

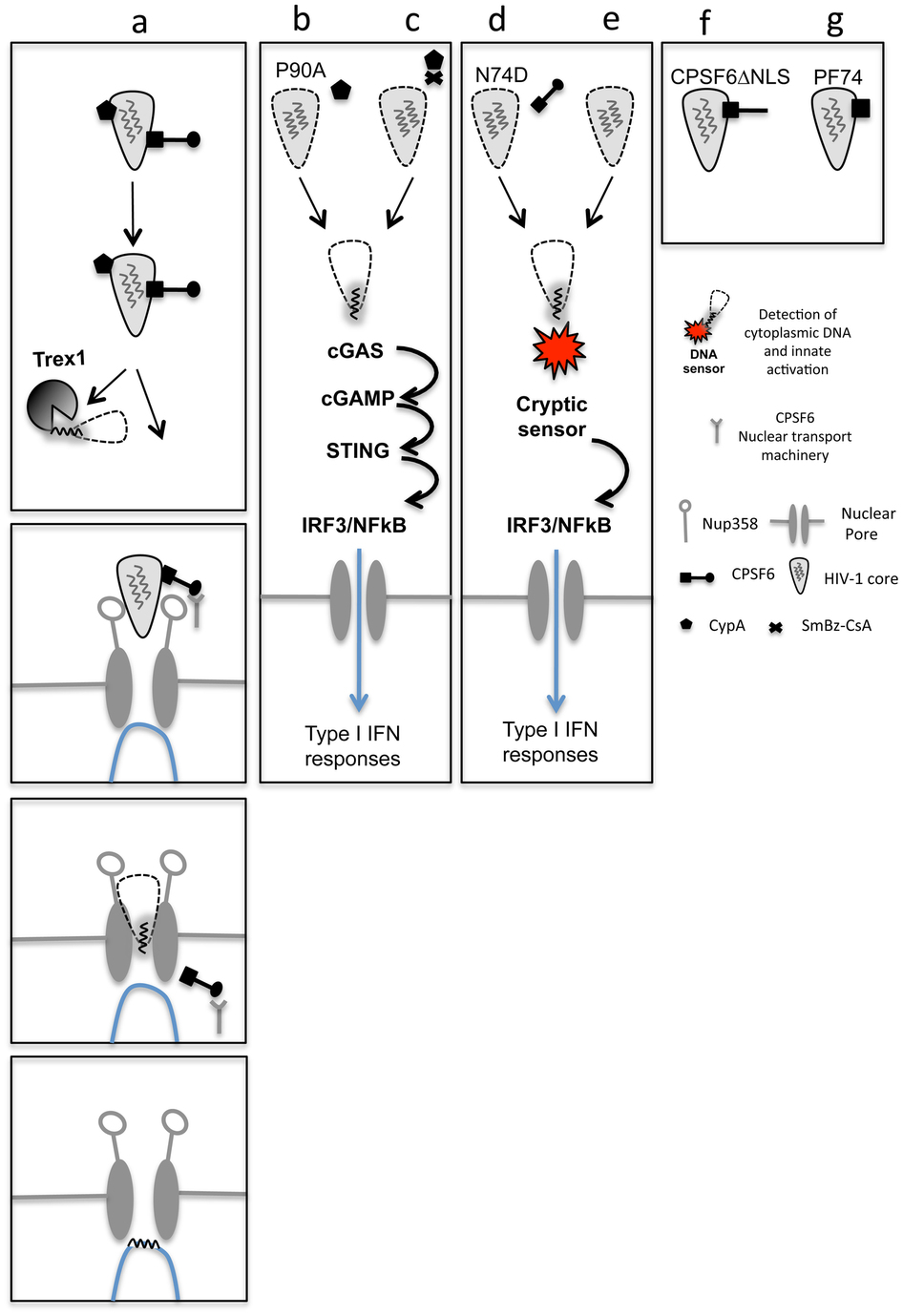
Abstract
HIV-1 is able to replicate in primary human macrophages without stimulating innate immunity despite reverse transcription of genomic RNA into double stranded DNA, an activity that might be expected to trigger innate pattern recognition receptors (PRRs). We hypothesized that, if correctly orchestrated HIV-1 uncoating and nuclear entry is important for evasion of innate sensors, then manipulation of specific interactions between HIV-1 capsid (CA) and host factors that putatively regulate these processes should trigger PRRs and stimulate type 1 interferon secretion. Here we show that HIV-1 CA mutants N74D and P90A, which are impaired for interaction with cofactors Cleavage and Polyadenylation Specificity Factor subunit 6 (CPSF6) and cyclophilins (Nup358 and CypA) respectively(1-2), cannot replicate in primary human monocyte derived macrophages (MDM) because they trigger innate sensors leading to nuclear translocation of NFκB and IRF3, the production of soluble type-1 interferon (IFN) and induction of an antiviral state. Depletion of CPSF6 with shRNA expression allows wild type virus to trigger innate sensors and interferon production. In each case, suppressed replication is rescued by IFN-receptor blockade demonstrating a role for IFN in restriction. IFN production is dependent on viral reverse transcription but not integration suggesting that a viral reverse transcription product comprises the HIV-1 pathogen associated molecular pattern (PAMP). Finally, we show that we can pharmacologically induce wild type HIV-1 infection to stimulate IFN secretion and an antiviral state using a non-immunosuppressive cyclosporine analogue. We conclude that HIV-1 has evolved to utilize CPSF6 and cyclophilins to cloak its replication allowing evasion of innate immune sensors and induction of a cell autonomous innate immune response in primary human macrophages (Extended Data Fig 1).
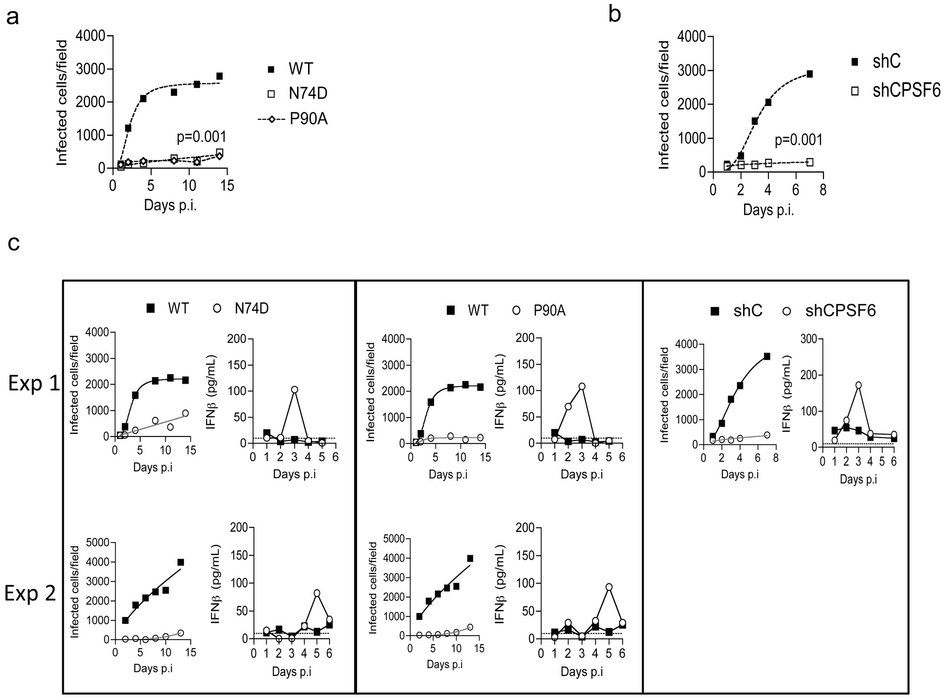
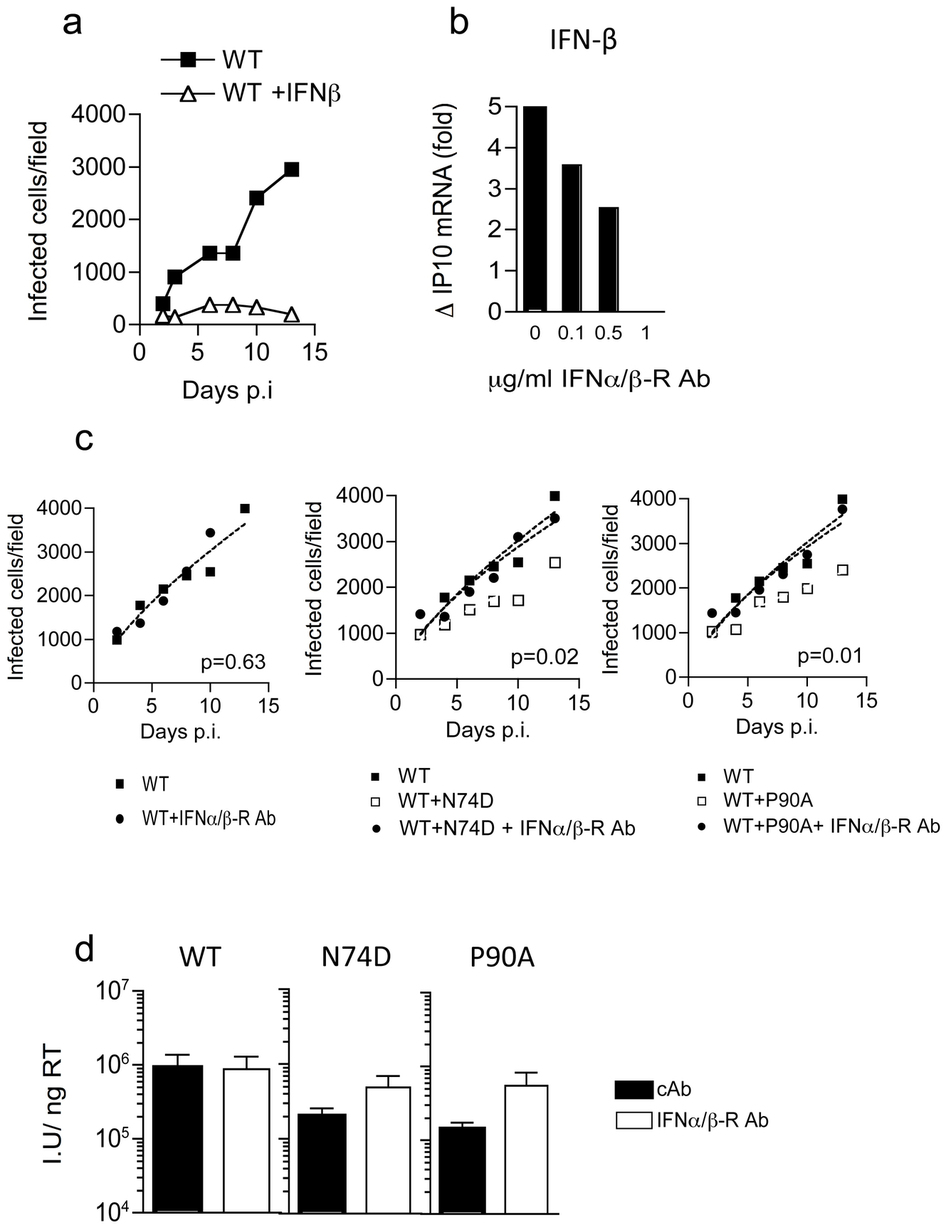
HIV-1 capsid (CA) mutant N74D cannot recruit CPSF6 and is insensitive to depletion of HIV-1 cofactors Nup358 and TNPO3 suggesting it may utilize alternate cofactors for nuclear entry1-3. Furthermore, unlike wild type (WT) HIV-1, HIV-1 N74D cannot replicate in monocyte-derived macrophages (MDM) (Fig. 1a, Extended Data Fig. 2)2,4. Remarkably, an inability to replicate was accompanied by a burst of IFN-β detected 2-5 days after low multiplicity infection (Fig. 1b, Extended Data Fig. 2). The antiviral activity of IFN-β (Extended Data Fig. 3a)5 was revealed by rescuing HIV-1 N74D, but not WT replication with antibody to the IFNα/β receptor α chain (IFNAR2) (Fig. 1c,d, Extended Data Fig. 3b). Co-infection of MDM with WT and HIV-1 N74D led to suppression of WT replication (Fig. 1e), which was also rescued by IFNAR2 antibody (Extended Data Fig. 3c). This demonstrated that sensitivity to IFN-mediated restriction was not limited to the mutant virus.
Figure 1. HIV-1 CPSF6 binding mutant CA N74D is restricted in MDM due to induction of Type-I IFN.

(a) Replication of WT HIV-1 or CA mutant N74D in MDM. (b) IFN-β levels in supernatants from (a). (c-d) Replication of HIV-1 CA N74D or WT HIV-1 with IFNAR2 or control antibody (cAb). (e) Replication of WT or WT plus CA N74D. Mean data and regression lines for biological replicates are shown in c-e. P values (2-way ANOVA) are given for (c-d) IFNAR2 blockade and (e) co-infection with CA mutant N74D. (f) Infection of MDM by HIV-1 measured at 48h (g) GAPDH normalized IP10 RNA levels expressed as fold change over untreated cells after infection with WT or HIV-1 mutants (Mean of 3 technical replicates ±SEM, f-g).
In contrast to the spreading infection assay in which HIV-1 N74D was completely suppressed, assessment of single-round infection in MDM with higher dose viral inocula, revealed only a 5-fold reduction of HIV-1 N74D infectivity as compared to WT (Fig. 1f). However, this reduction was also restored to WT levels by IFNAR2 blockade (Extended Data Fig. 3d). In this experiment we did not detect IFN-β, likely due to assay sensitivity, but interferon stimulated genes (ISGs) IP10, IFIT1 and CCL8, were induced following infection with HIV-1 N74D, but not WT virus (Extended Data Fig. 4a). ISG induction was confirmed by microarray transcriptional profiling of host responses to HIV-1 N74D, which showed expected enrichment for innate immune type -1 IFN pathways at a genome-wide level (Extended Data Fig. 4c,d). These findings, together with the time course of IFN release during spreading infection (Fig. 1b), suggest that multiple-round replication amplifies virus induced innate responses leading to high levels of IFN-β secretion and potent suppression of HIV-1 replication (Fig. 1a).
