

Abstract
Leigh syndrome (LS) is a severe neurodegenerative disorder with characteristic bilateral lesions, typically in the brainstem and basal ganglia. It usually presents in infancy and is genetically heterogeneous, but most individuals with mitochondrial complex IV (or cytochrome c oxidase) deficiency have mutations in the biogenesis factor SURF1. We studied eight complex IV-deficient LS individuals from six families of Lebanese origin. They differed from individuals with SURF1 mutations in having seizures as a prominent feature. Complementation analysis suggested they had mutation(s) in the same gene but targeted massively parallel sequencing (MPS) of 1,034 genes encoding known mitochondrial proteins failed to identify a likely candidate. Linkage and haplotype analyses mapped the location of the gene to chromosome 19 and targeted MPS of the linkage region identified a homozygous c.3G>C (p.Met1?) mutation in C19orf79. Abolishing the initiation codon could potentially still allow initiation at a downstream methionine residue but we showed that this would not result in a functional protein. We confirmed that mutation of this gene was causative by lentiviral-mediated phenotypic correction. C19orf79 was recently renamed PET100 and predicted to encode a complex IV biogenesis factor. We showed that it is located in the mitochondrial inner membrane and forms a ∼300 kDa subcomplex with complex IV subunits. Previous proteomic analyses of mitochondria had overlooked PET100 because its small size was below the cutoff for annotating bona fide proteins. The mutation was estimated to have arisen at least 520 years ago, explaining how the families could have different religions and different geographic origins within Lebanon.
Introduction
Leigh syndrome (LS [MIM 256000] or subacute necrotizing encephalomyelopathy) is a severe, progressive neurodegenerative disorder characterized by bilateral lesions in the brainstem, basal ganglia, thalamus, and spinal cord.1–3 Most often it presents in infancy with developmental delay, seizures, dysarthria, and ataxia, as well as episodes of lactic acidosis. It is caused by severe deficiency of ATP production in the mitochondria4 resulting from defects affecting mitochondrial oxidative phosphorylation (OXPHOS) or pyruvate metabolism.5 It is genetically heterogeneous, with mutations in almost 50 genes able to cause LS.5,6 The genes identified so far encode components or factors required for assembly and function of OXPHOS complexes I, II, IV, and V or maintenance or expression of mitochondrial DNA (mtDNA) or the pyruvate dehydrogenase complex.5–8 LS can be inherited because of mutations in the maternally inherited mtDNA, in an autosomal-recessive manner, or through X-linked inheritance.
Cytochrome c oxidase (COX) or OXPHOS complex IV (CIV) deficiency is one of the major causes of LS, although it can also cause a range of other clinical presentations. Functional CIV comprises a homodimer of 14 subunits, which is assembled via a multistep process. In addition to subunits, there are also four redox-active components (two copper centers and two heme a moieties) and three redox-inactive metal ions with unknown function (zinc, magnesium, and sodium ions).9,10 Studies done on yeast mutants with respiratory defects have identified more than 20 biogenesis factors required for CIV assembly.11–13 Not all of those yeast proteins have obvious human homologs.14 Identification of human CIV biogenesis factors has often involved homozygosity mapping and identification of candidate genes with potential pathogenic mutations, followed by bioinformatic or functional studies implicating the encoded protein in CIV biogenesis.
Mutations causing isolated CIV deficiency have been identified in four nuclear DNA (nDNA) and three mtDNA-encoded CIV subunits, as well as 11 other nDNA-encoded biogenesis factors that are required for the transcription, translation, and assembly of CIV holoenzyme.5,6,15–18 Of those genes, mutations in NDUFA4, which was recently shown to encode the 14th CIV subunit,19 SURF1 (MIM 185620), COX10 (MIM 602125), COX15 (MIM 603646), LRPPRC (MIM 607544), and TACO1 (MIM 612958) cause LS.2,3,15,20–24 SURF1 mutations are the most common cause of CIV deficiency,2,3 although the majority of CIV-deficient individuals do not yet have a molecular diagnosis. Individuals with SURF1 mutations usually present in infancy with typical LS symptoms and die before 10 years of age.25 SURF1 facilitates the early stages of CIV assembly,26 where its bacterial homologs were shown to mediate the assembly of heme moieties into COX1.27,28
Of the other CIV biogenesis factors, C2orf64 (recently renamed COA5) also has a role in an early stage of CIV assembly.17 COX10 and COX15 are involved in the mitochondrial heme a biosynthesis whereas SCO1 and SCO2 are required for the biogenesis of the CuA site of COX2.29 LRPPRC, TACO1, and C12orf62 (recently renamed COX14) regulate the transcription and translation of CIV subunits.18,21,30,31 ETHE1 is involved in the catabolism of sulfide and its mutation leads to isolated CIV deficiency in muscle and brain.32 The role of FASTKD2 in CIV biogenesis is not clear but its mutation causes isolated CIV deficiency.33
In this study, we identify PET100 as a CIV biogenesis factor in humans and characterize its location and role in mitochondria. We found PET100 (MIM 614770) mutations in ten Lebanese individuals with LS and isolated CIV deficiency. All affected individuals were homozygous for a founder mutation, which we estimate arose at least 520 years ago.
Subjects and Methods
The studies were approved by the human research ethics committee at the Royal Children’s Hospital, Melbourne. All samples and pedigrees were obtained as part of diagnostic investigations, and families provided informed consent.
Clinical Information
Individual A1 (family A, II-2 in Figure 1A) presented at 4 months of age with continuous crying and was encephalitic on admission to the emergency department. His development had been slow in all areas. A CT scan at that time showed bilateral symmetrical involvement of the putamen and thalamus. MRI scan at 5 months confirmed those findings and identified a lactate doublet on MRS. An MRI at 6 months showed marked progression of the disease with involvement of basal ganglia including the caudate and globus pallidus. There were also frontotemporal white matter changes. His blood lactate was elevated, ranging from 4.5 to 9.5 mmol/l (normal range [NR] < 2.5 mmol/l) and cerebrospinal fluid (CSF) lactate was raised to 6.2 mmol/l (NR < 2.5 mmol/l). From about 5 months he started having seizures, and toward the end was having 3–7 seizures/day, not responding to vigabatrin. He was also on nasogastric feeds from about 5 months and did not obviously fix and follow. He died at 8 months of age and in the last weeks he had breathing dysregularity with apneic episodes. He died at home, presumably of a prolonged apnea.
Figure 1.
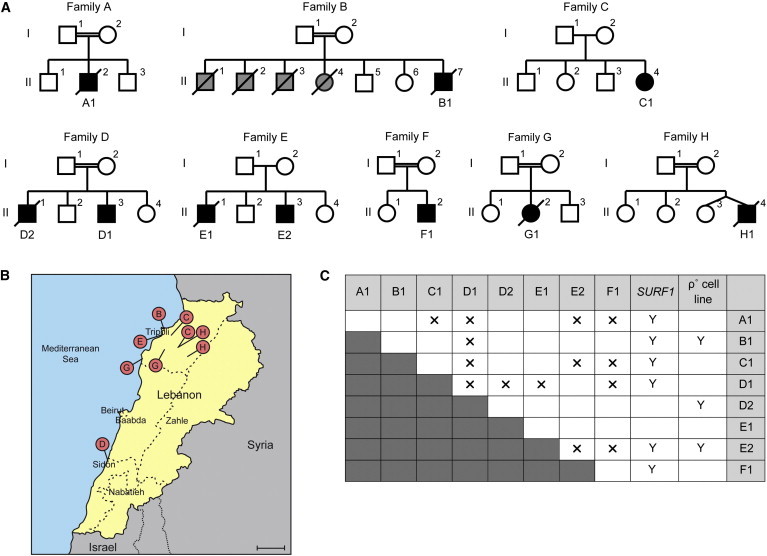
Ten PET100 Individuals from Eight Families of Lebanese Ancestry
(A) Pedigrees of the eight Lebanese families. Double horizontal line indicates consanguinity. Squares and circles represent males and females, respectively. Diamonds indicate gender is unknown. Black symbols represent affected individuals and diagonal lines denote deceased individuals. Grey symbols represent children who died without investigation and remain undiagnosed.
(B) A map of Lebanon with red pins marking the regions of origin of the parents in families B, C, D, E, G, and H. Scale bar represents 20 km.
(C) Summary of the complementation analysis based on galactose sensitivity of fused cell lines. “A1–F1” and “SURF1” indicate fibroblast cell lines from the Lebanese LS and SURF1 individuals, respectively. 143B ρ° cell line does not contain mtDNA. Y and X indicate positive and negative complementation in galactose media, respectively.
The parents of individual B1 (family B, II-7 in Figure 1A) came from Tripoli in Northern Lebanon. The first four children in the family were born in the 1970s in Lebanon and died within the first year of life without clear diagnoses but all had developmental delay and seizures. B1 was born at term after an uneventful pregnancy. He was irritable and inconsolable from about 6 weeks of age with developmental delay thought to be present from birth. At 2 months he was noted to have central hypotonia with peripheral hypertonia. He had seizures commencing at 6 months of age, which progressed to status epilepticus over the course of a few days. He reacted to loud sounds but there was generally a paucity of movement. The seizures settled with anticonvulsants but he continued to show gross developmental delay and failure to thrive. Cranial CT scan at 2 months was regarded as probably normal but repeat CT scan at 6 months showed extensive symmetrical low density white matter lesions with mild ventricular enlargement plus low density lesions in the heads of the caudate nuclei consistent with LS. Blood lactate was elevated to 7.0 mmol/l and CSF lactate levels were 3.3 and 4.0 mmol/l on two occasions. He died at home at 8 months of age.
