stage aPTT assay) and pharmacodynamic [whole blood clotting time (WBCT)] evaluations. After intravenous administration to haemophilic dogs, the plasma concentration at the first sampling point was comparable for turoctocog alfa and N8-GP, and the clearance was estimated to be 6.5 and 3.9 mL h−1kg−1 for turoctocog alfa and N8-GP respectively. Both turoctocog alfa and N8-GP were able to reduce the WBCT time to normal levels (<20 min), however, the reduced clearance was reflected in the WBCT, which returned to baseline at a later time point for N8-GP as compared with dogs dosed with turoctocog alfa. The clearance was 40% reduced for N8-GP as compared with turoctocog alfa. Simulations of a multiple dosing regimen in dogs, suggest that to maintain WBCT <20 min N8-GP can be dosed at reduced intervals, e.g. with 4 days between doses, whereas turoctocog alfa will have to be dosed with 2½ day between doses. Data thereby supports N8-GP as an alternative to standard rFVIII replacement therapy, with a more convenient dosing regimen.
Keywords: haemophilic dogs, N8-GP, NONMEM, pharmacokinetics, rFVIII, turoctocog alfa
Introduction
Haemophilia A is a congenital X-linked bleeding disorder caused by defective synthesis or by synthesis of dysfunctional FVIII molecules with an incidence of one in every 5 000 live male births. The bleeding disorder is characterized by excessive bleeding into various tissues and organs and if left untreated recurrent bleeding into the joints can lead to severe joint destruction and to resultant arthropathy. Treatment of haemophilia A is based on intravenous replacement therapy with recombinant FVIII or plasma-derived FVIII concentrates which can be given on demand following an acute bleed or as prophylaxis at regular intervals to prevent bleeding. Prophylaxis for severe haemophilia A is now recognized as the standard of care with optimal initiation very early in life and requires frequent intravenous injections (two to four times a week; 1,2). Nevertheless, high cost of products and the frequent intravenous access and side effects associated with central venous access devices compromises adherence and are barriers for an effective prophylactic treatment.
The mean half-life of FVIII in man is approximately 12 h and extension of the half-life could thus reduce the frequency of infusions and thereby improve compliance. Currently no long-acting FVIII is available on the market although several approaches to extend the half-life of FVIII are being investigated [3]. Novo Nordisk is developing a new FVIII replacement product, turoctocog alfa, which is a B-domain truncated rFVIII molecule [4]. Turoctacog alfa gives similar values in chromogenic and one-stage aPTT assay [5]. Furthermore, in animal models it has been demonstrated to have comparable haemostatic effect to Advate® [6,7], and in a clinical study it has been demonstrated to be bioequivalent to Advate® [8]. We have used site specific glyco-PEGylation as a means to prolong the half-life of FVIII. The N8-GP is a recombinant human FVIII molecule where a 40-kDa polyethylene glycol (PEG) is attached to a unique O-glycan in the truncated B-domain of turoctocog alfa [9]. When activated by thrombin, the B-domain containing the pegylation is cleaved off, thus generating active FVIIIa which is similar in structure to native FVIIIa [10]. In FVIII-KO mice, N8-GP has shown a prolonged half-life and prolonged duration of effect [6,11].
Dogs with congenital haemophilia A and B have been used extensively to evaluate the pharmacokinetic (PK) and pharmacodynamic (PD) behaviour of human coagulation factors, and studies have shown similarity between the haemophilic dogs and human patients with regard to both PK and PD profiles of coagulation factors and have been highly predictive of the efficacy and safety of replacement therapy in humans [12–16]. Ex vivo clot formation by whole blood clotting time (WBCT) and parameters of clot dynamics measured by thromboelastography (TEG) have been used as surrogate pharmacodynamic efficacy markers. We have recently reported that turoctocog alfa and Advate® exhibited similar PK/PD parameters, with a half-life of 7.7–11 h in haemophilia A dogs [7] which was similar to what was found for these two compounds in humans [8], suggesting that the dog as model also for rFVIII is predictive of what may be expected in a clinical setting.
