

Abstract
Optic nerve atrophy and hypoplasia can be primary disorders or can result from trans-synaptic degeneration arising from cerebral visual impairment (CVI). Here we report six individuals with CVI and/or optic nerve abnormalities, born after an uneventful pregnancy and delivery, who have either de novo heterozygous missense mutations in NR2F1, also known as COUP-TFI, or deletions encompassing NR2F1. All affected individuals show mild to moderate intellectual impairment. NR2F1 encodes a nuclear receptor protein that regulates transcription. A reporter assay showed that missense mutations in the zinc-finger DNA-binding domain and the putative ligand-binding domain decrease NR2F1 transcriptional activity. These findings indicate that NR2F1 plays an important role in the neurodevelopment of the visual system and that its disruption can lead to optic atrophy with intellectual disability.
Main Text
Optic nerve abnormalities are increasingly recognized as a cause of poor vision. The most frequently identified abnormalities consist of optic nerve atrophy or hypoplasia, due to either genetic or acquired causes.1 Optic nerve hypoplasia is often reported as part of a syndrome, for example, in conjunction with structural malformations of the brain or hypopituitarism, as observed in SOX2-related disorders (MIM 184429),2 or combined with anterior segmental defects of the eye, such as iris coloboma in PAX6-related diseases (MIM 307108).3 Optic nerve atrophy, however, can be part of a progressive syndromic or nonsyndromic disorder. The genetic defects underlying many syndromic forms of optic atrophy are known, but the molecular bases of nonsyndromic forms have been elucidated in only a few instances: mutations in OPA1 (MIM 605290) and OPA3 (MIM 606580) (autosomal-dominant inheritance),4,5 in TMEM126A (MIM 612988) (autosomal-recessive inheritance),6 and in mitochondrial DNA as part of the Leber Hereditary Optic Neuropathy (LHON) (MIM 535000).7 Both optic atrophy and hypoplasia can be caused by trans-synaptic degeneration as a part of cerebral visual impairment (CVI), a class of disorders of the projection and/or interpretation of visual input in the brain.8–10 Up to two-thirds of the cases of CVI are thought to be caused by perinatal damage, often the result of preterm birth, but genetic defects might play a significant role in the etiology of the remaining cases.11–13 The majority of persons with CVI exhibit additional clinical features, including intellectual disability (ID).11–14
We collected 56 individuals with CVI, all born after an uneventful pregnancy and birth, without any identifiable causes, and performed clinical, ophthalmological, and copy number variant investigations (see Table S1 available online). This study was approved by the Ethics Committee of the Radboud university medical center (Commissie Mensgebonden Onderzoek, regio Arnhem-Nijmegen), and written informed consent was obtained for all enrolled subjects. For 12 individuals and their parents, we performed exome sequencing by using a trio approach.15,16 Exome sequencing was done in ten of these trios through the Baylor-Hopkins Center for Mendelian Genomics; methods have been reported previously.17 In brief, 1 μg of genomic DNA from each individual was fragmented in a Covaris sonication system. Whole-exome targeted capture was performed in solution with the BCM-HGSC Core design. Sequencing was performed on the Illumina HiSeq 2000 platform (Illumina), producing an average of 9–10 Gb of raw data per sample. The sequence reads were mapped and aligned to the Human Genome Reference Assembly GRCh37/hg19 with the BCM-HGSC Mercury pipeline18 at an average depth of coverage of 114×. Variant calling was performed with the Atlas219 and SAMtools20 algorithms; variant annotation was performed with an intramurally developed annotation pipeline21 based on ANNOVAR22 and custom scripts to incorporate multiple databases to inform on identified variants. In the other two trios, we performed exome sequencing on a SOLiD 5500XL platform (Life Technologies) with the Agilent SureSelect All Exon V4 reagent for target enrichment (Agilent Technologies). The 50 bp paired-end reads were mapped to USCS Genome Browser GRCh37/hg19 Human Genome Reference Assembly, and variants and indels were called with Lifescope v2.1 (Life Technologies). There was an average coverage of 76×. After quality filtering (variant reads >15%) and exclusion of local and global variants (<1% occurrence), a de novo analysis was performed for all trios. Synonymous variants with no effect on splicing were excluded.