The parents of individual C1 (family C, II-4 in Figure 1A) were from nearby villages in northern Lebanon. There was no history of metabolic disease in the family, although C1’s older brother was suggested to have Rolandic seizures. C1 was born with good Apgar scores after a normal delivery. She presented at 3 months of age with episodes of stiffening and pallor. She was diagnosed as having reflux and responded well to cisapride. She had normal development when she was first seen in the clinic. At 4 months of age, she developed episodes of arm and leg extension with circumoral cyanosis. On examination, she was awake and active, but she did not fix or follow and had extremely poor head control. Head ultrasound showed bilateral subdural collections, which was confirmed by CT scan. Her later MRI result was consistent with LS. She had bilateral lamellar cataracts and screens for possible causes were performed but results were negative. She had low serum bicarbonate (14 mmol/l on arrival, NR > 19 mmol/l), elevated serum and CSF lactate levels (5.4 and 4.8 mmol/l, respectively), and elevated serum and CSF pyruvate levels (0.19 and 0.13 mmol/l, respectively, NR < 0.1 mmol/l). She is currently 15.5 years of age and in a near-vegetative state. She is fed by gastrostomy and has epilepsy, for which she receives multiple anticonvulsants, and marked thoracic scoliosis with reduced lung volume on the left with frequent respiratory infections.
Individual D1 (family D, II-3 in Figure 1A) had normal birth weight but was an irritable baby with poor head control and was slow to fix and follow. At 6 months of age, he had acute onset of dystonic posturing and probable generalized seizure activity. He showed good physical growth and no dysmorphic features but had marked hypotonia with minimal spontaneous activity. His plasma and CSF lactate levels were raised to 3.0 and 4.1 mmol/l, respectively. Cranial MRI showed symmetrical high-intensity signals in the putamen, thalamus, and medulla. At 10 years of age, he had profound mental retardation, microcephaly, spastic quadriplegia, short stature, and seizures. Now at the age of 24, he is in a chronic vegetative state with spastic quadriplegia and only very primitive communication abilities. He is fed via a gastrostomy tube. From the same family, individual D2 (family D, II-1 in Figure 1A) had seizures and delay in motor milestones at 4 months of age. On investigation at 5 months, his EEG showed right-sided focal patterns, and CT showed cerebral atrophy. Urine organic and amino acids at the time were normal. At 7 months old, he was readmitted to the hospital in status epilepticus and required ventilation. His lactate level was normal in the blood (1.2 mmol/l) but was raised to 3.8 mmol/l in the CSF. He died after elective extubation, with neuropathological findings of LS plus cerebral atrophy and recent myocardial infarction.
Individual E1 (family E, II-1 in Figure 1A) presented at 4 months of age with profound global developmental delay, hypotonia, and seizures. At 7 months of age, he had divergent strabismus and was unable to fix and follow. His cranial CT scan at 8 months was unremarkable. At 7 years, he had spastic quadraparesis and his height and weight were on the third centile. He developed kyphoscoliosis and dementia prior to his death at 16 years of age. From the same family, individual E2 (family E, II-3 in Figure 1A) had developmental delay, hypotonia, and a divergent squint at 9 months of age. Subsequently, he had failure to thrive and developed myoclonic/tonic seizures and vertical and horizontal nystagmus. His blood lactate levels were elevated to 4.3 and 5.4 mmol/l on two occasions. Epileptic discharges were shown by EEG. Cranial CT scan at 1 year of age showed cerebral atrophy with bilateral hyperlucent areas deep to the Sylvian fissures compatible with LS.
Individual F1 (family F, II-2 in Figure 1A) was born at full term after a normal delivery. He had been noted to be somewhat hypertonic and there were concerns regarding his visual attention. He was referred to the metabolic unit at 6 months of age when he was admitted with a history of viral meningitis. At that time, he had persistent elevated lactate in the blood (5–11 mmol/l) and CSF (5.8 mmol/l) and an abnormal CT scan with bilateral low attenuation signals in both lentiform nuclei. He was not dysmorphic and had mottled skin. His height and weight were on the 25th percentile and his head circumference was on the 3rd percentile. He was hypotonic with roving eye movements and no visual fixation. He later had seizures (commenced before 8 months old) with an infantile spasm pattern. At the age of 9 years his height, weight, and head circumference are all below the 3rd percentile. Seizures have become gelastic and generalized in nature. He has spastic quadraparesis, a bulbar palsy, and easy obtainable clonus and absent reflexes suggesting development of a peripheral neuropathy. He has a severe hearing deficit and visual impairment. There have been a number of hospital admissions for recurrent chest infections.
Individual G1 (family G, II-2 in Figure 1A) presented with developmental delay and hypotonia at 4 months of age after an uneventful prenatal/neonatal course. Her seizures were treated with nitrazepam and vigabatrin. Her head circumference was on the 90th centile, and her weight and length were on the 50th–75th centile. She was extremely hypotonic with significant head lag. She did not fix or follow or respond to visual confrontation. Her reflexes were brisk but equal in both upper and lower limbs. She frequently had myoclonic jerks. She had elevated lactic acid and alanine levels in the urine. Lactate level in her serum was mildly elevated (2.85 mmol/l). Her CSF lactate level was elevated to 5.6 mmol/l. Pyruvate level was within the normal range in blood (0.09 mmol/l) but was elevated in CSF (0.18 mmol/l). CT scan showed cortical atrophy and EEG showed abnormal spike and wave and nonclassic hypsarrhythmia. G1 died at 12 months of age.
The parents of individual H1 (family H, II-4 in Figure 1A) originated from two villages in northern Lebanon, which are about half an hour drive apart. H1 was born at 38 weeks with birth weight of 2.98 kg. He had hypoglycemia in the newborn period and required 4 days in the special care nursery. He presented at 4 months with marked irritability and a preceding history of motor delay. His blood and CSF lactate levels were elevated (6.3–11.1 mmol/l and 4.19 mmol/l, respectively). His blood pyruvate level was also elevated (0.17 and 0.3 mmol/l on two occasions). Urine organic acid analysis showed marked ketosis with elevated lactate and alanine. He had a twin sister who was not affected. Cranial MRI showed high-intensity signals in the putamen. He went on to have very poor neurological development with myoclonic spasms (treated with clobazam), cortical blindness, and feeding difficulties. He had a Nissen fundoplication and insertion of gastrostomy at 17 months, but his condition continued to progress and he died at 3.5 years of age.
Biochemical and Complementation Analyses
Spectrophotometric enzyme assays assessing mitochondrial OXPHOS activity in cultured fibroblasts and skeletal muscle or liver biopsies were performed as described previously.34 Pairs of cell lines were cocultured (5 × 104 cells of each cell line) in Dulbecco’s modified eagle medium until confluent in a 25 cm2 flask. Cells were fused by exposure to 1.5 ml Polyethylene glycol 1500 (Roche) for 70 s. 1 × 104 cells of each fused cell line were plated in each 0.75 cm2 well and allowed to recover overnight in glucose medium (serum-free medium supplemented with 5 mM glucose, 10% dialyzed FBS, and 1% PBS) before selective medium (serum-free medium lacking glucose and supplemented with 5 mM galactose, 10% dialyzed FBS, 1% PBS, and 5 μM sodium azide) was applied. Cell survival beyond 7 days inferred positive complementation.
Linkage and Haplotype Analysis
Genome-wide SNP genotyping of DNA extracted from fibroblasts of six affected individuals (A1, B1, C1, D1, E2, and F1) was performed with the Illumina Human1M-Duo BeadChips at the Australian Genome Research Facility (AGRF), Melbourne. We analyzed genotypes for a subset of 12,156 SNPs that were chosen to have high heterozygosity and lie in approximate linkage equilibrium.35 Inbreeding coefficients (F) were estimated with FEstim.36,37 Based on these results (Table S1 available online), hypothetical first cousin once removed inbreeding loops were added to pedigrees C and E; reported inbreeding loops were retained for the other pedigrees. Multipoint parametric linkage analysis under a fully penetrant recessive genetic model and accounting for consanguinity was performed with MERLIN.38 Haplotypes inferred to be “most likely” by MERLIN were plotted with HaploPainter.39
Sanger Sequencing
Sanger sequencing of candidate genes was performed with BigDye Terminators v3.1 Cycle Sequencing kit (Applied Biosystems) as per manufacturer’s protocol. Details of primers and conditions are available on request. DNA sequences from affected individuals were compared against the RefSeq sequence and/or the sequence of a healthy control that was sequenced in parallel. Variants present in the healthy control, or in dbSNP or ethnically matched controls with allele frequency of ≥0.005, or not predicted to cause a splicing defect40,41 were regarded as probably benign.
MitoExome Sequencing
The entire mtDNA and all exons of 1,381 nuclear genes in B1, C1, D1, and E2 were targeted for massively parallel sequencing (MPS). Subsequent analyses were restricted to the mtDNA and 1,034 nuclear genes with strong evidence of mitochondrial association.42 Methods for target selection, massively parallel sequencing, alignment and variant detection, and prioritization were described previously.42
Targeted MPS
The target region (chr19: 6,952,512–9,811,452) was captured with the Agilent SureSelect Target Enrichment system via 120 bp baits with 2× tiling frequency. DNA samples were prepared from fibroblasts of affected individuals and submitted to the AGRF, Melbourne, for capture and sequencing. Samples were indexed and sequenced in a single lane on the Illumina HiSeq 2000 platform with 100 bp single-end reads.