The objective of this study was to evaluate the pharmacokinetic (PK) and pharmacodynamic (PD) profiles of both turoctocog alfa and N8-GP in haemophilia A dogs, thereby evaluating which dosing frequency may be feasible of both products to keep the WBCT below 20 min, a level that recently has been demonstrated to reduce the frequency of or prevent spontaneous bleeding in the haemophilic dogs [17].
Materials and methods
Study design
The present study was conducted in six haemophilia A (HA) dogs from the haemophilia dog colony at the Francis Owen Blood Research Laboratory (FOBRL), University of North Carolina, Chapel Hill, NC, USA. The protocol was approved by the Institutional Animal Care and Use Committee at the University of North Carolina. The dogs included, have not previously been dosed with human FVIII. All bleeder animals were tested for pre-existing anti-FVIII neutralizing antibodies before being assigned to protocols. Due to antibody formation when dosing human VIII to dogs, it was not possible to design the study as a cross-over study; instead a parallel-group design was used. The weight of the dogs was in the range of 17– 21 kg, the baseline FVIII:C was below 0.01 U mL−1, and the WBCT was above 30 min for all dogs included (Table 1). The dose used was 125 U kg−1 for turoctocog alfa and N8-GP based on the one-stage aPTT assay (CLOT). Blood samples for FVIII concentration (FVIII:C) determined by one-stage aPTT assay one were collected at: 5, 15, 30 min, 1, 2, 3, 4, 6, 8,12, 24, 32, 48, 72, 80, 96, 104, 120, 144 and 168 h after dosing. Both WBCT and TEG evaluations were performed at the same sampling times (except: 15 min, 3, 6, 12 and 168 h after dosing).
Table 1.
Baseline characteristics of dogs dosed with turoctocog alfa and N8-GP. Turoctocog alfa N8-GP
| Turoctocog alfa
|
N8-GP
|
Normal Dogs [7] | |||||
|---|---|---|---|---|---|---|---|
| N04 | N02 | M76 | N32 | M70 | M71 | ||
| Sex | F | M | M | F | M | M | – |
| Weight (kg) | 17.5 | 18.4 | 21.6 | 17 | 19.2 | 20.6 | – |
| FVIII:C (U mL−1) | 0.005 | 0.005 | 0.005 | nd | 0.005 | 0.008 | 0.66–1.2 |
| WBCT (min) | 40 | 35 | 51.5 | 30.5 | 33–39 | 33.5 | 8–12 |
| TEG® | |||||||
| R-time (min) | >60 | >60 | >60 | >60 | >60 | 44 | 6–12 |
| Angle (degree) | na | na | na | na | na | 2.6 | ~68 |
na, not applicable, no signal on angle for deficient dogs; nd, not determined.
Compounds
Turoctocog alfa was available as clinical grade material, produced as previously described [8]. The N8-GP was produced by site-specific glyco-pegylation [9]. Briefly, the unique O-glycan in the truncated B-domain of turoctocog alfa was selected for glyco- PEGylation as the B-domain is released upon thrombin activation to produce FVIIIa with the same primary structure as endogenous FVIIIa. The initial step in the modification process is removal of all sialic acids from the glycans. The process then utilizes the ability of the ST3GalI sialic acid transferease to selectively transfer a single 40 kDa PEG modified sialic acid onto the O-glycan. The resulting product is isolated by a single ion exchange chromatography step which allows for separation of PEGylated and non- PEGylated material as well as removal of enzymes and excess 40 kDa PEG-sialic acid-CMP. All non-sialydated glycans are subsequently capped by treatment with excess CMP-SA and MBP-SBD-ST3GalIII before purification of the final product. The resulting N8-GP displayed high-affinity binding to human von Willebrand factor (VWF) with an apparent Kd in the sub-nM range and a specific activity similar to native FVIII when measured in a chromogenic assay whereas specific activity measured in a one-stage clot assay varied depending on the aPTT reagent used.
Assays
Except for the WBCT and TEG analysis all other assays were done in batch after the infusions and sampling were completed.
