

Abstract
Elevated body mass index (BMI) associates with cardiometabolic traits on observational analysis, yet the underlying causal relationships remain unclear. We conducted Mendelian randomization analyses by using a genetic score (GS) comprising 14 BMI-associated SNPs from a recent discovery analysis to investigate the causal role of BMI in cardiometabolic traits and events. We used eight population-based cohorts, including 34,538 European-descent individuals (4,407 type 2 diabetes (T2D), 6,073 coronary heart disease (CHD), and 3,813 stroke cases). A 1 kg/m2 genetically elevated BMI increased fasting glucose (0.18 mmol/l; 95% confidence interval (CI) = 0.12–0.24), fasting insulin (8.5%; 95% CI = 5.9–11.1), interleukin-6 (7.0%; 95% CI = 4.0–10.1), and systolic blood pressure (0.70 mmHg; 95% CI = 0.24–1.16) and reduced high-density lipoprotein cholesterol (−0.02 mmol/l; 95% CI = −0.03 to −0.01) and low-density lipoprotein cholesterol (LDL-C; −0.04 mmol/l; 95% CI = −0.07 to −0.01). Observational and causal estimates were directionally concordant, except for LDL-C. A 1 kg/m2 genetically elevated BMI increased the odds of T2D (odds ratio [OR] = 1.27; 95% CI = 1.18–1.36) but did not alter risk of CHD (OR 1.01; 95% CI = 0.94–1.08) or stroke (OR = 1.03; 95% CI = 0.95–1.12). A meta-analysis incorporating published studies reporting 27,465 CHD events in 219,423 individuals yielded a pooled OR of 1.04 (95% CI = 0.97–1.12) per 1 kg/m2 increase in BMI. In conclusion, we identified causal effects of BMI on several cardiometabolic traits; however, whether BMI causally impacts CHD risk requires further evidence.
Introduction
Over half a billion people worldwide are obese (defined as a body mass index [BMI] ≥ 30 kg/m2; MIM 601665),1 which negatively impacts multiple health outcomes.2 In the United States, two-thirds of adults are overweight or obese.2 Although lifestyle and behavioral factors have a strong role in the pathogenesis of obesity, genetic variation has also been shown to play a strong role; heritability estimates range from 40% to 85%.3
Large prospective population studies show that BMI strongly associates with coronary heart disease (CHD [MIM 607339]), type 2 diabetes (T2D [MIM 125853]), and all-cause mortality.4–9 BMI also associates with glycaemic traits10 (such as fasting glucose [MIM 612108]) and traits that are causally related to CHD, including blood pressure (MIM 145500) and blood lipids (MIM 604595).9,11,12 However, few randomized trials provide data that can precisely delineate the underlying causal relationships between BMI and cardiometabolic traits. This is important because observational associations between BMI and traits or disease events could arise from confounding (where an association does not reflect a causal relationship) and reverse causality (where the disease process alters the exposure of interest). Whether BMI causes adverse levels of traits or risk of outcomes is of critical importance given that BMI is modifiable. In terms of CHD events, a recent phase III randomized trial of weight loss for cardiovascular-disease prevention was terminated because of a lack of efficacy.13
An alternative and effective means of investigating whether an exposure causes an outcome is the use of genetic variants in the Mendelian randomization approach. This technique exploits the random allocation of genetic variants at gametogenesis, facilitating their use as instrumental variables (traits that can be used as proxies for the exposure but that are not affected by confounding) for investigating causality.14 The discrepancy between observational and randomized evidence to date motivated us to use a Mendelian randomization approach to investigate the role of BMI in cardiometabolic traits and events through instrumental variable analysis.
Subjects and Methods
We included up to 34,538 participants from eight cohorts that had been genotyped with the Human CVD BeadArray (Illumina), also termed the “IBC” or “CardioChip” array.15
Several studies using this array have been published and have confirmed previously established associations and identified new associations between SNPs and several phenotypes and disease outcomes, including coronary artery disease,16,17 plasma lipids,18 blood pressure,19,20 cardiomyopathy,21 T2D,22 and BMI.23
The eight cohorts used in the current study, listed in Table S1 (available online), are Atherosclerosis Risk in Communities (ARIC),24 the Cardiovascular Health Study (CHS),25 Coronary Artery Risk Development in Young Adults (CARDIA),26 the European Prospective Investigation into Cancer and Nutrition Netherlands (EPIC-NL),27 the Framingham Heart Study (FHS),28 Multinational Etoricoxib and Diclofenac Arthritis Long-term (MEDAL),29 the Multi-Ethnic Study of Atherosclerosis (MESA),30 and the Women’s Health Initiative (WHI).31
ARIC is a prospective population-based study of atherosclerosis and cardiovascular diseases in 15,792 men and women, including 11,478 non-Hispanic whites and 4,314 African Americans, drawn from four United States communities (suburban Minneapolis, Washington County, Forsyth County, and Jackson). CHS is a population-based longitudinal study of CHD and stroke in adults aged 65 years and older. The study design called for enrollment of 1,250 men and women in each of four communities: Forsyth County, Sacramento County, Washington County, and Pittsburgh. CARDIA is a prospective, multicenter investigation of the natural history and etiology of cardiovascular disease in African Americans and whites 18–30 years of age at the time of initial examination. The initial examination included 5,115 participants selectively recruited to represent proportionate racial, gender, age, and education groups from four communities: Birmingham, Chicago, Minneapolis, and Oakland. EPIC-NL is the Dutch contribution