In two persons (individuals 1 and 2) de novo missense mutations predicted to be pathogenic were identified in nuclear receptor subfamily 2, group F, member 1 gene, NR2F1 (chr5: 92921073 G>C; c.344G>C; p.Arg115Pro and chr5: 92921068 C>A; c.339C>A; p.Ser113Arg, respectively; Figure 1, Table S2) (MIM 132890, RefSeq accession number NM_005654.4). Both were situated in the DNA-binding domain. In addition to CVI, individual 1 had small optic discs with large excavations and ID; individual 2 manifested pale optic discs, small optic nerves on MRI, and developmental delay (Table 1; Table S3). We screened the remaining 44 CVI persons for mutations in NR2F1 by Sanger sequencing and identified a third case (individual 3) with a de novo missense mutation, predicted to be pathogenic, in the ligand-binding domain, chr5: 92923914 T>C; c.755T>C; p.Leu252Pro. She had excavated, pale optic discs and ID.
Figure 1.
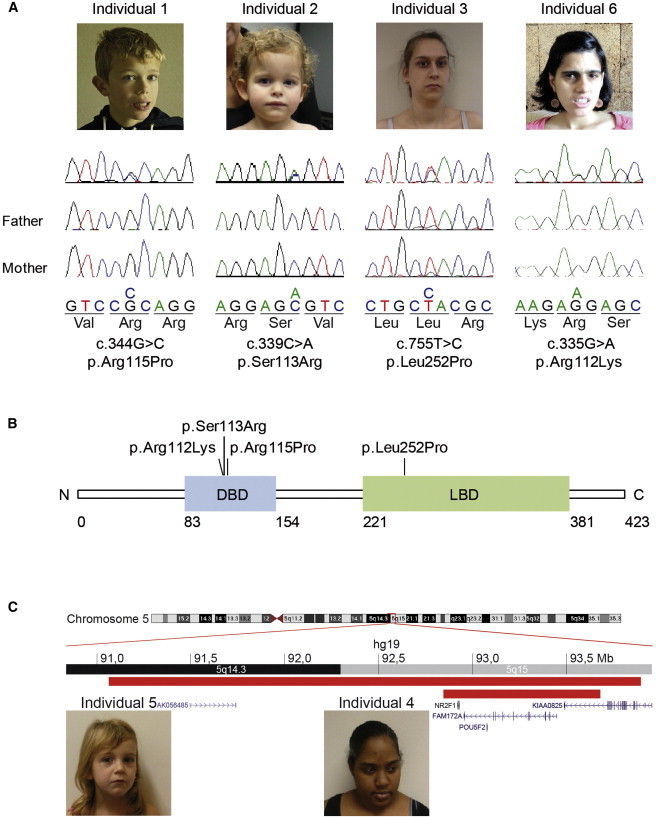
Phenotype and Genotype of the Individuals with Optic Atrophy
(A) Confirmation by Sanger sequencing of the de novo mutations in NR2F1 [NM_005654.4] found by exome sequencing (individuals 1, 2, and 6), the de novo mutation in NR2F1 found by cohort screening (individual 3), and segregation analyses. The pictures of the individuals show nonspecific facial dysmorphisms.
(B) Schematic representation of NR2F1, containing a DNA-binding domain (DBD) and a ligand-binding domain (LBD). The positions of the mutations identified in the affected individuals are indicated.
(C) Overview of the 5q14-q15 region encompassing the two overlapping microdeletions of individual 4 (lower red bar) and 5 (upper red bar) comprising NR2F1. The inheritance of the deletion in individual 4 was unknown, because the father was not available for testing and the deletion was on the paternal allele. The deletion of individual 5 was de novo. The pictures of the affected individuals show nonspecific facial dysmorphisms.
Table 1.
Genotype and Phenotype of the Affected Individuals
| Individual 1 | Individual 2 | Individual 3 | Individual 4 | Individual 5 | Individual 6 | |
|---|---|---|---|---|---|---|
| Defect in NR2F1 | c.344G>C p.Arg115Pro | c.339C>A p.Ser113Arg | c.755T>C p.Leu252Pro | Deletion (0.83 Mb) | Deletion (2.85 Mb) | c.335G>A p.Arg112Lys |
| Gender | Male | Female | Female | Female | Female | Female |
| Age | 12y | 2+4y | 18y | 24y | 4y | 35 y |
| Development | IQ 48 | DD | IQ 55–65 | IQ 61–74 | DD | IQ 52 |
| Behavioral abnormalities | - | - | - | - | - | OCD, autism |
| OFC (centile) | 98th | 55th | 55th | 99th | 50th | 13th |
| CVI | + | + | + | + | + | - |
| Optic disc abnormalities | Small and large excavation | Pale | Pale and large excavation | Partial pale | Small, pale and large excavation | Pale |
| MRI brain | Normal | Small optic nerve and chiasm | ND | ND | Normal | Normal |
DD, developmental delay; ND, not done; OCD, obsessive-compulsive disorder; OFC, occipital frontal head circumference.