WBCT
Whole blood clotting time was performed by a two-tube procedure at 28°C as previously described [7]. Briefly, one ml of whole blood collected with a 1 mL syringe was distributed equally between two siliconized tubes (Vacutainer™; Becton–Dickinson, Franklin Lakes, NJ, USA). The first tube was tilted every 30 s after an initial incubation of 1 min. After formation of the clot, the second tube was tilted and observed every 30 s. The endpoint was the clotting time of the second tube.
TEG
Blood was drawn at each selected time point and tested within 2 min after collection using Haemoscope TEG® 5000 Thromboelastograph Analyser (Haemonetics Corporation, Braintree, MA, USA) according to the manufacturers’ instruction. A 1 mL blood is collected and 360 μL placed in the cup after premixing 1 mL with Kaolin (provided by the manufacturer). The TEG recordings were allowed to proceed for approximately 90 min and the TEG® reaction time (R-time), maximum clot firmness (MA) and angle were determined.
One-stage aPTT assay (FVIII:C)
One-stage aPTT assay was determined using an ACL300R clot analyser (Instrumentation Laboratory, Milan, Italy). On the clot analyser, samples were diluted fivefold in HBS/BSA (20 mM hepes, 150 mM NaCl, pH 7.4 with 1% BSA). Human FVIII-deficient plasma with VWF (Siemens Healthcare Diagnostics Inc., Tarrytown, NY, USA) was used as substrate plasma and SynthA-Sil (Instrumentation Laboratory) as APTT reagent. Specific turoctocog alfa and N8-GP reference material calibrated against WHO Seventh International standard were used as standard when assaying plasma samples containing turoctocog alfa or N8-GP respectively. Standards and samples were prediluted in pooled FVIII-deficient canine plasma (FOBRL, UNC, Chapel Hill, NC, USA) as required, thereby keeping the amount of canine plasma constant in all samples and standards. The specific activity of N8-GP measured in a one-stage aPTT assay was reduced to various degrees depending on the aPTT reagent used. This may be an assay artefact common to PEGylated proteins as PEGylation of non-haemostatic proteins also has been reported to result in prolonged aPTT clotting [18].
Data analysis
The pharmacokinetic parameters for turoctocog alfa and N8-GP were assessed by non-linear mixed-effect modelling using the NONMEM program, version VII (GloboMax/ICON, Ellicott City, MD, USA), and the GNU fortran 95 compiler. The first order conditional (FOCE) were used for the modelling. In the modelling process the pharmacokinetic properties were based on FVIII:C results whereas the pharmacodynamics were only based on WBCT, pharmacokinetic and pharmacodynamic data were modelled simultaneously, all data were log-transformed prior to the modelling procedure. In the first step, one, two and three-compartmental models were tested to evaluate which model best described the FVIII:C plasma concentration vs. time data. The pharmacodynamics part of the model was a direct-effect model, different modifications were tested, e.g. with and without sigmoidicity factor. The goodness of fit was evaluated both comparing the objective function value and by graphical analysis of predicted vs. observed concentrations (distribution of points around the unity line, plots are not included in the manuscript) and by evaluation of observed concentration and the model fit vs. time, all plots were done using the R software package [19]. To describe the intra-individual error, additive, proportional and combined error models were tested. No between subject variability was included in the modelling process due to the relatively low number of dogs included in the study.
Simulations
The main objective of the simulations was to evaluate the differences observed between turoctocog alfa and N8-GP in a multiple dose setting, to better understand the pharmacokinetics and dynamics and also to investigate a feasible dosing interval in preclinical setting. A series of simulations were performed using the model and the estimated parameters. All simulations were performed using the NONMEM software; all plots were performed using the R software. The target of simulation was to keep the WBCT below 20 min as this value will essentially prevent bleeding in the dogs [17].
Results
Six dogs with haemophilia A were included in the study; turoctocog alfa and N8-GP were dosed to three dogs each at a dose of 125 U kg−1. After i.v. administration of turoctocog alfa and N8-GP, concentration plasma vs. time curves were best described by a two-compartmental model for both compounds (Fig. 1). The parameters include the inter-compartment flow rate (Q2), the volumes of the central and the peripheral compartment (V1 and V2), and the clearance (CL). Based on these primary parameters, both the distribution half-life (t½α) and the terminal half-life (t½β) were calculated (Table 2).
Fig. 1.