to the European Prospective Investigation into Cancer and Nutrition, recruited between 1993 and 1997, and consists of the Prospect cohort (a prospective population-based cohort of 17,357 women between 49 and 70 years of age and participating in breast cancer screening) and the Monitoring Project on Risk Factors for Chronic Diseases cohort (consisting of 22,654 men and women between the ages of 20 and 59 years at recruitment in three Dutch towns, Amsterdam, Maastricht, and Doetinchem). FHS is a longitudinal observational, community-based cohort initiated in 1948 in Framingham to prospectively investigate CVD and its risk factors. The children (and spouses of the children) of the original cohort, labeled the Offspring cohort, were recruited in 1971 and have been examined approximately every 4 years since. The MEDAL program was prospectively designed to pool data from three randomized, double-blind trials of etoricoxib versus diclofenac in arthritic individuals between June 2002 and May 2006 across 46 countries: the Etoricoxib versus Diclofenac Sodium Gastrointestinal Tolerability and Effectiveness (EDGE) study (ClinicalTrials.gov ID NCT00092703), EDGE II (ClinicalTrials.gov ID NCT00092742), and the MEDAL study (ClinicalTrials.gov ID NCT00250445). Recruited subjects were aged 50 years and older, had a clinical diagnosis of osteoarthritis (OA) or rheumatoid arthritis (RA), and were adjudged to require long-term therapy with a nonsteroidal anti-inflammatory drug or cylo-oxygenase-2 selective inhibitor. In total, 34,701 OA and RA subjects (7,111 in EDGE, 4,086 in EDGE II, and 23,504 in the MEDAL study), consisting of 24,913 (72%) OA subjects and 9,787 (28%) RA subjects, were followed for a mean duration of 18 months. The primary endpoint was thrombotic cardiovascular events. MESA is a multicenter prospective cohort study of the development of subclinical cardiovascular disease. A total of 6,814 women and men between the ages of 45 and 84 years were recruited for the first examination between 2000 and 2002. Participants were recruited in six United States cities (Baltimore, Chicago, Forsyth County, Los Angeles County, Northern Manhattan, and St. Paul). WHI is one of the largest (n = 161,808) studies of women’s health ever undertaken in the United States. There are two major components of WHI: a clinical trial that enrolled and randomized 68,132 women aged 50–79 into at least one of three placebo-control clinical trials (hormone therapy, dietary modification, and supplementation with calcium and vitamin D) and an observational study that enrolled 93,676 women of the same age range into a parallel prospective cohort study.
We restricted our analysis to individuals of European ancestry to avoid confounding by population admixture. The investigation was approved by the institutional review boards of each study, and written informed consent was obtained from all participants. ARIC, CHS, CARDIA, FHS, and MESA participated in this project as part of the NHLBI’s Candidate-Gene Association Resource Consortium.32
SNP Selection
A recent discovery BMI analysis including over 108,000 individuals (including cohorts in this analysis) and using the IBC CardioChip23 facilitated SNP selection for this analysis. All SNPs that met the array-wide threshold of p < 2.4 × 10−6 in the discovery analysis for BMI were included. Fourteen SNPs were identified, and their characteristics are listed in Table S2.
Construction of the Genetic Score
To increase precision, we weighted SNPs by the beta coefficients reported in the discovery paper. Because the original discovery paper presented estimates on the per SD scale,23 we transformed these to the native units (kg/m2) by multiplying the summary beta coefficients by the SD of the largest data set reported to date in individuals of European ancestry (1,462,958 individuals; SD = 4.7).4 We then summed the externally weighted value for each SNP in each individual to create a genetic score (GS). We did not impute missing genotype (or phenotype) values.
Data Handling
We had access to individual-level data for all participants in the studies and created a merged data set. We adjusted all analyses by study and restricted our data set to individuals with nonmissing data for BMI and the GS. Nonnormally distributed traits were natural-log transformed (resulting in a Gaussian distribution), and estimates from the analysis of these transformed traits were exponentiated and converted to a percent difference in the geometric mean. For one study with related individuals, only the eldest person in each family structure was included in the analysis (Table S1).
Cardiometabolic Traits and Outcomes
We investigated the following cardiometabolic traits and outcomes: waist circumference (used as a positive control for the BMI GS), fasting glucose, fasting insulin, C-reactive protein (CRP), interleukin-6 (IL-6), fibrinogen, high-density lipoprotein cholesterol (HDL-C), low-density lipoprotein cholesterol (LDL-C), triglycerides (TGs), systolic blood pressure (SBP), diastolic blood pressure (DBP), carotid intima medial thickness (cIMT), CHD, stroke, and T2D. The outcomes (CHD, stroke, and T2D) consisted of combined incident and prevalent cases and are defined in Table S3.
Genetic Association Analysis
The proportion of variance (R2) and the corresponding 95% confidence interval (CI) of the GS on BMI were estimated in each study, and values were combined across studies via fixed-effects meta-analysis. We quantified the first-stage F-statistic to inform on the strength of the genetic instrument.33 The association between the GS and cardiometabolic traits and events was estimated by linear and logistic regression, respectively. We also investigated the association between the GS and the traditional confounders age, gender, and smoking status.