We next measured ISG induction in HIV-1 N74D infected MDM when either DNA synthesis or integration were prevented by mutation of reverse transcriptase6 (RT) or integrase7 (IN) respectively. Infection by HIV-1 CA N74D, RT D185E double mutant did not stimulate IP-10 expression whereas infection with HIV-1 double mutant CA N74D, IN D116N induced IP10 expression comparable to the WT virus (Fig. 1g). These data suggest that the innate immune response in MDM depends on detection of the products of reverse transcription, not integration.
Given that CA mutation N74D prevents recruitment of CPSF61,3 we hypothesized that CPSF6 depletion would induce WT HIV-1 to trigger IFN responses in MDM. In fact, CPSF6 depletion by shRNA expression in MDM (Fig. 2a,b, Extended Data Fig. 2) completely abrogated HIV-1 replication (Fig. 2c) due to a burst of IFN-β. MDM expressing a non-targeting shRNA did not produce IFN-β on HIV-1 infection (Fig. 2d). The restrictive role of IFN was confirmed by rescue of infectivity with IFNAR2 antibody (Fig. 2e). Neither the IFNAR2 nor isotype control antibody had any effect on HIV-1 replication in control shRNA expressing MDM (Fig. 2f). Importantly, shRNA expression itself did not induce IFN-β production (Fig. 2g). We conclude that the defect in WT HIV-1 replication after CPSF6 depletion in MDM was largely due to type-1 IFN production. In line with observations made with HIV-1 N74D, CPSF6 depletion also reduced single round infectivity in MDM by a few fold, 3.5-fold versus 5-fold (Fig. 2h).
Figure 2. HIV-1 elicits a Type-1 IFN response that restricts replication in CPSF6 depleted MDM.

(a) Protocol schema. (b) CPSF6/actin detected at time of infection (c). HIV-1 replication in MDM expressing shRNA targeting CPSF6 or control shRNA (d) IFN-β levels in supernatants from (c). (e-f) Infection of CPSF6 depleted or MDM expressing control shRNA with IFNAR2 or control antibody (cAb). P values (2-way ANOVA) are given for the effect of (e) CPSF6 depletion or (f) control shRNA on biological replicates. (g) IFN-β produced from shRNA expressing MDM or IFN-β treated MDM. (h) Infection of MDM by HIV-1 measured at 48h on CPSF6 depleted or control shRNA expressing MDM (Mean of 3 technical replicates ±SEM).
The HIV-1 inhibitor PF-3450074 (PF74) binds CA and inhibits CPSF6 recruitment and HIV-1 replication1,3,8. As expected, PF74 completely blocked HIV-1 replication in MDM but did not induce soluble IFN-β secretion, nor was replication rescued by IFNAR2 blockade (Extended Data Fig. 5a-c). However, as reported, PF74 completely abrogated HIV-1 DNA synthesis (Extended Data Fig. 5d,e) 8,9. The fact that PF74 mimics CPSF6 binding to HIV-1 CA3 suggests that CPSF6 recruitment might prevent premature reverse transcription and innate recognition of viral DNA. To test this hypothesis, a human CPSF6 mutant deleted for its nuclear localization signal (CPSF6ΔNLS)10, was expressed in HeLa cells. Like PF74, human CPSF6ΔNLS blocked VSV-G HIV-1 GFP DNA synthesis and infectivity (Extended Data Fig. 5f,g). A CPSF6 mediated block to HIV-1 RT differs from previous observations showing no effect of CPSF6ΔNLS on HIV-1 RT but earlier work used a mouse CPSF6 cDNA with an alternate exon structure1,4. We hypothesize that HIV-1 has evolved to recruit CPSF6 to incoming HIV-1 CA and prevent premature DNA synthesis, which would otherwise trigger innate sensors (Extended Data Fig. 1).
Since HIV-1 N74D is unable to appropriately utilize nuclear pore components and has retargeted integration properties1-3,11, we hypothesized that the HIV-1 CA mutant P90A, which fails to interact with the cyclophilins CypA and nuclear pore component Nup358, and also has retargeted integration2, might also trigger innate sensors. Indeed, HIV-1 P90A infection of MDM induced IFN-β production and an antiviral state in both replication and single-round infectivity assays, which was rescued by IFN receptor antibody (Fig. 3a-f, Extended Data Fig. 2, 3c,d). We find MDM infection by HIV-1 N74D, P90A or WT were equally increased by SIVmac VLP encoding Vpx, suggesting that mutant viruses were not specifically Vpx sensitive (Extended Data Fig. 5h). Quantitative RT-PCR and whole genome profiling demonstrated ISG induction after HIV-1 P90A infection (Extended Data Fig. 4b-d). Consistently, double mutation of P90A and RT D185E, but not IN D116N, suppressed IP10 induction (Fig. 3g). We hypothesise that viral DNA produced by reverse transcription is the target for innate sensing of both HIV-1 CA mutants N74D and P90A in MDM.
Figure 3. HIV-1 CypA binding mutant CA P90A is restricted in MDM due to induction of Type-I IFN.

(a) Replication of WT HIV-1 or CA mutant P90A in MDM. (b) IFN-β levels in supernatants from (a). (c) Replication of HIV-1 CA P90A with IFNAR2 or control antibody (cAb). (d) as in Figure 1d. (e) Replication of WT or WT plus CA P90A. Mean data and regression lines are shown for biological replicates in c-e. P values (2-way ANOVA) are given for (c-d) IFNAR2 blockade and (e) co-infection with CA mutant P90A. (f) Infection of MDM by HIV-1 measured at 48h (g) GAPDH normalized IP10 RNA levels expressed as fold change over untreated cells after infection with WT or HIV-1 mutants (Mean of 3 technical replicates ±SEM, f-g).
We next considered the mechanism of HIV-1 mutant innate sensing. A recently identified cytosolic DNA sensor, cyclic GMP-AMP synthase (cGAS) that synthesizes the novel second messenger cGAMP12, has been shown to detect HIV-1 reverse transcribed DNA in human myeloid cells13. cGAMP production is detected by stimulator of interferon genes (STING) which transduces an innate signaling cascade leading to IRF3 activation and type-1 interferon production13. We used a biological assay to test for cGAMP production in MDM infected by HIV-1 N74D and P90A. Consistently, extracts from HIV-1 P90A CA mutant infected cells contained a benzonase and heat resistant component that activated an interferon sensitive promoter in a STING dependent way (Extended Data Fig. 6). Commercially prepared cGAMP validated the assay and acted as a positive control. Importantly, RNA purified from HIV-1 mutant infected cells was not immunostimulatory, as oppose to RNA from Sendai virus infected cells, which potently activated an IFN-β promoter, as expected for a RIG-I triggering virus. These data, support our hypothesis that HIV-1 DNA is the PAMP. Intriguingly, HIV-1 N74D did not stimulate in either of these assays suggesting that the two HIV-1 mutants activate independent DNA sensors. This possibility is consistent with the different integration site targeting preferences of the two mutants with HIV-1 N74D and P90A integrating into lower gene density or higher gene density regions of chromatin respectively, as compared to wild type virus2.
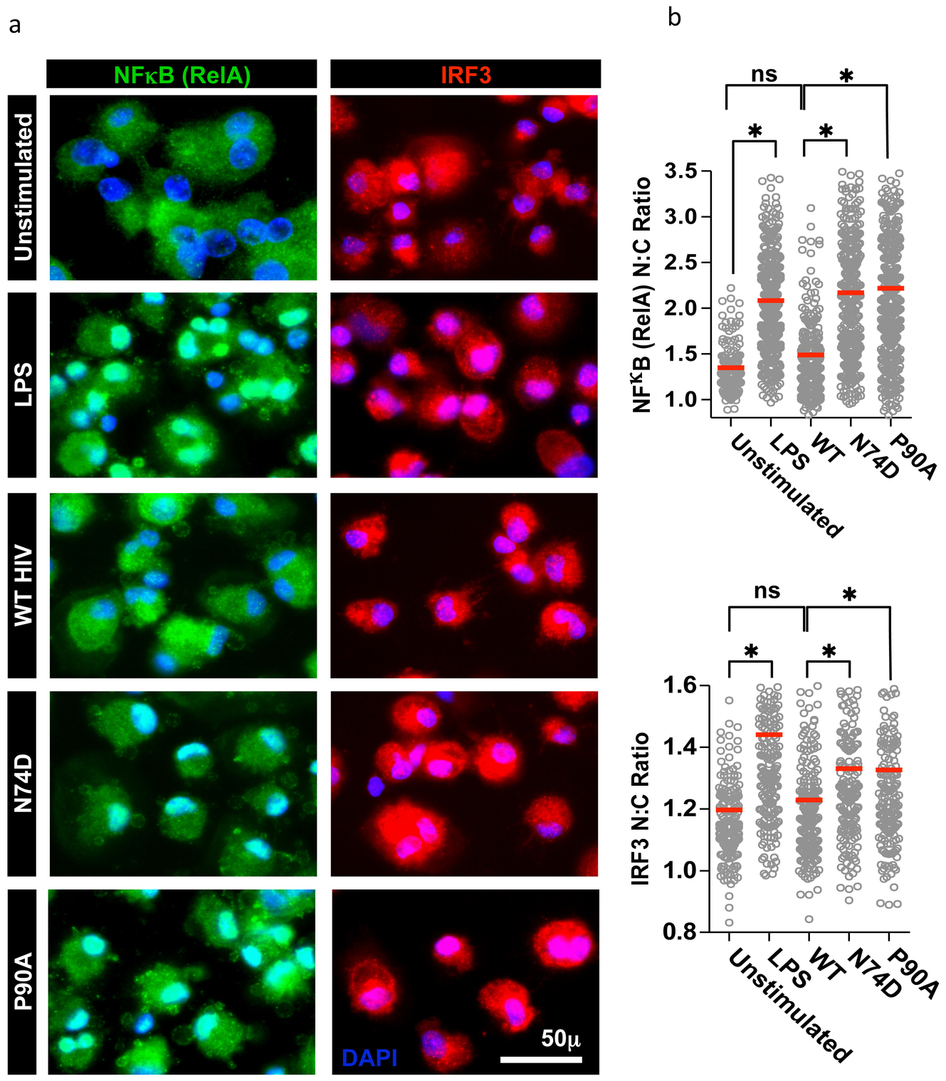
Immunofluorescent detection of NFκB and IRF3 revealed nuclear translocation of both transcription factors after exposure to either of the HIV-1 mutants but not WT virus (Fig. 4a-c, Extended Data Fig. 7). Concordantly, inhibition of NFκB activation with a peptide inhibitor of NEMO (IKK) rescued infectivity of both HIV-1 mutants in a dose dependent manner (Fig. 4b). Finally, we considered whether prevention of cofactor interaction using drugs could induce WT virus to trigger a cell autonomous innate immune response in the same way as mutant virus. We sought to phenocopy the HIV-1 P90A mutant by inhibiting cyclophilin recruitment using cyclosporine (Cs) or a non-immunosuppressive analogue of cyclosporine SmBz-CsA. SmBz-CsA is modified at the 3′-SAR position to include a methylphenyl-4-carboxylic acid group and therefore cannot inhibit calcineurin or affect T cell activation14 (Fig. 4d, 4e Extended Data table 1). Like Cs, SmBz-CsA inhibited recruitment of CypA, but not Nup358 Cyp, to HIV-1 CA (Extended Data Fig. 8e). Treatment of MDM with SmBz-CsA (Fig. 4f) or Cs (Extended Data Fig. 8a) completely suppressed WT HIV-1 replication and elicited IFN-β production (Fig. 4g, Extended Data Fig. 8b). Inhibited viral replication was rescued by IFNAR2 blockade, but not by control antibody (Fig. 4h, Extended Data Fig. 8c). After single-round infection in the presence of either drug, infection was 5-7 fold lower (Fig. 4i and Extended Data Fig. 8d). These data are consistent with observations of cyclosporine inhibition of Hepatitis C virus, in which innate immune responses are implicated15.
Figure 4. NFκB/IRF3 are activated by mutant HIV-1 and SmBz-CsA treatment causes WT HIV-1 to trigger innate responses.

(a, b) Mean (±SEM) nuclear:cytoplasmic ratios for NFκB or IRF3 in infected MDM (p<0.05, 2-way ANOVA). (c) Infection at 48h ±IKK inhibitor. (d, e) SmBz-CsA (green) complexed with CypA (gray), Cs (yellow) and calcineurin (orange/blue). (f) Replication of HIV-1 in MDM ±SmBz-CsA (g) IFN-β levels from (f). (h) MDM infected with WT HIV-1 plus SmBz-CsA and IFNAR2 antibody or cAb (mean data and regression lines). P value (2-way ANOVA) is given for IFNAR2 blockade of biological replicates. (i) Infection of MDM by WT HIV-1 ±SmBz-CsA at 48h (Mean of 3 technical replicates ±SEM).
Our findings demonstrate that human macrophages are able to detect HIV-1 infection and activate a cell autonomous innate immune signal, when specific interactions with HIV-1 cofactors are prevented by virus mutation (Fig. 1,3), depletion of cofactor expression (Fig. 2) or pharmacological inhibition of cofactor recruitment (Fig. 4). We envisage that appropriate interaction between CA and CPSF6/cyclophilins normally allows evasion of innate sensors and promotes HIV-1 infection. We propose a model in which CPSF6/CypA recruitment to CA suppresses premature viral DNA synthesis and thus innate triggering. Inhibition of DNA synthesis by CPSF6ΔNLS or the CPSF6 mimic PF74 support this possibility. In our model, nuclear entry of CPSF6 could release the virus enabling reverse transcription at the nuclear pore. The cytosolic exonuclease TREX1 degrades excess cytoplasmic DNA and prevents cGAS activation and IFN stimulation13,16 (Extended Data Fig. 8f-j). Our data suggest that DNA synthesized by HIV-1 mutants is insensitive to TREX1 degradation, either through nature or location, and in the case of HIV-1 CA P90A, is detected by cGAS leading to cGAMP production. In MDDC, CypA has been suggested to have a different role, acting to aid detection of HIV-1 by innate sensors during egress17. These observations suggest that HIV-1 may rely on cell type specific cofactor use to protect it from innate immune defenses. Intriguingly, both N74D and P90A CA mutants replicated in indicator cell lines GHOST and HeLa TZM-bl to WT levels (Extended Data Fig. 9). Replication was unaffected by IFN-receptor blockade and ISG expression was not induced illustrating that these cell lines cannot respond in the same way to HIV-1 infection. They also suggest that the only obstacle to HIV-1 CA mutant replication in MDM is due to induction of innate responses. Our observations facilitate the further study of the relationship between HIV-1 and innate immunity. We envisage therapeutics, or vaccine adjuvants, which induce virus to trigger potent cell autonomous innate immunity, IFN secretion and enhanced adaptive immune responses.
METHODS
Cells
Primary Monocyte-derived macrophages (MDM) were prepared from fresh blood from healthy volunteers as described5. The study was approved by the joint University College London/University College London Hospitals NHS Trust Human Research Ethics Committee and written informed consent was obtained from all participants. Briefly, peripheral blood mononuclear cells were isolated by Ficoll-Hypaque (Axis-Shield) density centrifugation. The isolated cells were washed with PBS and plated in RPMI (Invitrogen) supplemented with 10% heat-inactivated autologous human serum (HS) and 40ng/ml macrophage colony stimulating factor (M-CSF) (R&D systems). The medium was then refreshed after 3 days (RPMI 1640 with 10% HS), removing any remaining non-adherent cells. After 6 days, media was replenished with RPMI containing 5% type AB HS (Sigma-Aldrich). Replicate experiments were performed with cells derived from different donors.
GHOST18, a human osteosarcoma cell line stably expressing CD4, CCR5, CXCR4 and the green fluorescent protein (GFP) reporter gene under the control of the HIV-2 long terminal repeat were maintained in DMEM containing 10% heat-inactivated fetal calf serum (FCS), glutamine, antibiotics, G418 (500μg/ml), hygromycin (100μg/ml), and puromycin (1μg/ml) and were split twice a week. HEK293T cells were grown in DMEM (Invitrogen) supplemented with 10% FCS.
Reagents
Recombinant human interferon (IFN)-β (Merck Serono) was used at 10ng/ml, poly(I:C) (Sigma) was used at 10μg/ml, Cyclosporine (Sandoz) was used at 5μM. SmBz-CsA was synthesized as described14 and used at 10μM. PF74, a gift from Jason Chin, was synthesized as described3 and used at 10μM. Lipopolysaccaride (LPS) (Sigma) was used at 100ng/ml. Commercially prepared STING agonist, cyclic GMP-AMP (cGAMP), was purchased from Invivogen.
Plasmids
The CCR5-tropic Wild type NL4.3 (Ba-L Env) or NL4.3 (Ba-L Env) bearing CA mutations P90A or N74D were derived from an infectious clone of NL4.3 by cloning the Env gene from HIV-1 Ba-L between unique EcoR1 and BamH1 sites to replace the NL4.3 Env gene. ΔRT and ΔIN infectious clones were generated by making mutant RT D185E6, or IN D116N7 using site directed mutagenesis (Stratagene).
Short hairpin sequences were expressed from HIV-1 based shRNA expression vector HIVSiren2. CPSF6 shRNA target sequence was 5-CGAAGAGTTCAACCAGGAA-3; TREX1 shRNA target sequence was 5-CCAAGACCATCTGCTGTCA-3; CPSF6 was detected by western blot and TREX-1 was detected by RT-qPCR.
A human CPSF6 expression vector was prepared by PCR cloning the human CPSF6 ORF from cDNA (Superscript, Life Technologies), prepared from HeLa cells, into the MLV based gammaretroviral expression vector EXN19 using primers fwd 5-ATCGGAATTCATGGCGGACGGTGTGGACCACATAGACATTTAC-3 and rev 5-ATGCGCGGCCGCCTAACGATGACGATATTCGCGCTCTC-3, restriction sites underlined. The nuclear localization signal was removed from CPSF6 as described10 by deleting the C-terminal 50 amino acids by PCR using reverse primer ATGCGCGGCCGCTCATTCTCGTGATCTACTATGGTCCC and fwd primer as above. The resulting NLS mutant is defective for nuclear entry as described10 CPSF6ΔNLS was expressed in HeLa cells by gammaretroviral vector transduction as described3 and G418 selected pools of cells generated. Note that the human CPSF6 cDNA described herein differs from the murine cDNA described previously1,4,20 in that it represents the most common human CPSF6 isoform represented by GenBank accession number nm007007 and thus lacks exon 621-23.
Virus production
Virus particles were produced by transient transfection of HEK293T cells. 3.5μg of molecular clone DNA; for shRNA we used 1.5μg pHIVSIREN2 shRNA, 1μg p8.9124 and 1μg pMDG25 encoding VSV-G protein. For SIVmac-VLP we left out the genome plasmid and transfected 3μg pSIV3+26 and 1μg pMDG using 10μl Fugene 6 transfection reagent (Promega) as described6. HIV-1 GFP was produced by transfection of 293T with GFP encoding genome CSGW, packaging plasmid p8.91 and pMDG as described6. Virus supernatants were harvested 48hr, 72hr and 96hr post transfection. All virus suspensions were filtered and ultracentrifuged through a 20% sucrose buffer and resuspended in RPMI 1640 with 5% HS, for subsequent infection of MDM. All virus preparations were quantified by reverse transcriptase (RT) enzyme linked immunosorbant assay (ELISA) (Roche) except when doses were measured by p24 CA ELISA (National Cancer Institute at Frederick) where stated (Fig 1g, 3g). Viruses were also titrated on GHOST where described detecting infection by flow cytometry 72hrs post infection or HeLa TZM bl where infection was detected by CA staining as below.
Infection and stimulation
MDM were infected with 100 pg RT/well (MOI 0.2) in 48-well plates and subsequently fixed and stained using CA specific antibodies (EVA365 & 366 National Institute of Biological Standards AIDS Reagents Programme) and a secondary antibody linked to beta galactosidase, as described5. During the time course, supernatants were collected for IFN-β ELISA (PBL Interferon Source) according to manufacturer’s instructions. Anti-IFNα/β Receptor (PBL Interferon Source) or control IgG2A antibody (R&D systems) were added at 1μg/ml for 2hrs prior to infection and supplemented every 4 days. For inhibition of NFkB activation, a peptide inhibitor of NEMO (IKKγ) or control peptide (Imgenex), were added at either 50μM or 100μM for 12hrs prior to infection. For Agilent microarray analysis and RT-qPCR, MDM were infected with 1 ng RT/well (MOI 2) in 24-well plates. RNA was extracted 24 hours post infection (RNeasy, Qiagen) and subject to microarray analysis as described below. For shRNA transduction of MDM, day 3 differentiated cells were infected with shRNA (0.1 ng RT/ml), SIVmac-VLP (1 ng RT/ml) + 8μg/ml polybrene overnight.
Western blot analysis
CPSF6 expression was measured in extracted cell pellets by Western blot. Cells were lysed in Laemmli buffer then boiled before separation by SDS-PAGE as described previously3. After CPSF6 or STING detection membranes were stripped and reprobed for β-actin as a loading control. Antibodies used were CPSF6 (Abcam ab99347) STING (Abcam ab82960) and β-actin (Abcam ab6276)
Microarray analysis
Total RNA was purified from cell lysates collected in RLT buffer (Qiagen) using the RNEasy Mini kit (Qiagen). Samples were processed for Agilent microarrays as previously described5 and loess normalised data were analysed using the TM4 microarray software suite MeV v4.827. Pathway enrichment analysis of differentially expressed gene lists was performed using the online bioinformatics tool InnateDB28. Microarray data are available from the EBI Array Express repository (http://www.ebi.ac.uk/arrayexpress/) under accession no E-MTAB-1437.
Quantitative PCR
cDNA was synthesised using the Omniscript RT Kit (Qiagen) and quantitative (q)PCR of selected genes was performed using the following inventoried TaqMan assays (Applied Biosystems); CCL8 (Hs04187715_m1), and IFIT1 (Hs01911452_s1). IP-10 expression was quantified using: forward primer: TGAAATTATTCCTGCAAGCCAATT, reverse primer: CAGACATCTCTTCTCACCCTTCTTT, and probe: TGTCCACGTGTTGAGATCATTGCTACAATG. TREX1 expression was quantified using: forward primer: GCATCTGTCAGTGGAGACCA, reverse primer: AGATCCTTGGTACCCCTGCT, and probe: CACAACCAGGAACACTAGTCCCAGC. Expression levels of target genes were normalised to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as previously described5. To measure late RT products, total DNA was purified 9 hours post infection (QiaAmp, Qiagen) with DNAse treated virus (70U/ml DNAse (Affymetrix) in RQ1 buffer (Promega) for 37°C, 1 hour) and 500 ng were subjected to Taqman quantitative PCR using late RT primers and probe to detect provirus as described29. Cells were infected with virus that had been boiled for 2 minutes as a negative control. Infectivity was measured in parallel samples by intracellular p24 staining 48 hours post infection. Presented QPCR experiments are means of technical replicates and represent 3 biological replicates.
cGAMP reporter assay
MDM were infected with 1ng RT/well (MOI 2) in 24-well plates for 18hours. Cells were lysed in hypotonic buffer (10mM TRIS pH7.4, 10mM KCl and 1.5mM MgCl2). After freeze thaw, a proportion of cell extract was kept for stimulatory RNA reporter assay and the remaining was heated to 96°C for 10 min. Sonicated extracts were centrifuged (20,000g, 20 mins, 4°C), followed by Benzonase treatment (1U/ul Benzonase (Novagen), 2mM ATP, 37°C 90 mins). 4ul of lysate was introduced to reporter cells using Lipofectamine 2000 (Invitrogen). The reporter cells are L929 cells or L929 cells depleted of STING by transfecting (Oligofectamine, Invitrogen) a previously described STING siRNA 30. The L929 cells stably express Firefly Luciferase-driven by an interferon sensitive response element. Luciferase was read after 16 hrs using Steady-Glo (Promega) and a Luminometer.
Immunostimulatory RNA reporter assay
Cell extracts were subjected to Trizol extraction (Invitrogen) and the extracted RNA, plus a control plasmid encoding Renilla luciferase, was transfected into 293T cells expressing firefly luciferase driven by IFN-β promoter. Cells were transfected with 500ng of RNA extracted from macrophages and luciferase values were determine at 16 hours using Dual-Luciferase assay kit (Promega). IFN-β promoter activity (Firefly luciferase) was normalized by global transcription (Renilla luciferase) and fold induction compares normalized luciferase values against mock transfected reporter cells. As a positive control we infected MDM with Sendai virus, a gift from Steve Goodbourn, and purified immunostimulatory RNA. All transfections in this assay use Lipofectamine 2000 (Invitrogen).
Quantitative confocal immunofluorescence analysis of NF- κ B and IRF3 nuclear translocation
Nuclear:cytoplasmic ratios of NF- κ B RelA and IRF3 transcription factors were analyzed as previously described5 using a Hermes WiScan Cell Imaging System to analyse cells stained with rabbit polyclonal anti NF- κ B RelA (clone C-20) (Santa Cruz Biotechnology) or rabbit polyclonal anti IRF3 (clone FL 425) (Santa Cruz Biotechnology).
Protein expression, purification, crystallization, data collection, structure determination and refinement
CypA was expressed in Escherichia coli C41(DE3) cells (Lucigen) from tagless expression vector pOPT 3. Cells were grown overnight at 18°C before being harvested, sonicated and purified by SP ion-exchange chromatography (GE Healthcare) followed by gel filtration. Crystals of SmBz-CsA in complex with CypA were grown at 17 °C in sitting drops. Protein solution (1mM each of CypA and SmBz-CsA in 20 mM Tris pH 8, 50 mM NaCl, 1 mM DTT, 1 % DMSO) was mixed with reservoir solution (1 M LiCl, 0.1 M MES pH 6, 30 % w/v PEG 6000) in a 1:1 mix, producing 0.15 mm × 0.10 mm × 0.10 mm crystals within 24 h. Crystals were flash-frozen in liquid nitrogen before data collection using an in-house Mar-345 detector. Crystal data and diffraction statistics are provided in Extended Data table 1. Crystallographic analysis was performed using programs from the CCP4 suite31. Data were indexed and scaled in MOSFLM and SCALA, respectively. The structure of CsA:CypA (pdb 1CWA32) was used as a search model. Structures were refined in REFMAC and Coot31. Structural figures were created using PyMol (http://pymol.sourceforge.net/). PDB coordinates have been deposited under accession code 4IPZ.
Statistical methods
All data were normally distributed and analyzed for statistically significant differences between experimental groups by t-tests or 2-way ANOVA as indicated. Bar charts show mean ±SEM for experimental replicates in each case. Replication assays are presented for individual experiments, or where P values (2-way ANOVA) are given-replicate experiments. Individual or mean data points and non-linear regression lines are shown over time. Sample sizes for each experiment were based on pilot experiments to estimate the effect size and variance of the data.
Supplementary Material
a, Intact capsids recruit CypA and CPSF6 which direct the virus to the nucleus. CPSF6 interaction prevents premature DNA synthesis. Excess cytoplasmic DNA is degraded by TREX1. At the nuclear pore CPSF6 NLS-dependent dissociation from the virus allows reverse transcription to proceed. Reverse-transcribed DNA crosses the nuclear membrane and integrates. b, c, Disruption of CypA–CA interactions with either CA(P90A) mutation or cyclosporine treatment leads to detection of DNA reverse transcription product by cGAS initiating cGAMP production, STING activation, NF-κB/IRF3 nuclear localization, type I interferon secretion and initiation of an antiviral state. d, e, Disruption of CPSF6-CA interactions by N74D CA mutation, or depletion of CPSF6, leads to activation of a cryptic innate DNA sensor which also activates NF-κB/IRF3 nuclear localization. f, Disruption of CPSF6 engagement with the nuclear transport machinery by mutating its NLS prevents reverse transcription because the CPSF6 does not dissociate from the capsid at the nuclear pore. (g) PF74 mimics CPSF6 by inserting a phenyl ring into a CA pocket in the same position as CPSF6 and also prevents reverse transcription. Like CPSF6ΔNLS PF74 has no NLS and thus does not disengage from the core and therefore terminally prevents reverse transcription.
{kind=link}
a, MDM were infected with HIV-1 WT, CA N74D or CA(P90A) at low multiplicity. Cells were stained for Gag p24 at specific time points after infection and infected colonies counted. b, MDM transduced to express shRNA targeting CPSF6, or a scrambled control hairpin, were infected with wild-type NL4.3 (Ba-L Env) at low multiplicity. Cells were stained for Gag p24 at specific time points after infection and infected colonies counted. (a-b) P = 0.001, two-way ANOVA, for the effect of CA mutation or CPSF6 depletion. Data represent mean and nonlinear regression over time for 3 biological replicates. c, Individual experiments from two additional donors performed as experiments shown in Fig. 1a and b, and one additional donor performed as experiments in Fig. 2c and d. Each HIV-1 mutant is shown compared to wild-type virus data (WT) for comparison.
{kind=link}
a, In order to demonstrate that IFN is able suppress wild type HIV-1 replication, MDM were pretreated with 1 ng ml−1 of recombinant IFN-β for 2 h then infected with HIV-1 WT NL4.3 (BaL-Env) infection. Cells were stained for Gag p24 at specific time points post infection (p.i). b, In order to determine how much IFN-α/β receptor (IFNAR2) neutralizing antibody is required to neutralize an IFN response, MDM were pretreated with varying concentrations of anti-IFNAR2 antibody for 2 h then stimulated with 1 ng ml−1 of recombinant IFN-β for 24 h. IP10 gene expression levels were measured by qRT–PCR and normalized to GAPDH. Results are expressed as fold change of expression over untreated cells. 1 μg ml−1 of IFNAR2 antibody effectively neutralized 1 ng ml−1 recombinant IFN-β, and this dose was used in subsequent experiments. c, MDM were infected with WT or WT and CA mutants at low multiplicity in the presence of anti-IFNAR2 antibody. Cells were stained for Gag at specific time points after infection and infected colonies counted (P values are given for two-way ANOVA for the effect of IFNAR blockade). Data represent mean and nonlinear regression of biological replicate experiments over time. d, Infectious titres of WT and CA mutant viruses were determined on MDM measured by assay of p24 positive cells 48 h post infection. Cells were infected in the presence of anti-IFNAR2 antibody or isotype control antibody (cAb). Titres are expressed as infectious units per nanogram of reverse transcriptase activity determined by ELISA. Mean ±s.e.m. of titre determined at 3 doses (technical replicates).
{kind=link}
a, b, Selected IFN-stimulated genes significantly upregulated by HIV-1 CA mutant infection, as well as by IFN-β and poly(I:C) measured at 24 h shown as fold change in expression (Stim/Control) measured by qRT–PCR and normalized to GAPDH mRNA levels. c, The same RNA samples as a, b were subjected to expression array and are presented in an expression matrix illustrating fold change in gene expression. d, Upregulation of gene expression (mean >2 fold in 2 independent biological replicates) after infection of MDM by HIV-1 wild-type and mutants HIV-1 CA N74D and HIV-1 CA(P90A) as shown. 24 h after infection (MOI 2), total RNA was isolated and subject to expression array, see methods. Results were subject to pathway analysis using the online bioinformatics tool- InnateDB (http://www.innatedb.com). Type 1 IFN signalling was the most significantly over-represented pathway with IFN-β, PolyIC and both HIV-1 mutants, but not WT virus, based on the Reactome database (http://www.reactome.org). The proportion of genes in each list that map to this pathway and the p-value following Benjamini-Hochberg correction for multiple biological replicates are indicated.
{kind=link}
a, Infection of human MDM with WT NL4.3 (Ba-L Env) in the presence or absence of 10 μM PF74. Cells were stained for p24 at specific time points after infection and infected colonies counted. b, Supernatants collected from MDMs in a were assayed for soluble IFN-β levels by ELISA. c, MDM were infected at low multiplicity with WT HIV-1 in the presence of PF74 and IFNAR2 antibody or isotype cAb. d, Measurement of HIV-1 late reverse transcription product (LRT) in MDM infected for 9 h with two concentrations of WT HIV-1, in the presence or absence of 10 μM PF74. Cells infected with boiled virus served as a negative control for DNA contamination. e, A sample parallel to those in d was used to determine the number of infected cells by staining for p24 48 h post infection. f, Measurement of HIV-1 LRT product in HeLa cells that express CPSF6ΔNLS or empty vector (EV), infected for 9 h with VSV-G pseudotyped HIV-1 GFP. g, A parallel sample was used to determine the number of infected cells by flow cytometry 48 h post infection. h, Infectious titres of WT and CA mutant viruses were determined on MDM in the presence and absence of SIVmac-VLPs encoding the SAMHD1 antagonist Vpx. Titres are expressed as infectious units per nanogram of reverse transcriptase activity determined by ELISA. Mean ±s.e.m. of titre determined at 3 doses. Experiments represent 3 independent biological replicates.
{kind=link}
a, Immunoblot of extracts from L929 cells stably expressing Firefly Luciferase-driven by an interferon sensitive response element depleted of STING by siRNA or expressing control siRNA. b, L929 cells, control or STING depleted as in a, were treated with various concentrations of STING agonist cyclic GMP-AMP (cGAMP), and Luciferase activity was read after 16 h. c, MDM were infected with HIV-1 wild-type (WT) or mutant HIV-1 CA N74D or HIV-1 CA(P90A) for 18 h (MOI of 2), total cell extracts were isolated and were heat and benzonase treated and were applied to L929 cells as in a and Luciferase activity measured at 16 h (Mean of 4 ±s.e.m. of biological replicates). d, As c except RNA was extracted from the cell extracts and were transfected into 293T cells to measure IFN-β promoter driven Luciferase activity after 16 h. Sendai virus infection served as positive control for immuno-stimulatory RNA (Mean ±s.e.m. of biological replicates). c, * represents statistically significant difference between data sets (P < 0.05, t-test), NS represents non-significant differences.
{kind=link}
a, Confocal immunofluorescence microscopy was used to quantify nuclear translocation of NF-κB Rel A (green) and IRF3 (red) as a consequence of activation. Nuclear:cytoplasmic ratios of immunostaining were measured at single cell level by quantitation of NF-κB or IRF3 signal intensities inside and outside the nucleus (blue DAPI) as described in methods. b, Data for 500 single cell measurements are shown for unstimulated MDM and MDM stimulated for 2 h with lipopolysaccharide (LPS) (100 ng ml−1), or infected with wild-type (WT) HIV-1 or N74D or P90A CA mutants. Red lines represent the mean of each data set. * represent statistically significant differences between data sets (P< 0.01, t-test), ns represents non-significant differences (P > 0.05, t-test).
{kind=link}
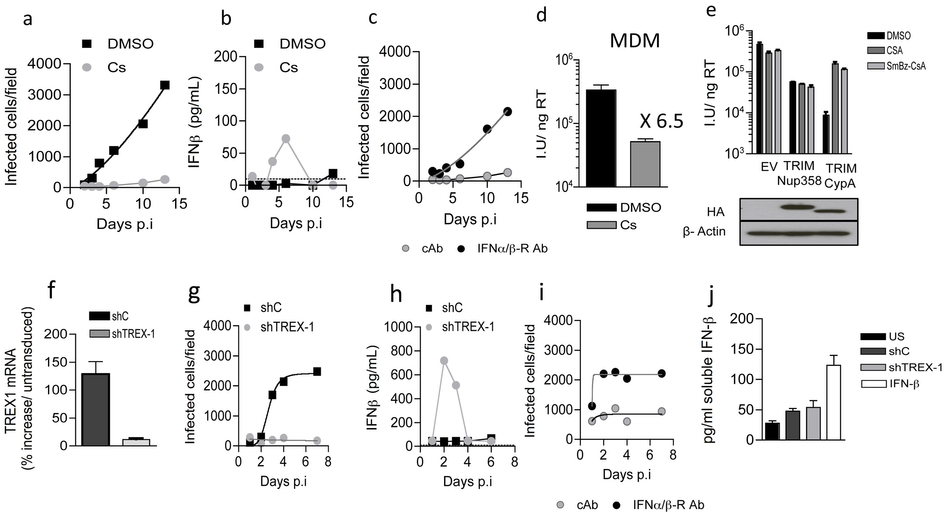
a, Infection of human MDM with WT NL4.3 (Ba-L Env) in the presence or absence of 5 μM cyclosporine. Cells were stained for p24 at specific time points after infection and infected colonies counted. b, Supernatants isolated from MDMs in a were assayed for soluble IFN-β levels by ELISA. c, MDM were infected at low multiplicity with WT HIV in the presence of cyclosporine and IFNAR2 antibody or isotype cAb. d, Infectious titre of WT HIV was determined on MDM in the presence of DMSO or cyclosporine at 48h post infection. The data are presented as infectious units (iu) per nanogram (ng) of reverse transcriptase (RT) measured by ELISA. e, To confirm that SmBz-CsA inhibits recruitment of cyclophilin A (CypA), but not Nup358 Cyp, to HIV-1 CA, we used the TRIMCyp restriction assay. HIV-1 GFP vector titer on CRFK cells expressing empty vector (EV), HA-tagged owl monkey TRIMCyp RBCC domain fused to human CypA (TRIMCypA) or human Nup358Cyp (TRIMNup358) in either DMSO, or 5 μM cyclosporine or 10 μM SmBz-CsA. Protein levels were measured by immunoblot detecting the HA tag with β-Actin as a loading control. In this assay both cyclosporine and SmBz-CsA inhibited CypA recruitment to CA and rescued VSV-G pseudotyped HIV-1 GFP infectivity from restriction by TRIMCypA. However, neither drug rescued HIV-1 infectivity from TRIMNup358, confirming cyclosporine specificity for CypA and not Nup358. f, TREX-1 expression was determined in MDM expressing TREX-1 specific shRNA or control shRNA by qRTPCR, normalized to GAPDH at the time of HIV-1 infection in g. g, Cells were stained for p24 at specific time points after WT HIV-1 infection of TREX-1 depleted and control shRNA expressing MDM and infected colonies counted. h, Supernatants isolated from MDM in g were assayed for soluble IFN-β by ELISA. i, Infection of TREX-1 depleted MDM with WT HIV-1 at low multiplicity in the presence of either IFNAR2 antibody or isotype cAb. Cells were stained for p24 at specific time points after infection and infected colonies counted. j, Hairpin transduced MDM were assayed for the production of soluble IFN-β levels before HIV-1 infection. Data are representative of 3 independent biological replicates. For HIV-1 replication assays in MDM data points and nonlinear regression lines over time are shown.
{kind=link}
GHOST (a-c) or HeLa TZM bl (d–f) indicator cell lines were infected with WT NL4.3 (Ba-L Env) or NL4.3 (Ba-L Env) bearing CA mutations N74D or P90A at low multiplicity (0.04). Replication was monitored by GFP expression (GHOST) or staining LacZ positive cells (HeLa TZM bl). Both mutants replicated well, slightly behind wild-type virus. Replication in GHOST (b) or HeLa TZM bl (e) was performed in the presence of IFNAR2 antibody or isotype cAb. Neither antibody had any effect on WT or mutant HIV-1 replication in GHOST or HeLa TZM bl indicator cell lines. c, f, Induction of ISGs IP10 and IFIT1 expression were measured by quantitative RT PCR after high multiplicity infection (MOI 2) by WT or CA mutant HIV-1 on GHOST (c) or HeLa TZM bl (f). 10 ng ml−1 of IFN-β treatment acted as a positive control. ISG expression levels were normalized to GAPDH and are expressed as fold change in expression over unstimulated cells (Mean of 3 replicates ± s.e.m.). Neither WT or CA mutant HIV-1 induced ISG expression in either cell line. g, HeLa TZM bl were infected WT or CA G89V at low multiplicity (0.04). Replication was monitored by staining LacZ positive cells. h, HIV-1 CA G89V replication in HeLa TZM bl was defective and not rescued by anti-IFNAR2 or isotype control antibodies. All results are representative of 2 biological replicates.
{kind=link}
{kind=link}
Acknowledgements
We are grateful to Jason W. Chin, Steve Goodbourn, KyeongEun Lee, Olga Perisic and Vineet KewalRamani for reagents and advice. This work was funded by Wellcome Trust Senior Fellowship 090940 to GJT, the Medical Research Council, an MRC Confidence in Concept Award to GJT and DS and the National Institute of Health Research UCL/UCLH Comprehensive Biomedical Research Centre.
Footnotes
Author Contributions: J.R, C.P.T, A.J.F, D.L.S., M.N. and G.J.T designed the study. J.R performed all experiments in MDM, C.P.T. performed cGAMP and immunostimulatory RNA assays, A.J.F performed CPSF6 experiments in HeLa cells. A.J.P solved the structure of SmBz-CsC in complex with CypA and L.H. performed the TRIMCyp experiments. J.R, A.J.F. A.J.P, L.H. D.A.J, L.C.J, M.N. and G.J.T analysed the data. A.J.F and C.B generated constructs and D.L.S. provided SmBz-CsA. J.R., M.N. and G.J.T. wrote the manuscript.
Author Information: Structural coordinates have been deposited under PDB accession code 4IPZ. Microarray data are available from the EBI Array Express repository (http://www.ebi.ac.uk/arrayexpress/) under accession no E-MTAB-1437. Reprints and permissions information is available at www.nature.com/reprints.
The authors declare that they have competing financial interests. JR, LCJ, DS and GJT are inventors on a patent claiming the anti-HIV activity of SmBzCsA.
REFERENCES
- 1.Lee K, et al. Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe. 2010;7:221–233. doi: 10.1016/j.chom.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schaller T, et al. HIV-1 capsid-cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS pathogens. 2011;7:e1002439. doi: 10.1371/journal.ppat.1002439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Price AJ, et al. CPSF6 Defines a Conserved Capsid Interface that Modulates HIV-1 Replication. PLoS pathogens. 2012;8:e1002896. doi: 10.1371/journal.ppat.1002896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ambrose Z, et al. Human immunodeficiency virus type 1 capsid mutation N74D alters cyclophilin A dependence and impairs macrophage infection. Journal of virology. 2012;86:4708–4714. doi: 10.1128/JVI.05887-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsang J, et al. HIV-1 infection of macrophages is dependent on evasion of innate immune cellular activation. AIDS. 2009;23:2255–2263. doi: 10.1097/QAD.0b013e328331a4ce. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Besnier C, Takeuchi Y, Towers G. Restriction of lentivirus in monkeys. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:11920–11925. doi: 10.1073/pnas.172384599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iyer SR, Yu D, Biancotto A, Margolis LB, Wu Y. Measurement of human immunodeficiency virus type 1 preintegration transcription by using Rev-dependent Rev-CEM cells reveals a sizable transcribing DNA population comparable to that from proviral templates. Journal of virology. 2009;83:8662–8673. doi: 10.1128/JVI.00874-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shi J, Zhou J, Shah VB, Aiken C, Whitby K. Small-molecule inhibition of human immunodeficiency virus type 1 infection by virus capsid destabilization. Journal of virology. 2011;85:542–549. doi: 10.1128/JVI.01406-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blair WS, et al. HIV capsid is a tractable target for small molecule therapeutic intervention. PLoS pathogens. 2010;6:e1001220. doi: 10.1371/journal.ppat.1001220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dettwiler S, Aringhieri C, Cardinale S, Keller W, Barabino SM. Distinct sequence motifs within the 68-kDa subunit of cleavage factor Im mediate RNA binding, protein-protein interactions, and subcellular localization. The Journal of biological chemistry. 2004;279:35788–35797. doi: 10.1074/jbc.M403927200. [DOI] [PubMed] [Google Scholar]
- 11.Ocwieja KE, et al. HIV integration targeting: a pathway involving Transportin-3 and the nuclear pore protein RanBP2. PLoS pathogens. 2011;7:e1001313. doi: 10.1371/journal.ppat.1001313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao D, et al. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science. 2013;341:903–906. doi: 10.1126/science.1240933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dube H, et al. A mitochondrial-targeted cyclosporin A with high binding affinity for cyclophilin D yields improved cytoprotection of cardiomyocytes. The Biochemical journal. 2012;441:901–907. doi: 10.1042/BJ20111301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu JP, Ye L, Wang X, Li JL, Ho WZ. Cyclosporin A inhibits hepatitis C virus replication and restores interferon-alpha expression in hepatocytes. Transplant infectious disease. 2011;13:24–32. doi: 10.1111/j.1399-3062.2010.00556.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yan N, Regalado-Magdos AD, Stiggelbout B, Lee-Kirsch MA, Lieberman J. The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat Immunol. 2010;11:1005–1013. doi: 10.1038/ni.1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Manel N, et al. A cryptic sensor for HIV-1 activates antiviral innate immunity in dendritic cells. Nature. 2010;467:214–217. doi: 10.1038/nature09337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morner A, et al. Primary human immunodeficiency virus type 2 (HIV-2) isolates, like HIV-1 isolates, frequently use CCR5 but show promiscuity in coreceptor usage. Journal of virology. 1999;73:2343–2349. doi: 10.1128/jvi.73.3.2343-2349.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang F, Hatziioannou T, Perez-Caballero D, Derse D, Bieniasz PD. Antiretroviral potential of human tripartite motif-5 and related proteins. Virology. 2006;353:396–409. doi: 10.1016/j.virol.2006.05.035. [DOI] [PubMed] [Google Scholar]
- 20.Lee K, et al. HIV-1 capsid-targeting domain of cleavage and polyadenylation specificity factor 6. Journal of virology. 2012;86:3851–3860. doi: 10.1128/JVI.06607-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ruepp MD, Schumperli D, Barabino SM. mRNA 3′ end processing and more--multiple functions of mammalian cleavage factor I-68. Wiley interdisciplinary reviews. RNA. 2011;2:79–91. doi: 10.1002/wrna.35. [DOI] [PubMed] [Google Scholar]
- 22.Yang Q, Gilmartin GM, Doublie S. The structure of human cleavage factor I(m) hints at functions beyond UGUA-specific RNA binding: a role in alternative polyadenylation and a potential link to 5′ capping and splicing. RNA biology. 2011;8:748–753. doi: 10.4161/rna.8.5.16040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hori T, et al. A carboxy-terminally truncated human CPSF6 lacking residues encoded by exon 6 inhibits HIV-1 cDNA synthesis and promotes capsid disassembly. Journal of virology. 2013;87:7726–7736. doi: 10.1128/JVI.00124-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zufferey R, Nagy D, Mandel RJ, Naldini L, Trono D. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nature Biotechnology. 1997;15:871–875. doi: 10.1038/nbt0997-871. [DOI] [PubMed] [Google Scholar]
- 25.Naldini L, et al. In vivo gene delivery and stable transduction of non-dividing cells by a lentiviral vector. Science. 1996;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 26.Negre D, et al. Characterization of novel safe lentiviral vectors derived from simian immunodeficiency virus (SIVmac251) that efficiently transduce mature human dendritic cells. Gene Ther. 2000;7:1613–1623. doi: 10.1038/sj.gt.3301292. [DOI] [PubMed] [Google Scholar]
- 27.Saeed AI, et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003;34:374–378. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]
- 28.Lynn DJ, et al. Curating the innate immunity interactome. BMC systems biology. 2010;4:117. doi: 10.1186/1752-0509-4-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Butler SL, Hansen MS, Bushman FD. A quantitative assay for HIV DNA integration in vivo. Nature medicine. 2001;7:631–634. doi: 10.1038/87979. [DOI] [PubMed] [Google Scholar]
- 30.Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Collaborative Computational Project, N The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 32.Mikol V, Kallen J, Pflugl G, Walkinshaw MD. X-ray structure of a monomeric cyclophilin A-cyclosporin A crystal complex at 2.1 A resolution. J Mol Biol. 1993;234:1119–1130. doi: 10.1006/jmbi.1993.1664. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
a, Intact capsids recruit CypA and CPSF6 which direct the virus to the nucleus. CPSF6 interaction prevents premature DNA synthesis. Excess cytoplasmic DNA is degraded by TREX1. At the nuclear pore CPSF6 NLS-dependent dissociation from the virus allows reverse transcription to proceed. Reverse-transcribed DNA crosses the nuclear membrane and integrates. b, c, Disruption of CypA–CA interactions with either CA(P90A) mutation or cyclosporine treatment leads to detection of DNA reverse transcription product by cGAS initiating cGAMP production, STING activation, NF-κB/IRF3 nuclear localization, type I interferon secretion and initiation of an antiviral state. d, e, Disruption of CPSF6-CA interactions by N74D CA mutation, or depletion of CPSF6, leads to activation of a cryptic innate DNA sensor which also activates NF-κB/IRF3 nuclear localization. f, Disruption of CPSF6 engagement with the nuclear transport machinery by mutating its NLS prevents reverse transcription because the CPSF6 does not dissociate from the capsid at the nuclear pore. (g) PF74 mimics CPSF6 by inserting a phenyl ring into a CA pocket in the same position as CPSF6 and also prevents reverse transcription. Like CPSF6ΔNLS PF74 has no NLS and thus does not disengage from the core and therefore terminally prevents reverse transcription.
a, MDM were infected with HIV-1 WT, CA N74D or CA(P90A) at low multiplicity. Cells were stained for Gag p24 at specific time points after infection and infected colonies counted. b, MDM transduced to express shRNA targeting CPSF6, or a scrambled control hairpin, were infected with wild-type NL4.3 (Ba-L Env) at low multiplicity. Cells were stained for Gag p24 at specific time points after infection and infected colonies counted. (a-b) P = 0.001, two-way ANOVA, for the effect of CA mutation or CPSF6 depletion. Data represent mean and nonlinear regression over time for 3 biological replicates. c, Individual experiments from two additional donors performed as experiments shown in Fig. 1a and b, and one additional donor performed as experiments in Fig. 2c and d. Each HIV-1 mutant is shown compared to wild-type virus data (WT) for comparison.
a, In order to demonstrate that IFN is able suppress wild type HIV-1 replication, MDM were pretreated with 1 ng ml−1 of recombinant IFN-β for 2 h then infected with HIV-1 WT NL4.3 (BaL-Env) infection. Cells were stained for Gag p24 at specific time points post infection (p.i). b, In order to determine how much IFN-α/β receptor (IFNAR2) neutralizing antibody is required to neutralize an IFN response, MDM were pretreated with varying concentrations of anti-IFNAR2 antibody for 2 h then stimulated with 1 ng ml−1 of recombinant IFN-β for 24 h. IP10 gene expression levels were measured by qRT–PCR and normalized to GAPDH. Results are expressed as fold change of expression over untreated cells. 1 μg ml−1 of IFNAR2 antibody effectively neutralized 1 ng ml−1 recombinant IFN-β, and this dose was used in subsequent experiments. c, MDM were infected with WT or WT and CA mutants at low multiplicity in the presence of anti-IFNAR2 antibody. Cells were stained for Gag at specific time points after infection and infected colonies counted (P values are given for two-way ANOVA for the effect of IFNAR blockade). Data represent mean and nonlinear regression of biological replicate experiments over time. d, Infectious titres of WT and CA mutant viruses were determined on MDM measured by assay of p24 positive cells 48 h post infection. Cells were infected in the presence of anti-IFNAR2 antibody or isotype control antibody (cAb). Titres are expressed as infectious units per nanogram of reverse transcriptase activity determined by ELISA. Mean ±s.e.m. of titre determined at 3 doses (technical replicates).
a, b, Selected IFN-stimulated genes significantly upregulated by HIV-1 CA mutant infection, as well as by IFN-β and poly(I:C) measured at 24 h shown as fold change in expression (Stim/Control) measured by qRT–PCR and normalized to GAPDH mRNA levels. c, The same RNA samples as a, b were subjected to expression array and are presented in an expression matrix illustrating fold change in gene expression. d, Upregulation of gene expression (mean >2 fold in 2 independent biological replicates) after infection of MDM by HIV-1 wild-type and mutants HIV-1 CA N74D and HIV-1 CA(P90A) as shown. 24 h after infection (MOI 2), total RNA was isolated and subject to expression array, see methods. Results were subject to pathway analysis using the online bioinformatics tool- InnateDB (http://www.innatedb.com). Type 1 IFN signalling was the most significantly over-represented pathway with IFN-β, PolyIC and both HIV-1 mutants, but not WT virus, based on the Reactome database (http://www.reactome.org). The proportion of genes in each list that map to this pathway and the p-value following Benjamini-Hochberg correction for multiple biological replicates are indicated.
a, Infection of human MDM with WT NL4.3 (Ba-L Env) in the presence or absence of 10 μM PF74. Cells were stained for p24 at specific time points after infection and infected colonies counted. b, Supernatants collected from MDMs in a were assayed for soluble IFN-β levels by ELISA. c, MDM were infected at low multiplicity with WT HIV-1 in the presence of PF74 and IFNAR2 antibody or isotype cAb. d, Measurement of HIV-1 late reverse transcription product (LRT) in MDM infected for 9 h with two concentrations of WT HIV-1, in the presence or absence of 10 μM PF74. Cells infected with boiled virus served as a negative control for DNA contamination. e, A sample parallel to those in d was used to determine the number of infected cells by staining for p24 48 h post infection. f, Measurement of HIV-1 LRT product in HeLa cells that express CPSF6ΔNLS or empty vector (EV), infected for 9 h with VSV-G pseudotyped HIV-1 GFP. g, A parallel sample was used to determine the number of infected cells by flow cytometry 48 h post infection. h, Infectious titres of WT and CA mutant viruses were determined on MDM in the presence and absence of SIVmac-VLPs encoding the SAMHD1 antagonist Vpx. Titres are expressed as infectious units per nanogram of reverse transcriptase activity determined by ELISA. Mean ±s.e.m. of titre determined at 3 doses. Experiments represent 3 independent biological replicates.
a, Immunoblot of extracts from L929 cells stably expressing Firefly Luciferase-driven by an interferon sensitive response element depleted of STING by siRNA or expressing control siRNA. b, L929 cells, control or STING depleted as in a, were treated with various concentrations of STING agonist cyclic GMP-AMP (cGAMP), and Luciferase activity was read after 16 h. c, MDM were infected with HIV-1 wild-type (WT) or mutant HIV-1 CA N74D or HIV-1 CA(P90A) for 18 h (MOI of 2), total cell extracts were isolated and were heat and benzonase treated and were applied to L929 cells as in a and Luciferase activity measured at 16 h (Mean of 4 ±s.e.m. of biological replicates). d, As c except RNA was extracted from the cell extracts and were transfected into 293T cells to measure IFN-β promoter driven Luciferase activity after 16 h. Sendai virus infection served as positive control for immuno-stimulatory RNA (Mean ±s.e.m. of biological replicates). c, * represents statistically significant difference between data sets (P < 0.05, t-test), NS represents non-significant differences.
a, Confocal immunofluorescence microscopy was used to quantify nuclear translocation of NF-κB Rel A (green) and IRF3 (red) as a consequence of activation. Nuclear:cytoplasmic ratios of immunostaining were measured at single cell level by quantitation of NF-κB or IRF3 signal intensities inside and outside the nucleus (blue DAPI) as described in methods. b, Data for 500 single cell measurements are shown for unstimulated MDM and MDM stimulated for 2 h with lipopolysaccharide (LPS) (100 ng ml−1), or infected with wild-type (WT) HIV-1 or N74D or P90A CA mutants. Red lines represent the mean of each data set. * represent statistically significant differences between data sets (P< 0.01, t-test), ns represents non-significant differences (P > 0.05, t-test).
a, Infection of human MDM with WT NL4.3 (Ba-L Env) in the presence or absence of 5 μM cyclosporine. Cells were stained for p24 at specific time points after infection and infected colonies counted. b, Supernatants isolated from MDMs in a were assayed for soluble IFN-β levels by ELISA. c, MDM were infected at low multiplicity with WT HIV in the presence of cyclosporine and IFNAR2 antibody or isotype cAb. d, Infectious titre of WT HIV was determined on MDM in the presence of DMSO or cyclosporine at 48h post infection. The data are presented as infectious units (iu) per nanogram (ng) of reverse transcriptase (RT) measured by ELISA. e, To confirm that SmBz-CsA inhibits recruitment of cyclophilin A (CypA), but not Nup358 Cyp, to HIV-1 CA, we used the TRIMCyp restriction assay. HIV-1 GFP vector titer on CRFK cells expressing empty vector (EV), HA-tagged owl monkey TRIMCyp RBCC domain fused to human CypA (TRIMCypA) or human Nup358Cyp (TRIMNup358) in either DMSO, or 5 μM cyclosporine or 10 μM SmBz-CsA. Protein levels were measured by immunoblot detecting the HA tag with β-Actin as a loading control. In this assay both cyclosporine and SmBz-CsA inhibited CypA recruitment to CA and rescued VSV-G pseudotyped HIV-1 GFP infectivity from restriction by TRIMCypA. However, neither drug rescued HIV-1 infectivity from TRIMNup358, confirming cyclosporine specificity for CypA and not Nup358. f, TREX-1 expression was determined in MDM expressing TREX-1 specific shRNA or control shRNA by qRTPCR, normalized to GAPDH at the time of HIV-1 infection in g. g, Cells were stained for p24 at specific time points after WT HIV-1 infection of TREX-1 depleted and control shRNA expressing MDM and infected colonies counted. h, Supernatants isolated from MDM in g were assayed for soluble IFN-β by ELISA. i, Infection of TREX-1 depleted MDM with WT HIV-1 at low multiplicity in the presence of either IFNAR2 antibody or isotype cAb. Cells were stained for p24 at specific time points after infection and infected colonies counted. j, Hairpin transduced MDM were assayed for the production of soluble IFN-β levels before HIV-1 infection. Data are representative of 3 independent biological replicates. For HIV-1 replication assays in MDM data points and nonlinear regression lines over time are shown.
GHOST (a-c) or HeLa TZM bl (d–f) indicator cell lines were infected with WT NL4.3 (Ba-L Env) or NL4.3 (Ba-L Env) bearing CA mutations N74D or P90A at low multiplicity (0.04). Replication was monitored by GFP expression (GHOST) or staining LacZ positive cells (HeLa TZM bl). Both mutants replicated well, slightly behind wild-type virus. Replication in GHOST (b) or HeLa TZM bl (e) was performed in the presence of IFNAR2 antibody or isotype cAb. Neither antibody had any effect on WT or mutant HIV-1 replication in GHOST or HeLa TZM bl indicator cell lines. c, f, Induction of ISGs IP10 and IFIT1 expression were measured by quantitative RT PCR after high multiplicity infection (MOI 2) by WT or CA mutant HIV-1 on GHOST (c) or HeLa TZM bl (f). 10 ng ml−1 of IFN-β treatment acted as a positive control. ISG expression levels were normalized to GAPDH and are expressed as fold change in expression over unstimulated cells (Mean of 3 replicates ± s.e.m.). Neither WT or CA mutant HIV-1 induced ISG expression in either cell line. g, HeLa TZM bl were infected WT or CA G89V at low multiplicity (0.04). Replication was monitored by staining LacZ positive cells. h, HIV-1 CA G89V replication in HeLa TZM bl was defective and not rescued by anti-IFNAR2 or isotype control antibodies. All results are representative of 2 biological replicates.