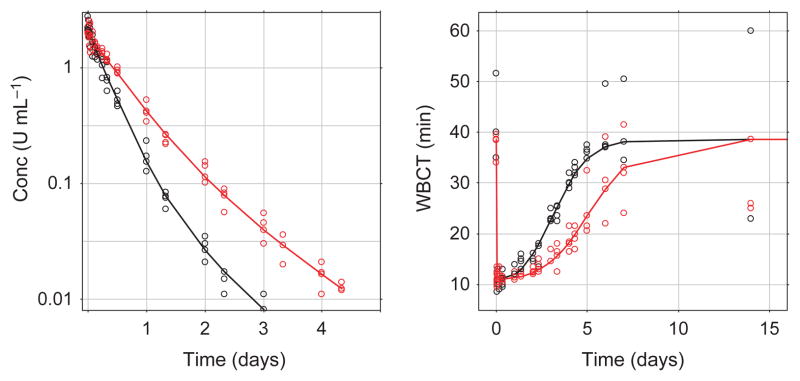
Left panel Plasma concentration based on FVIII:C vs. time, right panel whole blood clotting time (WBCT) vs. time of turoctocog alfa (black) and N8-GP (red); after intravenous administration of 125 U kg−1 to haemophilia dogs (n = 3). Lines represent model fit, open circles the individual observed plasma concentration levels (note the different scale on the x-axis).
Table 2.
Pharmacokinetic parameters based on non-linear mixed effects modelling after intravenous administration of 125 U kg−1 turoctocog alfa or N8-GP to severe haemophilia A dogs.
| Turoctocog alfa
|
N8-GP
|
|||
|---|---|---|---|---|
| Estimate | RSE% | Estimate | RSE% | |
| PK | ||||
| V1 (mL kg−1) | 59 | 7 | 63 | 7 |
| CL (mL h−1 kg−1) | 6.5 | 8 | 3.9 | 4 |
| V2 (mL kg−1) | 13 | 17 | 11 | 6 |
| Q (mL h−1 kg−1) | 0.66 | 10 | 0.43 | 29 |
| t½α (h) | 5.4 | – | 9.1 | – |
| t½β (h) | 16 | – | 22 | – |
| PD | ||||
| BASE (min) | 39 | 5 | 39 | 5 |
| EMAX (na) | 0.72 | 2 | 0.72 | 2 |
| EC50 (U mL−1) | 0.0059 | 15 | 0.0059 | 15 |
| Prop error | ||||
| PK (%) | 43 | 6 | 43 | 6 |
| PD (%) | 39 | 7 | 39 | 7 |
V1, volume of central compartment; CL, total clearance; V2, volume of peripheral compartment; Q, inter-compartmental flow; EMAX, the maximum effect (reduction in baseline levels); EC50, the potency of the compound (corresponding to the plasma concentration with half the maximum effect); BASE, WBCT baseline level; t½α, distribution half-life; t½β, terminal half-life; RSE%, relative standard error.
The PK and PD (WBCT) data were modelled simultaneously for turoctocog alfa and N8-GP. Plasma concentration was assumed to have a direct effect on WBCT using a standard sigmoid relationship:
where WBCT is the WBCT, BASE is the WBCT baseline before dosing, CP is the FVIII:C plasma concentration, EMAX is the maximum effect (defined to be between 0 and 1) and EC50 (potency) represents the plasma concentration that results in half the maximum effect. The applied model fitted the data well (Fig. 1), based on the PD vs. PK plot (Fig. 2), the PD parameters were assumed to be the same for turoctocog alfa and N8-GP. Adding a sigmoidicity factor to the PD relationship did not improve the model fit significantly. The intra-individual variation was best described by proportional error model for both the plasma concentration and for WBCT.
Fig. 2.

Whole blood clotting time (WBCT) plotted against the observed plasma concentration of turoctocog alfa (black) or N8-GP (red) after intravenous administration of 125 U kg−1 to haemophilic dogs (n = 3). Lines represent model fit, open circles the individual observed plasma concentration levels.