Pairwise Correlations between SNPs in BMI GS and Each Cardiometabolic Trait and Event
We investigated evidence of a genetic dose-response relationship by plotting pairwise associations of the pooled effect of each SNP on BMI against the corresponding value for each cardiometabolic trait and event. We tested for evidence of linearity through metaregression by using the “metareg” command in Stata v.13.1 (StataCorp, College Station).34 “Metareg” conducts a random-effects meta-regression that is in essence comparable to a linear regression analysis in which the linear association of BMI (the independent variable) is tested with each cardiometabolic trait (dependent variable). The analysis is weighted by the precision of the effect estimates.35 A small p value provides evidence of a linear relationship between BMI and the trait under investigation.
Instrumental Variable Analysis
To generate the causal estimate per 1 kg/m2 increase in BMI, we used instrumental variable analyses. For continuous traits, we used the two-stage least-squares estimator implemented in the “ivregress” command36 in Stata to generate the causal estimate per 1 kg/m2 increase in BMI, adjusted for study. For binary traits, we used the logistic control function estimator.37 For this, we first conducted a linear regression analysis with BMI as the dependent variable and the GS as the independent variable. We then incorporated the residuals from the first step into a logistic regression model of each binary trait on BMI by using robust standard errors, thereby adjusting for residuals from the first step.
Sensitivity Analyses
We conducted several sensitivity analyses to investigate whether the estimates obtained from instrumental variable analyses were influenced by adjustment for traits.
First, we adjusted the instrumental variable analysis for age and sex. Second, to investigate whether the association between BMI and LDL-C could be explained by lipid-lowering therapy, we adjusted for lipid-lowering therapy in the instrumental variable analysis. Third, to investigate whether the null estimate of BMI with CHD on instrumental variable analysis could be explained by the association with LDL-C, we repeated the analysis with adjustment for LDL-C.
To ensure that bias was not introduced by the combination of incident and prevalent cases, we investigated incident cases separately.
Finally, we created a second, stricter GS in which any SNP within a gene that has shown association with a trait unrelated to adiposity was excluded from the GS. This was based on information from the Catalog of Published Genome-Wide Association Studies.38 The SNPs used in the stricter GS are listed in Table S4. We tested the association between this stricter GS and the outcomes—T2D, CHD, and stroke.
Observational Analysis
We conducted minimally adjusted analyses (adjusted for age and sex) with BMI as the independent variable for each trait of interest (by using linear and logistic regression for continuous and binary traits, respectively). This was compared to the estimates derived from instrumental variable analysis.
Comparison of BMI GS to Findings from the Look AHEAD: Action for Health in Diabetes Randomized Trial
The Look AHEAD: Action for Health in Diabetes (Look AHEAD) trial13,39 was a multicenter trial in which 5,145 overweight participants with T2D were randomized to an intensive lifestyle intervention either to lose weight through reduced calorie intake and increased physical activity (the active arm) or to receive standard support and education (control arm). The trial ran for a median of 9.6 years and was terminated for futility when it failed to show an effect of the intervention on the primary outcome of a composite of cardiovascular death, nonfatal myocardial infarction or stroke, or hospitalization for angina.
In order to contextualize the findings from our Mendelian randomization analysis with that of the Look AHEAD trial, we scaled the effect of the instrumental variable estimates by the same magnitude of difference reported in the Look AHEAD trial (in which the active arm, compared to the control arm, achieved an average reduction in weight of 4 kg, equivalent to a 1.4 kg/m2 reduction in BMI). Data on all traits and outcomes were obtained from two publications.13,39 Given that findings for CRP were stratified by gender and lipid-lowering therapy,39 we combined them through fixed-effects meta-analysis to achieve an overall percent difference between intensive lifestyle intervention and usual care.
Comparison of Findings to Mendelian Randomization Studies
To place our findings into the context of previous studies, we searched PubMed from inception to November 3, 2013, by using the search term “(“Mendelian Randomization Analysis”[Mesh] OR “Mendelian randomization”[All Fields] OR “Mendelian randomization”[All Fields]) AND (“Body Mass Index”[Mesh] OR “bmi”[All Fields] OR “body mass index”[All Fields]).” This retrieved 54 studies, to which we added one study that was not identified by our search but that was referenced in a recent publication40 (Figure S1). A total of seven studies reported traits that overlapped with those reported in our analysis. We documented sample size, the number of alleles used for Mendelian randomization, and the power of the genetic instruments (measured as the proportion of variance [R2] of BMI explained by the genetic instrument or the first-stage F-statistic) and examined for consistency in the association and direction of the effect of instrumental variable estimates. In addition, we conducted a meta-analysis of the effect of a 1 kg/m2 increase in BMI on CHD risk by using fixed- and random-effects modeling implemented in the “metan” command41 in Stata, and we quantified between-study heterogeneity by using the I2 statistic.42
Results
A total of eight cohorts with up to 34,538 individuals were used in this study (Table S1). The mean age of study participants was 60 years (range = 17–100 years), and 65% of individuals were female. The mean BMI was 27.5 kg/m2 (range = 14.4–69.6 kg/m2). There were 4,407 T2D, 6,073 CHD, and 3,813 stroke cases recorded in the data sets.
The observed allele frequencies for the 14 SNPs comprising the GS (Table S2) were similar across the eight cohorts (Figure S2). The weighted GS was normally distributed in each of the eight studies, and SNPs showed concordant associations with BMI (Figures S3 and S4).