Reads were aligned to the UCSC Genome Browser hg19 reference genome via Novoalign (v. 2.07.09), with quality score calibration performed. Multi-mapping reads were removed; potential PCR duplicates were removed with Picard 1.47. Depth of coverage was calculated with the Genome Analysis Toolkit (GATK) (v. 1.0.5974).43,44
Variants were detected and genotyped with the mpileup and bcftools view utilities from SAMtools (v. 0.1.18),45,46 with low-confidence variants discarded by the vcfutils.pl script from the same package with default settings. Variants were annotated against UCSC KnownGene annotation,47 dbSNP 132,48 and 1000 Genomes (Nov 2010 and May 2011 releases)49 by ANNOVAR.50 Sequence variants of interest were verified with restriction fragment length polymorphism (RFLP) analysis and/or Sanger sequencing.
RFLP Analysis
PCR product amplified by forward primer 5′-gccagacccgttttctattg-3′ and reverse primer 5′-aacccactcctgtgtccact-3′ was digested for 5 hr with MboI (Promega) as per manufacturer’s protocol and resolved on 2% agarose gels.
Sequenom Genotyping Assay
A specifically designed multiplexed MALDI-TOF mass spectrometry (Sequenom) assay was used to genotype 81 unrelated Lebanese individuals for the c.3G>C PET100 mutation. These individuals largely comprised subjects referred for investigation of possible OXPHOS enzyme defects. Genotypes were called by the MassARRAY System Typer v. 4.0 software (Sequenom). Details of primers and conditions are available on request.
Evolutionary Conservation
Human PET100 (RefSeq accession number NP_001164626.1) was aligned with its orthologs in seven additional species: P. abelii (XP_002828599.1), M. mulatta (XP_001097045.1), B. taurus (XP_002688903.1), C. familiaris (XP_854300.2), M. musculus (NP_001182173.1), R. norvegicus (NP_001182174.1), and D. rerio (XP_003198224.1) via ClustalW2 software with default parameters.
Estimation of Mutation Age
Existing methods for estimating the age of a mutation51,52 depend on large sample sizes or knowing the population frequency of the mutation. We have developed a method53,54 based on the length of mutation-harboring ancestral segments by using ideas developed by McPeek and Strahs55 in the context of fine mapping with linkage disequilibrium.
Assuming the Haldane recombination model, it can be shown that the length (in Morgans) of an ancestral segment after t generations has a gamma(2,t) distribution. By using a sample of segment lengths, we derived a maximum likelihood estimate of the t parameter, then modified the estimate to become unbiased. The lengths were assumed to be statistically independent, which was equivalent to assuming that the sampled segments had independent recombination histories, a plausible assumption in fast-growing populations. Exact confidence intervals were obtained by deriving the sampling distribution of a pivotal quantity involving the estimate and the true mutation age by using properties of the gamma distribution. The large width of the reported interval was due to the small sample size. A generation time of 20 years was assumed to convert from age in generations to age in years.
The sample of ancestral segment lengths was obtained from the Illumina Human1M-Duo BeadChips SNP genotype data of A1, B1, C1, D1, E2, and F1. The lengths were defined by regions where there was continuous haplotype sharing between at least two individuals around the mutation, for the reason that such sharing (of hundreds of consecutive markers) could be plausibly explained only by assuming that the regions were identical by descent, i.e., they were ancestral segments. Since the sampled individuals had a recessive disease, as a result of consanguinity, each had a large region of homozygosity surrounding the mutation, and this removed the need to phase the SNP data: for obtaining the lengths, continuous haplotype sharing is equivalent to continuous sharing of identical homozygous genotypes. The ancestral segment lengths obtained were 3.06, 3.84, 4.65, 2.63, 3.51, and 3.84 cM.
Lentiviral Transduction
The open reading frame of PET100 alone or fused to a 3′ FLAG-tag was cloned into the 4-hydroxytamoxifen-inducible lentiviral vector, pF_5x_UAS_MCS_SV40_puroGEV16-W.56 PET100 viral particles were generated and cultured fibroblasts were transduced as described previously.57 Three independent transductions were performed and cells were harvested after 8–10 days selection with 1 μg/ml puromycin.
Immunofluorescence
D2 fibroblasts were transiently transfected with pcDNA3 (Invitrogen) containing PET100 fused with a 3′ FLAG-tag (PET100FLAG). Mitochondria were stained with MitoTracker Red, after which cells were fixed in 4% (w/v) paraformaldehyde and analyzed by immunostaining with the FLAG M2 antibody (Sigma). Nuclei were labeled with Hoechst dye.
In Vitro Protein Import into Isolated Mitochondria
Mitochondria were isolated from HEK293T cells and cultured fibroblasts as described previously.58 For in vitro transcription, PET100 and PET100Δ1-9 were amplified from cDNA with forward primers incorporating a SP6 promoter as described by Stojanovski et al.59 PCR products were transcribed with the mMessage mMachine SP6 kit (Ambion) according to manufacturer’s instructions, and transcripts were translated in rabbit reticulocyte lysate (Promega) in the presence of [35S]methionine/cysteine protein labeling mix (Perkin Elmer Life Sciences).59 Radiolabeled proteins were incubated with isolated mitochondria at 37°C for various times as indicated. Trypsin treatment, dissipation of ΔΨm, swelling, and carbonate extraction were performed according to Stojanovski et al.59
SDS-PAGE and Blue Native Gel Electrophoresis
Protein lysates were prepared from cultured fibroblasts as described previously.42 10 μg of each protein lysate was analyzed by SDS-PAGE as described previously.42 Isolated mitochondria (25–50 μg) were separated on 10%–16% polyacrylamide Tris-tricine gradient gels as described previously.60 BN-PAGE was performed as described previously.60 Membrane protein complexes (solubilized in 1% digitonin) were separated on a 4%–13% acrylamide-bisacrylamide BN-PAGE gel.
Immunoblotting
Primary antibodies used were against COX1, COX2 and SDHA (MitoSciences), NDUFA9, TOM70 and mt-HSP70 (rabbit polyclonal), TIM23 (BD Biosciences), and VDAC1 (Calbiochem). Where statistical significance was provided for the differences in protein levels, quantification of immunoblots was performed by densitometry measurement by ImageJ software followed by a two-way repeated-measures analysis of variance (ANOVA) for comparisons of groups and a post hoc analysis by the Bonferroni method to determine statistically significant differences.
Radiolabeling of mtDNA-Encoded Translation Products
Radiolabeling of mtDNA-encoded subunits was performed as previously described.61 In brief, cells were pretreated with 50 μg/ml chloramphenicol (Sigma) in Dulbecco’s modified eagle medium (DMEM, Invitrogen) containing 10% (v/v) FBS for 12 hr before labeling. To inhibit cytosolic translation, cells were subsequently incubated with 0.1 mg/ml cycloheximide (Sigma) for 15 min at 37°C in methionine-free DMEM (Invitrogen) containing 5% (v/v) dialyzed FBS (Thermo Scientific). The pulse was performed by addition of 20 μCi [35S]methionine/cysteine (EXPRE35S35S Protein Labeling Mix, PerkinElmer Life Sciences) for 2 hr, followed by the addition of 0.1 mM unlabeled methionine for 15 min. For the chase, media was removed and replaced with DMEM containing 10% (v/v) FBS and cells were cultured at 37°C for the times indicated. Mitochondria were isolated as described above and membrane protein complexes were solubilized in 1% Triton X-100 and separated on 4%–13% acrylamide-bisacrylamide BN-PAGE gels. For 2D-PAGE, gel slices were excised and subsequently separated on 10%–16% polyacrylamide Tris-tricine gradient gels as described previously.61
Results
Eight Lebanese Individuals with Isolated CIV Deficiency Had a Unique Form of LS and Belong to a Single Complementation Group
We studied eight Australian individuals of Lebanese ancestry with LS or Leigh-like encephalopathy and CIV deficiency (Table 1 and Figure 1A). Residual activity of CIV was similar to that of typical individuals with SURF1 mutations in skin fibroblasts and skeletal muscle (median values of 18% to 25%; for details see Table S2). Activities of OXPHOS complexes I and II in the eight Lebanese individuals and SURF1 individuals were comparable to normal controls, whereas complex III activity was comparable to normal controls in the eight Lebanese individuals and borderline low in SURF1 individuals (Table S2). The affected Lebanese individuals thus resembled SURF1 individuals in having an isolated systemic CIV defect but they differed from most SURF1 individuals in having global developmental delay, seizures, and an earlier age of onset (see Clinical Summaries and Table S3). They were from six families not known to be related (A–F). They had a range of religious backgrounds and geographic origins within Lebanon (Figure 1B). Four of the six families were known to be consanguineous.
Table 1.