The Poisson probability of identifying three de novo mutations by sequencing a cohort of 56 persons was calculated for NR2F1. Based on a de novo mutation rate of 1.18 × 10−8 per position23 and accounting for GC-content and gene size,24–26 the Poisson probability is p = 1.40 × 10−10, indicating that this is highly unlikely to have occurred by chance. This is further strengthened by the consistent clinical phenotype observed in the affected individuals.
Point mutations in NR2F1 have not yet been reported, but large deletions spanning NR2F1 and other genes have been associated recently with optic atrophy, visual motor integration deficit, visual perceptual disorder, and ID.27 Therefore, we searched for deletions of NR2F1 in both the CVI cohort and the intramural database of the Radboud university medical center, Nijmegen, Netherlands, that includes more than 10,000 array analyses from persons with intellectual disability and/or multiple congenital abnormalities. We identified a 5q15 deletion, arr 5q15(92845157–93679748)x1 (Human Genome Reference Assembly GRCh37/hg19) comprising NR2F1 with the CytoScan HD array (Affymetrix) in individual 4, part of the CVI cohort, who had pale optic discs and mild ID (Figure 1). The inheritance of the deletion is unknown, because the deletion was on the paternal allele, but the reportedly phenotypically normal father was not available for testing. We also identified a de novo deletion on chr5: 91064110–93896378 (Human Genome Reference Assembly CRCh37/hg19), arr SNP 5q14.3q15(SNP_A-1810903–>SNP_A-1788922)x1 in the intramural cohort (individual 5) with the 250k SNP array (Affymetrix). Individual 5 had developmental delay and ophthalmological examination showed poor visual acuity, CVI, in the right eye a small pale optic disc, and in the left eye a pale optic disc with a large excavation.
In parallel, a single additional individual with congenital optic atrophy and ID was enrolled through the Baylor-Hopkins Center for Mendelian Genomics study (individual 6, BAB3468, Figure 1; Table 1). This 35-year-old female also had autism spectrum disorder with marked obsessive-compulsive behaviors. The pregnancy and delivery had been uneventful (Table S1). Family history was negative for optic nerve atrophy, ID, developmental delay, and autism. The family was consented through a research protocol approved by the Institutional Review Board (IRB) of Baylor College of Medicine. DNA samples from the proband and her unaffected parents were submitted for exome sequencing by using a trio approach as described above at an average depth of coverage of 107× and with >91% of the target bases covered at a minimum of 20×. Inheritance of the identified variants was examined with the DeNovoCheck algorithm.16 Analysis of the variants identified in the proband under a sporadic de novo model of inheritance revealed a missense mutation (chr5: 92921064 G>A; c.335G>A; p.Arg112Lys) in NR2F1. This variant was situated in the DNA-binding domain and was predicted to be pathogenic.
As individual 6 manifested optic nerve atrophy without CVI, a cohort of mainly nonsyndromic persons (n = 37) with optic nerve abnormalities was screened for NR2F1 variants by Sanger sequencing (Table S4). In a 68-year-old male with optic atrophy, a missense mutation was identified, chr5:92924068 G>C; c.909G>C; p.Gln303His. However, this variant was also present in his unaffected sister and son and was interpreted as a nonpathogenic variant.
NR2F1 encodes a conserved orphan nuclear receptor protein (nuclear receptor subfamily 2, group F, member 1) that, activated by a yet unknown ligand, regulates transcription.28 It belongs to the nuclear receptor superfamily, of which several members already have been implicated in human disease: NR2E3 (MIM 604485) in retinitis pigmentosa,29 ESRRB (MIM 602167) in deafness,30 NR0B1 (MIM 300473) in congenital adrenal hypoplasia,31 and NR2F2 (MIM 107773) in congenital diaphragmatic hernia.32 NR2F1 has a classic nuclear receptor structure with two main domains, a functional DNA-binding domain (DBD) formed by two zinc-finger structural domains and a ligand-binding domain (LBD) with two highly conserved sequence regions.33 Interestingly, three of the four point mutations that we identified localize to the DBD of NR2F1, probably affecting the binding of NR2F1 to its transcriptional targets.