After intravenous administration to haemophilic dogs, the plasma concentration (FVIII:C activity) at the first sampling point (5 min) was comparable for turoctocog alfa and N8-GP being 2.35 ± 0.37 and 2.04 ± 0.06 U mL−1 (mean ± SD), the model predicted plasma concentrations to be 2.11 and 1.98 U mL−1 (no between subject variability included in the modelling), over time the plasma concentration levels declined at a slower rate for N8-GP as compared with turoctocog alfa. The clearance was estimated to be 6.5 and 3.9 mL h−1 kg−1 for turoctocog alfa and N8-GP respectively; the difference in clearance was reflected in the terminal half-life which estimated to be 22 h for N8-GP as compared to 16 h for turoctocog alfa (Fig. 1, Table 2). The maximum effect for both compounds was estimated to be 0.72, suggesting that both compounds were able to reduce the WBCT baseline by 72%.
The effect (PD) was monitored by assessment of whole blood clot formation using the WBCT and TEG, the clot time (R-time) and clot development (angle) at different time points (Figs 1 and 3). Similar to the FVIII:C plasma concentration, only limited variation is observed between the dogs, and at the first time point (5 min after dosing) the impaired clot formation was corrected by both turoctocog alfa and N8-GP to ranges obtained in normal dogs (i.e. WBCT for both turoctocog alfa and N8-GP were estimated to 11 min at the first sampling point). For all PD parameters it is clear that dogs dosed with N8-GP returned to baseline at a later time point than the dogs dosed with turoctocog alfa (Figs 1 and 2). To evaluate the PK-PD correlation, the PD parameter WBCT was plotted against the observed plasma concentration (Fig. 2). The plot suggests a direct relationship between PK and PD and in general only minor differences were observed between turoctocog alfa and N8-GP (Fig. 2). Furthermore, based on the PD vs. PK plot, it is evident that the one-stage clotting assay used for measuring the plasma concentration of the FVIII compounds is not sufficiently sensitive to follow the return of the PD parameters to the baseline value (Fig. 2).
Fig. 3.

Left panel R-time (min) vs. time, right panel angle vs. time of turoctocog alfa (black) and N8-GP (red) after intravenous administration of 125 U kg−1 to haemophilia dogs (n = 3). Lines represent mean concentration, open circles the individual observed plasma concentration levels.
Discussion
PEGylation has for long been established as an effective method of changing the clearance and thereby prolonging the half-life of different compounds [15,20–22]. N8-GP is a glyco-PEGylated version of turoctocog alfa where a PEG moiety is specifically attached to a unique O-glycosylation in the B-domain. Activation by thrombin will release the B-domain containing the PEG moiety, thus the activated form of N8-GP will be similar to native FVIIIa.
The pharmacokinetic of N8 was in the present study best described by a two-compartmental model; this is in agreement with previous publications based on clinical data [23,24]. However, the best model will most likely vary with the sampling schedule used in the study, and previously one compartment models have also been used to characterize the pharmacokinetic of VIII [25]. The terminal half-life of turoctocog alfa was estimated to be 16 h (Table 2), this is longer than the 8.9 h previously reported in dogs [7], the observed differences are most likely caused by differences in the methods used (two-compartmental pharmacokinetic analysis vs. non-compartmental analysis). In a clinical setting, the terminal half-life of VIII, based on a two-compartmental model, has previously been reported to be 12 and 19 h [23,24], which is in agreement with what reported in the present study.
The clearance of N8-GP was found to be reduced by 40% as compared to turoctocog alfa, the reduced clearance was also reflected in the terminal half-life of N8-GP, which was estimated to be 22 h which is 1.4-fold more than what observed for turoctocog alfa; the increase in the distribution half-life was 1.7-fold prolonged for N8-GP as compared with turoctocog alfa (9.1 vs. 5.4 h; Table 2). The observed prolongation in half-life is less than what has been reported for other glyco-PEGylated coagulation factors, e.g. for FIX attached to a 40 k PEG (N9-GP) a fivefold increase was observed in the terminal half-life [15], or rFVIIa attached to a 40 kDa PEG (N7-GP), where also an approximately fivefold prolongation in half-life was observed [20]. However, increase in half-life obtained in the present study corresponds with what reported for rFVIII using completely different prolongation principles, such as Fc-fusion [26] or targeted PEGylation to surface-exposed cysteines introduced in rFVIII via mutagenesis [27]. The reasons for not being able to obtain a more than a twofold prolongation in the half-life are not clear, but may be caused by both the complexity (e.g. VWF binding) and the size of FVIII as compared with the other molecules, which prohibit effective filtration in the kidneys, thus, the addition of a PEG group will therefore not impact kidney clearance. So far, it has to the author’s knowledge, not been possible to modify the FVIII protein to obtain a prolongation in the terminal half-life of beyond two.