Association between BMI GS and Waist Circumference
The GS associated with a BMI increase of 1.08 kg/m2 (95% CI = 0.95–1.21 per unit increase in weighted GS) and explained 0.8% (95% CI = 0.6%–1.0%) of its variance with a first-stage F-statistic of 237. We also identified associations between the GS and waist circumference (2.55 cm; 95% CI = 2.16–2.94), which served as a positive control.
Association between BMI GS and Age, Sex, and Smoking Status
No association between the BMI GS and smoking status (odds ratio [OR] of ever smoking per unit increase in weighted GS = 1.03; 95% CI = 0.97–1.08) or sex (OR of male gender = 1.03; 95% CI = 0.97–1.09) was observed. We identified a weak association between the BMI GS and reduced age (−0.19 years; 95% CI = −0.37 to −0.02).
Association between GS and Cardiometabolic Traits
The BMI GS showed strong associations with glycemic, inflammation, lipid, and blood-pressure traits. For each unit increase in GS, which associated with a BMI increase of 1.08 kg/m2, fasting glucose was 0.17 mmol/l (95% CI = 0.11–0.22) higher and fasting insulin was 8.4% (95% CI = 5.5–11.5) higher. The GS also showed strong association with CRP (11.8% higher; 95% CI = 7.8–15.9), IL-6 (7.4% higher; 95% CI = 4.2–10.7), and fibrinogen (0.9% higher; 95% CI = 0.2–1.6). Each unit increase in GS associated with a reduction in LDL-C and HDL-C of −0.04 mmol/l (95% CI = −0.06 to −0.01) and −0.016 mmol/l (95% CI = −0.030 to −0.005), respectively, and an increase in SBP and DBP of 0.73 mmHg (95% CI = 0.24–1.21) and 0.29 mmHg (95% CI = 0.02–0.55), respectively (Figure 1).
Figure 1.
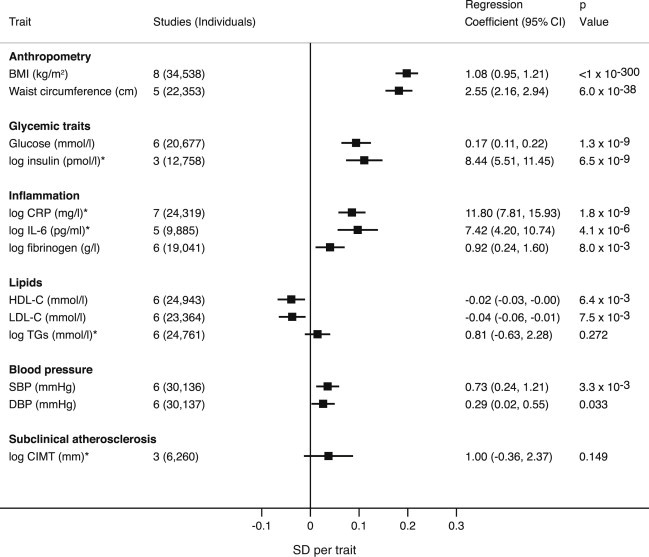
Association between the BMI GS and Cardiometabolic Traits
Effect estimates represent the beta (plotted) or regression (tabulated) coefficients (±95% CI) per 1-unit increase in weighted GS. The GS consisted of 14 SNPs taken from Guo et al.23 For log-transformed variables (marked by an asterisk), the regression coefficients are presented as a percent difference in the geometric mean.
We identified a significant association between the BMI GS and T2D risk (OR = 1.29; 95% CI = 1.20–1.39), but not CHD, stroke, or cIMT (Figures 1 and 2).
Figure 2.
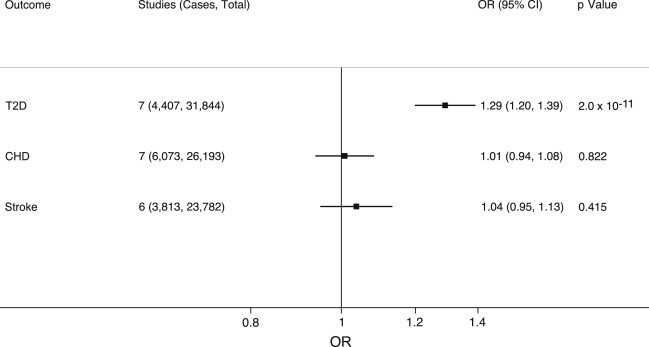
Association between the BMI GS and Cardiometabolic Events
Effect estimates represent the OR (±95% CI) for each outcome per 1-unit increase in weighted GS. The GS consisted of 14 SNPs (taken from Guo et al.23) that associated with a 1.08 kg/m2 increase in BMI (Figure 1).
Pairwise Associations between SNPs in the GS and Cardiometabolic Traits and Events
We found strong evidence of a positive genetic dose-response relationship (meaning that SNPs that associated more strongly with BMI tended to also associate more strongly with the cardiometabolic trait or outcome) between BMI SNPs and the following traits: fasting glucose, fasting insulin, IL-6, and T2D (Figures S5 and S6).
Causal Analysis of BMI on Cardiometabolic Traits
In instrumental variable (causal) analysis, for every 1 kg/m2 increase in BMI, fasting glucose increased by 0.18 mmol/l (95% CI = 0.12–0.24) and fasting insulin increased by 8.5% (95% CI = 5.9–11.1).