To investigate the functional effects of our identified point mutations in NR2F1, we established a reporter assay. In this assay, luciferase is under control of an NR2F1 activated promoter and cells are cotransfected with this plasmid and either wild-type (WT) or mutant Nr2f1 constructs. The luciferase activity reflects the transcriptional activity of the constructs. The expression plasmids of the mouse Nr2f1 - COUP-TFI, and the NGFI-A (−168/+33) promoter luciferase reporter in pXP2 were previously described.34,35 All point mutations were generated with the QuickChange site-directed mutagenesis kit (Agilent). For luciferase assays, HEK293T cells were plated in 24-well plates (1 × 105 cells per well) 24 hr prior to transfection. Cells were transfected with 5 ng of reporter per well with Lipofectamine 2000 following the manufacturer’s instructions. Forty-eight hr after transfection, cells were harvested and luciferase activity was measured by the luciferase assay system (Promega). As indicated in Figure 2, all DNA-binding domain mutants (p.Arg112Lys, p.Ser113Arg, and p.Arg115Pro) and the ligand-binding domain mutant (p.Leu252Pro) lost their ability to fully activate NR2F1 - COUP-TFI reporter. This result supports our contention that persons who have these mutations do not have fully functional NR2F1 - COUP-TFI product.
Figure 2.
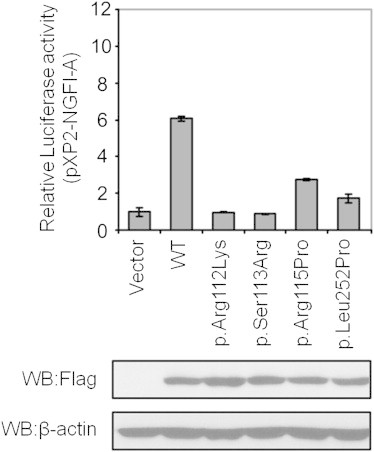
In Vitro Luciferase Reporter Assay for NR2F1 Point Mutations
Effects of the Nr2f1 (COUP-TFI) mutations on activation of an NGFI-A promoter-driven reporter. Top shows that pXP2-NGFI-A was cotransfected with 5 ng FLAG-tagged Nr2f1 (COUP-TFI) WT, FLAG-tagged Nr2f1 (COUP-TFI) mutations or empty vector into HEK293T cells as indicated. The cell sample transfected with pXP2-NGFI-A and vector served as the control, and its value (relative luciferase activity) was set as 1. The relative luciferase activities of the other samples are normalized to this control. Data are means ± SD (n = 3). Bottom shows immunoblot analysis of Nr2f1 (COUP-TFI) expression in transfection. Results indicate similar expression of WT and mutant Nr2f1 (COUP-TFI) constructs.
Additionally, the function of NR2F1 has been investigated in mice. The mouse ortholog, Nr2f1, is expressed in various tissues throughout development, with important functions in cell-fate determination, differentiation, migration, and survival, and an apparently important role in organogenesis.33 In the developing nervous system, it is mainly expressed at high levels in the optic nerve, thalamus, and pallium (future cortex).36–38 Studies of Nr2f1 knockout mice indicate that the protein is important for neurodevelopment, including oligodendrocyte differentiation (partly in vitro experiment),36 cortical patterning,39–41 and guidance of thalamocortical axons,42 as well as eye and optic nerve development.35,43 Furthermore, overexpression of Nr2f1 in mouse neuronal differentiating teratocarcinoma PCC7 cells antagonized neuronal differentiation induced by retinoic acid signaling.44 Given our results from the in vitro reporter assays and because deletions of NR2F1 give rise to a similar phenotype, we hypothesize that the heterozygous mutations identified in our affected individuals cause loss of function and that the observed phenotypes are due to haploinsufficiency. Interestingly, it appears that the latter are restricted to brain and optic nerves, because no other major organ abnormalities are observed in our cohort of patients with either NR2F1 point mutations or deletions. This might be due directly to levels of NR2F1 reduced below a threshold necessary during brain development or indirectly via the target genes it regulates for specific expression in brain development. Brown et al. proposed haploinsufficiency of NR2F1 as the cause of deafness and multiple congenital anomalies in an individual with NR2F1 deletion and paracentric inversion inv(5)(q15q33.2).45 The absence of deafness and multiple congenital anomalies in the affected individuals 4 and 5 suggests that these might be caused by the more complex chromosomal rearrangement identified in the reported person, although phenotypic variability cannot be ruled out at this time.
In addition to the optic nerve abnormalities, five out of six individuals were diagnosed with CVI. This potentially could reflect thalamocortical axon projection disorders, as reported in Nr2f1 knockout mice.42 Until now, optic atrophy in combination with optic disc excavation was thought to be the result of perinatal damage.46–48 We showed, however, that optic disc excavations can also be present in individuals with a genetic cause of CVI (individuals 1, 3, and 5). NR2F1 investigation should, therefore, also be considered in preterm children with CVI.
In conclusion, four individuals with optic atrophy and CVI of unknown cause had heterozygous de novo point mutations in NR2F1, and two persons with a similar phenotype had deletions comprising NR2F1. Our findings indicate that NR2F1 has an important role in the development of the visual system and that haploinsuffiency can lead to optic atrophy with intellectual impairment.
Acknowledgments
We are grateful to the individuals involved and their families for their support and cooperation. This work has been supported by grants from Stichting ODAS that contributed through UitZicht (to F.N.B. and F.P.M.C.), Vereniging Bartiméus-Sonneheerdt (5781251 to F.N.B. and F.P.M.C.), and the Netherlands Organisation for Health Research and Development (ZON-MW grant 917-86-319 to B.B.A.d.V). Additionally, this study was accomplished in part through the Center for Mendelian Genomics research effort funded by The National Institutes of Health (NIH) and supported by the National Human Genome Research Institute grant U54HG006542 to the Baylor-Hopkins Center for Mendelian Genomics. C.P.S. is a recipient of a Clinical Scientist Development Award by the Doris Duke Charitable Foundation. His work is generously supported by the Stanford and Joan Alexander Family. M.-J.T. and S.Y.T. are supported by NIH grants (DK45641 and HL114539). W.W. is supported by a K23NS078056 grant from the NINDS. R.A.L. is a Senior Scientific Investigator of Research to Prevent Blindness, New York.
Contributor Information
Bert B.A. de Vries, Email: Bert.deVries@radboudumc.nl.
Christian P. Schaaf, Email: schaaf@bcm.edu.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
References
- 1.Rahi J.S., Cable N., British Childhood Visual Impairment Study Group Severe visual impairment and blindness in children in the UK. Lancet. 2003;362:1359–1365. doi: 10.1016/S0140-6736(03)14631-4. [DOI] [PubMed] [Google Scholar]
- 2.Kelberman D., Rizzoti K., Avilion A., Bitner-Glindzicz M., Cianfarani S., Collins J., Chong W.K., Kirk J.M., Achermann J.C., Ross R. Mutations within Sox2/SOX2 are associated with abnormalities in the hypothalamo-pituitary-gonadal axis in mice and humans. J. Clin. Invest. 2006;116:2442–2455. doi: 10.1172/JCI28658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Azuma N., Yamaguchi Y., Handa H., Tadokoro K., Asaka A., Kawase E., Yamada M. Mutations of the PAX6 gene detected in patients with a variety of optic-nerve malformations. Am. J. Hum. Genet. 2003;72:1565–1570. doi: 10.1086/375555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alexander C., Votruba M., Pesch U.E., Thiselton D.L., Mayer S., Moore A., Rodriguez M., Kellner U., Leo-Kottler B., Auburger G. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 2000;26:211–215. doi: 10.1038/79944. [DOI] [PubMed] [Google Scholar]
- 5.Reynier P., Amati-Bonneau P., Verny C., Olichon A., Simard G., Guichet A., Bonnemains C., Malecaze F., Malinge M.C., Pelletier J.B. OPA3 gene mutations responsible for autosomal dominant optic atrophy and cataract. J. Med. Genet. 2004;41:e110. doi: 10.1136/jmg.2003.016576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hanein S., Perrault I., Roche O., Gerber S., Khadom N., Rio M., Boddaert N., Jean-Pierre M., Brahimi N., Serre V. TMEM126A, encoding a mitochondrial protein, is mutated in autosomal-recessive nonsyndromic optic atrophy. Am. J. Hum. Genet. 2009;84:493–498. doi: 10.1016/j.ajhg.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wallace D.C., Singh G., Lott M.T., Hodge J.A., Schurr T.G., Lezza A.M., Elsas L.J., 2nd, Nikoskelainen E.K. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science. 1988;242:1427–1430. doi: 10.1126/science.3201231. [DOI] [PubMed] [Google Scholar]
- 8.Uggetti C., Egitto M.G., Fazzi E., Bianchi P.E., Zappoli F., Martelli A., Lanzi G. Transsynaptic degeneration of lateral geniculate bodies in blind children: in vivo MR demonstration. AJNR Am. J. Neuroradiol. 1997;18:233–238. [PMC free article] [PubMed] [Google Scholar]
- 9.Jacobson L.K., Dutton G.N. Periventricular leukomalacia: an important cause of visual and ocular motility dysfunction in children. Surv. Ophthalmol. 2000;45:1–13. doi: 10.1016/s0039-6257(00)00134-x. [DOI] [PubMed] [Google Scholar]
- 10.Dutton G.N., Jacobson L.K. Cerebral visual impairment in children. Semin. Neonatol. 2001;6:477–485. doi: 10.1053/siny.2001.0078. [DOI] [PubMed] [Google Scholar]
- 11.Fazzi E., Signorini S.G., Bova S.M., La Piana R., Ondei P., Bertone C., Misefari W., Bianchi P.E. Spectrum of visual disorders in children with cerebral visual impairment. J. Child Neurol. 2007;22:294–301. doi: 10.1177/08830738070220030801. [DOI] [PubMed] [Google Scholar]
- 12.Khetpal V., Donahue S.P. Cortical visual impairment: etiology, associated findings, and prognosis in a tertiary care setting. J. AAPOS. 2007;11:235–239. doi: 10.1016/j.jaapos.2007.01.122. [DOI] [PubMed] [Google Scholar]
- 13.Huo R., Burden S.K., Hoyt C.S., Good W.V. Chronic cortical visual impairment in children: aetiology, prognosis, and associated neurological deficits. Br. J. Ophthalmol. 1999;83:670–675. doi: 10.1136/bjo.83.6.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boonstra N., Limburg H., Tijmes N., van Genderen M., Schuil J., van Nispen R. Changes in causes of low vision between 1988 and 2009 in a Dutch population of children. Acta Ophthalmol. (Copenh.) 2012;90:277–286. doi: 10.1111/j.1755-3768.2011.02205.x. [DOI] [PubMed] [Google Scholar]
- 15.Vissers L.E., de Ligt J., Gilissen C., Janssen I., Steehouwer M., de Vries P., van Lier B., Arts P., Wieskamp N., del Rosario M. A de novo paradigm for mental retardation. Nat. Genet. 2010;42:1109–1112. doi: 10.1038/ng.712. [DOI] [PubMed] [Google Scholar]
- 16.de Ligt J., Willemsen M.H., van Bon B.W., Kleefstra T., Yntema H.G., Kroes T., Vulto-van Silfhout A.T., Koolen D.A., de Vries P., Gilissen C. Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 2012;367:1921–1929. doi: 10.1056/NEJMoa1206524. [DOI] [PubMed] [Google Scholar]
- 17.Lupski J.R., Gonzaga-Jauregui C., Yang Y., Bainbridge M.N., Jhangiani S., Buhay C.J., Kovar C.L., Wang M., Hawes A.C., Reid J.G. Exome sequencing resolves apparent incidental findings and reveals further complexity of SH3TC2 variant alleles causing Charcot-Marie-Tooth neuropathy. Genome Medicine. 2013;5:57. doi: 10.1186/gm461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bainbridge M.N., Wiszniewski W., Murdock D.R., Friedman J., Gonzaga-Jauregui C., Newsham I., Reid J.G., Fink J.K., Morgan M.B., Gingras M.C. Whole-genome sequencing for optimized patient management. Sci. Transl. Med. 2011;3:re3. doi: 10.1126/scitranslmed.3002243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shen Y., Wan Z., Coarfa C., Drabek R., Chen L., Ostrowski E.A., Liu Y., Weinstock G.M., Wheeler D.A., Gibbs R.A., Yu F. A SNP discovery method to assess variant allele probability from next-generation resequencing data. Genome Res. 2010;20:273–280. doi: 10.1101/gr.096388.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gonzaga-Jauregui C., Lotze T., Jamal L., Penney S., Campbell I.M., Pehlivan D., Hunter J.V., Woodbury S.L., Raymond G., Adesina A.M. Mutations in VRK1 Associated With Complex Motor and Sensory Axonal Neuropathy Plus Microcephaly. JAMA Neurol. 2013;70(12):1491–1498. doi: 10.1001/jamaneurol.2013.4598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Conrad D.F., Keebler J.E., DePristo M.A., Lindsay S.J., Zhang Y., Casals F., Idaghdour Y., Hartl C.L., Torroja C., Garimella K.V., 1000 Genomes Project Variation in genome-wide mutation rates within and between human families. Nat. Genet. 2011;43:712–714. doi: 10.1038/ng.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bainbridge M.N., Hu H., Muzny D.M., Musante L., Lupski J.R., Graham B.H., Chen W., Gripp K.W., Jenny K., Wienker T.F. De novo truncating mutations in ASXL3 are associated with a novel clinical phenotype with similarities to Bohring-Opitz syndrome. Genome Medicine. 2013;5:11. doi: 10.1186/gm415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Antonarakis S.E. Human Genome Sequence and Variation. In: Speicher M., Antonarakis S.E., Motulsky A.G., editors. Vogel and Motulsky’s Human Genetics. Springer-Verlag; Berlin Heidelberg, Germany: 2010. pp. 31–53. [Google Scholar]
- 26.Sanders S.J., Murtha M.T., Gupta A.R., Murdoch J.D., Raubeson M.J., Willsey A.J., Ercan-Sencicek A.G., DiLullo N.M., Parikshak N.N., Stein J.L. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Al-Kateb H., Shimony J.S., Vineyard M., Manwaring L., Kulkarni S., Shinawi M. NR2F1 haploinsufficiency is associated with optic atrophy, dysmorphism and global developmental delay. Am. J. Med. Genet. A. 2013;161A:377–381. doi: 10.1002/ajmg.a.35650. [DOI] [PubMed] [Google Scholar]
- 28.Sagami I., Tsai S.Y., Wang H., Tsai M.J., O’Malley B.W. Identification of two factors required for transcription of the ovalbumin gene. Mol. Cell. Biol. 1986;6:4259–4267. doi: 10.1128/mcb.6.12.4259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haider N.B., Jacobson S.G., Cideciyan A.V., Swiderski R., Streb L.M., Searby C., Beck G., Hockey R., Hanna D.B., Gorman S. Mutation of a nuclear receptor gene, NR2E3, causes enhanced S cone syndrome, a disorder of retinal cell fate. Nat. Genet. 2000;24:127–131. doi: 10.1038/72777. [DOI] [PubMed] [Google Scholar]
- 30.Collin R.W., Kalay E., Tariq M., Peters T., van der Zwaag B., Venselaar H., Oostrik J., Lee K., Ahmed Z.M., Caylan R. Mutations of ESRRB encoding estrogen-related receptor beta cause autosomal-recessive nonsyndromic hearing impairment DFNB35. Am. J. Hum. Genet. 2008;82:125–138. doi: 10.1016/j.ajhg.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muscatelli F., Strom T.M., Walker A.P., Zanaria E., Récan D., Meindl A., Bardoni B., Guioli S., Zehetner G., Rabl W. Mutations in the DAX-1 gene give rise to both X-linked adrenal hypoplasia congenita and hypogonadotropic hypogonadism. Nature. 1994;372:672–676. doi: 10.1038/372672a0. [DOI] [PubMed] [Google Scholar]
- 32.Ishii S., Tsai S.Y., Tsai M.-J. Role of COUP-TFII in Congenital Diaphragmatic Hernia. In: Bradshaw R.A., Dennis E., editors. Handbook of Cell Signaling. Academic Press; Oxford: 2009. pp. 2021–2026. [Google Scholar]
- 33.Lin F.J., Qin J., Tang K., Tsai S.Y., Tsai M.J. Coup d’Etat: an orphan takes control. Endocr. Rev. 2011;32:404–421. doi: 10.1210/er.2010-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pipaón C., Tsai S.Y., Tsai M.J. COUP-TF upregulates NGFI-A gene expression through an Sp1 binding site. Mol. Cell. Biol. 1999;19:2734–2745. doi: 10.1128/mcb.19.4.2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tang K., Xie X., Park J.I., Jamrich M., Tsai S., Tsai M.J. COUP-TFs regulate eye development by controlling factors essential for optic vesicle morphogenesis. Development. 2010;137:725–734. doi: 10.1242/dev.040568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yamaguchi H., Zhou C., Lin S.C., Durand B., Tsai S.Y., Tsai M.J. The nuclear orphan receptor COUP-TFI is important for differentiation of oligodendrocytes. Dev. Biol. 2004;266:238–251. doi: 10.1016/j.ydbio.2003.10.038. [DOI] [PubMed] [Google Scholar]
- 37.Qiu Y., Cooney A.J., Kuratani S., DeMayo F.J., Tsai S.Y., Tsai M.J. Spatiotemporal expression patterns of chicken ovalbumin upstream promoter-transcription factors in the developing mouse central nervous system: evidence for a role in segmental patterning of the diencephalon. Proc. Natl. Acad. Sci. USA. 1994;91:4451–4455. doi: 10.1073/pnas.91.10.4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qiu Y., Pereira F.A., DeMayo F.J., Lydon J.P., Tsai S.Y., Tsai M.J. Null mutation of mCOUP-TFI results in defects in morphogenesis of the glossopharyngeal ganglion, axonal projection, and arborization. Genes Dev. 1997;11:1925–1937. doi: 10.1101/gad.11.15.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou C., Tsai S.Y., Tsai M.J. COUP-TFI: an intrinsic factor for early regionalization of the neocortex. Genes Dev. 2001;15:2054–2059. doi: 10.1101/gad.913601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Armentano M., Chou S.J., Tomassy G.S., Leingärtner A., O’Leary D.D., Studer M. COUP-TFI regulates the balance of cortical patterning between frontal/motor and sensory areas. Nat. Neurosci. 2007;10:1277–1286. doi: 10.1038/nn1958. [DOI] [PubMed] [Google Scholar]
- 41.Faedo A., Tomassy G.S., Ruan Y., Teichmann H., Krauss S., Pleasure S.J., Tsai S.Y., Tsai M.J., Studer M., Rubenstein J.L. COUP-TFI coordinates cortical patterning, neurogenesis, and laminar fate and modulates MAPK/ERK, AKT, and beta-catenin signaling. Cereb. Cortex. 2008;18:2117–2131. doi: 10.1093/cercor/bhm238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou C., Qiu Y., Pereira F.A., Crair M.C., Tsai S.Y., Tsai M.J. The nuclear orphan receptor COUP-TFI is required for differentiation of subplate neurons and guidance of thalamocortical axons. Neuron. 1999;24:847–859. doi: 10.1016/s0896-6273(00)81032-6. [DOI] [PubMed] [Google Scholar]
- 43.Satoh S., Tang K., Iida A., Inoue M., Kodama T., Tsai S.Y., Tsai M.J., Furuta Y., Watanabe S. The spatial patterning of mouse cone opsin expression is regulated by bone morphogenetic protein signaling through downstream effector COUP-TF nuclear receptors. J. Neurosci. 2009;29:12401–12411. doi: 10.1523/JNEUROSCI.0951-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Neuman K., Soosaar A., Nornes H.O., Neuman T. Orphan receptor COUP-TF I antagonizes retinoic acid-induced neuronal differentiation. J. Neurosci. Res. 1995;41:39–48. doi: 10.1002/jnr.490410106. [DOI] [PubMed] [Google Scholar]
- 45.Brown K.K., Alkuraya F.S., Matos M., Robertson R.L., Kimonis V.E., Morton C.C. NR2F1 deletion in a patient with a de novo paracentric inversion, inv(5)(q15q33.2), and syndromic deafness. Am. J. Med. Genet. A. 2009;149A:931–938. doi: 10.1002/ajmg.a.32764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jacobson L., Hellström A., Flodmark O. Large cups in normal-sized optic discs: a variant of optic nerve hypoplasia in children with periventricular leukomalacia. Arch. Ophthalmol. 1997;115:1263–1269. doi: 10.1001/archopht.1997.01100160433007. [DOI] [PubMed] [Google Scholar]
- 47.Jacobson L., Hård A.L., Svensson E., Flodmark O., Hellström A. Optic disc morphology may reveal timing of insult in children with periventricular leucomalacia and/or periventricular haemorrhage. Br. J. Ophthalmol. 2003;87:1345–1349. doi: 10.1136/bjo.87.11.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ruberto G., Salati R., Milano G., Bertone C., Tinelli C., Fazzi E., Guagliano R., Signorini S., Borgatti R., Bianchi A., Bianchi P.E. Changes in the optic disc excavation of children affected by cerebral visual impairment: a tomographic analysis. Invest. Ophthalmol. Vis. Sci. 2006;47:484–488. doi: 10.1167/iovs.05-0529. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.