The effect of both turoctocog alfa and N8-GP was clearly demonstrated on all pharmacodynamic parameters; both compounds were able to normalize both the angle and R-time measured in TEG, and the WBCT (Figs 1 and 2). However, N8-GP was able to normalize the pharmacodynamic parameters for a longer period of time than that observed for turoctocog alfa. By plotting the observed effect on WBCT against the observed plasma concentration (Fig. 2), it is evident that the relationship between turoctocog alfa and N8-GP is almost the same suggesting similar potency and effect of turoctocog alfa and N8-GP; which also has been indicated in other animal studies [11]. The good correlation between the pharmacokinetics and pharmacodynamics of turoctocog alfa and N8-GP is not unexpected as the active form of the two compounds is structurally similar. It should, however, be noted, that the FVIII:C assay is not sufficiently sensitive to allow a comparison at the low values obtained when the baseline levels are reached, in theory there could appear to be differences at lower concentration levels (Fig. 2). Based on these observations, indicating that the potency and the effect of turoctocog alfa and N8-GP is the same, the observed difference in the pharmacodynamic parameters of turoctocog alfa and N8-GP is mainly caused by the difference in the pharmacokinetic characteristics of the two compounds (Figs 1 and 3).
Modelling the pharmacokinetics and pharmacodynamics simultaneously using the suggested model, provided a nice model fit (Fig. 1); the main difference was in the clearance value which was estimated to be 6.5 and 3.9 mL min−1 kg−1 for turoctocog alfa and N8-GP respectively (Table 2). The estimated model parameters were used to perform a series of simulations. To have a target for the simulations, we used a WBCT below 20 min which seems to prevent spontaneous bleeds in the haemophilia A dogs [17]. Based on the simulation of a multiple dose regimen of 75 U kg−1 in dogs, it is clear that turoctocog alfa dosed every 2 or 3 days will be able to maintain WBCT below the target level of 20 min, this seems to be in agreement with the currently used dosing interval in haemophilia A patients [1]. When simulating the N8-GP in a multiple dose setting, the dosing interval in dogs may be expanded to once every fourth day and still maintain the WBCT below 20 min (Fig. 4). Currently no clinical data are available for N8-GP, but present data suggest that the longer half-life of N8-GP may enable a significant prolongation of the current available dosing interval using regular FVIII products, and thereby have the potential of increasing convenience and compliance for the patients.
Fig. 4.
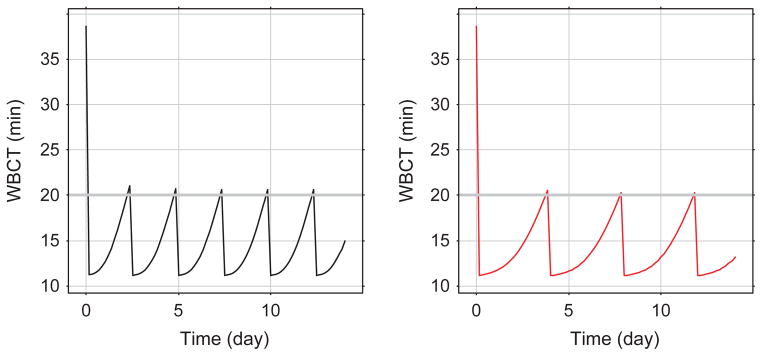
Simulation of whole blood clotting time (WBCT) in a multiple dose setting. Turoctocog alfa dosed every 2½ days is plotted on left panel (black line), N8 N8-GP dosed every fourth day is plotted on right panel (red line) both at a dose of 75 U kg−1. The grey horizontal line on both plots represents the target WBCT of 20 min.
Footnotes
Author’s contribution
HA: wrote manuscript, study design, performed pharmacokinetic analysis, evaluation on results; HRS: study design, evaluation of results, review of manuscript; HP and ENO: performed assays, reviewed manuscript; EPM and NAF: performed animal experiments, reviewed manuscript; TCN: study design, animal experiments, reviewed manuscript; ME: wrote manuscript, study design, evaluated results.
Disclosures
HA, HRS, HP, ENO and ME are all employed by Novo Nordisk A/S. EPM, NAF and TCN received support from Novo Nordisk A/S to perform the animal studies.
References
- 1.Blanchette VS. Prophylaxis in the haemophilia population. Haemophilia. 2010;16 (Suppl 5):181–8. doi: 10.1111/j.1365-2516.2010.02318.x. [DOI] [PubMed] [Google Scholar]
- 2.Manco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357:535–44. doi: 10.1056/NEJMoa067659. [DOI] [PubMed] [Google Scholar]
- 3.Saenko EL, Pipe SW. Strategies towards a longer acting factor VIII. Haemophilia. 2006;12(Suppl 3):42–51. doi: 10.1111/j.1365-2516.2006.01260.x. [DOI] [PubMed] [Google Scholar]
- 4.Thim L, Vandahl B, Karlsson J, et al. Purification and characterization of a new recombinant factor VIII (N8) Haemophilia. 2010;16:349–59. doi: 10.1111/j.1365-2516.2009.02135.x. [DOI] [PubMed] [Google Scholar]
- 5.Viuff D, Barrowcliffe T, Saugstrup T, Ezban M, Lillicrap D. International comparative field study of N8 evaluating factor VIII assay performance. Haemophilia. 2011;17:695–702. doi: 10.1111/j.1365-2516.2010.02481.x. [DOI] [PubMed] [Google Scholar]
- 6.Elm T, Karpf DM, Ovlisen K, et al. Pharmacokinetics and pharmacodynamics of a new recombinant FVIII (N8) in haemophilia A mice. Haemophilia. 2012;18:139–45. doi: 10.1111/j.1365-2516.2011.02608.x. [DOI] [PubMed] [Google Scholar]
- 7.Karpf DM, Kjalke M, Thim L, et al. Pharmacokinetics and ex vivo whole blood clot formation of a new recombinant FVIII (N8) in haemophilia A dogs. Haemophilia. 2011;17:e963–8. doi: 10.1111/j.1365-2516.2011.02580.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martinowitz U, Bjerre J, Brand B, et al. Bioequivalence between two serum-free recombinant factor VIII preparations (N8 and ADVATE®) – an open-label, sequential dosing pharmacokinetic study in patients with severe haemophilia A. Haemophilia. 2011;17:854–9. doi: 10.1111/j.1365-2516.2011.02495.x. [DOI] [PubMed] [Google Scholar]
- 9.Stennicke HR, Kjalke M, Karpf D, et al. A Novel B-Domain O-glycoPEGylated FVIII (N8-GP) Demonstrates Full Efficacy and Prolonged Effect in Hemophilic Mice Models. 2012 doi: 10.1182/blood-2012-01-407494. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stennicke R, Kjalke M, Moeller F, Johansen P, Oevli Sen K, Karpf D. Design of a novel long-acting recombinant coagulation factor VIII. Haemophilia. 2010;16(Suppl 4):40. [Google Scholar]
- 11.Elm T, Ezban M, Tranholm M, Stennicke H, Holmberg H. Glycopegylated rFVIII (N8-GP) has Prolonged Haemostatic Effect in Haemophilia A Mice. J Thromb Haemost. 2011;9(Suppl 2):116–116. doi: 10.1111/j.1538-7836.2011.04252.x. Special Issue: SI. [DOI] [PubMed] [Google Scholar]
- 12.Agersø H, Kristensen NR, Østergaard H, et al. Clearance of rFVIIa and NN1731 after intravenous administration to Beagle dogs. Eur J Pharm Sci. 2011;42:578–83. doi: 10.1016/j.ejps.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 13.Knudsen LB, Nielsen PF, Huusfeldt PO, et al. Potent derivatives of glucagon-like peptide-1 with pharmacokinetic properties suitable for once daily administration. J Med Chem. 2000;43:1664–9. doi: 10.1021/jm9909645. [DOI] [PubMed] [Google Scholar]
- 14.McCarthy K, Stewart P, Sigman J, et al. Pharmacokinetics of recombinant factor IX after intravenous and subcutaneous administration in dogs and cynomolgus monkeys. Thromb Haemost. 2002;87:824–30. [PubMed] [Google Scholar]
- 15.Østergaard H, Bjelke JR, Hansen L, et al. Prolonged half-life and preserved enzymatic properties of factor IX selectively PEGylated on native N-glycans in the activation peptide. Blood. 2011;118:2333–41. doi: 10.1182/blood-2011-02-336172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schaub R, Garzone P, Bouchard P, et al. Preclinical studies of recombinant factor IX. Semin Hematol. 1998;35(2 Suppl 2):28–32. [PubMed] [Google Scholar]
- 17.Sabatino DE, Lange AM, Altynova ES, et al. Efficacy and safety of long-term prophylaxis in severe hemophilia A dogs following liver gene therapy using AAV vectors. Mol Ther. 2011;19:442–9. doi: 10.1038/mt.2010.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smolen J, Landewe RB, Mease P, et al. Efficacy and safety of certolizumab pegol plus methotrexate in active rheumatoid arthritis: the RAPID 2 study. A randomised controlled trial. Ann Rheum Dis. 2009;68:797–804. doi: 10.1136/ard.2008.101659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.R Development Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; 2009. Available at: http://www.R-project.org. [Google Scholar]
- 20.Holmberg H, Elm T, Karpf D, et al. GlycoPEGylated rFVIIa (N7-GP) has a prolonged hemostatic effect in hemophilic mice compared with rFVIIa. J Thromb Haemost. 2011;9:1070–2. doi: 10.1111/j.1538-7836.2011.04252.x. [DOI] [PubMed] [Google Scholar]
- 21.Harris JM, Martin NE, Modi M. Pegylation: a novel process for modifying pharmacokinetics. Clin Pharmacokinet. 2001;40:539–51. doi: 10.2165/00003088-200140070-00005. [DOI] [PubMed] [Google Scholar]
- 22.Harris JM, Chess RB. Effect of pegylation on pharmaceuticals. Nat Rev Drug Discov. 2003;2:214–21. doi: 10.1038/nrd1033. [DOI] [PubMed] [Google Scholar]
- 23.Bjorkman S, Folkesson A, Berntorp E. In vivo recovery of factor VIII and factor IX: intra- and interindividual variance in a clinical setting. Haemophilia. 2007;13:2–8. doi: 10.1111/j.1365-2516.2006.01401.x. [DOI] [PubMed] [Google Scholar]
- 24.Bolon-Larger M, Chamouard V, Bressolle F, Boulieu R. A limited sampling strategy for estimating individual pharmacokinetic parameters of coagulation factor VIII in patients with hemophilia A. Ther Drug Monit. 2007;29:20–6. doi: 10.1097/FTD.0b013e3180311384. [DOI] [PubMed] [Google Scholar]
- 25.Karafoulidou A, Suarez E, Anastasopoulou I, et al. Population pharmacokinetics of recombinant factor VIII:C (ReFacto) in adult HIV-negative and HIV-positive haemophilia patients. Eur J Clin Pharmacol. 2009;65:1121–30. doi: 10.1007/s00228-009-0699-3. [DOI] [PubMed] [Google Scholar]
- 26.Dumont JA, Kamphaus GD, Fraley C, et al. Factor VIII-Fc fusion protein shows extended half-life and hemostatic activity in hemophilia A dogs. ASH. 2009;119:3024– 30. doi: 10.1182/blood-2011-08-367813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mei B, Pan C, Jiang H, et al. Rational design of a fully active, long-acting PEGylated factor VIII for hemophilia A treatment. Blood. 2010;116:270–9. doi: 10.1182/blood-2009-11-254755. [DOI] [PubMed] [Google Scholar]