Clinical and Biochemical Characteristics of Individuals with PET100 Mutation
| Subject ID | Sex | Age of Onset | Age of Death (Current Age) | Clinical Presentation | % CIV Activitya | Relationship between Parents | Religious Background of Parents |
|---|---|---|---|---|---|---|---|
| A1 | M | 4 months | 0.7 years | LS | 17 (F), 19 (M), 22 (L) | first cousins | Christian |
| B1 | M | 1.5 months | 0.7 years | DD, FTT, seizures | 24 (F) | second cousins | Sunni Muslim |
| C1 | F | 3 months | (15.5 years) | LS | 10 (F), 21 (M) | unrelated, from nearby villages | Maronite Catholic |
| D1 | M | 6 months | (24 years) | LS, dystonia, seizures | 27 (F) | first cousins | Maronite Catholic |
| D2 | M | 4 months | 0.6 years | LS, seizures | 22 (F) | ||
| E1 | M | 4 months | 16 years | DD, FTT, seizures | 29 (F) | unrelated | Maronite Catholic |
| E2 | M | 9 months | (27 years) | LS, seizures | 25 (F) | ||
| F1 | M | 6 months | (9.7 years) | LS, seizures | 34 (F) | first cousins | Muslim |
| G1 | F | 4 months | 1 year | DD, seizures | 12 (F), 39 (L) | first cousins once removed | Christian Orthodox |
| H1 | M | 4 months | 3.5 years | LS, seizures | 51 (F), 48 (M), 21 (L) | second cousins | Maronite Catholic |
Abbreviations are as follows: LS, Leigh syndrome; DD, developmental delay; FTT, failure to thrive.
CIV activity is expressed as percent of control mean value for CIV activity relative to citrate synthase activity in fibroblasts (F), skeletal muscle (M), or liver (L).
Cells cultured in galactose media are forced to produce energy via OXPHOS,62 and those with severe OXPHOS defects die within 7 days. We performed pair-wise cell fusions between the cell lines of the Lebanese individuals and none survived beyond 7 days in galactose media (Figure 1C). In contrast, fusion with cell lines containing a SURF1 defect or lacking mtDNA (ρ0) resulted in positive complementation (Figure 1C). The Lebanese individuals thus belonged to a single complementation group, suggesting a common defective gene.
Linkage and Haplotype Analyses Identified a Candidate Region on Chromosome 19
We genotyped one affected individual from each family via Illumina Human1M-Duo arrays. Estimated inbreeding coefficients revealed undocumented inbreeding in families C and E, and families B and F were more consanguineous than reported (Table S1). Although families C and E were not known to be consanguineous, the parents within each of the families originated from the same geographical area.
We performed parametric linkage analysis simultaneously for six affected individuals via a fully penetrant recessive genetic model. A unique linkage peak at chr19: 7,251,842–8,468,337 (UCSC Genome Browser build hg19) was identified, with a maximum heterogeneity LOD score of 7.53 (Figure S1). All families showed linkage to this region (Figure 2A). All six affected individuals were homozygous within this region and shared identical homozygous genotypes for a smaller region (chr19: 7,251,842–7,813,268) (Figure 2B), suggesting they might share a mutation inherited from a common ancestor. There were 49 genes within the broader linkage region, 22 of which were within the smaller linkage region.
Figure 2.
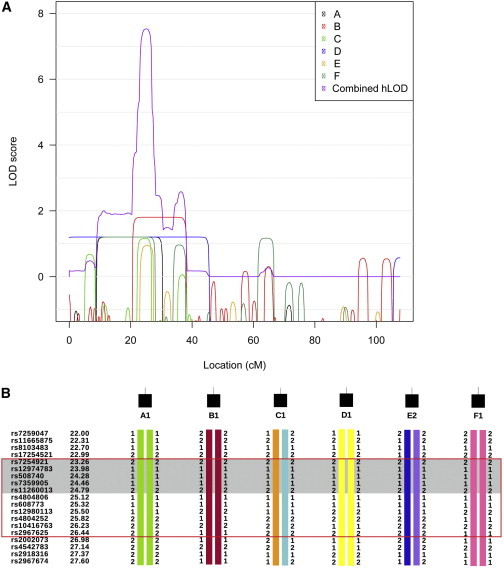
Linkage and Haplotype Anlayses of Affected Individuals from Families A–F
(A) The contribution of each family to the combined hLOD score peak on chromosome 19. LOD scores of individual families were plotted in black, red, green, blue, orange, and dark aqua, respectively. The combined hLOD score was plotted in purple.
(B) Haplotypes for each affected individuals in the region under the chromosome 19 linkage peak. “1” and “2” are arbitrary labels to indicate the two different alleles of each SNP. Different colors indicate alleles inherited from different ancestors. Some markers used for linkage analysis are not displayed for conciseness. All affected individuals were homozygous by state from 23.14 to 26.89 cM (red box) and shared identical homozygous alleles from 23.14 to 25.05 cM (shaded in gray).
Targeted MPS of Candidate Region Identified a Mutation in C19orf79
Prior to performing linkage analysis, we Sanger sequenced SURF1, SCO2 (MIM 604272), TACO1, and 12 nDNA-encoded CIV subunit genes. We also sequenced 11 candidate genes with known or predicted mitochondrial association from chr19 in at least three of the eight affected individuals (Table S4). Furthermore, the entire mtDNA and 1,034 nuclear genes encoding the known “MitoExome” were analyzed for four affected individuals by MPS.42 No putative pathogenic variant was identified.
Targeted MPS of the candidate region was performed for one affected individual from each family. The median percentage of targeted bases covered by at least two reads was 92% across the six individuals (Table S5). A total of 542 variants were detected within the chr19: 7,251,842–7,813,268 region of shared homozygosity, of which 407 remained after the removal of low-confidence variants. Of these variants, 95 were rare (absent from the 1000 Genomes data set or present with <0.01 alternate allele frequency); of these, three variants were predicted to alter splicing or protein sequence (Table S6). All six cases were homozygous for the alternate allele for only one of these three variants: c.3G>C in PET100 (RefSeq NM_001171155.1). The mutation segregated with disease in all families and was predicted to abolish the translation initiation codon (p.Met1?; Figure S2). It was absent from public databases48,49 but was homozygous in 2/81 ethnically matched individuals screened. The 81 “controls” were derived from individuals of Lebanese ancestry who had mostly been referred for analysis of a suspected OXPHOS defect. Of these, three individuals had isolated CIV deficiency and were without a previous molecular diagnosis. Two of the three (G1 and H1, Figure 1A) were homozygous for the c.3G>C mutation and segregation of the mutation with disease was confirmed in both additional families (Figure S2). The third individual lacked the c.3G>C mutation. In addition, we sequenced the PET100 coding region in three non-Lebanese individuals with CIV deficiency and without previous molecular diagnosis. No pathogenic mutation was identified.
The c.3G>C Mutation in PET100 Was Estimated to Be at Least 520 Years Old
The age of the c.3G>C PET100 mutation was estimated (to the nearest generation) as 43 generations (95% confidence interval of 26, 83). Assuming an average generation time of 20 years, the mutation was estimated to be 860 years old (95% confidence interval of (520, 1,660) years).
PET100 Is Localized to the Mitochondrial Inner Membrane
PET100 encodes a 73 amino acid protein, which was not known or predicted to have mitochondrial association42,64–66 until it was recently identified as the human ortholog of yeast Pet100p,67 a CIV biogenesis factor in the mitochondrial inner membrane.68 Szklarczyk and colleagues recently demonstrated that reanalysis of raw mass spectrometry data of isolated mouse heart mitochondria66 identified PET100 peptides, indicative of mitochondrial association.67 Our immunofluorescence analysis of FLAG-tagged human PET100 (PET100FLAG) also revealed mitochondrial localization (Figure 3A). To investigate the submitochondrial localization, we used in vitro imported [35S]PET100. The protein is shown to associate with mitochondria (Figure 3B, lanes 2–4), and after treatment with trypsin, a portion was in a protease protected location (Figure 3B, lanes 5 and 6). Furthermore, this accumulation was dependent on the mitochondrial membrane potential for its accumulation inside mitochondria (Figure 3B, lanes 4 and 7), typical of proteins located in the mitochondrial inner membrane or matrix.69,70 PET100 lacks a cleavable N-terminal presequence because the imported protein has the same size as the translated form (Figure 3B, lanes 1–4). Imported [35S]PET100 was digested with trypsin only after mitochondrial swelling, like TIM23, which is exposed to the intermembrane space (Figure 3B, lanes 8–10). The control outer membrane protein, TOM70, was susceptible to trypsin digestion, whereas matrix-located NDUFA9 was protected as expected. PET100 contains a predicted transmembrane anchor67 (Figure 3D) and remained predominantly in the membrane fraction after alkaline extraction (Figure 3B), unlike the peripherally associated mtHSP70. These observations lead to the conclusion that PET100 is located in the mitochondrial inner membrane and exposed to the intermembrane space.
Figure 3.
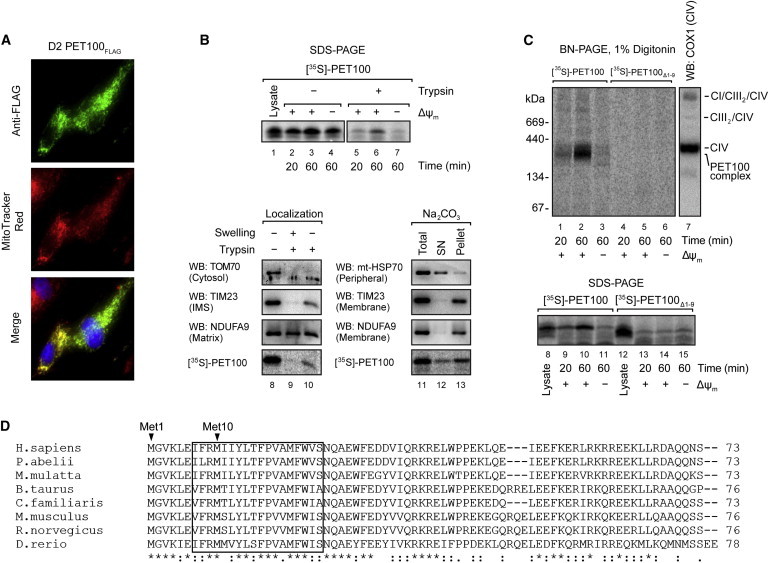
Mitochondrial Localization and Assembly of PET100
(A) D2 fibroblasts expressing PET100FLAG were stained with MitoTracker Red (red), fixed, and immunostained for the FLAG epitope (green). Nuclei were labeled with Hoechst (blue). Colocalization of the expressed PET100FLAG in the mitochondria (yellow) is shown in the merged image.
(B) Top: The presence of membrane potential (ΔΨm) was required for the import of 35S-labeled PET100 into isolated HEK293T mitochondria, protecting it from digestion by externally added trypsin. Untreated input lysate is shown for comparison. 10% of input lysate used in the import was loaded in lane 1. Bottom: [35S]PET100 imported into HEK293T mitochondria was digested by trypsin only after hypo-osmotic mitochondrial swelling (left) and remained in the membrane (Pellet) fraction after alkaline extraction (Na2CO3) (right). Abbreviations are as follows: SN, supernatant; WB, immunoblotting.
(C) BN-PAGE (top) or SDS-PAGE (bottom) followed by phosphorimaging showed that [35S]PET100Δ1-9 was incapable of assembly into the ∼300 kDa complex (lanes 4–6) and was not imported into mitochondria (lanes 13–15). Abbreviations are as follows: CI, complex I; CIII, complex III; CIV, complex IV.
(D) Sequence alignment of human PET100 with its homologs in seven additional vertebrate species. The PET100 in the Lebanese LS individuals (if present) was predicted to lack the first nine amino acid residues, which are highly conserved in vertebrate species. Asterisk (∗), colon (:), and period (.) indicate that the amino acids are identical, strongly similar, and weakly similar, respectively, across the aligned species. The transmembrane domain predicted from the human protein67 is boxed.
PET100 Assembles into a 300 kDa Complex
Pet100p interacts with yeast CIV subunits VII, VIIa, and VIII (human COX7A, COX6C, and COX7C, respectively)68 whereas TAP-tagged PET100 interacts with COX7A2 in HEK293 cells.67 BN-PAGE analysis showed that imported PET100 assembles into a ∼300 kDa complex that accumulates over time and is dependent on the mitochondrial membrane potential (Figure 3C). This complex is distinct from the endogenous CIV (Figure 3C, lane 7). Its molecular weight is higher than predicted from the known interactions, implying that it contains additional subunits to those listed above.
Mutant PET100 Does Not Form a Complex and Is Not Imported into the Mitochondria
The c.3G>C mutation in PET100 was predicted to abolish the initiation codon, which could potentially lead to translation initiation at the next methionine located at residue 10 (p.Met10) within the predicted transmembrane domain67 (Figure 3D). This mutant protein (PET100Δ1-9) was incapable of assembly into the ∼300 kDa complex (Figure 3C). Further analysis showed [35S]PET100Δ1-9 was not imported into mitochondria (Figure 3C). Therefore, we conclude that the N terminus of PET100 is essential for its mitochondrial localization.
The Import and Assembly of [35S]PET100 into Mitochondria of Affected Individuals
Because PET100Δ1-9 is not imported into mitochondria, it is likely that mitochondria of PET100 individuals lack a pre-existing ∼300 kDa complex. To determine whether [35S]PET100 is capable of de novo assembly, we imported the full-length protein into mitochondria of PET100 individuals. An increased efficiency of complex assembly and import was observed in cells from the PET100 individuals compared with cells from a healthy control or SURF1 individual (Figure 4A). [35S]PET100 also assembled into a number of very high molecular weight complexes in the PET100 and SURF1 individuals (Figure 4A). Interestingly, in the SURF1 individual, [35S]PET100 was imported but not assembled into the ∼300 kDa complex, instead assembling into a ∼250 kDa complex (Figure 4A). This suggests that PET100 interacts with components that also rely on SURF1 for their biogenesis (Figure 4A).
Figure 4.
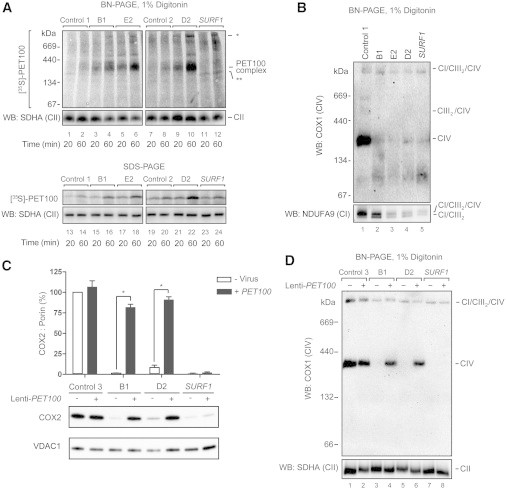
The Import of 35S-Labeled PET100 or Overexpression of PET100 cDNA in Fibroblasts from Affected Individuals
(A) Samples treated with trypsin and analyzed by BN-PAGE (top) or SDS-PAGE (bottom) and phosphorimaging showed that [35S]PET100 was imported (lanes 13–22) and assembled (lanes 1–10) into the ∼300 kDa complex with an increased efficiency in the fibroblast mitochondria of affected individuals. In SURF1 fibroblast mitochondria, [35S]PET100 was imported (lanes 23–24) but not assembled into the ∼300 kDa complex (lanes 11–12) and instead, assembled into a ∼250 kDa complex (∗∗). [35S]PET100 additionally assembled into a number of very high molecular weight complexes in mitochondria of affected individuals (∗). Immunoblot detection of SDHA, a complex II (CII) subunit, was used as loading control.
(B) BN-PAGE and immunoblotting showed that CIV holoenzyme was barely detectable in the fibroblasts of affected individuals. The PET100 individuals also had CI/CIII2 supercomplex and a small amount of CI/CIII2/CIV supercomplex.
(C) Lentiviral-mediated overexpression of wild-type PET100 restored COX2 levels in the PET100 individuals but not the SURF1 individuals (SURF1). Top: Bar graph showing protein levels of COX2 relative to VDAC1 (loading control) normalized to control, before or after transduction. The bars: mean of three biological replicates. Error bars: ± 1 standard error of the mean (SEM). ∗p < 0.001. Bottom: A representative blot of SDS-PAGE immunoblot analysis of COX2 and VDAC1 from whole cell lysates before (-) and after (+) transduction.
(D) Lentiviral-mediated overexpression of wild-type PET100 restored CIV assembly in the PET100 individuals but not in the SURF1 individual.
Mitochondria of PET100 Individuals Lacked CIV Holoenzyme
Given the homology of PET100 to yeast Pet100p, we hypothesized that individuals carrying mutant PET100 would harbor defects in CIV biogenesis. BN-PAGE analysis of fibroblasts from PET100 individuals revealed a significant loss of CIV holoenzyme (Figure 4B). Affected cells harbored a small amount of CI/CIII2/CIV supercomplex and a population of supercomplex lacking CIV (CI/CIII2).
Overexpression of PET100 Restored COX2 Levels and CIV Assembly in the Affected Lebanese Individuals
Lentiviral-mediated overexpression of PET100 in cultured fibroblasts significantly increased COX2 levels (Figure 4C) and restored CIV assembly (Figure 4D) in the affected Lebanese individuals but not in the SURF1 individual. This confirmed that the PET100 mutation caused the CIV deficiency in the affected Lebanese individuals.
Assembly of mtDNA-Encoded Subunits in Affected Mitochondria
To investigate the consequences of PET100 loss, we analyzed the assembly of newly synthesized mtDNA-encoded subunits into their respective OXPHOS complexes. The mtDNA-encoded subunits efficiently assembled into complexes I, III, and V in control and D2 fibroblasts. However, almost no assembled CIV was detected in D2 (Figure 5A). We extended this analysis by using two-dimensional (2D) BN-PAGE (Figure 5B). Before the chase, COX2 and COX3 were present in an ∼80 kDa subcomplex, whereas COX1 was found in an ∼100 kDa subcomplex (CIVI), demonstrating their translation and entry into the CIV assembly pathway in control and D2 mitochondria (Figure 5B, 0 hr chase). In the control these subunits were chased into mature CIV (Figure 5B, 3 and 24 hr chase), but in D2 they remain stalled in various subcomplexes. COX2 and COX3 remain in their ∼80 kDa subcomplex, whereas COX1 is stalled in the CIVI subcomplex and an additional subcomplex slightly larger than mature CIV. Interestingly, we observed significant turnover of COX2 and COX3 in D2 (Figure 5B, 24 hr chase), consistent with the barely detectable level of COX2 in mitochondria of PET100 individuals via immunoblotting.
Figure 5.
