

Abstract
Trafficking of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) is regulated by specific interactions with other proteins and by post-translational mechanisms, such as phosphorylation. We have found that the type II cGMP-dependent protein kinase (cGKII) phosphorylates GluA1 (formerly GluR1) at S845, augmenting the surface expression of AMPARs at both synaptic and extrasynaptic sites. Activation of cGKII by 8-Br-cGMP enhances the surface expression of GluA1, whereas its inhibition or suppression effectively diminished the expression of this protein at the cell surface. In granule cells, NMDA receptor activation (NMDAR) stimulates nitric oxide and cGMP production, which in turn activates cGKII and induces the phosphorylation of GluA1, promoting its accumulation in the plasma membrane. GluA1 is mainly incorporated into calcium permeable AMPARs as exposure to 8-Br-cGMP or NMDA activation enhanced AMPA-elicited calcium responses that are sensitive to NASPM inhibition. We summarize evidence for an increase of calcium permeable AMPA receptors downstream of NMDA receptor activation that might be relevant for granule cell development and plasticity.
Keywords: calcium permeable AMPA receptors, NMDA receptors, Nitric oxide signalling, cGMP signalling, cGKII
1. Introduction
AMPA glutamate receptors (AMPARs) are hetero-tetrameric cation channels generated by the combinatorial assembly of four subunits, GluA1-GluA4 [1–3]. The specific properties of the channels are defined by their subunit composition and for example, the inclusion of one or more GluA2 subunits in the channel makes it impermeable to Ca2+ [4]. Physiological responses of AMPARs can be modulated by the differential expression of their subunits and by their number at the cell surface, which largely depends on neuronal development [5]. The ubiquitous expression and efficient editing of GluA2 within neurons ensures that calcium impermeable AMPARs predominate in excitatory neurons. However, Ca2+ permeable AMPARs lacking GluA2 are transiently expressed by principal neurons early in development, and they can be assembled and incorporated into synapses under certain conditions [6–10]. Indeed, cerebellar granule cells express all four AMPAR subunits at early developmental stages and their expression is increased by reducing calcium entry [11].
Besides AMPARs, cerebellar granule cells also express N-methyl-D-aspartate (NMDA) glutamate receptors (NMDARs) [5,11]. The stimulation of NMDARs is essential for the survival and differentiation of these cells during development, which play a primary role in neuroplasticity [12,13]. The formation of NO is one consequence of NMDAR stimulation [14], due to the anchoring of neuronal nitric oxide synthase (nNOS) to the NMDARs through the postsynaptic density-95 protein (PSD-95), and calcium entry through these receptors activates nNOS. The major physiological receptor for NO is soluble guanylyl cyclase (sGC), which synthesizes the cGMP that can drive specific cellular responses through the regulation of many proteins, including cGMP-regulated phosphodiesterases, cGMP-gated ion channels and cGMP-dependent serine/threonine kinases [15]. The main targets of cGMP are kinases, which can transduce these signals to a vast array of other proteins through phosphorylation. Two types of cGMP-dependent protein kinases (cGKI and cGKII) have been identified in mammalian tissues [16,17], as well as two splice variants of cGKI with different amino-terminal domains (cGKIα and cGKIβ) [18,19]. In the brain, earlier studies demonstrated that the distribution of cGKIα was very restricted, being found primarily in cerebellar Purkinje cells and rarely at other sites [20–23]. By contrast, cGKII mRNA was found in a multitude of brain regions [24] where it is strongly expressed and can be activated by the increase in cGMP produced by NO donors [25]. Furthermore, the key enzymes in cGMP metabolism and signalling are strongly expressed in the cerebellum, which is an area of considerable plasticity during development [26]. These enzymes are developmentally regulated in cerebellar granule cells, and maturation of the NO system is important for both granule cell survival and acquisition of a mature phenotype [27–31].
In the mouse hippocampus, it was recently reported that cGKII binds to the C-terminus domain (CTD) of GluA1 in a cGMP-dependent manner, which enables cGKII to phosphorylate S845 of GluA1 and increases the amount of GluA1 at the plasma membrane [32]. This activity of cGKII provides a new mechanism to enhance the levels of GluA1 at the plasma membrane, which complements the increase of GluA1 at the cell surface induced by PKA. Because the NMDAR regulates NO production by nNOS and hence, it controls cGMP levels and cGKII activity, this pathway provides a novel mechanism for NMDAR and NO to control the accumulation of GluA1 at the plasma membrane. The molecular mechanisms that regulate GluA1 levels at the cellular surface are complex and they involve interactions with scaffolding proteins [33,34], as well as a series of phosphorylation steps at several Ser residues in the GluA1 CTD [35]. The CTD of GluA1 is phosphorylated at S831 by both CaMKII and PKC [35–37], at S845 by PKA and cGKII [32,38], and at S818 by PKC [39]. Phosphorylation of S845 in GluA1 is required, although not sufficient, for GluA1 synaptic insertion during LTP in the hippocampus [40]. Moreover, phosphorylation of S845 by PKA and/or cGKII has been shown to augment the delivery of AMPARs to extrasynaptic sites, as well as priming the receptor for synaptic insertion in hippocampal neurons [32,41,42]. Cerebellar granule cells express cGKII and GluA1 at early developmental stages [29,10] and hence, we have investigated whether GluA1 surface expression may be regulated by cGKII activation in cerebellar granule cells during this period of development.
2. Materials and Methods
2.1 Primary cerebellar granule neuron cultures
All procedures conducted at the Universidad Complutense, Madrid, relating to the care of animals were carried out in accordance with our institute’s ethical guidelines for animal experiments and the regulations established in the European Council Directive (86/609/EEC). Primary cultures of dissociated cerebellar neurons were established from the cerebellum of 7-day-old (P7) male or female Wistar albino rat pups following the protocol described previously [28]. The meninges and blood vessels were cleaned from the cerebellum, which was placed in a tube containing Papain (Worthington, Lakewood NJ, USA) and triturated to isolate the granule cells. The digestion was then stopped by adding ovomucoid solution and the cerebellar neurons were diluted in Neurobasal A supplemented with B27 (Invitrogen, Life Technologies, Madrid, Spain), 20 mM KCl, glutamax (Invitrogen, Life Technologies, Madrid, Spain) and a stabilized antibiotic antimycotic solution (Sigma-Aldrich, Madrid, Spain). The cells were seeded onto poly-Lysine coated coverslips and placed in 6-well tissue culture plates. The cultures were maintained at 37°C in a humidified incubator in 5% CO2 and after 24 h in culture, 10 μM cytosine-β-D-arabinofuranoside (Sigma-Aldrich, Madrid, Spain) was added to restrict glial cell growth. All the experiments were performed on cells that had been cultured for 7 or 9 days in vitro (DIV).
2.2 Antibodies
The antibodies used in this study were: a purified rabbit polyclonal anti-GluA1 (Ref. PC246, Calbiochem, Millipore Ibérica, Madrid, Spain); rabbit polyclonal anti-GluA1 (Ref. 07-660, Upstate, Millipore Ibérica, Madrid, Spain); monoclonal rabbit anti-phospho-GluA1 (Ser845), clone EPR2148 (Ref. 04-1073, Millipore Ibérica, Madrid, Spain); mouse anti-GluA1-NT (N-terminus) clone RH95 (Ref.MAB2263, Millipore Iberica); monoclonal mouse anti-synapsin I (Ref. 106 001, Synaptic Systems, Göttingen, Germany); rabbit anti-GluA1 polyclonal antibody (Ref. PC246, Calbiochem); monoclonal mouse anti β-tubulin, clone D66 (Ref.T0198, Sigma-Aldrich, Madrid, Spain); monoclonal mouse anti-MAP2 (Ref. 188 101, Synaptic Systems, Göttingen, Germany), monoclonal mouse anti β-actin clone AC15 (Ref. AM4302, Life technologies USA, NY).
2.3 Treatments
Cultured cells 7–9 DIV were incubated for 1 hour in HBM (NaCl 14mM; KCl 5mM; NaHCO3 5mM; NaH2PO4 1.2 mM; MgCl2 1mM; Glucose 10mM; HEPES 10mM; CaCl2 1.33 mM) to allow them to re-polarize their membranes before they were subjected to the distinct treatments: 8Br-cGMP (500 μM: BioLog, Bremen, Germany) for 20 min; NMDA (50 μM: Ascent Scientific, Abcam Biochemicals) plus glycine (10 μM: Scientific, Abcam Biochemicals) in a magnesium free solution for 10 minutes; KT5823 (2 μM: Calbiochem, Merck Chemicals, Spain), a selective inhibitor of protein kinase G, for 30 min; L-NMMA (10 μM: Cayman Chemical, Ann Arbor, MI, USA), a relatively non-selective inhibitor of all NOS isoforms, for 60 min; NPLA (1 μM: Cayman Chemical, Ann Arbor, MI, USA) a potent and selective inhibitor of neuronal nitric oxide synthase (nNOS); Angeli’s salt (2 μM: Cayman Chemical, Ann Arbor, MI, USA), a nitric oxide donor, for 10 min; FK409 (10 μM: Cayman Chemical, Ann Arbor, MI, USA), a nitric oxide donor, for 10 min. When KT5823 administration was combined with 8-Br-cGMP or NMDA plus Glycine, the latter was added for the last 20 or 10 minutes of the KT5823 incubation, respectively. When L-NMMA or NPLA administration was combined with NMDA plus Glycine, the latter was added for the last 10 minutes of the NOS inhibitors incubation.
Similar treatments were used for the calcium imaging experiments, in which AMPA (50 μM: Ascent Scientific, Abcam Biochemicals, Cambridge, UK) and Cyclothiazide (10 μM: Ascent Scientific, Abcam Biochemicals, Cambridge, UK) were applied by perfusion over 10 seconds in a perfusion solution (NaCl 140mM; KCl 15mM; CaCl2 1.33mM; MgCl2 1mM; TES 10mM; Glucose 10mM). 1-Naphthylacetylspermine trihydrochloride (NASPM, 10 μM: Tocris Bioscience, Bristol, UK), a selective antagonist of Ca2+ permeable AMPARs was applied along with AMPA by switching the perfusion solution. At the end of each experiment, a KCl (50mM) solution was applied for 10 seconds as a control of the neurons’ activity.
2.4 Real-time RT-PCR reactions
GluA1 mRNA was quantified as described previously [11], performing real-time RT-PCR with predesigned primers and MGB probes from applied Biosystems for GluA1 (Rn00709588, lot 493728). Kits with MGB probes to amplify rat GAPDH were used as an endogenous control (Applied Biosystems). PCR reactions were followed in a 9800 Fast Thermal Cycler (Applied Biosystems) with TaqMan Gold PCR reagents. The results are expressed as 50-ΔCT, where ΔCT is the difference between the CT for the GluA1 amplification curve and that for the GAPDH amplification curve considered as a house-keeping control gene.
2.5 Western blotting
To assess the phosphorylation of GluA1, 7–9 DIV cerebellar granule cells were transferred to an HBM solution for 1 hour at 37 °C and then subjected to the different treatments as indicated in the text. The cells were then homogenized in RIPA buffer containing EDTA (5 mM), protease inhibitors and phosphatase inhibitors (Pierce, Thermo Fisher Scientific Inc, Rockford, IL, USA). Equal amounts of total protein for each sample were separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) on 8 or 10% gels and transferred electrophoretically to nitrocellulose membranes (GE Healthcare, Barcelona, Spain) as described previously [29]. The membranes were then probed with the appropriate primary antibodies: rabbit polyclonal anti-GluA1 (1:1000: Upstate, Millipore), monoclonal rabbit anti-phospho-GluA1 (Ser845, 1:3000: Millipore), anti-β tubulin (1:5000: Sigma-Aldrich). After several washes, the antibodies were detected with the corresponding IRD-labelled secondary antibody diluted (1:15000: Li-Cor Biosciences, Lincoln, Nebraska USA) and the blots were scanned in an Odyssey Infrared imaging system, comparing the immunoreactive proteins by measuring the fluorescent intensity by densitometry. The β-tubulin signal was used to normalize for loading differences and the data was quantified with the Odyssey 2.0 software, normalizing the final results to the control.
2.6 Immunocytochemistry
Two different protocols were used to specifically label the surface GluA1 of granule cells:
Cerebellar granule cells (control or transfected) were plated on polylysine-coated coverslips at a density of 100,000 cells/coverslip. The attached cells were rinsed twice with PBS, fixed with 4% formaldehyde in PBS for 15 minutes at room temperature, rinsed briefly with PBS twice and then blocked with PBS containing 10 % normal donkey serum for 1 hour at 37°C. The cells were then incubated overnight at 4°C under non-permeabilizing conditions with a rabbit anti-GluA1 antiserum that recognizes amino acids 271–285, corresponding to the N-terminal domain (Calbiochem, Millipore Ibérica, Madrid, Spain), at a dilution of 1:200 in PBS containing 5 % normal donkey serum [10]. A secondary donkey anti-rabbit Alexa Fluor 488 conjugated antibody (Life Technologies), was used at 1:200 to stain surface GluA1 again under non-permeabilizing conditions. After washing several times with PBS, the cells were permeabilized for 6 minutes with 0.1% Triton X-100 in PBS and incubated for 1 hour at 37°C with PBS containing 10% normal donkey serum and then overnight at 4°C with a mouse anti-MAP2 antibody (Synaptic Systems, Göttingen, Germany) diluted 1:1,000 in PBS containing 5 % normal donkey serum and 0.05% Triton X-100. The coverslips were then incubated for 1 hour at 37°C with donkey anti-mouse Alexa Fluor 594 diluted 1:300, and following several washes in PBS, one wash with PBS with 0.1% Triton X-100 and a last rinse in ultrapure water, the coverslips were mounted with prolong antifade with DAPI (Life Technologies). The cells were viewed on a Nikon Diaphot microscope equipped with a 100×1.3 (NA) oil immersion fluor objective, a mercury lamp light source and fluorescein or rhodamine Nikon filter sets. Images were obtained using a slow-scan CCD camera (Hamamatsu C4880) operating at 16-bits, and the camera output was stored on a computerized imaging system (Kinetic Imaging, Ltd.). To control the staining and confirm the specificity of the antibodies used, the cells were incubated with the secondary antibodies alone, in the absence of primary antibodies.
The cell surface GluA1 receptors were labelled by incubating live cultures for 20 min at 37 °C with rabbit NT-GluA1 (20 μg/ml: Calbiochem, Millipore Ibérica, Madrid, Spain) in HBM solution as described previously [32]. Cultured neurons were then fixed with 4% paraformaldehyde and stained with a rabbit 594-Alexa Fluor secondary antibody (Life Technologies) under non-permeabilizing conditions. Cells were then permeabilized with 0.1% Saponin (Sigma-Aldrich) in PBS, blocked with 10% donkey serum in PBS containing 0.05% Saponin for 1 hour at 37°C and incubated overnight at 4 °C with mouse anti-synapsin (Synaptic Systems; Göttingen, Germany) diluted 1:800 in PBS containing 5% donkey serum and 0.05% Saponin. Afterwards, the cells were stained for 1 hour at 37°C with a mouse 488-Alexa Fluor polyclonal secondary antiserum (Molecular probes, Invitrogen) and following several washes in PBS, the coverslips were rinsed with deionized water to remove the salts, mounted and viewed as before.
2.7 Imaging analysis
For quantitative analysis, all the images were acquired with identical settings of brightness and contrast. For the siRNA experiments (see Figure 6), the ROIs (regions of interest) were selected with Igor Pro software having established standard parameters to select only specific antibody stained clusters. The automatic threshold with the Isodata algorithm [43] was then used to convert the image into a binary mask that included all the fluorescence emissions above background, and spurious ROIs were removed manually [30]. The staining intensities were then normalized to the MAP2 labelling (internal control for quantitative analysis) and used to compare signal intensities between the control and treated cells.
Figure 6. cGKII silencing decreases GluA1 surface expression.

Cerebellar granule cells at 5 DIV were transfected with the cGKII siRNA and immunostained at 8 DIV with the specific rabbit NT-GluA1 antiserum and a mouse anti-MAP2 antibody. (A) Representative images of GluA1/MAP2 immunolabelling in control (scrambled siRNA) and cGKII silenced cells. (B and E) Cumulative probability plots and bar chart for the GluA1/MAP2 mean fluorescence intensity (**p<0.001, Mann–Whitney and Kolmogorov–Smirnov tests): Control 878 ROIs 0.27±0.07; scrambled siRNA 3,365 ROIs 0.26±0.03; cGKII siRNA 2,716 ROIs 0.06±0.01. (D) Cumulative probability plots of the mean MAP2 fluorescence intensity (a.u.) of cells also labelled with anti-GluA1 (MAP2 mean intensity 60.7±0.54, 58.7±1, 59.2±1.5 for control, scrambled siRNA and cGKII siRNA respectively: p>0.05, Mann–Whitney and Kolmogorov–Smirnov tests).
In experiments where GluA1 were labelled in living neurons (see Figures 4 and 5), the images were processed with ImageJ and background subtraction was carried out with the ImageJ plugin, based on the rolling ball radius algorithm [44] and applying a radius of 5 pixels for all the images. In all cases, the staining intensities of the ROIs were measured as grey-scale values (ranging from 0 to 255) using the ImageJ software. Similarly, the integrated density value was calculated as the sum of the grey values of each of the different pixels defined in a given ROI.
Figure 4. Activation of cGKII by cGMP causes an increase of cellular surface GluA1.

(A) Cerebellar granule cells maintained for 8 DIV were subjected to different treatments as shown: incubation with 8Br-cGMP (500 μM) for 20 minutes or inhibition with KT5823 during 30 minutes followed by incubation with a GluA1-NT antibody (20 μg/ml) for 20 minutes as indicated in the Methods section. Images represent the immunostaining for synapsin I and GluA1, the merged image and an amplification of each condition. (B) Density of GluA1 clusters in dendrites (clusters/μm). ROI’s of the specific antibody were selected for each condition from at least 15 fields from 2 different experiments. (C) Analysis of the fraction of GluA1 clusters apposed to synapsin I puncta, either upon stimulation or inhibition (n=30 fields from 2 independent experiments). (D) Cumulative probability plots for GluA1 mean fluorescence intensity (mean values: control 19.4±0.06; 8Br-cGMP 23.1±0.11; KT5823 16.4±0.03; 8Br-cGMP+KT 16.7±0.06, a.u.). In B and C the data are represented as the mean ± SEM and analyzed with an unpaired, two-sample t test (*p<0.001). The Komolgorov-Smirnov test was used for the cumulative probability. Scale bars: 10 μm.
Figure 5. NMDA dependent production of NO causes GluA1 accumulation at plasma membrane.

(A) Cerebellar granule cells maintained for 8 DIV were treated for 10 minutes with NMDA (50 μM) + glycine (10μM), or for 30 minutes with KT5823, followed by incubation with a GluA1-NT antibody (20 μg/ml) over 20 minutes. The images represent the immunostaining of synapsin I and GluA1, the merged image and an amplification of each condition. (B) The density of GluA1 clusters in dendrites (clusters/μm), selecting ROI’s of the specific antibody from at least 15 fields from 2 different experiments for each condition. (C) Analysis of the fraction of GluA1 clusters apposed to synapsin I puncta, either upon stimulation or inhibition (n=30 fields from two independent experiments with different cell cultures). (D) Cumulative probability plots of GluA1 mean fluorescence intensity (mean values: control 19.7±0.04; 8Br-cGMP 23.4±0.08; KT5823 16.2±0.06; 8Br-cGMP+KT 16.9±0.07, a.u.). In B and C, the data are represented as the mean ± SEM and analyzed using unpaired, two-sample t test (*p<0.001). For the cumulative probability the Komolgorov-Smirnov test was used. Scale bars: 10 μm.
When indicated, the GluA1 cluster density was calculated by dividing the overall number of clusters by the whole neurite length in each field, measuring the total neurite length with the NeuronJ plugin of ImageJ [45]. The total number of ROI’s for each field, corresponding to the total number of clusters, was divided by the corresponding total length and the result was expressed as clusters/μm.
The GluA1-Synapsin apposition analysis was performed using the ImageJ software to make a mask for the two proteins, selecting only the clusters that were considered to be specific and merging the two masks. GluA1-Synapsin spots were considered to be apposed if the distance between them was smaller or equal to 1 pixel [46]. These regions were used to determine the percentage of postsynaptic clusters apposed to a presynapse. Quantitative fluorescence data were exported from ImageJ to Origin Pro 8 software for further analysis and presentation.
2.8 Co-immunoprecipitation
The primary antibodies used for immunoprecipitation were a: rabbit polyclonal cGMP dependent kinase II antiserum (Antibodies-online GmbH, Aachen, Germany); a mouse anti-GluA1-NT (N-terminus) clone RH95 (ref.MAB2263: Millipore-Iberica), and a rabbit anti-GluA1 polyclonal antiserum (ref. PC246: Calbiochem). The secondary antibodies used were horseradish peroxidase-conjugated anti-rabbit IgG or anti-mouse IgG TrueBlot™ (1:1000: eBioscience, San Diego, CA). The proteins recovered were resolved by SDS-PAGE on 7.5% polyacrylamide gels and transferred to polyvinylidene difluoride membranes using a semi-dry transfer system, which were then probed with the antibody indicated and a horseradish peroxidase-conjugated secondary antibody. The immunoreactive bands were visualized by chemiluminescence (Pierce) and detected in a LAS-3000 imaging system (FujiFilm Life Science, Woodbridge, CT). For immunoprecipitation, membranes from the rat (P14) cerebellum were solubilized for 30 min on ice in radio immunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl [pH 7.4], 100 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.2% SDS and 1 mM EDTA). The solubilized preparation was then centrifuged at 13,000×g for 30 min. and the supernatant (1mg/ml) was processed for immunoprecipitation, each step of which was carried out with constant rotation at 0–4 °C. The supernatant was incubated overnight with the antibody indicated, before 50 μl of TrueBlot™ anti-rabbit Ig IP or anti-mouse Ig IP beads (eBioscience) were added and the mixture was again incubated overnight. Subsequently, the beads were washed twice with ice-cold RIPA buffer, twice with ice cold RIPA buffer diluted 1:10 in TBS (50 mM Tris-HCl [pH 7.4], 100 mM NaCl) and once with TBS, and they were aspirated to dryness with a 28-gauge needle. Subsequently, 100 μl of SDS-PAGE sample buffer (0.125 M Tris HCl [pH 6.8], 4% SDS, 20% glycerol, 0.004% bromphenol blue) was added to each sample, the immune complexes were dissociated by adding fresh dithiothreitol (DTT, 50 mM final concentration) and heating to 100°C for 10 min, and the proteins were resolved and detected as described above.
2.9 Fractionation of the cerebellum
Fourteen day old wild-type and homozygous cGKII knockout (cGKII−/−) [47] mice were anesthetized with CO2, sacrificed in compliance with New York University Medical Center’s Institutional Animal Care and Use Committee “Principles of Laboratory Animal Care” (NIH publication number 85–23), and the cerebellum was extracted and rapidly frozen in liquid nitrogen. After genotyping, we purified the brain fractions and PSDs, adapting a protocol described by Jordan et al. [48]. The cerebella were homogenized in 1 ml of solution A (0.32 mM Sucrose, 1 mM NaHCO3, 1 mM MgCl2 and 0.5 mM CaCl2) and part of the tissue was stored as the whole cell fraction. An aliquot of the homogenates was diluted 1:10 (weight/volume) with solution A and centrifuged at 1,000×g for 10 min. The supernatant was saved, and the pellet was re-homogenized in solution A and then further centrifuged at 1,000×g for 10 min. The supernatants were then spun at 15,000×g for 30 min to obtain the crude P2 fraction, and the pellet was resuspended in solution B (0.32 M Sucrose, 1mM NaHCO3). At this time a gradient of two different sucrose concentrations was prepared (5.5 ml of 1M sucrose on the top; 5.5 ml of 1.2 M sucrose on the bottom), and 1 ml of the sample was layered onto this gradient and centrifuged at 82,500×g for 2 hours. After the first centrifugation, the pellet was diluted five-fold in solution B and spun at 150,000×g for 30 minutes to obtain the synaptoneurosomal fraction. Part of the pellet was resuspended in a Tris/SDS solution at 4 °C and rocked for 30 minutes before storing this synaptoneurosome fraction. The other part was resuspended in a solution containing Triton X-100 1%, 0.32 M sucrose and 12 mM TrisHCl to obtain the crude PSD fraction. We applied a further 15,000×g centrifugation for 30 minutes, and the pellet resuspended in a Tris/SDS solution (12 mM TrisHCl and 4% SDS). Finally, the proteins were separated by SDS-PAGE, detected with the GluA1 and anti-S845-PO4 GluA1 antibodies, then normalized to the levels of beta-actin as a loading control and compared to the WT.
2.10 cGKII silencing
An Accell siRNA (1 μM) designed to selectively silence cGKII (target sequence: CUUUCAAGGACAAUAAAUA) or a negative control (random non-silencing sequence: Dharmacon, Thermo Fisher Scientific Inc., Rockford, IL USA) were added to the cells after 4 DIV in Accell medium supplemented with B27 (Invitrogen, Life Technologies), 20 mM KCl, 0.5 mM glutamine and the stabilized antibiotic antimycotic solution (Sigma-Aldrich). After 72 hours, the cells were fixed and immunocytochemistry was performed by the same protocol used in the imaging experiments described above. The silencing of cGKII was verified by measuring its mRNA levels by real time PCR 24 hours after transfection using the TaqMan® Gene Expression Assay Rn00435938_m1 for cGKII (Applied Biosystems, Life Technologies; Madrid, Spain) and Rn01775763_g1 for GAPDH as an endogenous control, as described previously [28].
2.11 Measurement of [Ca2+] in individual cerebellar granule cells
The calcium responses in individual ROIs along the dendrites were measured according to a previously described method [10]. Cerebellar granule cells were plated on polylysine-coated coverslips at a density of 100,000 cells/coverslip and they were maintained in culture for 7 DIV. In primary culture, cerebellar granule cells establish functional excitatory contacts with other granule cells. These cells were then incubated with FM1-43 dye (10 μM: Life Technologies) in high potassium buffer (NaCl 95mM, KCl 50mM, CaCl2 1.33mM, MgCl2 1mM, glucose 10mM and TES 10mM [pH 7.4]) for 2 minutes, in order to stain synaptic boutons. Then, they were loaded with Fura-2 acetoxy methylester (3 μM, Fura-2 AM) for 55 min at 37 °C in TES buffer (NaCl 140mM, KCl 5mM, CaCl2 1.33mM, MgCl2 1mM, glucose 10mM and TES 10mM [pH 7.4]) and stimulated as appropriate. At the beginning of each experiment an image of FM1-43 loaded cells was taken, excitation at 479-nm was provided by a monochromator and the emitted light was collected using a fluoresceinisothiocyanate (FITC) filter. Images were taken every second with a Nikon microscope equipped with a Nikon 40×1.3 (NA) oil-immersion objective. For Fura-2 measurements alternative excitation at 350-nm and 380-nm was performed and emitted light was collected through a band-pass filter centred at 510 nm. The video images were obtained using an iXonEM + EMCCD camera (ANDOR™ Technology) operating at 16-bits, and the output from the camera was stored on a computerized imaging system (Andor Software; Belfast, Northern Ireland). The image of FM1-43 loaded cells was used to identify the ROIs with Igor Pro software in order to create a binary mask of randomly selected fields. This mask was superimposed on Fura-2 images and the calcium concentration was measured in these areas, analysing only the regions that clearly overlap with a dendrite. Fura-2 experiments were analysed using the Ratio-plus plug-in of ImageJ software. The background of the two channels was subtracted choosing areas not illuminated by the fluorescent dye and the 350nm/380nm ratio was processed and represented using Origin 8.0 software.
2.12 Statistical analysis
The data was analyzed with the Statgraphic, OriginPro 7.5 or SigmaPlot 10 statistical software and the specific test applied in each case is indicated in the figure legends or the text. The data is shown as the mean ± SEM: * indicates p<0.05; ** indicates p<0.01; and *** p<0.001. ImageJ, Lucida Analyse 4 (Kinetic Imaging) and Soft Imaging Viewer (Soft Imaging System) software were used for image processing. The difference was considered statistically significant when p<0.05 with a confidence limit of 95%.
3. Results
3. 1 cGKII phosphorylates GluA1 at S845 in vivo
When maintained for several DIV, rat cerebellar granule cells express GluA1 mRNA and protein (Fig. 1A), in which residues S818, S831 and S845 are major phosphorylation sites and substrates for different protein kinases [35,38]. When cerebellar granule cells were incubated with 8-Br-cGMP (500 μM), an increase in S845 phosphorylation was detected using a phosphopeptide-specific Ab (Fig. 1B and 1D). Conversely, incubation with KT5823 (2 μM), a cGKII inhibitor, reduced the phosphorylation of this protein and prevented the increase produced by 8-Br-cGMP. In none of these conditions was the total amount of GluA1 in the cells altered. Incubation with NMDA plus glycine also enhanced S845 phosphorylation (Fig. 1C and 1E), which was similarly abolished by pre-treating the cells with KT5823 (2 μM), confirming that NMDAR requires cGKII activity to induce the phosphorylation of GluA1 (Fig. 1C and 1E). We concluded that cGKII activation under the regulation of the NMDAR and NO leads to S845 phosphorylation in these neurons.
Figure 1. GluA1 is expressed in cultured cerebellar granule cells and it is phosphorylated at S845 by cGMP dependent protein kinase.

(A) Expression of GluA1 (mRNA and protein) during the development of cerebellar granule cells in culture. Total RNA was extracted from freshly isolated cells (FIC), and cells 7 and 14 DIV. Equal amounts of mRNA were used for Reverse Transcription and then real time PCR reactions were performed with the specific primers described in the Methods. The results correspond to the mean ± SEM of four experiments performed in triplicate and they are expressed as 50-ΔCT. Total protein extracts (15 μg) obtained from cells 3, 7 and 14 DIV were resolved by electrophoresis and transferred to nitrocellulose membranes, which were probed with a rabbit polyclonal anti-GluA1 N-terminal specific antiserum. (B and C) Cells were incubated with the compounds indicated (see the Methods section: NMDA implies NMDA 50 μM + glycine 10 μM). Total GluA1 was assayed with a rabbit anti-GluA1 antiserum, phosphorylated GluA1 at S845 was assayed with a rabbit PO-S845-GluA1 antiserum and β-tubulin was used as a loading control. (D and E) The diagrams show the quantification of several experiments, where the phospho-specific signal was normalized to the total amount of GluA1 and the final values were normalized to the controls.(D, Control: 1; 8Br-cGMP: 1.30±0.03; KT5823: 0.79±0.04; 8Br-cGMP+KT: 0.75±0.08; E, Control: 1; NMDA: 1.24±0.02; KT5823: 0.63±0.05; NMDA+KT: 0.82±0.09). The data are represented as the mean ± SEM and analyzed with an unpaired, two-sample t test (*p < 0.05, n = 7).
We examined whether the level of GluA1 or its phosphorylaton at S845 was modified in the cerebellum of homozygous cGKII knockout mice (cGKII−/−, KO: 47), comparing distinct cerebellar cell fractions from wild type (WT) and cGKII−/− mice (see Methods). The amount of GluA1 was similar in the whole homogenate in both WT and KO mice (Fig. 2A and B), whereas there was clearly less GluA1 in the P2 fraction of the KO mice. Similarly, there was a reduction in the amount of GluA1 in the synaptoneurosome and PSD fraction from the KO mice (Fig. 2A and B), which was probably responsible for the small reduction in S845-GluA1 phosphorylation observed in these fractions (Fig. 2C and D), as the pGluA1/GluA1 ratio remained constant in all the subcellular fractions analyzed (Fig. 2E).
Figure 2. Reduced GluA1 in the synaptic fraction of cGKII KO (cGKII−/−) mice.

Subcellular fractions of the cerebellum from 14 day old WT and cGKII−/− mice were analyzed in immunoblots. P2: crude membranes; syn: synaptoneurosome; PSD: postsynaptic density. (B) Quantification of GluA1 immunoreactivity normalized to β-actin and finally, to WT values (*p < 0.05, n = 3). (C) Cerebellar fractions probed in immunoblots with a rabbit PO-S845-GluA1 antiserum. (D) Quantification of GluA1 phosphorylation in the synaptoneurosomal and crude PSD fractions. (E). Ratio between the phosphorylated and total GluA1 in the synaptic fractions. Data are represented as the mean ± SEM and analyzed using unpaired, two-sample t test (n=3; *p<0.05).
3.2 Interaction of GluA1 and cGKII in cerebellar membranes
To assess the physiological relevance of the interaction between GluA1 and cGKII, co-immunoprecipitation experiments were performed on rat (P14) cerebellar homogenates. The anti-GluA1 antibody immunoprecipitated a protein of around 100 kDa from soluble cerebellum extracts that as demonstrated previously [32], corresponded to GluA1 (Fig. 3, IB: anti-GluA1, lane 4). Similarly, an anti-cGKII antibody immunoprcipitated a protein of around 85 kDa that appeared to correspond to cGKII (Fig. 3, IB: anti-cGKII, lane 2: 32). Moreover, the anti-GluA1 antibody was able to co-immunoprecipitate cGKII (Fig. 3, IB: anti-cGKII, lane 4) from these extracts and the anti-cGKII antibody GluA1 (Fig. 3, IB: anti-GluR1, lane 2). These protein bands were not evident when an irrelevant mouse or rabbit IgG was used for immunoprecipitation (Fig. 3, lane 1 and 3, respectively), confirming the specificity of the reaction. Accordingly, these results suggest that GluA1 and cGKII might assemble into stable protein-protein complexes in the rat cerebellum, which survive the conditions required for solubilisation and co-immunoprecipitation, suggesting that these oligomeric complexes might be physiologically relevant in vivo.
Figure 3. Co-immunoprecipitation of GluR1 and cGKII from the rat cerebellum.
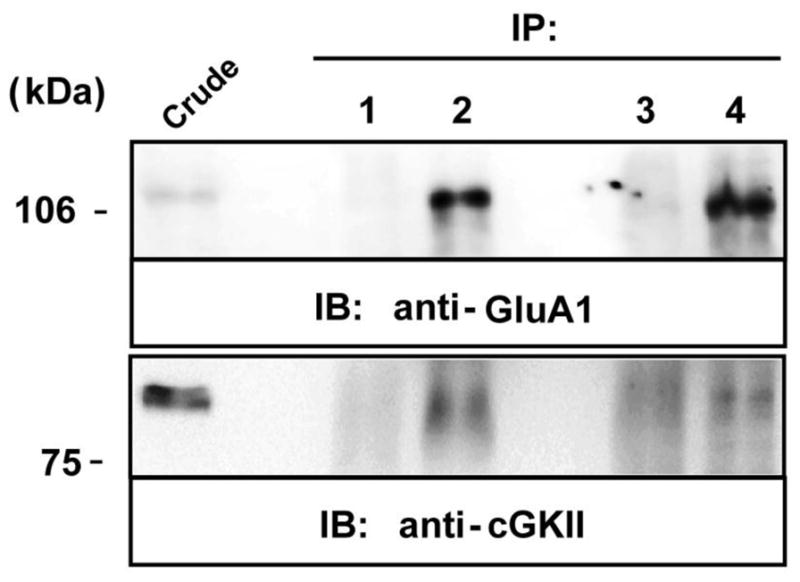
Solubilized extracts of the rat (P14) cerebellum were immunoprecipitated using a mouse anti-flag antibody (2 μg, lane 1), rabbit anti-cGKII polyclonal antiserum (2 μg, lane 2), rabbit anti-HA antiserum (2 μg, lane3), a rabbit anti-GluA1 polyclonal antiserum (2 μg, lane 4) or a mouse anti-GluA1 monoclonal antibody (2mg, lane 4 of cGKII IB). Extracts (Crude) and immunoprecipitates (IP) were analyzed in immunoblots probed with a rabbit anti-GluA1 polyclonal antiserum (1 μg/ml) and rabbit anti-cGKII (1 μg/ml) antiserum. HRP-conjugated anti-rabbit IgG and anti-mouse TrueBlot™ (1:1000) antibodies were used as secondary antibodies to avoid IgG cross-reactivity. The immunoreactive bands were visualized by chemiluminescence.
3.3 Activation of cGKII augments the GluA1 at the cell surface
Exposing granule neurons in culture to 8-Br-cGMP (500 μM) caused a significant increase in the density of GluA1 clusters at the cell surface (cluster/μm length of dendrite), from 0.27 ± 0.012 in control cells to 0.36 ± 0.016 (p<0.01: Fig. 4A and 4B). This increase was prevented by preincubation with KT5823 (2 μM, added 10 min before 8-Br-cGMP; 0.18 ± 0.014: Fig. 4A and 4B), suggesting that cGKII mediates the cGMP-dependent accumulation of GluA1 at the plasma membrane. Moreover, the mean fluoresence intensity of the clusters was also greater in cells treated with 8-Br-cGMP (control: 22.50±0.55 a.u.; 8-Br-cGMP: 29±0.74 a.u., p<0.01), although both the surface GluA1 cluster density (0.19±0.02) and the mean labelling intensity (15±0.32 a.u.) decreased significantly when these neurons were incubated with KT5823 (2 μM: Fig. 4A, 4B and 4C), again indicating that this signalling pathway is active in control conditions [29]. Taken together all these results indicate that the phosphorylation of S845 is required for the cGMP-dependent increase in GluA1 surface expression.
Since PKA only increases surface GluA1 at extrasynaptic sites [41,42], we assayed the surface location of GluA1 following 8-Br-cGMP application to determine if cGKII produces a similar effect. We measured synaptic GluA1 by quantifying the proportion of GluA1 clusters apposed to the synaptic marker, synapsin I (see Methods section). In control conditions, surface 22±0.43 % of GluA1 clusters were apposed to synapsin I (Fig. 4C) and 8-Br-cGMP did not change the percentage of synaptic GluA1 puncta apposed to synapsin I (22±0.74%; p>0.05: Fig. 4C). Neither the density nor the intensity of the synapsin I posive puncta changed in any of the experimental conditions (results not shown). Since the total GluA1 positive cluster increased in cells incubated with 8-Br-cGMP, although the percentage of apposition was equal the absolute value also increased. These results indicate that the new receptors are likely to be incorporated into both synaptic and extrasynaptic areas of the membrane. Conversely, a reduction in plasma membrane GluA1 was induced in the presence of KT5823, as there was a clear reduction in the GluA1 clusters apposed to synapsin I both in control conditions and in cells treated with 8-Br-cGMP (15.6±0.49 %; p<0.05).
The stimulation of cells with NMDA and glycine also produced an increase in GluA1 cluster density and intensity similar to that produced by 8-Br-cGMP (Fig. 5A and B). Moreover, KT5823 reduced this increase in cluster density, counteracting the effect of NMDA. Indeed, while the slight increase in the intensity of GluA1 puncta produced by NMDA was reduced by KT5823 (Fig. 5D), NMDA did not modify the percentage of GluA1 positive puncta apposed to synapsin I, although KT5823 did reduce this parameter (Fig. 5C).
We assumed that the cGMP-dependent activation of GKII depends on NO produced by NOS upon NMDA receptor activation. In order to more directly confirm that, we analyzed whether two different NOS inhibitors (L-NMMA and NPLA) were able to abolish the effect of NMDA receptor activation on plasma membrane GluA1 increase. As it shown in the supplementary figure S1 these two inhibitors suppressed the NMDA effect and also reduced the control values as KT5823 did. On the other hand, when cells were incubated for ten minutes with either 10 μM FK-409 or 2 μM Angeli’s salt the cell surface GluA1 immunoreactivity increased (Fig. S2). These results confirm the involvement of NO in the NMDA stimulated signaling that triggers GluA1 phosphorylation and incorporation into plasma membrane.
3.4 cGKII silencing decreases surface GluA1
Cerebellar granule cells were transfected with a specific siRNA to silence cGKII, which effectively reduced the amount of cGKII mRNA to 22±0.003 % that of the control levels 48 hours after transfection (as determined by real time PCR). There was a clear reduction in plasma membrane GluA1 in these cells (Fig. 6A, B and C), without any change in total GluA1 or GluA4 (results not shown). The non-silencing siRNA used as a control did not modify the levels of GluA1 in the membrane, ruling out any non-specific effects of siRNA transfection. Moreover, the labelling of MAP2, used as an internal labelling control, did not change in any of the experimental conditions (Fig. 6D).
3.5 The surface increase in GluA1 is accompanied by an increase in calcium permeable AMPARs
To examine whether the increase in GluA1 subunits at the plasma membrane affected the response to AMPA stimulation, we measured the AMPA-elicited calcium increases in cell processes. Since FURA2-AM produces homogeneous labelling and in order to analyze calcium responses in discrete regions, we labelled synaptic boutons at the beginning of the experiment with the styryl dye FM1-43. After washing the excess dye, a picture was obtained (Fig. 7A) to use as a mask that could be superimposed onto the FURA-2 images and that served to analyze the calcium responses in dendritic ROIs the nearest possible to the synapses. Taking advantage of the selective blockage of calcium permeable AMPAR by 1-naphthyl acetyl spermine (NASPM), a synthetic analogue of Joro spider toxin (JSTX), we analyzed the contribution of calcium permeable AMPARs to each individual response in these regions. Different responses were observed in terms of the capacity of NASPM to block the increase in calcium elicited by AMPA (Fig. 7B). The response to AMPA (50 μM) plus cyclothiazide (10 μM) was enhanced by pretreatment with 8-Br-cGMP or NMDA in 72% and 55% of the ROIs respectively, while it decreased in the presence of the cGKII inhibitor, KT5823 or NOS inhibitors (Fig. 7C and D). A dampened response to AMPA was also observed in cGKII knockdown cells and in these cells, KT5823 did not produce any additional effect (Fig. 7C). In cells in which the AMPA response was increased by exposure to 8-Br-cGMP or NMDA, the increase in calcium elicited by AMPA was almost completely inhibited by NASPM (Fig. 7E and 7F). Conversely, in cells preincubated with either the cGKII inhibitor or NOS inhibitors or in which cGKII was silenced, conditions in which GluA1 subunits are removed from the plasma membrane, the inhibition produced by NASPM was clearly diminished (Fig. 7E and 7F).
Figure 7. The [Ca2+] in individual cerebellar granule cells.

(A) Scheme of the experiment and representative pictures showing Fura 2-AM, FM1-43 fluorescence and the binary mask obtained from the FM1-43 image (scale bar: 10μm). (B) Representative AMPA elicited calcium responses and their inhibition by NASPM: AMPA (50 μM) + cyclothiazide (10 μM: 1st stimulus), AMPA (50 μM) + cyclothiazide (10 μM) in the presence of NASPM (10 μM: 2nd stimulus), and KCl (50 mM) used as a control of depolarization (3rd stimulus). (C) Δ(F/F0) of the AMPA response for each condition expressed as the mean ± SEM (control, 1,363 ROIs analyzed from 9 experiments; 8Br-cGMP, 823 ROIs from 6 experiments; NMDA, 341 ROIs from 5 experiments; KT5823, 817 ROIs from 9 experiments; 8Br+KT, 315 ROIs from 6 experiments; NMDA+KT, 454 ROIs from 5 experiments; cGKII KD, 1,138 ROIs from 4 experiments; cGKII KD+KT, 882 ROIs from 4 experiments). (D) Δ(F/F0) of the AMPA response for each condition expressed as the mean ± SEM (control, 220 ROIs analyzed from 2 experiments; NMDA, 140 ROIs from 2 experiments; L-NMMA, 130 ROIs from 2 experiments; NMDA+L-NMMA, 145 ROIs from 2 experiments; NPLA, 200 ROIs from 2 experiments; NPLA+ NMDA, 108 ROIs from 2 experiments). (E) Cumulative probability plots of the percentage of inhibition produced by NASPM of the AMPA-induced calcium responses in the different experimental conditions (p<0.05, Kolmogorov–Smirnov tests). (F) Cumulative probability plots of the percentage of inhibition produced by NASPM of the AMPA-induced calcium responses in the different experimental conditions (p<0.05, Kolmogorov–Smirnov tests). Data from C and D are represented as the mean ± SEM and the statistical significance was analyzed using an unpaired, two-sample t test and one way ANOVA followed by Bonferroni’s test (*p<0.05).
4. Discussion
We show here that cGKII phosphorylates GluA1 at S845 in cerebellar granule cells, thereby enhancing its insertion into the plasma membrane and the formation of functional calcium permeable receptors sensitive to NASPM. Moreover, stimulation of the NMDAR activates cGKII, also increasing GluA1 phosphorylation and its plasma membrane insertion. Notably, a cGKII siRNA that effectively silenced the production of cGKII mRNA reduces the GluA1 in the plasma membrane, and in the cerebellar PSD fraction of cGKII KO mice, less S845 pGluA1 and total GluA1 accumulates than in the same fraction of WT mice. Moreover, the co-immunoprecipitation of GluA1 and cGKII from cerebellar membranes isolated from 14 day old rats suggest that these proteins might assemble into stable complexes in the rat cerebellum, as described in hippocampus [32]. Since we did not stimulate cGMP production in our preparation, our results are consistent with the constitutive presence of this complex and that the cGKII activation would probably lead to the phosphorylation of GluA1 and its incorporation into the plasma membrane. These results demonstrate the existence of a GluA1 trafficking pathway regulated by NO and cGKII that leads to an increase in calcium permeable AMPARs at the cell surface and, as a consequence, enhanced Ca2+ entry in response to cell stimulation.
The enhancement of GluA1 membrane insertion by NMDA-NO-cGKII signalling has been demonstrated previously in hippocampal and cortical neurons, where there is evidence that this mechanism is relevant in LTP [32]. An important mechanism underlying NMDAR-dependent LTP involves the insertion of GluA1 into synapses [49,50] and while S845 phosphorylation by cGKII enhances the insertion of GluA1 into the membrane at extrasynaptic sites, this may prime the system for subsequent potentiation by a tetanic stimulation [32]. Conversely, it has also been shown that activation of the NO-cGMP-cGKI pathway increases both GluA1 and synaptophysin puncta in hippocampal neurons, suggesting that such signalling can act presynaptically and postsynaptically to form new synaptic contacts [51].
There is growing evidence of a significant overlap between signalling pathways involved in differentiation at early developmental stages and those that later mediate plasticity in mature neurons [52,53]. Because cerebellar granule cells express GluA1 early in development, this mechanism might help strengthen existing connections or the formation of new ones during the functional development triggered by NO and cGMP.
In this sense, both 8-Br-cGMP and NMDA induce an increase in the density of synapsin puncta [30] and GluA1 clusters, effects that are counteracted and reversed by KT5823. The increase in cluster density was accompanied by an increase in immunofluorescence intensity compatible with the presence of more protein in these clusters, while conversely, the reduced cluster density provoked by exposure to KT5823 was accompanied by a reduction in their mean fluorescence. In 7 DIV cerebellar granule cells, GluA1 is mainly located extrasynaptically since less than 25% of GluA1 clusters are juxtaposed to synapsin puncta. Both 8-Br-cGMP and NMDA increased the GluA1 cluster apposed to synapsin I positive puncta although they did not augmented the percentages, however KT5823 significantly diminished the proportion of GluA1 clusters juxtaposed to synapsin puncta. Hence, S845 dephosphorylation caused a loss of plasma membrane GluA1, a reduction that may also occur in synapses. S845 is dephosphorylated during hippocampal LTD where the removal of GluA1 containing AMPAR from synapses occurs [54]. Moreover, in the cGKII KO mouse there is less S845-pGluA1 in the PSD fraction than in WT mice, which reflected a reduction in total GluA1. Although GluA1-S845 is also a substrate for PKA [38], the results presented here indicate that cGKII plays a central role in regulating the level of GluA1 at synapses in the cerebellum.
Calcium permeable AMPARs are expressed early in development [55,56] and although their role has yet to be fully elucidated, it has been suggested that they may mediate localized and regulated Ca2+ entry that is pivotal in the development and maturation of neurons [56–58]. The presence of calcium permeable AMPARs in the membrane of granule neurons and their up-regulation by cGKII activation might contribute to the activity- and NO-dependent survival and differentiation of neurons [28–31,59]. The role of the NO-cGMP pathway in cerebellar development and function has been firmly demonstrated. The activity-dependent maturation of granule cells in vivo is triggered by mossy fibre inputs [60] and mossy fibre stimulation causes a significant NMDAR- and NOS-dependent release of NO in the granular layer of rat cerebellar slices [61]. NO synthesized downstream of NMDAR activation plays a central role in neuronal activity, mediating important events like neurotransmitter release, activity-dependent survival and long-term potentiation [13,62,63]. Since NO is a diffusible signal, it may not only affect the synapse where it is produced but also, neighbouring synapses and cells [64]. In culture, cerebellar granule cells establish synapses with themselves and their development is also affected by the NO and cGMP pathway. While NO negatively regulates precursor proliferation through a cGMP-independent mechanism [65], it also triggers differentiation and maturation by a cGMP-dependent one [30,31,66].
In summary, our data clearly demonstrate that at early developmental stages, cGKII regulates calcium permeable AMPAR density at the plasma membrane of cerebellar granule cells through the phosphorylation of S845 from GluA1. It remains to be elucidated to what extent this mechanism is relevant for the development and maturation of these cells.
Supplementary Material
(A) Cerebellar granule cells maintained for 8 DIV were treated for 10 minutes with NMDA (50 μM) + glycine (10 μM), for 60 minutes with either L-NMMA (10 μM) or NPLA (1 μM) or 60 minutes with NOS inhibitor plus NMDA (50 mM) + glycine (10 μM) added the latest 10 minutes. Then cells were fixed and labelled with a GluA1-NT antibody (15 μg/ml) over 120 minutes at 37 °C. The images represent the immunostaining of synapsin I and GluA1, the merged image and an amplification of each condition. (B) The density of GluA1 clusters in dendrites (clusters/μm), selecting ROI’s of the specific antibody from at least 15 fields from 2 different experiments for each condition. (C) Analysis of the fraction of GluA1 clusters apposed to synapsin I puncta, either upon stimulation or inhibition (n=30 fields from two independent experiments). (D) Cumulative probability plots of GluA1 mean fluorescence intensity (mean values: control 18.51±0.06; NMDA 21.44±0.05; L-NMMA 8.58±0.06; L-NMMA+NMDA 10.18±0.07; NPLA 7.91±0.04; NPLA+NMDA 8.66±0.06, a.u.). In B and C, the data are represented as the mean ± SEM and analyzed using unpaired, two-sample t test (*p<0.001). For the cumulative probability the Komolgorov-Smirnov test was used. Scale bars: 10 μm.
(A) Cerebellar granule cells maintained for 8 DIV were treated for 10 minutes with either Angeli’s salt (2 μM) or FK409 (10 μM) or maintained in control conditions. Then cells were fixed and labelled with a GluA1-NT antibody (15 μg/ml) over 120 minutes at 37 °C. The images represent the immunostaining of synapsin I and GluA1, the merged image and an amplification of each condition. (B) The density of GluA1 clusters in dendrites (clusters/μm), selecting ROI’s of the specific antibody from at least 15 fields from 2 different experiments for each condition. (C) Analysis of the fraction of GluA1 clusters apposed to synapsin I puncta, either upon stimulation or inhibition (n=30 fields from two independent experiments). (D) Cumulative probability plots of GluA1 mean fluorescence intensity (mean values: control 8.47±0.06; Angeli’s salt 13.12±0.08; FK409 13.40±0.07, a.u.). In B and C, the data are represented as the mean ± SEM and analyzed using unpaired, two-sample t test (*p<0.001). For the cumulative probability the Komolgorov-Smirnov test was used. Scale bars: 10 μm.
Research highlights.
Developing rat cerebellar granule cells express GluA1 AMPARs at early developmental stages
cGKII phosphorylates GluA1 at Ser845 and augments the GluA1 at the cell surface
NMDAR stimulation leads to cGKII activation and GluA1 phosphorylation.
cGKII inhibition or knockdown leads to a decrease of GluA1 expression in the synaptic membranes both in vivo and in vitro.
The surface increase in GluA1 is accompanied by an increase in calcium permeable AMPARs
Acknowledgments
This study was financed by grants from the Ministerio de Economía y Competitividad (BFU2009-07092/BFI), the Comunidad de Madrid (S2011/BMD-2349), the UCM (GR35/10-A-920307) and the US National Institute of Mental Health, (5R01MH067229). Salvatore Incontro was supported by a fellowship from the former Ministerio de Ciencia e Innovación. We thank Dr M. Sefton for editorial assistance and Ma Carmen Zamora for technical assistance.
Abbreviations
- AMPA
α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate
- AMPAR
AMPA receptor
- Angeli’s salt
disodium diazem-1-ium-1,2,2-triolate
- CaCMKII
Ca2+-calmodulin kinase II
- cGK
cGMP-dependent protein kinase
- cGKI
cGMP-dependent protein kinase type I
- cGKII
cGMP-dependent protein kinase type II
- CTD
C-terminus domain
- DAPI
4′,6-diamino-phenylindole dihydrochloride
- DIV
days in vitro
- FIC
freshly isolated cells
- FITC
fluoresceinisothiocyanate
- FK409
4-ethyl-2E-(hydroxyimino)-5-nitro-3E-hexenamide
- GAPDH
glyceraldehyde phosphate dehydrogenase
- IRD
near-infrared fluorescent dye
- JSTX
Joro spider toxin
- KA
kainate
- L-NMMA
L-NG-Monomethyl Arginine citrate
- LTD
long term depression
- LTP
long term potentiation
- MAP2
microtubule-associated protein 2
- NASPM
1-naphthyl acetyl spermine (NASPM)
- NMDA
N-methyl-D-aspartate
- NMDAR
NMDA receptor
- nNOS
neuronal nitric oxide synthase
- NO
nitric oxide
- NPLA
Nω-Propyl-L-Arginine
- PKA
cAMP dependent protein kinase
- PKC
protein kinase C
- PSD-95
postsynaptic density-95 protein
- ROI
region of interest
- sGC
soluble guanylyl cyclase
- siRNA
small interfering RNA
- TES
N-Tris (hydroxymethyl)methyl-2-aminoethanesulfonic acid
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- 2.Borges K, Dingledine R. AMPA receptors: Molecular and functional diversity. Prog Brain Res. 1998;116:153–170. doi: 10.1016/s0079-6123(08)60436-7. [DOI] [PubMed] [Google Scholar]
- 3.Wenthold RJ, Roche KW. The organization and regulation of non-NMDA receptors in neurons. Prog Brain Res. 1998;116:133–152. doi: 10.1016/s0079-6123(08)60435-5. [DOI] [PubMed] [Google Scholar]
- 4.Seeburg PH, Higuchi M, Sprengel R. RNA editing of brain glutamate receptor channels: mechanism and physiology. Brain Res Brain Res Rev. 1998;26:217–229. doi: 10.1016/s0165-0173(97)00062-3. [DOI] [PubMed] [Google Scholar]
- 5.Losi G, Prybylowski K, Fu ZY, Luo JH, Vicini S. Silent synapses in developing cerebellar granule neurons. J Neurophysiol. 2002;87:1263–1270. doi: 10.1152/jn.00633.2001. [DOI] [PubMed] [Google Scholar]
- 6.Condorelli DF, Dell’Albani P, Aronica E, Genazzani AA, Casabona G, Corsaro M, Balázs R, Nicoletti F. Growth conditions differentially regulate the expression of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionate (AMPA) receptor subunits in cultured neurons. J Neurochem. 1993;61:2133–2139. doi: 10.1111/j.1471-4159.1993.tb07451.x. [DOI] [PubMed] [Google Scholar]
- 7.Kamboj SK, Swanson GT, Cull-Candi SG. Intracellular spermine confers rectification on rat calcium-permeable AMPA and kainate receptors. J Physiol. 1995;486:297–303. doi: 10.1113/jphysiol.1995.sp020812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumar SS, Bacci A, Kharazia V, Huguenard JR. A developmental switch of AMPA receptor subunits in neocortical pyramidal neurons. J Neurosci. 2002;22:3005–3015. doi: 10.1523/JNEUROSCI.22-08-03005.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eybalin M, Caicedo A, Renard N, Ruel J, Puel JL. Transient Ca2+-permeable AMPA receptors in postnatal rat primary auditory neurons. Eur J Neurosci. 2004;20:2981–2989. doi: 10.1111/j.1460-9568.2004.03772.x. [DOI] [PubMed] [Google Scholar]
- 10.Incontro S, Ramírez-Franco J, Sánchez-Prieto J, Torres M. Membrane depolarization regulates AMPA receptor subunits expression in cerebellar granule cells in culture. Biochim Biophys Acta. 2011;1813:14–26. doi: 10.1016/j.bbamcr.2010.10.016. [DOI] [PubMed] [Google Scholar]
- 11.Garthwaite J, Garthwaite G, Hajos F. γ-Aminobutyric acid affects the developmental expression of neuron-associated proteins in cerebellar granule cell culture. J Neurochem. 1986;46:1256–1262. doi: 10.1111/j.1471-4159.1986.tb00647.x. [DOI] [PubMed] [Google Scholar]
- 12.Balazs R, Jorgensen OS, Hack N. N-Methyl-D-aspartate promotes the survival of cerebellar granule cells in culture. Neuroscience. 1988;27:437–451. doi: 10.1016/0306-4522(88)90279-5. [DOI] [PubMed] [Google Scholar]
- 13.Contestabile A. Roles of NMDA receptor activity and nitric oxide production in brain development. Brain Res Rev. 2000;32:476–509. doi: 10.1016/s0165-0173(00)00018-7. [DOI] [PubMed] [Google Scholar]
- 14.Garthwaite J, Boulton CL. Nitric oxide signalling in the central nervous system. Annu Rev Physiol. 1995;57:683–706. doi: 10.1146/annurev.ph.57.030195.003343. [DOI] [PubMed] [Google Scholar]
- 15.Lincoln TM, Cornwell TL. Intracellular cyclic GMP receptor proteins. FASEB J. 1993;7:328–338. doi: 10.1096/fasebj.7.2.7680013. [DOI] [PubMed] [Google Scholar]
- 16.Francis SH, Corbin JD. Structure and function of cyclic nucleotide-dependent protein kinases. Annu Rev Physiol. 1994;56:237–272. doi: 10.1146/annurev.ph.56.030194.001321. [DOI] [PubMed] [Google Scholar]
- 17.Vaandrager AB, Tilly BC, Smolenski A, Schneider-Rasp S, Bot AG, Edixhoven M, Scholte BJ, Jarchau T, Walter U, Lohmann SM, Poller WC, de Jonge HR. cGMP stimulation of cystic fibrosis transmembrane conductance regulator Cl- channels co-expressed with cGMP-dependent protein kinase type II but not type I beta. J Biol Chem. 1997;272:4195–4200. doi: 10.1074/jbc.272.7.4195. [DOI] [PubMed] [Google Scholar]
- 18.Sandberg M, Natarajan V, Ronander I, Kalderon D, Walter U, Lohmann SM, Jahnsen T. Molecular cloning and predicted full-length amino acid sequence of the type I beta isozyme of cGMP-dependent protein kinase from human placenta. Tissue distribution and developmental changes in rat. FEBS Lett. 1989;255:321–329. doi: 10.1016/0014-5793(89)81114-7. [DOI] [PubMed] [Google Scholar]
- 19.Wernet W, Flockerzi V, Hofmann F. The cDNA of the two isoforms of bovine cGMP-dependent protein kinase. FEBS Lett. 1989;251:191–196. doi: 10.1016/0014-5793(89)81453-x. [DOI] [PubMed] [Google Scholar]
- 20.Lohmann SM, Walter U, Miller PE, Greengard P, De Camilli P. Immunohistochemical localization of cyclic GMP-dependent protein kinase in mammalian brain. Proc Natl Acad Sci USA. 1981;78:653–657. doi: 10.1073/pnas.78.1.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.El-Husseini AE, Williams J, Reiner PB, Pelech S, Vincent SR. Localization of the cGMP-dependent protein kinases in relation to nitric oxide synthase in the brain. J Chem Neuroanat. 1999;17:45–55. doi: 10.1016/s0891-0618(99)00023-x. [DOI] [PubMed] [Google Scholar]
- 22.Kleppisch T, Pfeifer A, Klatt P, Ruth P, Montkowski A, Fässler R, Hofmann F. Long-term potentiation in the hippocampal CA1 region of mice lacking cGMP-dependent kinases is normal and susceptible to inhibition of nitric oxide synthase. J Neurosci. 1999;19:48–55. doi: 10.1523/JNEUROSCI.19-01-00048.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arancio O, Antonova I, Gambaryan S, Lohmann SM, Wood JS, Lawrence DS, Hawkins RD. Presynaptic role of cGMP-dependent protein kinase during long-lasting potentiation. J Neurosci. 2001;21:143–149. doi: 10.1523/JNEUROSCI.21-01-00143.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.El-Husseini AE, Bladen C, Vincent SR. Molecular characterization of a type II cyclic GMP-dependent protein kinase expressed in the rat brain. J Neurochem. 1995;64:2814–2817. doi: 10.1046/j.1471-4159.1995.64062814.x. [DOI] [PubMed] [Google Scholar]
- 25.De Vente J, Asan E, Gambaryan S, Markerink-van Ittersum M, Axer H, Gallatz K, Lohmann SM, Palkovits M. Localization of cGMP-dependent protein kinase type II in rat brain. Neuroscience. 2001;108:27–49. doi: 10.1016/s0306-4522(01)00401-8. [DOI] [PubMed] [Google Scholar]
- 26.Lein ES, Hawrylyc MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, Chen L, Chen L, Chen TM, Chin MC, Chong J, Crook BE, Czaplinska A, Dang CN, Datta S, Dee NR, Desaki AL, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- 27.Ciani E, Virgili M, Contestabile A. Akt pathway mediates a cGMP-dependent survival role of nitric oxide in cerebellar granule neurons. J Neurochem. 2002;81:218–228. doi: 10.1046/j.1471-4159.2002.00857.x. [DOI] [PubMed] [Google Scholar]
- 28.Jurado S, Sánchez-Prieto J, Torres M. Differential expression of nitric oxide-sensitive guanylyl cyclase subunits during the development of rat cerebellar granule cells: regulation via N-methyl-D-aspartate receptors. J Cell Sci. 2003;116:3165–3175. doi: 10.1242/jcs.00620. [DOI] [PubMed] [Google Scholar]
- 29.Jurado S, Sánchez-Prieto J, Torres M. Elements of the nitric oxide/cGMP pathway expressed in cerebellar granule cells: Biochemical and functional characterisation. Neurochem Int. 2004;45:833–843. doi: 10.1016/j.neuint.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 30.López-Jiménez ME, Bartolomé-Martín D, Sánchez-Prieto J, Torres M. Suppression of beta1 subunit of NO-dependent guanylyl cyclase expression impairs neurite outgrowth and synaptogenesis in granule cells. Cell Death Differ. 2009;16:1266–1278. doi: 10.1038/cdd.2009.57. [DOI] [PubMed] [Google Scholar]
- 31.López-Jiménez ME, González JC, Lizasoain I, Sánchez-Prieto J, Hernández-Guijo JM, Torres M. Functional cGMP-gated channels in cerebellar granule cells. J Cell Physiol. 2012;227:2252–2263. doi: 10.1002/jcp.22964. [DOI] [PubMed] [Google Scholar]
- 32.Serulle Y, Zhang S, Ninan I, Puzzo D, McCarthy M, Khatri L, Arancio O, Ziff EB. A GluR1-cGKII interaction regulates AMPA receptor trafficking. Neuron. 2007;56:670–688. doi: 10.1016/j.neuron.2007.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leonard AS, Davare MA, Horne MC, Garner CC, Hell JW. SAP97 is associated with the alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor GluR1 subunit. J Biol Chem. 1998;273:19518–19524. doi: 10.1074/jbc.273.31.19518. [DOI] [PubMed] [Google Scholar]
- 34.Shen L, Liang F, Walensky LD, Huganir RL. Regulation of AMPA receptor GluR1 subunit surface expression by a 4.1N-linked actin cytoskeletal association. J Neurosci. 2000;20:7932–7940. doi: 10.1523/JNEUROSCI.20-21-07932.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boehm J, Malinow R. AMPA receptor phosphorylation during synaptic plasticity. Biochem Soc Trans. 2005;33:1354–1356. doi: 10.1042/BST0331354. [DOI] [PubMed] [Google Scholar]
- 36.Barria A, Muller D, Derkach V, Griffith LC, Soderling TR. Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science. 1997;276:2042–2045. doi: 10.1126/science.276.5321.2042. [DOI] [PubMed] [Google Scholar]
- 37.Mammen AL, Kameyama K, Roche KW, Huganir RL. Phosphorylation of the alpha-amino-3-hydroxy-5-methylisoxazole4-propionic acid receptor GluR1 subunit by calcium/calmodulin-dependent kinase II. J Biol Chem. 1997;272:32528–32533. doi: 10.1074/jbc.272.51.32528. [DOI] [PubMed] [Google Scholar]
- 38.Roche KW, O’Brien RJ, Mammen AL, Bernhardt J, Huganir RL. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron. 1996;16:1179–1188. doi: 10.1016/s0896-6273(00)80144-0. [DOI] [PubMed] [Google Scholar]
- 39.Boehm J, Kang MG, Johnson RC, Esteban JA, Huganir RL, Malinow R. Synaptic incorporation of AMPA receptors during LTP is controlled by a PKC phosphorylation site on GluR1. Neuron. 2006;51:213–225. doi: 10.1016/j.neuron.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 40.Esteban JA, Shi SH, Wilson C, Nuriya M, Huganir RL, Malinow R. PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat Neurosci. 2003;6:136–143. doi: 10.1038/nn997. [DOI] [PubMed] [Google Scholar]
- 41.Oh MC, Derkach VA, Guire ES, Soderling TR. Extrasynaptic membrane trafficking regulated by GluR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. J Biol Chem. 2006;281:752–758. doi: 10.1074/jbc.M509677200. [DOI] [PubMed] [Google Scholar]
- 42.Sun X, Zhao Y, Wolf ME. Dopamine receptor stimulation modulates AMPA receptor synaptic insertion in prefrontal cortex neurons. J Neurosci. 2005;25:7342–7351. doi: 10.1523/JNEUROSCI.4603-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ridler TW, Calvard S. Picture thresholding using an iterative selection method, IEEE Trans System. Man and Cybernetics. 1978;8:630–632. [Google Scholar]
- 44.Sternberg SR. Biomedical image processing. Computer. 1983;16:22–34. [Google Scholar]
- 45.Meijering E, Jacob M, Sarria JCF, Steiner P, Hirling H, Unser M. Design and validation of a tool for neurite tracing and analysis in fluorescence microscopy. Cytometry Part A. 2004;58A:167–176. doi: 10.1002/cyto.a.20022. [DOI] [PubMed] [Google Scholar]
- 46.Mondin M, Labrousse V, Hosy E, Heine M, Tessier B, Levet F, Poujol C, Blanchet C, Choquet D, Thoumine O. Neurexin-Neuroligin Adhesions Capture surface diffusing AMPA Receptors through PSD-95 Scaffolds. J Neurosci. 2011;31:13500–13515. doi: 10.1523/JNEUROSCI.6439-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pfeifer A, Aszódi A, Seidler U, Ruth P, Hofmann F, Fässler R. Intestinal secretory defects and dwarfism in mice lacking cGMP-dependent protein kinase II. Science. 1996;274:2082–2086. doi: 10.1126/science.274.5295.2082. [DOI] [PubMed] [Google Scholar]
- 48.Jordan BA, Fernholz BD, Boussac M, Xu C, Grigorean G, Ziff EB, Neubert TA. Identification and verification of novel rodent postsynaptic density proteins. Mol Cell Proteomics. 2004;3:857–871. doi: 10.1074/mcp.M400045-MCP200. [DOI] [PubMed] [Google Scholar]
- 49.Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- 50.Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 51.Wang HG, Lu FM, Jin I, Udo H, Kandel ER, de Vente J, Walter U, Lohmann SM, Hawkins RD, Antonova I. Presynaptic and postsynaptic roles of NO, cGK, and RhoA in long-lasting potentiation and aggregation of synaptic proteins. Neuron. 2005;45:389–403. doi: 10.1016/j.neuron.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 52.Frank CL, Tsai LH. Alternative functions of core cell cycle regulators in neuronal migration, neuronal maturation, and synaptic plasticity. Neuron. 2009;62:312–326. doi: 10.1016/j.neuron.2009.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Knafo S, Esteban JA. Common pathways for growth and for plasticity. Curr Opin Neurobiol. 2012;22:1–7. doi: 10.1016/j.conb.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 54.Lee HK, Kameyama K, Huganir RL, Bear MF. NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron. 1998;21:151–162. doi: 10.1016/s0896-6273(00)80632-7. [DOI] [PubMed] [Google Scholar]
- 55.Cull-Candy S, Kelly L, Farrant M. Regulation of Ca2+-permeable AMPA receptors: synaptic plasticity and beyond. Curr Opin Neurobiol. 2006;16:288–297. doi: 10.1016/j.conb.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 56.Whitney NP, Peng H, Erdmann NB, Tian C, Monaghan DT, Zheng JC. Calcium-permeable AMPA receptors containing Q/R-unedited GluR2 direct human neural progenitor cell differentiation to neurons. FASEB J. 2008;22:2888–2900. doi: 10.1096/fj.07-104661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nakanishi S, Okazawa M. Membrane potential-regulated Ca2+ signalling in development and maturation of mammalian cerebellar granule cells. J Physiol. 2006;575:389–395. doi: 10.1113/jphysiol.2006.113340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Okazawa M, Abe H, Katsukawa M, Iijima K, Kiwada T, Nakanishi S. Role of calcineurin signalling in membrane potential-regulated maturation of cerebellar granule. J Neurosci. 2009;29:2938–2947. doi: 10.1523/JNEUROSCI.5932-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Contestabile A. Role of nitric oxide in cerebellar development and function: focus on granule neurons. Cerebellum. 2012;11:50–61. doi: 10.1007/s12311-010-0234-1. [DOI] [PubMed] [Google Scholar]
- 60.Hamori J, Somogyi J. Differentiation of cerebellar mossy fiber synapses in the rat: a quantitative electron microscope study. J Com Neurol. 1983;220:365–377. doi: 10.1002/cne.902200402. [DOI] [PubMed] [Google Scholar]
- 61.Maffei A, Prestori F, Shibuki K, Rossi P, Taglietti V, D’Angelo E. NO enhances presynaptic currents during cerebellar mossy fiber-granule cell LTP. J Neurophysiol. 2003;90:2478–2483. doi: 10.1152/jn.00399.2003. [DOI] [PubMed] [Google Scholar]
- 62.Bredt DS, Snyder SH. Transient nitric oxide synthase neurons in embryonic cerebral cortical plate, sensory ganglia, and olfactory epithelium. Neuron. 1994;13:301–313. doi: 10.1016/0896-6273(94)90348-4. [DOI] [PubMed] [Google Scholar]
- 63.Holscher C. Nitric oxide, the enigmatic neuronal messenger: its role in synaptic plasticity. Trends Neurosci. 1997;20:298–303. doi: 10.1016/s0166-2236(97)01065-5. [DOI] [PubMed] [Google Scholar]
- 64.Wood J, Garthwaite J. Models of the diffusional spread of nitric oxide: implications for neuronal nitric oxide signalling and its pharmacological properties. Neuropharmacology. 1994;33:1235–1244. doi: 10.1016/0028-3908(94)90022-1. [DOI] [PubMed] [Google Scholar]
- 65.Ciani E, Calvanese V, Crochemore C, Bartesaghi R, Contestabile A. Proliferation of cerebellar precursor cells is negatively regulated by nitric oxide in the new born rat. J Cell Sci. 2006;119:3161–3170. doi: 10.1242/jcs.03042. [DOI] [PubMed] [Google Scholar]
- 66.Ciani E, Guidi S, Bartesaghi R, Contestabile A. Nitric oxide regulates cGMP-dependent cAMP-responsive element binding protein phosphorylation and Bcl-2 expression in cerebellar neurons: implication for a survival role of nitric oxide. J Neurochem. 2002;82:1282–1289. doi: 10.1046/j.1471-4159.2002.01080.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Cerebellar granule cells maintained for 8 DIV were treated for 10 minutes with NMDA (50 μM) + glycine (10 μM), for 60 minutes with either L-NMMA (10 μM) or NPLA (1 μM) or 60 minutes with NOS inhibitor plus NMDA (50 mM) + glycine (10 μM) added the latest 10 minutes. Then cells were fixed and labelled with a GluA1-NT antibody (15 μg/ml) over 120 minutes at 37 °C. The images represent the immunostaining of synapsin I and GluA1, the merged image and an amplification of each condition. (B) The density of GluA1 clusters in dendrites (clusters/μm), selecting ROI’s of the specific antibody from at least 15 fields from 2 different experiments for each condition. (C) Analysis of the fraction of GluA1 clusters apposed to synapsin I puncta, either upon stimulation or inhibition (n=30 fields from two independent experiments). (D) Cumulative probability plots of GluA1 mean fluorescence intensity (mean values: control 18.51±0.06; NMDA 21.44±0.05; L-NMMA 8.58±0.06; L-NMMA+NMDA 10.18±0.07; NPLA 7.91±0.04; NPLA+NMDA 8.66±0.06, a.u.). In B and C, the data are represented as the mean ± SEM and analyzed using unpaired, two-sample t test (*p<0.001). For the cumulative probability the Komolgorov-Smirnov test was used. Scale bars: 10 μm.
(A) Cerebellar granule cells maintained for 8 DIV were treated for 10 minutes with either Angeli’s salt (2 μM) or FK409 (10 μM) or maintained in control conditions. Then cells were fixed and labelled with a GluA1-NT antibody (15 μg/ml) over 120 minutes at 37 °C. The images represent the immunostaining of synapsin I and GluA1, the merged image and an amplification of each condition. (B) The density of GluA1 clusters in dendrites (clusters/μm), selecting ROI’s of the specific antibody from at least 15 fields from 2 different experiments for each condition. (C) Analysis of the fraction of GluA1 clusters apposed to synapsin I puncta, either upon stimulation or inhibition (n=30 fields from two independent experiments). (D) Cumulative probability plots of GluA1 mean fluorescence intensity (mean values: control 8.47±0.06; Angeli’s salt 13.12±0.08; FK409 13.40±0.07, a.u.). In B and C, the data are represented as the mean ± SEM and analyzed using unpaired, two-sample t test (*p<0.001). For the cumulative probability the Komolgorov-Smirnov test was used. Scale bars: 10 μm.