A 1 kg/m2 increase in BMI increased CRP by 12.0% (95% CI = 8.0–16.2), IL-6 by 7.0% (95% CI = 4.0–10.1), and fibrinogen by 0.9% (95% CI = 0.3%–1.6%). A 1 kg/m2 increase in BMI increased SBP by 0.70 mmHg (95% CI = 0.24–1.16). For these glycemic, inflammatory, and blood-pressure traits, the instrumental variable estimates were all directionally concordant and, in general, of similar magnitude to estimates derived from observational analyses (Figure 3 and Table 1).
Figure 3.
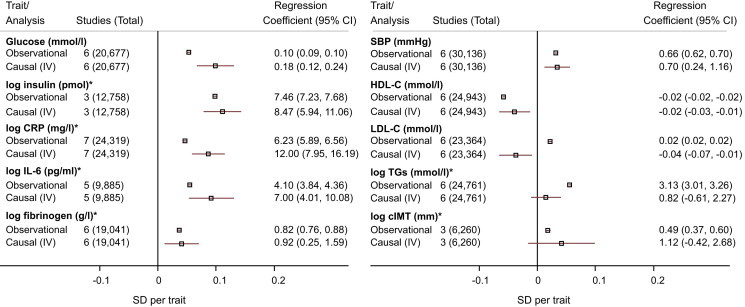
Observational and Instrumental Variable Estimates of the Effect of BMI on Cardiometabolic Traits
Effect estimates represent the beta (plotted) or regression (tabulated) coefficients (±95% CI) per 1 kg/m2 increase in BMI. Observational estimates were adjusted for age, sex, and study. For log-transformed variables (marked by an asterisk), the summary estimates are presented as a percent difference in the geometric mean. Causal estimates were derived from instrumental variable (IV) analysis.
Table 1.
Estimates of the Causal Relationship between BMI and Cardiometabolic Traits and Events
| Studies (Individuals) | Regression Coefficienta(95% CI) | |
|---|---|---|
| Metabolic Traits | ||
| Fasting glucose (mmol/l) | 6 (20,677) | 0.18 (0.12–0.24) |
| Fasting insulin (% difference)b | 3 (12,758) | 8.47 (5.94–11.06) |
| Inflammation Traits | ||
| CRP (% difference)b | 7 (24,319) | 12.00 (7.95–16.19) |
| IL-6 (% difference)b | 5 (9,885) | 7.00 (4.01–10.08) |
| Fibrinogen (% difference)b | 6 (19,041) | 0.92 (0.25–1.59) |
| Blood-Pressure Traits | ||
| SBP (mmHg) | 6 (30,136) | 0.70 (0.24–1.16) |
| DBP (mmHg) | 6 (30,137) | 0.28 (0.03–0.52) |
| Lipid Traits | ||
| HDL-C (mmol/l) | 6 (24,943) | −0.02 (−0.03 to −0.01) |
| LDL-C (mmol/l) | 6 (23,364) | −0.04 (−0.07 to −0.01) |
| TGs (% difference)b | 6 (24,761) | 0.82 (−0.61–2.27) |
| Surrogate Marker of CHD | ||
| cIMT (% difference)b | 3 (6,260) | 1.12 (−0.42–2.68) |
| Events | Studies (Cases/ Individuals) | ORa(95% CI) |
| T2D | 7 (4,407/31,844) | 1.27 (1.18–1.36) |
| CHD | 7 (6,073/26,193) | 1.01 (0.94–1.08) |
| Stroke | 6 (3,813/23,782) | 1.03 (0.95–1.12) |
These estimates were derived from instrumental variable analysis using the GS (consisting of 14 SNPs). Abbreviations are as follows: CHD, coronary heart disease; CI, confidence interval; cIMT, carotid intima medial thickness; CRP, C-reactive protein; DBP, diastolic blood pressure; HDL-C, high-density lipoprotein cholesterol; IL-6, interleukin-6; LDL-C, low-density lipoprotein cholesterol; OR, odds ratio; SBP, systolic blood pressure; T2D, type 2 diabetes, and TG, triglyceride.
Estimates are per 1 kg/m2 increase in BMI.
Percent difference in geometric mean.
A 1 kg/m2 increase in BMI reduced HDL-C by −0.02 mmol/l (95% CI = −0.03 to −0.01) and LDL-C by −0.04 mmol/l (95% CI = −0.07 to −0.01). Although the estimate for HDL-C was concordant with the observational estimate, the estimate for LDL-C was directionally opposite.
The point estimates for the instrumental variable estimates for TGs and cIMT were both directionally concordant with observational estimates; however, unlike those of the observational estimates, the 95% CIs of the instrumental variable estimates included the null value (Figure 3 and Table 1).
Causal Analysis of BMI on T2D, CHD, and Stroke
For T2D, a 1 kg/m2 increase in BMI resulted in an OR of 1.27 (95% CI = 1.18–1.36), which was similar to (but greater in magnitude than) the estimate obtained from observational analysis (OR = 1.13; 95% CI = 1.12–1.13). We did not identify evidence of a causal relationship between BMI and CHD (OR = 1.01; 95% CI = 0.94–1.08); however, the 95% CI overlapped the estimates from observational analysis (OR = 1.02; 95% CI = 1.01–1.02; Figure 4). Neither the instrumental variable analysis nor the observational analysis identified an association between BMI and stroke (Table 1). Similar findings were yielded when CHD and stroke were restricted to incident-only cases (Table S5).
Figure 4.
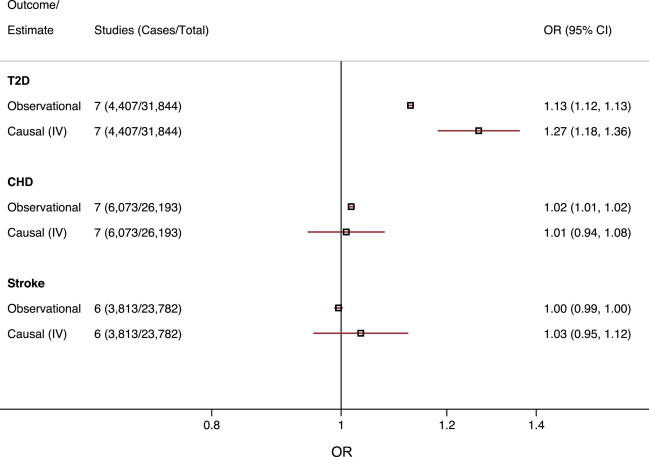
Observational and Instrumental Variable Estimates of the Effect of BMI on Cardiometabolic Events
Effect estimates represent the OR (±95% CI) per 1 kg/m2 increase in BMI. Observational estimates were adjusted for age, sex, and study. Causal estimates were derived from instrumental variable (IV) analysis.
Sensitivity Analyses
Adjustment of the instrumental variable analysis for age and gender did not alter any of the estimates (Table S6), nor did adjustment of the instrumental variable analysis for CHD events by LDL-C (OR of CHD per 1 kg/m2 increase in BMI = 1.04; 95% CI = 0.96–1.14) (Table S6). When we used a stricter GS including SNPs that were more specific to BMI, the findings of the instrumental variable analysis for T2D, CHD, and stroke remained unaltered (Table S7).
When we investigated the association between CHD and the 14 individual SNPs constituting the BMI GS, the SNPs were evenly balanced around the null estimate (the OR point estimates were <1, 1, and >1 for six, one, and seven SNPs, respectively), and not one of the 14 SNPs showed individual association with CHD (all p values > 0.05). The heterogeneity between the estimates was low (I2 = 0%), and the summary estimate derived from the fixed-effects model was numerically identical to the estimate derived from the random-effects model in the meta-analysis (Figure S7). This was in contrast to the association between the individual SNPs and T2D, for which the majority of SNPs (11 of 14) had a point estimate concordant with a positive association and five showed individual association at p < 0.05.
Comparison of Findings from the GS to the Look AHEAD Trial
When we compared findings from the recently published Look AHEAD trial to a comparable difference in BMI by using the GS, the estimates from both the GS and the trial were concordant for all traits (Table 2).
Table 2.
Randomized Evidence of a Causal Relationship between BMI and Cardiometabolic Traits and Events from Genetic and Trial Data
| Mendelian Randomization Estimate Using GS (14 SNPs) in Healthy Individuals (per 1.4 kg/m2Reduction in BMI) | Look AHEAD Trial in Overweight or Obese Individuals with T2D (Intensive Lifestyle Intervention versus Diabetes Support and Education)a | ||||||
|---|---|---|---|---|---|---|---|
| Cardiometabolic Traits | Individuals | Mean Difference (95% CI) | p Value | Individualsb | Mean Difference (95% CI) | p Value | Concordant |
| SBP (mmHg) | 30,136 | −1.01 (−1.67 to −0.34) | 0.003 | 5,145 | −1.9 (−2.6 to −1.1) | <0.05 | yes |
| DBP (mmHg) | 30,137 | −0.39 (−0.75 to −0.04) | 0.03 | 5,145 | −0.1 (−0.5–0.3) | 0.72 | yesc |
| LDL-C (mmol/l)d | 23,364 | 0.05 (0.01–0.09) | 0.01 | 5,145 | 0.04 (0.01–0.08) | <0.05 | yes |
| HDL-C (mmol/l)d | 24,943 | 0.02 (0.01–0.04) | 0.004 | 5,145 | 0.03 (0.02–0.05) | <0.05 | yes |
| TGs (% difference) | 24,761 | −1.16 (−3.15–0.87) | 0.26 | 5,145 | −1 (−4–1) | 0.26 | yes |
| CRP (% difference) | 24,319 | −14.96 (−19.32 to −10.36) | 1.6 × 10−9 | 5,145 | −30.1 (−52.4 to −7.7) | 0.008 | yes |
| Cardiometabolic Events | Events/Individuals | OR (95% CI) | p Value | Events/Individuals | OR (95% CI) | p Value | |
| CHD | 6,073/26,193 | 0.99 (0.90–1.09) | 0.79 | 354/5,145 | 0.84 (0.68–1.04) | 0.11 | yes |
| Stroke | 3,813/23,782 | 0.95 (0.85–1.07) | 0.42 | 165/5,145 | 1.05 (0.77–1.42) | 0.78 | yes |
Abbreviations are as follows: CHD, coronary heart disease; CI, confidence interval; CRP, C-reactive protein; DBP, diastolic blood pressure; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; OR, odds ratio; SBP, systolic blood pressure; T2D, type 2 diabetes, and TG, triglyceride.
Compared to the control arm (randomized to diabetes support and education), the intervention arm (randomized to intensive lifestyle intervention) in the Look AHEAD trial experienced a BMI reduction of 1.4 kg/m2. In order to provide a comparable estimate, we estimated the Mendelian randomization instrumental variable for the same magnitude of difference of BMI.
The precise numbers of individuals contributing to each trait are not reported.
Although the estimate from the Look AHEAD trial was not statistically significant for DBP, the direction of effect was consistent with that obtained from the Mendelian randomization analysis.
LDL-C and HDL-C were converted from mg/dl to mmol/l by multiplication by 0.02586.
Comparison of Findings to Published Studies
We identified seven previous Mendelian randomization studies that investigated BMI and cardiometabolic events (Figure S1).40,43–48 Consistent with the findings we report, all prior Mendelian randomization studies identified causal effects of BMI on levels of fasting insulin, CRP, SBP, DBP, and risk of T2D. In contrast, there was a discrepancy for fasting glucose, IL-6, LDL-C, TGs, cIMT, and CHD (Table S8).
For CHD, we conducted a meta-analysis of instrumental variable estimates from three studies (including this report) with a total of 27,465 CHD events in 219,423 individuals. This yielded a pooled OR estimate of 1.05 (95% CI = 1.00–1.10) for fixed-effects models and 1.04 (95% CI = 0.97–1.12) for random-effects models and moderate between-study heterogeneity (I2 = 50%) (Figure 5).
Figure 5.
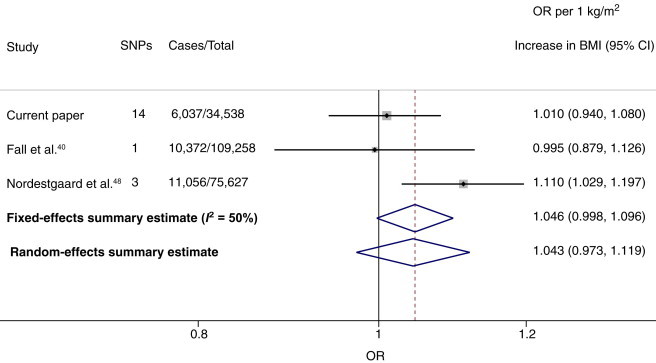
Meta-analysis of Studies Investigating the Effect of BMI on CHD through Mendelian Randomization Analysis
Effect estimates represent the OR (±95% CI) per 1 kg/m2 increase in BMI on the odds of CHD.
Discussion
We used a Mendelian randomization approach to investigate the causal role of BMI on a wide range of cardiometabolic traits. Our analysis, which combined 14 BMI SNPs in order to maximize power,49 supports the importance of BMI in regulating cardiometabolic traits and T2D risk, but the precise causal relationship between BMI and CHD and stroke is less clear.
The general similarity between the estimates obtained from causal (instrumental variable) analysis and those from a minimally adjusted observational analysis for the continuous cardiometabolic traits is striking and indicates that there is little unmeasured confounding in the observational estimates (i.e., our findings suggest that the observed relationships of BMI are very close to the causal estimates rather than arising from bias or confounding). Our findings suggest that reductions in BMI are likely to result in reduced blood pressure, inflammation, fasting glucose and insulin, and risk of T2D and therefore improve the cardiometabolic milieu. Furthermore, our instrumental variable estimates provide a direct quantification of the effect of a 1 kg/m2 alteration in BMI on these cardiometabolic traits. These findings are an important enhancement to previous Mendelian randomization studies and randomized trials that have evaluated the effect of lifestyle intervention on risk of T2D.50,51
When we compared the estimates from the instrumental variable analysis to observational (nongenetic) estimates for clinical events, which can be affected by bias or confounding, we found concordant estimates for T2D risk and stroke; however, this was not the case for CHD. No single SNP in the GS showed association with CHD, and the heterogeneity between the SNPs was low. Therefore, the null association with CHD was a general characteristic shared by all SNPs in the BMI GS. That said, the observational estimate of the effect of BMI on risk of CHD was of small magnitude (OR = 1.02; 95% CI = 1.01–1.03 per 1 kg/m2 increase in BMI), and the 95% CI overlapped the estimate from instrumental variable analysis, meaning that we might have had insufficient power to identify a small effect of BMI on CHD if it were present.
In support of our data is the concordance between our Mendelian randomized estimates for all traits (including CHD) and the clinical-trial randomized estimates from the Look AHEAD trial.13 Combining the Mendelian randomization estimates from published studies with our own gave a total sample size of 219,423 individuals, including 27,465 CHD events, and yielded an estimate that is inconclusive (the OR for a 1 kg/m2 increase in BMI on risk of CHD was 1.04; 95% CI = 0.97–1.12 with considerable between-study heterogeneity). This highlights the need for further large studies using randomized evidence to resolve this important question.
BMI is a complex phenotype, and SNPs that associate with subphenotypes of BMI could potentially have important roles in CHD; this requires further investigation using SNPs specific to refined adiposity phenotypes. Studies included in this analysis contributed to the original discovery analysis,23 and thus use of overlapping studies for discovery and Mendelian randomization analysis could theoretically result in model overfitting. Our instrumental variable (causal) analysis made the assumption of a linear relationship14 between BMI and cardiometabolic traits and events. This is at odds with some published observational studies.4 Specifically, observational studies have suggested that individuals with low BMI might have a higher mortality rate than individuals with normal BMI, creating an apparent J-shaped curve. However, debate persists as to whether this J-shaped association reflects a true protective effect of moderate BMI (compared to low BMI) or whether it is an artifact arising from concurrent illness in some individuals in the low-BMI group and thus appears as a protective effect of normal BMI (a form of reverse causality).52
Additional assumptions of Mendelian randomization14 are (1) that the GS associates with the exposure of interest (in this case, BMI, which we provide very strong evidence for), (2) that the GS only associates with the outcomes through BMI, and (3) that the GS is not influenced by confounding factors. In this regard, the use of multiple SNPs in the GS should in theory enhance specificity of the GS for BMI (reducing the possibility that the GS has pleiotropic effects, i.e., effects nonspecific to BMI). It remains theoretically plausible that a subset of SNPs in the GS also affect glycemic traits (independently of BMI), which could potentially violate the instrumental variable assumptions. Against this is the general concordance between our findings and those of previous Mendelian randomization studies using different genetic variants (Table S8) and the Look AHEAD trial (Table 2). Confounding should be minimized from the randomized inheritance of the alleles used for generating the GS. A further advantage of using multiple SNPs in a GS for Mendelian randomization analysis is that it can increase statistical power by explaining a greater proportion of variance of BMI. Despite this, only a small proportion (0.8%) of the variance of BMI was explained. However, the proportion of variance of drugs on their target phenotypes is also small; for example, blood-pressure-lowering drugs explain only a small proportion of the variance of SBP (∼2% in our data set), yet there is indisputable evidence of the causal role of SBP in CHD from phase III randomized controlled trials.53 Finally, Mendelian randomization mitigates the bias that can arise from nondifferential measurement error in BMI.54
In conclusion, our findings quantify the causal relationships between BMI and cardiometabolic traits and outcomes. With the use of a GS derived from 14 SNPs for Mendelian randomization, an increase in BMI resulted in increased fasting glucose and insulin, SBP, inflammation, and risk of T2D. However, we did not identify a causal effect between BMI and CHD. Whether a reduction in BMI impacts risk of CHD events requires further evidence from appropriately designed randomized studies.
Contributor Information
Michael V. Holmes, Email: michael.holmes@uphs.upenn.edu.
Brendan J. Keating, Email: bkeating@mail.med.upenn.edu.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Catalog of Published Genome-Wide Association Studies, http://www.genome.gov/gwastudies/
ClinicalTrials.gov, http://clinicaltrials.gov/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
References
- 1.Global Health Observatory (GHO). (2013). Obesity: Situation and Trends. World Health Organization, http://www.who.int/gho/ncd/risk_factors/obesity_text/en/index.html.
- 2.Lewis C.E., McTigue K.M., Burke L.E., Poirier P., Eckel R.H., Howard B.V., Allison D.B., Kumanyika S., Pi-Sunyer F.X. Mortality, health outcomes, and body mass index in the overweight range: a science advisory from the American Heart Association. Circulation. 2009;119:3263–3271. doi: 10.1161/CIRCULATIONAHA.109.192574. [DOI] [PubMed] [Google Scholar]
- 3.Hebebrand J., Volckmar A.L., Knoll N., Hinney A. Chipping away the ‘missing heritability’: GIANT steps forward in the molecular elucidation of obesity - but still lots to go. Obes Facts. 2010;3:294–303. doi: 10.1159/000321537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berrington de Gonzalez A., Hartge P., Cerhan J.R., Flint A.J., Hannan L., MacInnis R.J., Moore S.C., Tobias G.S., Anton-Culver H., Freeman L.B. Body-mass index and mortality among 1.46 million white adults. N. Engl. J. Med. 2010;363:2211–2219. doi: 10.1056/NEJMoa1000367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zheng W., McLerran D.F., Rolland B., Zhang X., Inoue M., Matsuo K., He J., Gupta P.C., Ramadas K., Tsugane S. Association between body-mass index and risk of death in more than 1 million Asians. N. Engl. J. Med. 2011;364:719–729. doi: 10.1056/NEJMoa1010679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Poirier P., Giles T.D., Bray G.A., Hong Y., Stern J.S., Pi-Sunyer F.X., Eckel R.H. Obesity and cardiovascular disease: pathophysiology, evaluation, and effect of weight loss. Arterioscler. Thromb. Vasc. Biol. 2006;26:968–976. doi: 10.1161/01.ATV.0000216787.85457.f3. [DOI] [PubMed] [Google Scholar]
- 7.Reeves G.K., Pirie K., Beral V., Green J., Spencer E., Bull D., Million Women Study Collaboration Cancer incidence and mortality in relation to body mass index in the Million Women Study: cohort study. BMJ. 2007;335:1134. doi: 10.1136/bmj.39367.495995.AE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Renehan A.G., Tyson M., Egger M., Heller R.F., Zwahlen M. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet. 2008;371:569–578. doi: 10.1016/S0140-6736(08)60269-X. [DOI] [PubMed] [Google Scholar]
- 9.Whitlock G., Lewington S., Sherliker P., Clarke R., Emberson J., Halsey J., Qizilbash N., Collins R., Peto R., Prospective Studies Collaboration Body-mass index and cause-specific mortality in 900 000 adults: collaborative analyses of 57 prospective studies. Lancet. 2009;373:1083–1096. doi: 10.1016/S0140-6736(09)60318-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bhargava S.K., Sachdev H.S., Fall C.H., Osmond C., Lakshmy R., Barker D.J., