

Abstract
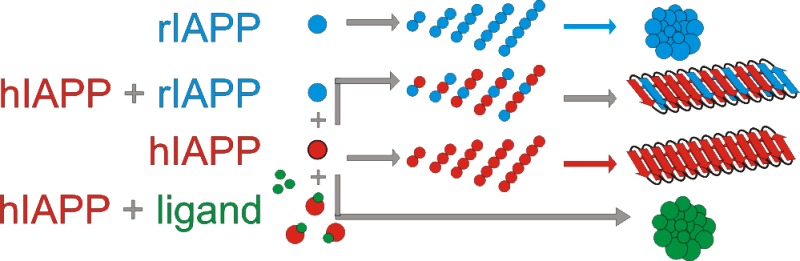
The molecular mechanisms by which different proteins assemble into highly ordered fibrillar deposits and cause disease remain topics of debate. Human amylin (also known as islet amyloid polypeptide/hIAPP) is found in vivo as amyloid deposits in the pancreatic islets of sufferers of type II diabetes mellitus, and its self-aggregation is thought to be a pathogenic factor in disease and to contribute to the failure of islet transplants. Here, electrospray ionization-ion mobility spectrometry-mass spectrometry (ESI-IMS-MS) has been used to monitor oligomer formation from IAPP. The detection, identification and characterization of oligomers from both human and rat amylin (rIAPP) are described. Oligomers up to and including hexamers have been detected for both peptides. From ESI-IMS-MS derived collision cross sections (CCS), these species are shown to be elongated in conformation. Collision-induced dissociation (CID-MS/MS) revealed differences in the gas-phase stability of the oligomers formed from hIAPP and rIAPP, which may contribute to their differences in amyloid propensity. Using ESI-IMS-MS, the mode of inhibition of amyloid formation from hIAPP using small molecules or co-incubation with rIAPP was also investigated. We show that the polyphenolic compounds epigallocatechin gallate (EGCG) and silibinin bind to specific conformers within a dynamic ensemble of hIAPP monomers, altering the progress of oligomerization and fibril assembly. Hetero-oligomer formation also occurs with rIAPP but leads only to inefficient inhibition. The results indicate that although different small molecules can be effective inhibitors of hIAPP self-assembly, their modes of action are distinct and can be distinguished using ESI-IMS-MS.
Introduction
Amyloid disorders are characterized by the aberrant aggregation of proteins or peptides into amyloid fibrils.1 In each case, normally soluble proteins or peptides that may be folded, partially folded, or intrinsically disordered embark on alternative aggregation energy landscapes2 leading to the formation of β-sheet-rich fibrillar assemblies that can be characterized by the binding of dyes such as Congo red or thioflavin T (ThT).3,4 The identity of the toxic species associated with amyloid diseases is widely debated as a result of the difficulty of separating, identifying, and individually characterizing these heterogeneous and transient intermediates of the assembly process.
Human islet amyloid polypeptide (hIAPP), also known as amylin, is a highly amyloidogenic 37-residue peptide hormone produced by the β-cells of the pancreas. It is produced, stored, and co-secreted with insulin and plays a role in the control of gastric emptying, glucose homeostasis, and suppression of glucagon release.5,6 In its monomeric state, hIAPP is a soluble, intrinsically disordered polypeptide but forms islet amyloid in cases of type-2 diabetes mellitus (T2DM).5,7 Islet amyloid formation leads to β-cell dysfunction, death, and reduction in β-cell mass8,9 and contributes to the failure of islet cell transplantation.5 Amyloid formation by IAPP is highly sequence-specific.10 hIAPP forms amyloid readily at neutral pH, while rat IAPP (rIAPP) does not, despite differing in sequence at only six out of 37 amino acid positions (Figure 1a). Significantly, five of these amino acid substitutions are located within residues 20–29, three of which are Pro residues in rIAPP, leading to supposed disruption of secondary structure formation.11 Despite numerous studies on the conformational properties, membrane binding, and aggregation of IAPP,5,10,12 important challenges remain in revealing the mechanism of amyloid formation of hIAPP, particularly in the characterization of oligomeric intermediates, which would enable detailed studies of the mechanisms of assembly and the effects of known inhibitors on the aggregation process.13,14
Figure 1.
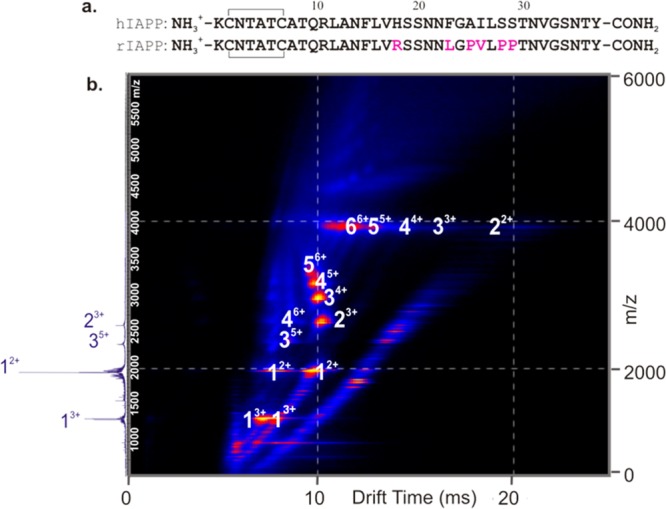
hIAPP forms an array of oligomeric species during fibril formation. (a) Comparison of hIAPP and rIAPP sequences. Both peptides have a disulfide bridge between Cys-2 and Cys-7 and have an amidated C-terminus. Residues that differ from those of the human peptide are colored pink in the rat sequence. (b) ESI-IMS-MS driftscope plot of the hIAPP oligomers present 2 min after diluting the monomer to a final peptide concentration of 50 μM in 20 mM ammonium acetate, pH 6.8, 37 °C, 600 rpm. ESI-IMS-MS driftscope plots show IMS drift time versus m/z versus intensity (z = square root scale), and the corresponding mass spectrum is shown on the left-hand side. Numbers adjacent to peaks denote oligomer order, with the positive charge state of each oligomer ions in superscript. The ESI mass spectrum shows the 2+ and 3+ charge state ions of hIAPP monomer (labeled 1) and minor amounts of dimer and trimer (labeled 2 and 3, respectively).
Most conventional biophysical techniques used in the study of amyloid systems, including CD, FTIR spectroscopy, and fluorescence-based assays, are limited to providing data relating to a global average of species within heterogeneous mixtures. Previous analytical ultracentrifugation studies,15 conducted at pH 4.9 where aggregation is very slow, and 19F NMR studies16 have failed to detect low order oligomeric species for hIAPP, possibly due to the low population, or heterogeneous and/or transient nature of such species. By contrast, photoinduced cross-linking has identified oligomeric states, including monomer through hexamer.17 Ion mobility spectrometry-mass spectrometry (IMS-MS) has the unique advantage of being capable of resolving complex mixtures of species present in solution, including transiently populated states and even isobaric species without requiring their prior separation.18−21 IMS-MS has been utilized previously to provide insights into the oligomerization pathways of other intrinsically unstructured amyloid-related peptides and proteins including amyloid-β (Aβ) peptide20,22 and α-synuclein,21,23 which are associated with Alzheimer’s disease and Parkinson’s disease, respectively. In the case of Aβ40 and Aβ42, oligomeric species up to and including 16-mer and 12-mer, respectively, have been identified, and their rotationally averaged collision cross-sectional areas (CCS) were measured using either positive or negative ion IMS-MS.20,22 For α-synuclein, the conformeric states of monomers were determined and the pH dependence of dimer formation was demonstrated.23 IMS-MS has also provided structural insights into the hIAPP monomer24 and dimer,25 as well as the self-assembly of a series of short synthetic peptides derived from an amyloidogenic sequence (20–29) of hIAPP.26 Using IMS-MS and replica-exchange molecular dynamics (REMD) simulations, Dupuis et al. proposed that full-length monomeric hIAPP occupies both a helix–coil conformation and an extended β-hairpin conformation that has a significantly (18%) larger collision cross-section (CCS).24 These extended hairpin structures were not observed for rIAPP, leading to the conclusion that this conformer is an amyloidogenic precursor. Dupuis et al. also investigated dimeric structures of hIAPP, using IMS-MS, reporting that the dimers observed are significantly more extended than those formed by rIAPP, suggestive of a higher percentage of β-sheet content that is formed from extended β-hairpin-containing monomers.25 An early conformational transition to β-sheet-rich conformers was proposed, therefore, as a first step in hIAPP self-assembly, by contrast with other reports that have suggested a transition to amyloid via helix-rich oligomers.27−29 None of these studies, however, reported the detection or identification of higher order species.