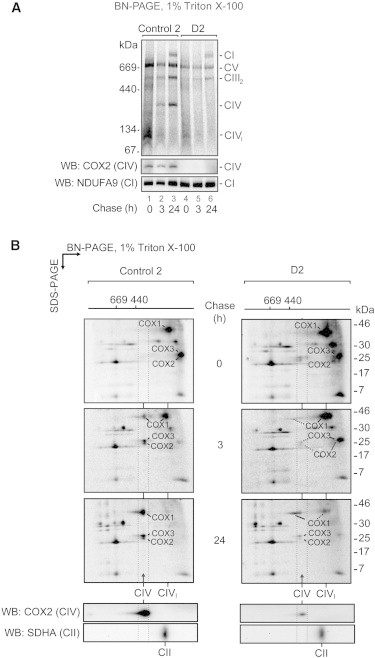
Assembly of mtDNA-Encoded Subunits in D2 Mitochondria
(A) Assembly of newly synthesized mtDNA-encoded subunits into OXPHOS complexes was studied by pulse/chase labeling with [35S]methionine/cysteine in whole cells, followed by isolation of mitochondria and analysis by BN-PAGE and phosphorimaging. The CIV holoenzyme was not assembled in D2 mitochondria. The bottom panel shows the steady-state levels of mature complexes IV and I by immunoblotting. Abbreviations are as follows: CIVI, complex IV subcomplex; CV, complex V.
(B) 2D-PAGE analysis of labeled mtDNA-encoded subunits from D2 revealed a blocked entry of COX1, COX2, and COX3 into the CIV holoenzyme, the stalling of these subunits in various subcomplexes, and an increased turnover of COX2 and COX3. The bottom panel shows the steady-state levels of mature complexes IV and II by immunoblotting. The positions of mtDNA-encoded CIV subunits COX1, COX2, and COX3 are indicated.
Discussion
CIV deficiency is one of the most common and best studied OXPHOS disorders. A minority of molecularly diagnosed affected individuals have mutations in CIV subunits, with most having mutations in CIV biogenesis factors.5,6,15–18 Here we describe ten Australian affected individuals from eight families with a homozygous PET100 mutation causing CIV deficiency. Their clinical presentations were similar to but distinct from SURF1 LS and all had Lebanese ancestry. Half of the affected individuals died in infancy or early childhood, and the others survived for periods up to 24 years or more. This variable survival could relate to the effect of modifier genes or to the inherent variation in conditions like LS, where acute life-threatening decompensations can be triggered by events such as intercurrent illnesses. The affected individuals were shown to belong to a single genetic complementation group by cell fusion studies. Linkage and haplotype analyses identified a linkage peak on chromosome 19 and MPS of the linkage region prioritized PET100 as a top candidate gene. All ten affected individuals were homozygous for a c.3G>C mutation predicted to abolish the first methionine residue. Expressing the wild-type and mutant PET100 proteins in fibroblasts of affected individuals showed that only the wild-type protein could rescue CIV assembly, providing definitive proof of causation.
Similar to the recently described mitochondrial CIV biogenesis factors COA5 and COX14,17,18 PET100 is a relatively small protein (9.1 kDa). This small size meant that PET100 was overlooked as a component of the mammalian mitochondrial proteome, because previous proteomic analyses of mouse mitochondria did not annotate most proteins smaller than ∼10 kDa.66 Knowledge of the mitochondrial proteome (MitoCarta proteins66) has been valuable in identifying other genes in which mutations cause disease, because it allows filtering out of 95% of “nonmitochondrial” proteins from whole-exome data sets. MitoCarta was originally predicted to be more than 85% complete66 and thus is expected to miss a subset of mitochondrial proteins. In this instance we identified PET100 as a result of the combined power of detailed biochemical phenotyping, complementation, linkage, and haplotype analyses on multiple families, enabling highly effective filtering of MPS variants.
The gene was originally termed C19orf79 before being renamed earlier this year after its yeast ortholog, Pet100. Identification of the distant homology between the C19orf79/PET100 product and yeast Pet100p by Szklarczyk and colleagues further enhanced the likelihood that this was the correct candidate gene.67 Our discovery of a PET100 mutation causing CIV deficiency demonstrates its essential role in OXPHOS function. Nevertheless, further investigation is required to elucidate its exact function. Its yeast homolog, Pet100p, is not required for the synthesis and mitochondrial localization of CIV subunits or the biogenesis of heme a and mitochondrial cytochromes,68,71 and it is involved in a later step of CIV assembly.68,70 We showed that whereas the mtDNA-encoded CIV subunits in D2 mitochondria were translated, their assembly into the mature CIV was impaired, leading to increased turnover. Taken together, these results suggest that PET100 acts in the intermediate stage of CIV assembly, probably coordinating the biogenesis of one or more nDNA-encoded subunits.
To our knowledge this is only the second instance of a founder mutation causing CIV deficiency. Mutations in LRPPRC result in LS, French-Canadian type (LSFC [MIM 220111]), which is also known as Saguenay-Lac-Saint-Jean CIV deficiency.20 All reported LRPPRC individuals originated from the northern Québec region in Canada, particularly the Saguenay-Lac-Saint-Jean region.72 Most LRPPRC individuals are homozygous for a founder mutation, c.1061C>T,72 which is estimated to have been introduced into the French-Canadian population in the 17th century from Europe.73 We estimated that the c.3G>C PET100 mutation first occurred at least 520 years ago. The parents of our PET100 individuals originated from different regions in Lebanon and had different religious backgrounds, which is consistent with c.3G>C being a relatively old mutation. To date, this mutation has been identified only in individuals with a Lebanese ancestral background and further studies are warranted into its prevalence in Lebanon. Like individuals with SURF1 and LRPPRC mutations, the clinical presentation of those with PET100 mutations appears to be relatively homogeneous and mutation analysis should be considered in individuals with LS or Leigh-like encephalopathy and CIV deficiency.
Acknowledgments
We thank C. Zetzer, K. Reed, H. Dahl, and S. White for assisting with complementation and linkage analyses and S. Tregoning, W. Salter, K. Canavan, S. Smith, and W. Hutchison for technical assistance with cell culture, enzyme, and molecular analyses. A. Boneh, M. Nash, A. Mansour, and G. Wise are thanked for referral of affected individuals or clinical review. We thank the subjects and their families for their involvement and V.K. Mootha and S.E. Calvo for their collaboration by sequencing known MitoExome genes and for helpful discussion. The authors would like to thank the NHLBI GO Exome Sequencing Project and its ongoing studies, which produced and provided exome variant calls for comparison: the Lung GO Sequencing Project (HL-102923), the WHI Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926), and the Heart GO Sequencing Project (HL-103010). This work was supported by the Australian National Health and Medical Research Council via Project (M.T.R., D.R.T.) and Program (M.B.) grants, a Career Development Fellowship (M.McK.), a Principal Research Fellowship (D.R.T.), and the Independent Research Institute Infrastructure Support Scheme (M.B., K.R.S.). Other support came from a Melbourne Research Scholarship (S.C.L.), a Future Fellowship from the Australian Research Council (M.B.), a Pratt Foundation scholarship (K.R.S.), the James and Vera Lawson Trust (M.McK.) and the Victorian Government’s Operational Infrastructure Support Program (D.R.T., M.McK., M.B., and K.R.S.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Berkeley Drosophila Genome Project NNSplice 0.9, http://www.fruitfly.org/seq_tools/splice.html
Human Splicing Finder, http://www.umd.be/SSF/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
Picard, http://picard.sourceforge.net/
UCSC Genome Browser, http://genome.ucsc.edu
Accession Numbers
The dbSNP accession number for the TRAPPC5 (c.548G>C) variant reported in this paper is rs398122420. The dbSNP accession number for the FLJ22184 (c.2294G>A) variant reported in this paper is rs398122421.
References
- 1.Leigh D. Subacute necrotizing encephalomyelopathy in an infant. J. Neurol. Neurosurg. Psychiatry. 1951;14:216–221. doi: 10.1136/jnnp.14.3.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tiranti V., Hoertnagel K., Carrozzo R., Galimberti C., Munaro M., Granatiero M., Zelante L., Gasparini P., Marzella R., Rocchi M. Mutations of SURF-1 in Leigh disease associated with cytochrome c oxidase deficiency. Am. J. Hum. Genet. 1998;63:1609–1621. doi: 10.1086/302150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu Z., Yao J., Johns T., Fu K., De Bie I., Macmillan C., Cuthbert A.P., Newbold R.F., Wang J., Chevrette M. SURF1, encoding a factor involved in the biogenesis of cytochrome c oxidase, is mutated in Leigh syndrome. Nat. Genet. 1998;20:337–343. doi: 10.1038/3804. [DOI] [PubMed] [Google Scholar]
- 4.Dahl H.-H.M. Getting to the nucleus of mitochondrial disorders: identification of respiratory chain-enzyme genes causing Leigh syndrome. Am. J. Hum. Genet. 1998;63:1594–1597. doi: 10.1086/302169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kirby D.M., Thorburn D.R. Approaches to finding the molecular basis of mitochondrial oxidative phosphorylation disorders. Twin Res. Hum. Genet. 2008;11:395–411. doi: 10.1375/twin.11.4.395. [DOI] [PubMed] [Google Scholar]
- 6.Tucker E.J., Compton A.G., Thorburn D.R. Recent advances in the genetics of mitochondrial encephalopathies. Curr. Neurol. Neurosci. Rep. 2010;10:277–285. doi: 10.1007/s11910-010-0112-8. [DOI] [PubMed] [Google Scholar]
- 7.Hong Y.S., Kerr D.S., Craigen W.J., Tan J., Pan Y., Lusk M., Patel M.S. Identification of two mutations in a compound heterozygous child with dihydrolipoamide dehydrogenase deficiency. Hum. Mol. Genet. 1996;5:1925–1930. doi: 10.1093/hmg/5.12.1925. [DOI] [PubMed] [Google Scholar]
- 8.Matthews P.M., Marchington D.R., Squier M., Land J., Brown R.M., Brown G.K. Molecular genetic characterization of an X-linked form of Leigh’s syndrome. Ann. Neurol. 1993;33:652–655. doi: 10.1002/ana.410330616. [DOI] [PubMed] [Google Scholar]
- 9.Muramoto K., Hirata K., Shinzawa-Itoh K., Yoko-o S., Yamashita E., Aoyama H., Tsukihara T., Yoshikawa S. A histidine residue acting as a controlling site for dioxygen reduction and proton pumping by cytochrome c oxidase. Proc. Natl. Acad. Sci. USA. 2007;104:7881–7886. doi: 10.1073/pnas.0610031104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stiburek L., Hansikova H., Tesarova M., Cerna L., Zeman J. Biogenesis of eukaryotic cytochrome c oxidase. Physiol. Res. 2006;55(Suppl 2):S27–S41. doi: 10.33549/physiolres.930000.55.S2.27. [DOI] [PubMed] [Google Scholar]
- 11.Fontanesi F., Soto I.C., Horn D., Barrientos A. Assembly of mitochondrial cytochrome c-oxidase, a complicated and highly regulated cellular process. Am. J. Physiol. Cell Physiol. 2006;291:C1129–C1147. doi: 10.1152/ajpcell.00233.2006. [DOI] [PubMed] [Google Scholar]
- 12.McEwen J.E., Ko C., Kloeckner-Gruissem B., Poyton R.O. Nuclear functions required for cytochrome c oxidase biogenesis in Saccharomyces cerevisiae. Characterization of mutants in 34 complementation groups. J. Biol. Chem. 1986;261:11872–11879. [PubMed] [Google Scholar]
- 13.Tzagoloff A., Dieckmann C.L. PET genes of Saccharomyces cerevisiae. Microbiol. Rev. 1990;54:211–225. doi: 10.1128/mr.54.3.211-225.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fernández-Vizarra E., Tiranti V., Zeviani M. Assembly of the oxidative phosphorylation system in humans: what we have learned by studying its defects. Biochim. Biophys. Acta. 2009;1793:200–211. doi: 10.1016/j.bbamcr.2008.05.028. [DOI] [PubMed] [Google Scholar]
- 15.Pitceathly R.D., Rahman S., Wedatilake Y., Polke J.M., Cirak S., Foley A.R., Sailer A., Hurles M.E., Stalker J., Hargreaves I., UK10K Consortium NDUFA4 mutations underlie dysfunction of a cytochrome c oxidase subunit linked to human neurological disease. Cell Rep. 2013;3:1795–1805. doi: 10.1016/j.celrep.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Indrieri A., van Rahden V.A., Tiranti V., Morleo M., Iaconis D., Tammaro R., D’Amato I., Conte I., Maystadt I., Demuth S. Mutations in COX7B cause microphthalmia with linear skin lesions, an unconventional mitochondrial disease. Am. J. Hum. Genet. 2012;91:942–949. doi: 10.1016/j.ajhg.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huigsloot M., Nijtmans L.G., Szklarczyk R., Baars M.J., van den Brand M.A., Hendriksfranssen M.G., van den Heuvel L.P., Smeitink J.A., Huynen M.A., Rodenburg R.J. A mutation in C2orf64 causes impaired cytochrome c oxidase assembly and mitochondrial cardiomyopathy. Am. J. Hum. Genet. 2011;88:488–493. doi: 10.1016/j.ajhg.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weraarpachai W., Sasarman F., Nishimura T., Antonicka H., Auré K., Rötig A., Lombès A., Shoubridge E.A. Mutations in C12orf62, a factor that couples COX I synthesis with cytochrome c oxidase assembly, cause fatal neonatal lactic acidosis. Am. J. Hum. Genet. 2012;90:142–151. doi: 10.1016/j.ajhg.2011.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Balsa E., Marco R., Perales-Clemente E., Szklarczyk R., Calvo E., Landázuri M.O., Enríquez J.A. NDUFA4 is a subunit of complex IV of the mammalian electron transport chain. Cell Metab. 2012;16:378–386. doi: 10.1016/j.cmet.2012.07.015. [DOI] [PubMed] [Google Scholar]
- 20.Mootha V.K., Lepage P., Miller K., Bunkenborg J., Reich M., Hjerrild M., Delmonte T., Villeneuve A., Sladek R., Xu F. Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proc. Natl. Acad. Sci. USA. 2003;100:605–610. doi: 10.1073/pnas.242716699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weraarpachai W., Antonicka H., Sasarman F., Seeger J., Schrank B., Kolesar J.E., Lochmüller H., Chevrette M., Kaufman B.A., Horvath R., Shoubridge E.A. Mutation in TACO1, encoding a translational activator of COX I, results in cytochrome c oxidase deficiency and late-onset Leigh syndrome. Nat. Genet. 2009;41:833–837. doi: 10.1038/ng.390. [DOI] [PubMed] [Google Scholar]
- 22.Antonicka H., Leary S.C., Guercin G.H., Agar J.N., Horvath R., Kennaway N.G., Harding C.O., Jaksch M., Shoubridge E.A. Mutations in COX10 result in a defect in mitochondrial heme A biosynthesis and account for multiple, early-onset clinical phenotypes associated with isolated COX deficiency. Hum. Mol. Genet. 2003;12:2693–2702. doi: 10.1093/hmg/ddg284. [DOI] [PubMed] [Google Scholar]
- 23.Oquendo C.E., Antonicka H., Shoubridge E.A., Reardon W., Brown G.K. Functional and genetic studies demonstrate that mutation in the COX15 gene can cause Leigh syndrome. J. Med. Genet. 2004;41:540–544. doi: 10.1136/jmg.2003.017426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bugiani M., Tiranti V., Farina L., Uziel G., Zeviani M. Novel mutations in COX15 in a long surviving Leigh syndrome patient with cytochrome c oxidase deficiency. J. Med. Genet. 2005;42:e28. doi: 10.1136/jmg.2004.029926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wedatilake Y., Brown R., McFarland R., Yaplito-Lee J., Morris A.A., Champion M., Jardine P.E., Clarke A., Thorburn D.R., Taylor R.W. SURF1 deficiency: a multi-centre natural history study. Orphanet J. Rare Dis. 2013;8:96. doi: 10.1186/1750-1172-8-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tiranti V., Jaksch M., Hofmann S., Galimberti C., Hoertnagel K., Lulli L., Freisinger P., Bindoff L., Gerbitz K.D., Comi G.P. Loss-of-function mutations of SURF-1 are specifically associated with Leigh syndrome with cytochrome c oxidase deficiency. Ann. Neurol. 1999;46:161–166. doi: 10.1002/1531-8249(199908)46:2<161::aid-ana4>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 27.Bundschuh F.A., Hannappel A., Anderka O., Ludwig B. Surf1, associated with Leigh syndrome in humans, is a heme-binding protein in bacterial oxidase biogenesis. J. Biol. Chem. 2009;284:25735–25741. doi: 10.1074/jbc.M109.040295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith D., Gray J., Mitchell L., Antholine W.E., Hosler J.P. Assembly of cytochrome-c oxidase in the absence of assembly protein Surf1p leads to loss of the active site heme. J. Biol. Chem. 2005;280:17652–17656. doi: 10.1074/jbc.C500061200. [DOI] [PubMed] [Google Scholar]
- 29.Leary S.C., Sasarman F., Nishimura T., Shoubridge E.A. Human SCO2 is required for the synthesis of CO II and as a thiol-disulphide oxidoreductase for SCO1. Hum. Mol. Genet. 2009;18:2230–2240. doi: 10.1093/hmg/ddp158. [DOI] [PubMed] [Google Scholar]
- 30.Ruzzenente B., Metodiev M.D., Wredenberg A., Bratic A., Park C.B., Cámara Y., Milenkovic D., Zickermann V., Wibom R., Hultenby K. LRPPRC is necessary for polyadenylation and coordination of translation of mitochondrial mRNAs. EMBO J. 2012;31:443–456. doi: 10.1038/emboj.2011.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sasarman F., Brunel-Guitton C., Antonicka H., Wai T., Shoubridge E.A., LSFC Consortium LRPPRC and SLIRP interact in a ribonucleoprotein complex that regulates posttranscriptional gene expression in mitochondria. Mol. Biol. Cell. 2010;21:1315–1323. doi: 10.1091/mbc.E10-01-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tiranti V., Viscomi C., Hildebrandt T., Di Meo I., Mineri R., Tiveron C., Levitt M.D., Prelle A., Fagiolari G., Rimoldi M., Zeviani M. Loss of ETHE1, a mitochondrial dioxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy. Nat. Med. 2009;15:200–205. doi: 10.1038/nm.1907. [DOI] [PubMed] [Google Scholar]
- 33.Ghezzi D., Saada A., D’Adamo P., Fernandez-Vizarra E., Gasparini P., Tiranti V., Elpeleg O., Zeviani M. FASTKD2 nonsense mutation in an infantile mitochondrial encephalomyopathy associated with cytochrome c oxidase deficiency. Am. J. Hum. Genet. 2008;83:415–423. doi: 10.1016/j.ajhg.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kirby D.M., Crawford M., Cleary M.A., Dahl H.H., Dennett X., Thorburn D.R. Respiratory chain complex I deficiency: an underdiagnosed energy generation disorder. Neurology. 1999;52:1255–1264. doi: 10.1212/wnl.52.6.1255. [DOI] [PubMed] [Google Scholar]
- 35.Bahlo M., Bromhead C.J. Generating linkage mapping files from Affymetrix SNP chip data. Bioinformatics. 2009;25:1961–1962. doi: 10.1093/bioinformatics/btp313. [DOI] [PubMed] [Google Scholar]
- 36.Leutenegger A.L., Labalme A., Genin E., Toutain A., Steichen E., Clerget-Darpoux F., Edery P. Using genomic inbreeding coefficient estimates for homozygosity mapping of rare recessive traits: application to Taybi-Linder syndrome. Am. J. Hum. Genet. 2006;79:62–66. doi: 10.1086/504640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leutenegger A.L., Prum B., Génin E., Verny C., Lemainque A., Clerget-Darpoux F., Thompson E.A. Estimation of the inbreeding coefficient through use of genomic data. Am. J. Hum. Genet. 2003;73:516–523. doi: 10.1086/378207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 39.Thiele H., Nürnberg P. HaploPainter: a tool for drawing pedigrees with complex haplotypes. Bioinformatics. 2005;21:1730–1732. doi: 10.1093/bioinformatics/bth488. [DOI] [PubMed] [Google Scholar]
- 40.Desmet F.-O., Hamroun D., Lalande M., Collod-Béroud G., Claustres M., Béroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37:e67. doi: 10.1093/nar/gkp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reese M.G., Eeckman F.H., Kulp D., Haussler D. Improved splice site detection in Genie. J. Comput. Biol. 1997;4:311–323. doi: 10.1089/cmb.1997.4.311. [DOI] [PubMed] [Google Scholar]
- 42.Calvo S.E., Compton A.G., Hershman S.G., Lim S.C., Lieber D.S., Tucker E.J., Laskowski A., Garone C., Liu S., Jaffe D.B. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci. Transl. Med. 2012;4:18ra10. doi: 10.1126/scitranslmed.3003310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li H., Ruan J., Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fujita P.A., Rhead B., Zweig A.S., Hinrichs A.S., Karolchik D., Cline M.S., Goldman M., Barber G.P., Clawson H., Coelho A. The UCSC Genome Browser database: update 2011. Nucleic Acids Res. 2011;39(Database issue):D876–D882. doi: 10.1093/nar/gkq963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sherry S.T., Ward M.H., Kholodov M., Baker J., Phan L., Smigielski E.M., Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.The 1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Colombo R. Dating mutations. In: Cooper D.N., Kehrer-Sawatzki H., editors. Handbook of Human Molecular Evolution. John Wiley & Sons, Ltd; Chichester: 2008. p. 138. [Google Scholar]
- 52.Slatkin M., Rannala B. Estimating allele age. Annu. Rev. Genomics Hum. Genet. 2000;1:225–249. doi: 10.1146/annurev.genom.1.1.225. [DOI] [PubMed] [Google Scholar]
- 53.Corbett M.A., Schwake M., Bahlo M., Dibbens L.M., Lin M., Gandolfo L.C., Vears D.F., O’Sullivan J.D., Robertson T., Bayly M.A. A mutation in the Golgi Qb-SNARE gene GOSR2 causes progressive myoclonus epilepsy with early ataxia. Am. J. Hum. Genet. 2011;88:657–663. doi: 10.1016/j.ajhg.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boissé Lomax L., Bayly M.A., Hjalgrim H., Møller R.S., Vlaar A.M., Aaberg K.M., Marquardt I., Gandolfo L.C., Willemsen M., Kamsteeg E.-J. ‘North Sea’ progressive myoclonus epilepsy: phenotype of subjects with GOSR2 mutation. Brain. 2013;136:1146–1154. doi: 10.1093/brain/awt021. [DOI] [PubMed] [Google Scholar]
- 55.McPeek M.S., Strahs A. Assessment of linkage disequilibrium by the decay of haplotype sharing, with application to fine-scale genetic mapping. Am. J. Hum. Genet. 1999;65:858–875. doi: 10.1086/302537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yeap Y.Y., Ng I.H., Badrian B., Nguyen T.V., Yip Y.Y., Dhillon A.S., Mutsaers S.E., Silke J., Bogoyevitch M.A., Ng D.C. c-Jun N-terminal kinase/c-Jun inhibits fibroblast proliferation by negatively regulating the levels of stathmin/oncoprotein 18. Biochem. J. 2010;430:345–354. doi: 10.1042/BJ20100425. [DOI] [PubMed] [Google Scholar]
- 57.Calvo S.E., Tucker E.J., Compton A.G., Kirby D.M., Crawford G., Burtt N.P., Rivas M., Guiducci C., Bruno D.L., Goldberger O.A. High-throughput, pooled sequencing identifies mutations in NUBPL and FOXRED1 in human complex I deficiency. Nat. Genet. 2010;42:851–858. doi: 10.1038/ng.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Johnston A.J., Hoogenraad J., Dougan D.A., Truscott K.N., Yano M., Mori M., Hoogenraad N.J., Ryan M.T. Insertion and assembly of human tom7 into the preprotein translocase complex of the outer mitochondrial membrane. J. Biol. Chem. 2002;277:42197–42204. doi: 10.1074/jbc.M205613200. [DOI] [PubMed] [Google Scholar]
- 59.Stojanovski D., Pfanner N., Wiedemann N. Import of proteins into mitochondria. Methods Cell Biol. 2007;80:783–806. doi: 10.1016/S0091-679X(06)80036-1. [DOI] [PubMed] [Google Scholar]
- 60.McKenzie M., Lazarou M., Thorburn D.R., Ryan M.T. Analysis of mitochondrial subunit assembly into respiratory chain complexes using Blue Native polyacrylamide gel electrophoresis. Anal. Biochem. 2007;364:128–137. doi: 10.1016/j.ab.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 61.McKenzie M., Lazarou M., Ryan M.T. Chapter 18 Analysis of respiratory chain complex assembly with radiolabeled nuclear- and mitochondrial-encoded subunits. Methods Enzymol. 2009;456:321–339. doi: 10.1016/S0076-6879(08)04418-2. [DOI] [PubMed] [Google Scholar]
- 62.Robinson B.H., Petrova-Benedict R., Buncic J.R., Wallace D.C. Nonviability of cells with oxidative defects in galactose medium: a screening test for affected patient fibroblasts. Biochem. Med. Metab. Biol. 1992;48:122–126. doi: 10.1016/0885-4505(92)90056-5. [DOI] [PubMed] [Google Scholar]
- 64.Claros M.G., Vincens P. Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur. J. Biochem. 1996;241:779–786. doi: 10.1111/j.1432-1033.1996.00779.x. [DOI] [PubMed] [Google Scholar]
- 65.Guda C., Guda P., Fahy E., Subramaniam S. MITOPRED: a web server for the prediction of mitochondrial proteins. Nucleic Acids Res. 2004;32(Web Server issue):W372–W374. doi: 10.1093/nar/gkh374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pagliarini D.J., Calvo S.E., Chang B., Sheth S.A., Vafai S.B., Ong S.-E., Walford G.A., Sugiana C., Boneh A., Chen W.K. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Szklarczyk R., Wanschers B.F., Cuypers T.D., Esseling J.J., Riemersma M., van den Brand M.A., Gloerich J., Lasonder E., van den Heuvel L.P., Nijtmans L.G., Huynen M.A. Iterative orthology prediction uncovers new mitochondrial proteins and identifies C12orf62 as the human ortholog of COX14, a protein involved in the assembly of cytochrome c oxidase. Genome Biol. 2012;13:R12. doi: 10.1186/gb-2012-13-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Church C., Goehring B., Forsha D., Wazny P., Poyton R.O. A role for Pet100p in the assembly of yeast cytochrome c oxidase: interaction with a subassembly that accumulates in a pet100 mutant. J. Biol. Chem. 2005;280:1854–1863. doi: 10.1074/jbc.M410726200. [DOI] [PubMed] [Google Scholar]
- 69.Chacinska A., Koehler C.M., Milenkovic D., Lithgow T., Pfanner N. Importing mitochondrial proteins: machineries and mechanisms. Cell. 2009;138:628–644. doi: 10.1016/j.cell.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mick D.U., Fox T.D., Rehling P. Inventory control: cytochrome c oxidase assembly regulates mitochondrial translation. Nat. Rev. Mol. Cell Biol. 2011;12:14–20. doi: 10.1038/nrm3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Church C., Chapon C., Poyton R.O. Cloning and characterization of PET100, a gene required for the assembly of yeast cytochrome c oxidase. J. Biol. Chem. 1996;271:18499–18507. doi: 10.1074/jbc.271.31.18499. [DOI] [PubMed] [Google Scholar]
- 72.Debray F.-G., Morin C., Janvier A., Villeneuve J., Maranda B., Laframboise R., Lacroix J., Decarie J.-C., Robitaille Y., Lambert M. LRPPRC mutations cause a phenotypically distinct form of Leigh syndrome with cytochrome c oxidase deficiency. J. Med. Genet. 2011;48:183–189. doi: 10.1136/jmg.2010.081976. [DOI] [PubMed] [Google Scholar]
- 73.Morin C., Mitchell G., Larochelle J., Lambert M., Ogier H., Robinson B.H., De Braekeleer M. Clinical, metabolic, and genetic aspects of cytochrome C oxidase deficiency in Saguenay-Lac-Saint-Jean. Am. J. Hum. Genet. 1993;53:488–496. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.