A wealth of studies has assessed the ability of small molecules to interfere with the progress of fibril formation from many amyloid-prone peptides and proteins in vitro.29−31 Despite these analyses, the precise mechanisms of inhibition remain elusive. Currently, there are no clinically approved inhibitors of hIAPP assembly. In addition, the mode of action of small molecule inhibitors of hIAPP aggregation is not well understood. Electrospray ionization (ESI)-IMS-MS has been utilized previously to identify inhibitors of β-2-microglobulin (β2m) amyloid formation.32 Using a similar approach, we aimed here to detect, identify, and characterize oligomeric intermediates of hIAPP aassembly and, thereby, to determine the mode of action of two known small molecule inhibitors of hIAPP amyloid formation, epigallocatechin gallate (EGCG)33 and silibinin.34 In addition, we used ESI-IMS-MS to investigate the moderately effective inhibition caused by mixing hIAPP and rIAPP.11 EGCG is one of the most effective amyloid inhibitors known35 and has generated considerable attention given its ability to inhibit or modulate amyloid formation from a diverse range of polypeptides. Despite the plethora of these studies33,35−40 and the interest they have generated, the mode of action of EGCG is not well understood. It is thought to direct some, but not all, amyloidogenic proteins into off-pathway aggregates.33,35
Here, using ESI-IMS-MS, we demonstrate the presence of high order oligomers formed by both hIAPP and rIAPP and use this technique to determine the populations, shapes, and gas-phase stabilities of these co-populated, transient species. The results reveal significant differences in the stability of otherwise similarly organized oligomers of these peptides. In addition, the binding of the small molecule inhibitors, EGCG and silibinin, to specific conformers within the dynamic ensemble of hIAPP species has also been ascertained, and the progress of oligomer formation and fibril assembly and disassembly was monitored. The results indicate that ligand binding either arrests oligomerization (silibinin) or diverts hIAPP onto new assembly routes resulting in the formation of amorphous aggregates without a buildup of oligomeric species (EGCG). The results presented provide evidence for the mode of action of these two effective small molecule inhibitors of hIAPP amyloid formation and reveal why rIAPP is such a poor inhibitor of hIAPP assembly.
Experimental Section
Sample Preparation for MS
hIAPP and rIAPP were synthesized and purified as described previously.41 Lyophilized peptide samples were dissolved in dimethyl sulfoxide (DMSO) at a final concentration of peptide of 5 mM. After 24 h of incubation at 25 °C, stock solutions were diluted 100-fold into 20 mM ammonium acetate, pH 6.8, to a final peptide concentration of 50 μM for MS analysis. The final concentration of DMSO was 1% (v/v). All samples were incubated at 37 °C in 96-well plates with or without agitation (600 rpm). Experiments were repeated at a range of IAPP concentrations, from 10 μM to 100 μM. As expected, there is a concentration-dependence of the rate of fibril formation and oligomer consumption as measured using ESI-IMS-MS. However, the oligomers formed are of the same mass and CCS under each condition.
ESI-(IMS)-MS Analysis
A Synapt HDMS quadrupole time-of-flight mass spectrometer (Micromass UK Ltd., Waters Corpn., Manchester, U.K.), equipped with a Triversa (Advion Biosciences, Ithaca, NY, U.S.) automated nano-ESI interface, was used for these analyses. Positive ion mode was chosen because IAPP has a pI of >9 and therefore should be predominantly positively charged at near-neutral pH. The instrument has a traveling-wave IMS device situated in-between the quadrupole and the time-of-flight analyzers and has been described in detail elsewhere.42 Samples were analyzed by positive ionization nanoESI (nESI) with a capillary voltage of 1.7 kV and a nitrogen nebulizing gas pressure of 0.8 psi. The following instrumental parameters were used: cone voltage 30–100 V; source temperature 60 °C; backing pressure 4.0 mBar; ramped traveling wave height 7–20 V; traveling wave speed 400 m/s; IMS nitrogen gas flow 20 mL/min; IMS cell pressure 0.55 mBar. The cone voltage was optimized to transmit the higher order peptide oligomers; the CCS of the oligomers did not alter over the range 30–100 V. Data were processed by use of MassLynx v4.1 and Driftscope software supplied with the mass spectrometer. The m/z scale was calibrated with aq CsI cluster ions. CCSs were estimated by use of an IMS-MS calibration.43 Calibration of the drift time cross-section function was achieved by analysis of the denatured proteins equine cytochrome c and horse heart myoglobin (10 μM in 50:40:10, v/v/v, acetonitrile/water/acetic acid44), whose CCS values had been predetermined elsewhere by use of conventional ion mobility measurements.44
The collision cross-sectional areas (Ω) of the analytes were then obtained after calibration from their corrected drift times according to eq 1:43
| 1 |
This step also includes an adjustment for the mass and charge of the protein ions, where Ω is the calibrated collision cross-section, A is the calibration determined constant, tD′ is the absolute drift time (corrected), z is the charge state of the ion, mion is the mass of the ion, and mgas is the mass of the target gas used in the IMS cell. The exponential factor B is determined experimentally.43
Collision induced dissociation (CID) MS/MS experiments were performed using the quadrupole analyzer to select isobaric m/z ions representing the dimer and tetramer, separating these ions in the IMS device and performing CID in the transfer collision cell prior to the time-of-flight analyzer (the pressure in the collision cell was kept constant). Increasing collision energy was applied to the transfer collision cell in 10 V increments from 10 to 100 V until the oligomers ions were completely dissociated into monomer ions. CID energies are displayed as center of mass energy (eV) by use of the eq 2:
| 2 |
where z is the charge state of the ion, EMS is the collision energy used (eV), Margon is the mass of argon the collision gas, and Mion is the mass of the analyte of interest. Note that the equation is missing a multiplication factor of e, elementary charge (1.602 × 10–19 C). This has been omitted as conversion of the zeVMS from Joules to electron volts is facilitated by division of e.
Fibril Depolymerization
A mixed sample containing a 1:1 molar ratio of hIAPP:rIAPP was prepared by diluting 5 mM stock solutions of each peptide in DMSO 100-fold into 20 mM ammonium acetate, pH 6.8, to a final concentration of each peptide of 50 μM in 1% (v/v) DMSO for MS analysis. After 5 days of incubation at 37 °C and 600 rpm, mixed samples were centrifuged in a Beckman ultracentrifuge at 300,000g for 45 min. Fibrillar samples in the pellet were depolymerized by incubation in 100% (v/v) HFIP for 24 h. Samples were air-dried and then redissolved in 50:40:10 (v/v/v) acetonitrile/water/acetic acid, and fibril constituent peptides were identified by ESI-MS.
Analysis of Ligand Binding to Monomeric hIAPP
hIAPP (50 μM) was dissolved in 20 mM ammonium acetate (pH 6.8) containing 5, 50, or 500 μM EGCG or silibinin in 1% (v/v) DMSO. For analysis of these samples by nESI-MS, a sampling cone voltage of 30 V was used to preserve protein–ligand interactions, and a backing pressure of 3.0 mbar was applied. Data were acquired over the range m/z 400–8,000. For IMS-MS experiments, the wave height was ramped from 7 to 20 V at a speed of 300 m/s.
Thioflavin T Fluorescence Assays
Samples were prepared in a 96-well plate (Corning Costar 3915, Corning Life Sciences, Amsterdam, The Netherlands) sealed with clear sealing film and were incubated in a FLUOstar OPTIMA plate reader (BMG Labtech, Aylesbury, Bucks, U.K.) for 5 days at 37 °C with or without agitation (600 rpm). Samples had a volume of 100 μL containing 100 μM ThT and 50 μM peptide in 20 mM ammonium acetate, pH 6.8 and a 1% (v/v) final concentration of DMSO. Fluorescence was excited at 440 nm, and emission intensity was measured at 485 nm. Turbidity measurements were performed by monitoring the apparent absorbance at 635 nm using a NEPHALOstar Galaxy reader (BMG Labtech, Aylesbury, Bucks, UK) and 96-well clear plates sealed with clear sealing film. For these experiments, the samples were prepared as described above, except that ThT was not included. In each experiment readings were taken every 5 min, from 2 min to 50 h. In parallel, samples were analyzed at different time points (but in the absence of ThT) using ESI-IMS-MS. Each experiment under each condition was repeated a minimum of three times with at least triplicate measurements for each condition, and representative traces are shown.
Transmission Electron Microscopy (TEM)
The TEM images of each 50 μM peptide solution were acquired after 5 days of incubation at 37 °C on a CM10 microscope (Phillips, Electron Optics, Amsterdam, The Netherlands). Carbon grids were prepared by irradiating under UV light for 30 min and stained with 4% (w/v) uranyl acetate solution as described previously.45
Results
hIAPP Forms an Array of Oligomers Early in Amyloid Assembly
To understand the aggregation process of amyloid-prone peptides and proteins, oligomeric intermediates need to be identified and characterized in detail. Here, by exploiting the separative and investigative powers of MS coupled with IMS and CID-MS/MS, we describe higher order oligomeric states populated by hIAPP and rIAPP and elucidate their CCS, growth mechanism, and relative gas-phase stability. The rat and human peptides were each dissolved in 100% DMSO to remove any preformed aggregates46 and diluted 100-fold into 20 mM ammonium acetate, pH 6.8, and the distribution of oligomeric species was analyzed immediately and at various time points after dilution, using ESI-IMS-MS. The data obtained (Figure 1b) showed that oligomers up to and including hexamers are formed within 2 min of dilution of hIAPP into buffer. ESI-IMS-MS allows co-populated oligomeric ions with the same m/z to be separated and identified individually (e.g., dimer3+ and tetramer6+) (Figure 1b). Multiple charge states, predominantly doubly and triply charged, and different conformers, both compact and expanded for species of the same mass, are observed for the hIAPP monomer at this pH. For example, the monomer 2+ ions occupy two distinct peaks in the IMS-MS spectrum (Figure 1b) with drift times of 7.6 and 10.6 ms. These represent two conformations of this charge state, which have CCSs that differ by a significant ∼15%. Although less well-resolved, the monomer 3+ ions also exhibit two distinct conformations, having drift times of 6.0 and 8.0 ms, a ∼14% difference in their CCS (Figure 1b). Oligomers of hIAPP also populate a range of charge states and conformations, for each of which the CCS was determined (Supporting Information, Table 1).
Previous studies have shown that increased ionic strength increases the rate of aggregation of many proteins and peptides, including hIAPP.47 To investigate whether the presence of oligomers of hIAPP correlates with the ability of the peptide to assemble into amyloid fibrils, the ionic-strength dependency of the oligomers of hIAPP observed using ESI-IMS-MS was investigated and compared with the rate of fibril formation, measured using ThT fluorescence. Under the conditions employed, an enhanced rate of amyloid formation is observed as the ionic strength is increased from 20 to 500 mM, consistent with previous studies47 (Figure 2a). Gross fibril morphology was unchanged with the buffer conditions employed (a representative TEM image is shown (Figure 2b)). A more rapid decrease in oligomer signal intensity over time was also detected with increasing ionic strength (using ESI-IMS-MS, Figure 2c), consistent with the oligomers observed being involved in assembly into amyloid.
Figure 2.

Dependence of hIAPP oligomer and fibril formation on ionic strength. (a) ThT fluorescence intensity of hIAPP (50 μM peptide, 37 °C, 600 rpm) in 20 mM (squares/purple), 100 mM (triangles/orange), or 500 mM (circles/green) ammonium acetate buffer, pH 6.8. (b) Representative negative stain TEM image of hIAPP fibrils after 5 days in 100 mM buffer (37 °C, 600 rpm) (scale bar = 100 nm). (c) ESI-IMS-MS driftscope plots of hIAPP oligomers present at t = 2 min (left-hand side) and t = 5 h (right-hand side) at different ionic strengths (20, 100, and 500 mM). (d) CCSs of hIAPP oligomers measured using ESI-IMS-MS plotted versus oligomer order showing that oligomers have the same CCS regardless of ionic strength: 20 mM (squares), 100 mM (triangles), and 500 mM (circles). The orange dashed line represents a globular fit based on the average density of a protein (0.44 Da/Å3),49 the purple dashed line represents a linear growth model,48 and the green dashed line represents an isotropic growth model.48
To probe the properties of the oligomers formed from hIAPP in more detail, their CCSs were estimated from the ESI-IMS-MS arrival time distributions and compared with CCSs estimated for theoretical models including a fit assuming isotropic growth,48 a globular fit based on the average density of a protein under similar conditions (0.44 Da/Å3),49,50 and a model that assumes growth in a single dimension48 (note that more detailed molecular models cannot be generated for the underlying structures of the oligomers detected, given the lack of information about the type, location, or extent of secondary structure in these species). CCS determination (Experimental Section) suggests that hIAPP oligomers ≥4-mer in size adopt an extended conformation and that the CCS of the different oligomers observed are independent of ionic strength (Figure 2d). However, the relative population of compact vs expanded monomeric and dimeric conformers is altered with increasing ionic strength (discussed below).
CID Reveals Differences in Gas-Phase Stability between hIAPP and rIAPP Oligomers and Monomers
For rIAPP, which does not form ordered amyloid fibrils when incubated at near neutral pH,5 a surprisingly similar array of oligomers was observed using ESI-IMS-MS compared with that detected for hIAPP (Figure 3a, Supporting Information Figures S1a and S2). Akin to the results observed for hIAPP, multiple conformers are observed for rIAPP monomer and oligomers, albeit at different relative intensities compared with those observed for hIAPP. Accordingly, hIAPP consistently occupies a greater proportion of more expanded conformers than rIAPP (Supporting Information Figure S1b). To investigate the relative stabilities of the different monomeric conformers of hIAPP and rIAPP, the dependence of the ion arrival time distribution versus increasing the trap energy (used to effect CID) was examined (Supporting Information, Figure S1c). The results of these experiments showed that hIAPP monomer 3+ ions unfold at lower trap collision energies than those required for the rIAPP monomer, with hIAPP more readily converting to expanded conformers at lower trap voltages (Supporting Information Figure S1c).
Figure 3.
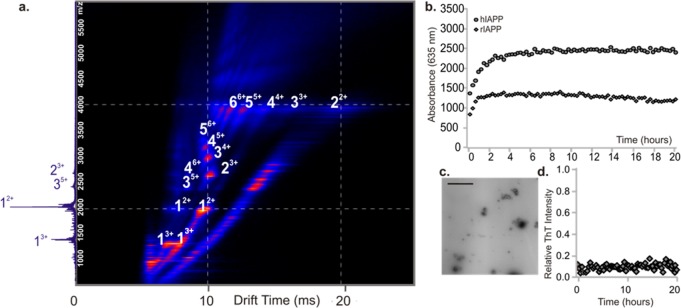
Oligomers formed from rIAPP resemble those of hIAPP. (a) ESI-IMS-MS driftscope plot of rIAPP oligomers present at 2 min after dilution into 20 mM ammonium acetate buffer, pH 6.8 to a final peptide concentration of 50 μM. The number adjacent to each peak denotes oligomer order with charge state of the oligomer in superscript. (b) Aggregation of rIAPP (diamonds) and hIAPP (circles) monitored using turbidity at 635 nm. In both cases, 50 μM peptide was incubated in 20 mM ammonium acetate buffer, pH 6.8 (37 °C, 600 rpm). (c) Negative stain TEM image of rIAPP aggregates after 5 days of incubation (37 °C, 600 rpm); scale bar = 100 nm. (d) ThT fluorescence intensity of rIAPP (50 μM peptide, 37 °C, pH 6.8, 600 rpm). The data are normalized to the signal intensity of a hIAPP fibril formation end point at the same peptide concentration.
Although rIAPP has been reported previously not to form amyloid fibrils,5 turbidity measurements (Figure 3b) and TEM images (Figure 3c) showed that under the conditions employed here (20 mM ammonium acetate, pH 6.8, 1% DMSO (v/v)), rIAPP forms small globular aggregates that scatter light but do not bind ThT (Figure 3d). The rIAPP oligomers observed are similar in CCS, as well as order and mass, to those of hIAPP (Supporting Information Table S1 and Figures S1a and S3). These results indicate, therefore, that the presence of oligomers of similar numbers of subunits and CCS cannot account for the very different amyloidogenic properties of the two sequences.
Next, the gas-phase stabilities of oligomers of hIAPP and rIAPP were probed to investigate whether their gas-phase dissociation can be related to the differences in the ability of these sequences to form amyloid. These oligomers are not amenable to solution-phase stability assays, since they are so lowly populated and co-populated with each other and the monomeric species. Accordingly, CID-MS/MS was utilized to provide a side-by-side comparison of the gas-phase stabilities of the two peptides in different oligomeric forms. In this experiment, ions of specific m/z were selected by the quadrupole analyzer, followed by IMS separation of the different oligomers contributing to this m/z and then sequential fragmentation of the oligomer ions in the transfer collision cell immediately prior to the time-of-flight analyzer. The data revealed that hIAPP dimers are significantly less stable than rIAPP dimers in the gas phase. hIAPP dimers were found to dissociate into monomer at an energy (0.4 eV) that is lower than that required for the equivalent rIAPP dimers, which only began to dissociate at 0.7 eV (Figure 4a). Oligomeric species, including tetramer ions with six charges, also showed subtle differences in stability for the two sequences. The hIAPP tetramer6+ ions begin to dissociate at 0.3 eV, whereas rat tetramer6+ ions remain fully associated until an energy of 0.5 eV is applied (Supporting Information Figure S4a,b). The significant difference in the gas-phase stability of the dimer ions of hIAPP and rIAPP could be related to the capability of hIAPP to access amyloidogenic conformations more easily than its rIAPP counterpart. Consistent with this view, the expanded conformers of the hIAPP monomer and dimer ions became increased in population, and the dimer 3+ ions become less stable in the gas phase as the ionic strength of the buffer used is increased (Figure 4b). This supports the hypothesis that peptide gas-phase stability is inversely correlated with the amyloid competence of IAPP, given that both the hIAPP monomer and its oligomers are less stable than their counterparts for the rat sequence and that high ionic strength destabilizes oligomers and increases the rate of amyloid fibril formation for hIAPP.
Figure 4.
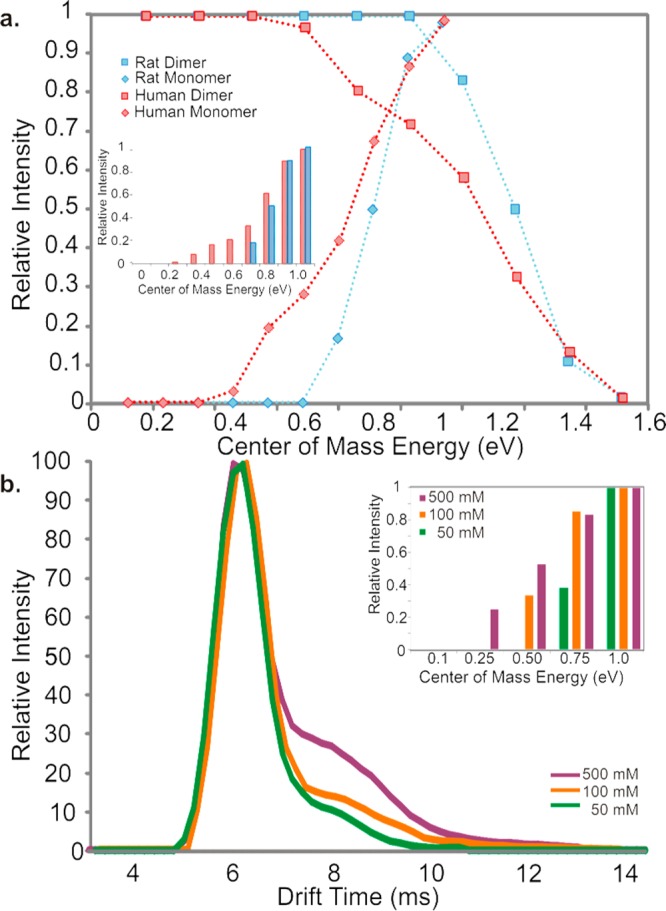
Differences between rIAPP and hIAPP dimer and monomer stabilities in the gas phase measured using collision induced dissociation (CID). (a) CID MS/MS of rIAPP (blue) and hIAPP (red) dimers (50 μM peptide, 20 mM ammonium acetate buffer, pH 6.8). Relative intensity of the 3+ dimer ions (squares) of each peptide is plotted versus increasing ion-accelerating voltage into the transfer T-wave collision cell (CID). Monomer ion intensity (diamonds) increases as dimers (squares) dissociate. Bar chart (inset) showing the appearance of hIAPP (red) and rIAPP (blue) monomer from dissociation of dimer ions with increasing CID voltage. (b) Arrival time distributions (ATDs) of 3+ hIAPP monomer ions 2 min after dissolving into 50 mM (purple), 100 mM (orange), or 500 mM (green) ammonium acetate, pH 6.8. Bar chart (inset) showing relative intensity of monomer ions with increasing ion-accelerating voltage at different ionic strengths: 50 mM (green), 100 mM (orange), and 500 mM (purple) (25 μM peptide, 50/100/500 mM ammonium acetate buffer, pH 6.8).
Probing the Mechanism of Inhibition of hIAPP Fibril Formation with Small Molecules
Having identified the prefibrillar species populated by hIAPP, the mode of action of two known small molecule inhibitors of hIAPP fibril formation was investigated using ESI-IMS-MS. In these experiments, hIAPP (50 μM) was incubated at 37 °C in 20 mM ammonium acetate buffer, pH 6.8, in the presence of EGCG33,35 or silibinin34 at molar ratios of small molecule:hIAPP of 0.1:1, 1:1, or 10:1. The oligomer populations were characterized subsequently using ESI-IMS-MS. To complement the gas-phase analyses, fibril formation was monitored by ThT fluorescence, and peptide aggregates were characterized using negative stain EM. The primary objectives here were to determine how the presence (or binding) of each small molecule affects the distribution of monomeric conformers and populations of oligomers and whether such changes can be correlated with the inhibition of hIAPP amyloid formation.
1. Action of EGCG on Fibril Formation of hIAPP Probed by ESI-IMS-MS
EGCG, a polyphenol flavanol found in green tea, has shown promise as a generic anti-amyloid agent as this small molecule has been found to redirect aggregating proteins onto alternative pathways35 or to maintain them in a native-like state.37 It has also been shown to promote remodeling of mature amyloid fibrils,33 with recent work of Kelly et al. demonstrating that hydrophobic binding to the amyloid fibril by EGCG is a significant step in this process.40 Despite being proposed as a potent inhibitor of amyloid formation for several proteins and peptides including hIAPP,36 α-synuclein, and Aβ42,35 the mode of action of EGCG remains elusive. To identify the effects of EGCG on hIAPP aggregation, the binding capabilities of individual monomeric conformers of hIAPP for EGCG were investigated. The results revealed that EGCG binds to monomeric hIAPP (∼10% monomer remains bound in the gas phase) (Figure 5a) (note that estimating Kd values of binding events by MS was not feasible in this instance, given the difficulty of maintaining quantitative amounts of bound ligand in the gas phase). The presence of the small molecule also alters the equilibrium of monomer charge states present. The monomer 3+ ions are more highly populated in samples incubated with EGCG (compare Figures 1b and 5a), with a change in ratio of 3+:2+ monomer charge states in the spectra changing from ∼1:2 to ∼1:1.2 in the presence of the ligand. It is possible that the changes in monomer charge state distribution observed may arise from direct binding of EGCG to the monomer that causes conformational changes and hence alters the profile of the charge state distribution, despite the ligand dissociating in the gas phase. Alternatively, changes in the charge state distribution may arise from gas-phase dissociation of ligand-bound higher order oligomeric species. In addition to its effects on the monomeric conformers of hIAPP, EGCG binding inhibits assembly of the peptide into higher order oligomers and fibrils (Figure 5b–d). Both of the observed charge states (2+ and 3+) of the hIAPP monomer (Figure 5a) and indeed each monomeric conformer (both the expanded and the compact form of each charge state) (Supporting Information Figure S5) are bound by EGCG, with one or two molecules binding to one doubly charged monomer and up to three EGCG molecules binding to the triply charged monomer when EGCG is added in an 10-fold molar excess over hIAPP (Figure 5a). Binding of EGCG to both the 2+ and 3+ monomer ions was also observed at a stoichiometry of 1:1 hIAPP:EGCG, albeit at a lower intensity (data not shown). The low levels of binding observed, despite complete inhibition of fibrillation, are consistent with hydrophobic interactions playing a role in the binding interface36 and may help toward explaining the ability of EGCG to inhibit a wide range of natively unfolded polypeptides, including IAPP mutants.37,38
Figure 5.

Inhibition of hIAPP fibril formation by EGCG. (a) Positive ion ESI mass spectrum showing binding of EGCG (added at 500 μM to 50 μM peptide) to both the 2+ (orange) and 3+ (purple) charge state ions of hIAPP monomer. Stoichiometry of binding is shown by color: 1:1 inhibitor molecule bound to an IAPP monomer is highlighted in green, 2:1 in pink, and 3:1 in blue. EGCG is shown as an inset. (b) ThT fluorescence intensity of hIAPP (black) (50 μM peptide, 20 mM ammonium acetate buffer, pH 6.8, 37 °C, 600 rpm) with increasing EGCG:hIAPP molar ratios: 0.1:1 (pink), 1:1 (orange), and 10:1 (blue). (c) ESI-IMS-MS driftscope plots of hIAPP oligomers formed in the presence of (i) 0.1:1, (ii) 1:1, and (iii) 10:1 molar ratios of EGCG:peptide monomer at t = 5 h. The number of EGCG molecules bound to each species is shown as a colored dot. (d) Negative stain TEM images of hIAPP incubated with (i) 0.1:1, (ii) 1:1, and (iii) 10:1 molar ratios of EGCG for 5 days (37 °C, 600 rpm). (iv) hIAPP fibrils alone and aggregates formed when a 10-fold molar excess of EGCG:hIAPP is added to preformed hIAPP fibrils after 5 h (v) and 24 h (vi). Scale bar is 100 nm.
ESI-IMS-MS data (Figure 5c) reveal that incubation with increasing molar ratios of EGCG:hIAPP prevents the appearance of higher order oligomers in a dose-dependent manner. At 0.1:1 and 1:1 molar ratios of EGCG:hIAPP, monomer through tetramer species of hIAPP are observed (Figure 5c), but no pentamer or hexamer (which were observed in the absence of the small molecule (Figure 1b)). At a 10:1 molar ratio of EGCG:hIAPP, no oligomers are observed (Figure 5c). Under the latter conditions, there is no increase in ThT fluorescence (Figure 5b), and TEM images (Figure 5d, panel iii) do not show any aggregated material, indicating complete inhibition of amorphous aggregate and amyloid formation. These results suggest that EGCG may trap amyloidogenic oligomers as low order species and differentially stabilize distinct monomeric charge states of hIAPP with the result that amyloid fibrils cannot form. Alternatively, EGCG may divert low order oligomers of hIAPP onto other aggregation pathways that result in the formation of aggregates that are incapable of forming amyloid. In the latter scenario, the oligomers that form must be of low abundance, low ionization propensity, or too heterogeneous to be detected by ESI-IMS-MS.
Remodeling of Mature IAPP Fibrils by EGCG
In addition to its ability to remodel oligomers,35 EGCG has been shown previously to remodel amyloid fibrils formed by a range of polypeptides, including hIAPP.35,36 Having determined the effect of EGCG on the aggregation reaction of hIAPP, the effect of this small molecule on fibril depolymerization was studied also, using TEM to monitor fibril loss and ESI-IMS-MS to investigate how fibril remodeling by EGCG influences oligomer populations. Fibril samples formed by incubation of hIAPP at pH 6.8 for 5 days were mixed with a 10-fold molar excess of EGCG, and the subsequent loss of fibrils was monitored by TEM and ESI-IMS-MS at various time points after addition of the small molecule. The results of these experiments are shown in Figure 5d (panels iv–vi) and in Supplementary Figure S6a. Under the conditions employed, clumping of hIAPP fibrils followed by remodeling into amorphous aggregates over a 24 h period was observed (Figure 5d, panels iv–vi). This is accompanied by a subtle increase in intensity of the signal arising from monomer ions, measured using ESI-IMS-MS, compared with the same sample prior to addition of EGCG (Supporting Information Figure S6a). There is, however, an absence of higher order oligomers indicating that fibril remodeling by EGCG does not result in the reformation of higher order oligomeric species.
2. Action of Silibinin on Fibril Formation of hIAPP Probed by ESI-IMS-MS
Silibinin, a flavonol extracted from seeds of the herb milk thistle, has been proposed as a potential therapeutic for amyloidosis associated with T2DM.34 The interaction of this inhibitor with hIAPP was also probed using ESI-IMS-MS. The resulting data (Figure 6a) revealed that silibinin binds to the 3+ charge state ions of monomeric hIAPP (∼20% remaining bound in the gas phase), with no detectable binding to the 2+ charge state ions of the monomer. This may be due to selectivity of the small molecule or the low abundance of the 2+ charge state in the presence of the small molecule. In the latter case, the intensity of ligand-bound ions may fall below the levels of detection. Like EGCG, but more obviously, the presence of excess silibinin alters the equilibrium of the two different charge states of monomeric hIAPP, favoring the population of the triply charged monomer ions, changing the ratio of 3+:2+ monomer charge state ions in the spectra from ∼1:2 without silibinin to ∼3:1 in the presence of the small molecule. In addition, analysis using ESI-IMS-MS indicates that the presence of substoichiometric ratios of silibinin:hIAPP prevents hIAPP oligomer formation (Figure 6b). More specifically, binding of silibinin is observed only to the expanded conformer of the 3+ monomer ions (Figure 6c). This conformer (CCS = ∼790 Å2), adopted by the hIAPP monomer, but not the rIAPP monomer (Supporting Information Figure S1b,ii), has been proposed previously as the on-pathway, amyloid-competent conformation, the formation of which precedes the generation of extended dimers and β-sheet-rich oligomers.24,25 There is no detectable binding of silbinin to the rIAPP monomer when incubated under identical conditions (Supporting Information Figure S7). Specific binding to the expanded monomer conformation of hIAPP is thus consistent with the ability of silibinin to inhibit amyloid formation. ThT data (Figure 6d) showed that aggregation of hIAPP decreases significantly as the concentration of silibinin is increased, although in a 10-fold molar excess of silibinin a weak positive ThT signal is still observed (Figure 6d), likely due to the presence of aggregates not visible by ESI-IMS-MS. TEM images indicated that incubation of monomeric hIAPP with silibinin for 5 days leads to few aggregates, with none visible by TEM when a 10-fold molar excess of silibinin:hIAPP is used (Figure 6e, panels i–iii).
Figure 6.

Inhibition of hIAPP fibril formation by silibinin. (a) Positive ion ESI mass spectra showing binding of silibinin molecules (added at 500 μM to 50 μM peptide) to the 3+ monomer ions (purple) and absence of binding to the 2+ monomer ions (orange) at a molar ratio of silibinin:hIAPP of 10:1. The structure of silibinin is inset. (b) ESI-IMS-MS driftscope plots showing the lack of hIAPP oligomers in the presence of 0.1:1, 1:1, and 10:1 molar ratios of silibinin:hIAPP at t = 5 h. (c) Arrival time distribution of 3+ monomer ions shows two conformers are present (tD = 6 and 8 ms). Silibinin binds to the extended conformer of the 3+ monomer ions (pink) but does not bind to the compact conformer (blue). (d) ThT fluorescence intensity of hIAPP (black) (50 μM peptide, 20 mM ammonium acetate buffer, pH 6.8, 37 °C, 600 rpm) with increasing silibinin:hIAPP molar ratios: 0.1:1 (pink), 1:1 (orange), and 10:1 (blue). (e) Negative stain TEM images of hIAPP incubated with (i) 0.1:1, (ii) 1:1, and (iii) 10:1 molar ratios of silibinin:hIAPP for 5 days (37 °C, 600 rpm). Lower panels show hIAPP fibrils alone (iv) and aggregates formed when a 10-fold molar excess of silibinin:hIAPP is added to preformed hIAPP fibrils after 5 h (v) and 24 h (vi). Scale bar is 100 nm.
Remodeling of Mature IAPP Fibrils by Silibinin
Although silibinin has been shown previously to inhibit fibril formation by hIAPP,34 the interaction of this small molecule with preformed fibrils had not been reported. Preformed hIAPP fibrils were incubated, therefore, with a 10-fold molar excess of silibinin, and the effect of addition of the small molecule was measured using TEM and ESI-IMS-MS. The results showed that addition of silbinin causes depolymerization of preformed fibrils over a 24 h time period during which time fibrils decrease in length (Figure 6e, panels iv–vi). During fibril depolymerization, ESI-IMS-MS revealed a simultaneous increase in low-order oligomeric species (Supporting Information Figure S6b), suggesting that depolymerization may occur via oligomer release. Alternatively, monomer release may occur and initiate rapid reformation of oligomers in solution. Oligomers reformed upon fibril depolymerization are comparable in CCS to those formed during amyloid formation (data not shown).
Formation of Hetero-oligomers and Heterofibrils of hIAPP and rIAPP
A range of peptide inhibitors has been developed against hIAPP.51−54 However, the lack of structural information available on the intermediates of aggregation has made it difficult to rationalize why some are more effective than others. Despite rIAPP having been used previously as the basis for design of an FDA approved therapy to treat T2DM,55 this peptide is only moderately effective in inhibiting hIAPP aggregation when added at equimolar concentrations.11 A two-dimensional infrared spectroscopy study revealed that rIAPP can become amyloid-competent in the presence of hIAPP, converting from a natively disordered rIAPP monomer into an ordered β-sheet-rich complex with hIAPP.56 To determine why the rIAPP peptide is an ineffective inhibitor of hIAPP assembly and to identify how the hIAPP-rIAPP complex forms, ESI-IMS-MS was used to study the oligomeric structures present in a mixture (1:1 molar ratio) of the two peptides.
By contrast with the decrease in oligomer populations of hIAPP observed in the presence of the two small molecule inhibitors studied here, oligomers up to and including hexamer were formed upon incubation of the hIAPP and rIAPP (Figure 7a), consistent with results observed when each peptide was incubated in isolation. The presence of rIAPP did not prevent fibril formation of hIAPP under the conditions of these experiments (Figure 7b), consistent with previous reports suggesting that the rat peptide must be present in an ∼10-fold molar excess to inhibit hIAPP amyloid formation noticeably.11,56 The fibrils formed from the mixed sample were of similar morphology to those of hIAPP incubated alone (Figure 7c). Mixed oligomers were observed using ESI-IMS-MS with m/z values corresponding to all-hIAPP, all-rIAPP, and oligomers containing a mixture of hIAPP and rIAPP monomer subunits. The triply charged dimer ions (Figure 7a, inset), for example, have an m/z of 2608, corresponding to one hIAPP monomer (3904 Da) and one rIAPP monomer (3921 Da), carrying three positive charges. Mixed trimers and higher order oligomers were observed also, as well as homo-oligomers of both peptides. The ability to form mixed oligomers rationalizes why rIAPP is inefficient at inhibiting hIAPP amyloid formation. After 5 days, the fibrils formed were ultracentrifuged and depolymerized by incubation in 100% HFIP for 24 h with agitation. Samples were then air-dried and resuspended in denaturing solvent (50:40:10 acetonitrile/water/acetic acid (v/v/v)). The resulting mass spectra (Figure 7d, panel i) showed the presence of both hIAPP and rIAPP monomer subunits in the aggregate pellet, confirming the presence of both peptides in the fibrillar state, with approximately twice as many hIAPP monomer units being incorporated into the fibrils compared with rIAPP monomers under the conditions employed. Both monomers were also found in the supernatant of the original mixed sample, with rIAPP monomers being in excess here (Figure 7d, panel ii). The CID-MS/MS method of interrogation of oligomer stability was used to probe the stability of the mixed oligomers formed from hIAPP and rIAPP by subjecting the heterodimer and heterotetramer ions to increasing transfer collision cell voltages to promote gas-phase dissociation. Interestingly, the hetero-oligomers exhibited gas-phase stabilities between those of homo-oligomers of hIAPP and rIAPP of the same mass, being less stable than rIAPP oligomers but more stable than hIAPP oligomers (Supporting Information Figure S8).
Figure 7.

Lack of hIAPP inhibition by rIAPP. (a) ESI-IMS-MS driftscope plot of oligomers present at t = 2 min in a mixed sample of hIAPP and rIAPP at a 1:1 molar ratio (50 μM final peptide concentration, 20 mM ammonium acetate buffer, pH 6.8, 25 °C). Extracted driftscope peak shows masses corresponding to a mixed dimer of one hIAPP monomer plus one rIAPP monomer (inset). (b) ThT fluorescence intensity during agitation of hIAPP:rIAPP 1:1 (50 μM final peptide concentration, 20 mM ammonium acetate buffer, pH 6.8, 37 °C, 600 rpm). Data for three replicates are shown. (c) Negative stain TEM image of 1:1 hIAPP: rIAPP fibrils after 5 days. Scale bar = 100 nm. (d) ESI mass spectra of depolymerized fibrils showing the presence of both hIAPP and rIAPP monomer constituents in the pellet (i) and supernatant (ii) following ultracentrifugation.
Taken together, therefore, the ESI-IMS-MS and CID-MS/MS data suggest that differences in gas-phase stability of monomer and low order oligomers between hIAPP and rIAPP sequences could be related to their differences in amyloid propensity. In addition, small molecule binding to hIAPP prevents fibril formation by disfavoring oligomer formation via binding to monomers. As a result, the aggregation pathway is diverted to alternative routes that result in the formation of monomers and/or amorphous aggregates.
Discussion
Identifying and characterizing the structures and dynamics of prefibrillar oligomers is vital for understanding the mechanisms of protein aggregation in amyloid disease, identifying the specific culprits of toxicity, and designing therapeutics to prevent aggregation. Here, using ESI-IMS-MS, the high order oligomers of hIAPP and rIAPP have been characterized for the first time, and their structural organization and relative gas-phase stabilities were compared. Additionally, the binding and mode of action of two small molecule inhibitors of hIAPP have been determined, and the relative ineffective inhibition of hIAPP by rIAPP has been investigated. Previous IMS-MS studies into the structure of hIAPP monomer and dimer have suggested that a conformational transition to an extended structure is likely as an early step in amyloid formation.24,25 In the study presented here, the population of monomer through hexamer has been revealed for both peptides. These oligomers likely assemble via monomer addition (since every species from monomer to hexamer is observed). In the case of hIAPP, further monomer association into higher order oligomers is not observed, suggesting that fibril formation occurs without measurable population of larger oligomeric intermediates or that higher order species are too lowly populated, too transient, or too heterogeneous to be detected by ESI-IMS-MS. The oligomeric states observed may be “on-pathway” to fibril formation or alternatively may be off-pathway. In the latter case, conformational changes occurring due to the oligomers’ relative lack of structural stability may facilitate amyloid assembly compared with their counterparts in rIAPP.
CCS estimations on hIAPP species indicate that the early oligomers formed are elongated rather than globulamers, akin to the oligomers observed during β2m assembly into amyloid.50 Of interest, early oligomers of rIAPP, which are similar in size and CCS to those of hIAPP, are formed when the peptide is incubated under conditions identical to those used to analyze hIAPP but are significantly more stable in the gas phase as judged by CID experiments. Although solution-phase stability cannot be directly inferred from gas-phase stability,57,58 the difference in stability observed may rationalize the difference in aggregation propensity of the two peptides, assuming that a conformational change is necessary for fibril formation that is impeded for the more stable rat peptide. The ability of the two peptides to form mixed oligomeric species on pathway to heterofibrils in vitro has been revealed using ESI-IMS-MS and has provided an explanation as to the inefficient inhibition of hIAPP amyloid formation by rIAPP. The study highlights the pitfalls associated with designing peptide inhibitors based on amino acid sequence, given that rIAPP in principle would be expected to be a good inhibitor of hIAPP since it combines a recognition motif with a β-sheet breaker unit.11
Having characterized the oligomeric species of hIAPP, the binding of two potent hIAPP amyloid inhibitors, EGCG and silibinin, were studied using ESI-IMS-MS. Negative controls were performed using molecules that do not have an inhibitory effect on hIAPP amyloid formation (e.g., rifampicin59 and benzimidazole); these do not to bind hIAPP or inhibit oligomerization (data not shown). We show that both EGCG and silibinin are able to block oligomer and fibril formation from hIAPP when added prior to amyloid assembly, despite showing different binding characteristics and having different effects on the equilibrium of species present. By contrast with the observations made at high ionic strength, in which oligomers are formed and then lost from the ensemble rapidly as they elongate into fibrils, the presence of these small molecules inhibits formation of oligomeric species from monomer subunits. In both scenarios, the observation is the same (lack of persistence of signal arising from oligomeric ions). However, the interpretation of the molecular events that lead to the observed absence of oligomeric species is fundamentally different. EGCG was found to bind to both observed charge states of the hIAPP monomer, inhibiting early oligomer formation in a dose-dependent manner and preventing fibril formation. By contrast, silibinin was detected to bind only to the most expanded conformer of the monomer 3+ ions within the dynamic ensemble of intrinsically disordered monomeric conformations, blocking oligomerization and fibrillation. The latter result supports the notion of the role of the extended state of the monomer 3+ species in the fibril assembly mechanism of hIAPP because the hIAPP conformer bound by silibinin is absent in rIAPP. Binding to monomer may inhibit assembly directly or may create a structure that associates with other monomers to generate non-amyloidogenic aggregates. The fact that both EGCG and silibinin were found to be effective as amyloid inhibitors at substoichiometric concentrations suggests that the small molecules may bind to oligomers, as well as the monomer. Due to the low intensity of oligomers in the presence of small molecules, however, binding of small molecules to these species could not be observed by ESI-IMS.
Both EGCG and silibinin alter the equilibrium of the monomeric charge states of hIAPP present, with the addition of each small molecule leading to increased population of the triply charged monomer ions. We propose that binding to these extended species prevents their self-assembly into amyloid. Previous studies have shown that altering the equilibration between different monomeric conformers can divert β2m to alternative assembly pathways,32 leading to formation of spherical aggregates rather than highly ordered β-sheet rich fibrillar assemblies. For hIAPP, these alternative aggregation pathways are presumably kinetically or thermodynamically unfavorable in the absence of EGCG and silibinin but made feasible upon ligand binding to unfolded hIAPP monomers. The fact that both of these small molecules are capable of disaggregating hIAPP amyloid fibrils in distinguishable manners is also of interest. EGCG remodels fibrils into amorphous aggregates without reformation of low order oligomers, while silibinin depolymerizes fibrils and re-establishes the prefibrillar array of monomer plus early oligomeric species observed by ESI-IMS-MS.
It is widely accepted that early oligomeric states are key to protein self-assembly and subsequent amyloid disease, and determination of which species are on-pathway to amyloid assembly is vital. Here we have used ESI-IMS-MS to show that specific, lowly populated hIAPP monomeric conformers are capable of binding small molecule ligands. In addition we have used this technique to identify a range of extended oligomeric assemblies. We have assessed the effect of ligand binding on each individual species within a heterogeneous mixture of peptide monomers and their conformers populated prior to fibril formation. Our experimental results provide support for a route to hIAPP amyloid fibrils via formation of elongated oligomers. We propose that reduced stability (relative to nonamyloidogenic rIAPP) is related to the ability of hIAPP to form extended monomer and oligomeric conformations that have increased amyloid propensity compared with their more stable counterparts. Binding to and stabilizing monomers, thereby preventing their polymerization, could be key to the mechanism of inhibition of amyloid by EGCG and silibinin.
Conclusions
The data presented herein, and previously by Bowers and co-workers,24,25 demonstrate that formation of extended structures is likely key in early amylin amyloid formation. Small molecule binding to these extended structures, which results in alteration of the distribution of conformers present, leads to inhibition of oligomerization and fibrillation by hIAPP. We hypothesize that stability plays a role in the sequence specificity of IAPP amyloid formation, given that oligomers of rIAPP (that differs at only six amino acid positions compared with hIAPP) are similar in size and shape to those of hIAPP but are significantly more stable in the gas phase. In order for amyloid systems to be fully understood, all species on the energy landscape, both “on-pathway” and “off-pathway”, must be identified. We cannot determine unequivocally whether the oligomers detected here using MS are on- or off-pathway. Whichever scenario is correct, our data are consistent with a lower stability corresponding to an enhanced rate and/or ability to form amyloid fibrils. Oligomers either may be on-pathway to fibril formation and require conformational changes to progress assembly or alternatively may undergo conformational changes allowing off-pathway species to enter the aggregation pathway to amyloid. Additionally, we demonstrate that the mixing of the hIAPP and rIAPP sequences leads to copolymerization into amyloid, hence explaining the lack of inhibition of hIAPP fibril formation by rIAPP.
Acknowledgments
L.M.Y. is funded by a Biotechnology and Biological Sciences Research Council CASE studentship (Grant Number BB/I015361/1) sponsored by Micromass UK Ltd/Waters Corpn, Manchester, U.K. The Synapt HDMS mass spectrometer was purchased with funds from the Biotechnology and Biological Sciences Research Council through its Research Equipment Initiative scheme (BB/E012558/1). This work was also supported by a grant from the United States National Institutes of Health GM078114. We thank all members of the Ashcroft, Radford, and Raleigh groups for helpful discussions.
Supporting Information Available
Table of CCS values of hIAPP and rIAPP oligomers and eight supplementary figures. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
§ Structural Biology Program, Skirball Institute of Biomolecular Medicine, New York University School of Medicine, New York, NY, USA.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Sipe J. D.; Benson M. D.; Buxbaum J. N.; Ikeda S.; Merlini G.; Saraiva M. J.; Westermark P.; Nomenclature Committee of the International Society of Amyloidosis. . Amyloid 2012, 19, 167–70. [DOI] [PubMed] [Google Scholar]
- Vendruscolo M.; Knowles T. P.; Dobson C. M. Cold Spring Harbor Perspect. Biol. 2011, 3, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiti F.; Dobson C. M. Annu. Rev. Biochem. 2006, 75, 333–66. [DOI] [PubMed] [Google Scholar]
- Eichner T.; Radford S. E. Mol. Cell 2011, 43, 8–18. [DOI] [PubMed] [Google Scholar]
- Westermark P.; Andersson A.; Westermark G. T. Physiol. Rev. 2011, 91, 795–826. [DOI] [PubMed] [Google Scholar]
- Westermark P. Upsala J. Med. Sci. 2011, 116, 81–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper G. J.; Willis A. C.; Clark A.; Turner R. C.; Sim R. B.; Reid K. B. Proc. Natl. Acad. Sci. U.S.A. 1987, 84, 8628–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzo A.; Razzaboni B.; Weir G. C.; Yankner B. A. Nature 1994, 368, 756–60. [DOI] [PubMed] [Google Scholar]
- Konarkowska B.; Aitken J. F.; Kistler J.; Zhang S.; Cooper G. J. FEBS J. 2006, 273, 3614–24. [DOI] [PubMed] [Google Scholar]
- Westermark P.; Engstrom U.; Johnson K. H.; Westermark G. T.; Betsholtz C. Proc. Natl. Acad. Sci. U.S.A. 1990, 87, 5036–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao P.; Meng F.; Abedini A.; Raleigh D. P. Biochemistry 2010, 49, 872–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillay K.; Govender P. Biomed. Res. Int. 2013, 2013, 826706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao P.; Marek P.; Noor H.; Patsalo V.; Tu L. H.; Wang H.; Abedini A.; Raleigh D. P. FEBS Lett. 2013, 587, 1106–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abedini A.; Schmidt A. M. FEBS Lett. 2013, 587, 1119–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaiana S. M.; Ghirlando R.; Yau W. M.; Eaton W. A.; Hofrichter J. Biophys. J. 2008, 94, L45–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y.; Brender J. R.; Hartman K.; Ramamoorthy A.; Marsh E. N. Biochemistry 2012, 51, 8154–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng B.; Liu X.; Gong H.; Huang L.; Chen H.; Zhang X.; Li C.; Yang M.; Ma B.; Jiao L.; Zheng L.; Huang K. J. Agric. Food Chem. 2011, 59, 13147–55. [DOI] [PubMed] [Google Scholar]
- Woods L. A.; Radford S. E.; Ashcroft A. E. Biochim. Biophys. Acta 2013, 1834, 1257–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams D. M.; Pukala T. L. Mass Spectrom. Rev. 2013, 32, 169–87. [DOI] [PubMed] [Google Scholar]
- Bernstein S. L.; Dupuis N. F.; Lazo N. D.; Wyttenbach T.; Condron M. M.; Bitan G.; Teplow D. B.; Shea J. E.; Ruotolo B. T.; Robinson C. V.; Bowers M. T. Nat. Chem. 2009, 1, 326–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlad C.; Iurascu M. I.; Slamnoiu S.; Hengerer B.; Przybylski M. Methods Mol. Biol. 2012, 896, 399–412. [DOI] [PubMed] [Google Scholar]
- Kłoniecki M.; Jabłonowska A.; Poznański J.; Langridge J.; Hughes C.; Campuzano I.; Giles K.; Dadlez M. J. Mol. Biol. 2011, 407, 110–24. [DOI] [PubMed] [Google Scholar]
- Bernstein S. L.; Liu D.; Wyttenbach T.; Bowers M. T.; Lee J. C.; Gray H. B.; Winkler J. R. J. Am. Soc. Mass Spectrom. 2004, 15, 1435–43. [DOI] [PubMed] [Google Scholar]
- Dupuis N. F.; Wu C.; Shea J. E.; Bowers M. T. J. Am. Chem. Soc. 2009, 131, 18283–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuis N. F.; Wu C.; Shea J. E.; Bowers M. T. J. Am. Chem. Soc. 2011, 133, 7240–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young L.; Ndlovu H.; Knapman T.; Harris S.; Radford S.; Ashcroft A. Imt. J. Ion Mobility Spectrom. 2013, 16, 29–39. [Google Scholar]
- Kumar S.; Miranker A. D. Chem. Commun. (Cambridge) 2013, 49, 4749–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson J. A.; Loria J. P.; Miranker A. D. J. Mol. Biol. 2009, 393, 383–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porat Y.; Abramowitz A.; Gazit E. Chem. Biol. Drug Des. 2006, 67, 27–37. [DOI] [PubMed] [Google Scholar]
- Feng B. Y.; Toyama B. H.; Wille H.; Colby D. W.; Collins S. R.; May B. C.; Prusiner S. B.; Weissman J.; Shoichet B. K. Nat. Chem. Biol. 2008, 4, 197–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitken J. F.; Loomes K. M.; Konarkowska B.; Cooper G. J. Biochem. J. 2003, 374, 779–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods L. A.; Platt G. W.; Hellewell A. L.; Hewitt E. W.; Homans S. W.; Ashcroft A. E.; Radford S. E. Nat. Chem. Biol. 2011, 7, 730–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng F.; Abedini A.; Plesner A.; Verchere C. B.; Raleigh D. P. Biochemistry 2010, 49, 8127–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng B.; Gong H.; Li X.; Sun Y.; Zhang X.; Chen H.; Liu X.; Zheng L.; Huang K. Biochem. Biophys. Res. Commun. 2012, 419, 495–9. [DOI] [PubMed] [Google Scholar]
- Ehrnhoefer D. E.; Bieschke J.; Boeddrich A.; Herbst M.; Masino L.; Lurz R.; Engemann S.; Pastore A.; Wanker E. E. Nat. Struct. Mol. Biol. 2008, 15, 558–66. [DOI] [PubMed] [Google Scholar]
- Cao P.; Raleigh D. P. Biochemistry 2012, 51, 2670–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson S. A.; Ecroyd H.; Dehle F. C.; Musgrave I. F.; Carver J. A. J. Mol. Biol. 2009, 392, 689–700. [DOI] [PubMed] [Google Scholar]
- Bieschke J.; Russ J.; Friedrich R. P.; Ehrnhoefer D. E.; Wobst H.; Neugebauer K.; Wanker E. E. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 7710–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezai-Zadeh K.; Arendash G. W.; Hou H.; Fernandez F.; Jensen M.; Runfeldt M.; Shytle R. D.; Tan J. Brain Res. 2008, 1214, 177–87. [DOI] [PubMed] [Google Scholar]
- Palhano F. L.; Lee J.; Grimster N. P.; Kelly J. W. J. Am. Chem. Soc. 2013, 135, 7503–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marek P.; Woys A. M.; Sutton K.; Zanni M. T.; Raleigh D. P. Org. Lett. 2010, 12, 4848–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles K.; Pringle S. D.; Worthington K. R.; Little D.; Wildgoose J. L.; Bateman R. H. Rapid Commun. Mass Spectrom. 2004, 18, 2401–14. [DOI] [PubMed] [Google Scholar]
- Smith D. P.; Knapman T. W.; Campuzano I.; Malham R. W.; Berryman J. T.; Radford S. E.; Ashcroft A. E. Eur. J. Mass. Spectrom. 2009, 15, 113–30. [DOI] [PubMed] [Google Scholar]
- Valentine S. J.; Counterman A. E.; Clemmer D. E. J. Am. Soc. Mass Spectrom. 1999, 10, 1188–211. [DOI] [PubMed] [Google Scholar]
- Platt G. W.; Routledge K. E.; Homans S. W.; Radford S. E. J. Mol. Biol. 2008, 378, 251–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abedini A.; Singh G.; Raleigh D. P. Anal. Biochem. 2006, 351, 181–6. [DOI] [PubMed] [Google Scholar]
- Marek P. J.; Patsalo V.; Green D. F.; Raleigh D. P. Biochemistry 2012, 51, 8478–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleiholder C.; Dupuis N. F.; Wyttenbach T.; Bowers M. T. Nat. Chem. 2011, 3, 172–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzen K.; Olia A. S.; Uetrecht C.; Cingolani G.; Heck A. J. J. Mol. Biol. 2008, 379, 385–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D. P.; Radford S. E.; Ashcroft A. E. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 6794–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L. M.; Tatarek-Nossol M.; Velkova A.; Kazantzis A.; Kapurniotu A. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 2046–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng F.; Raleigh D. P.; Abedini A. J. Am. Chem. Soc. 2010, 132, 14340–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapurniotu A.; Schmauder A.; Tenidis K. J. Mol. Biol. 2002, 315, 339–50. [DOI] [PubMed] [Google Scholar]
- Gilead S.; Gazit E. Angew. Chem., Int. Ed. 2004, 43, 4041–4. [DOI] [PubMed] [Google Scholar]
- Hollander P. A.; Levy P.; Fineman M. S.; Maggs D. G.; Shen L. Z.; Strobel S. A.; Weyer C.; Kolterman O. G. Diabetes Care 2003, 26, 784–90. [DOI] [PubMed] [Google Scholar]
- Middleton C. T.; Marek P.; Cao P.; Chiu C. C.; Singh S.; Woys A. M.; de Pablo J. J.; Raleigh D. P.; Zanni M. T. Nat. Chem. 2012, 4, 355–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyachenko A.; Goldflam M.; Vilaseca M.; Giralt E. Biopolymers 2010, 94, 689–700. [DOI] [PubMed] [Google Scholar]
- Marcoux J.; Robinson C. V. Structure 2013, 21, 1541–50. [DOI] [PubMed] [Google Scholar]
- Meng F.; Marek P.; Potter K. J.; Verchere C. B.; Raleigh D. P. Biochemistry 2008, 47, 6016–24. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.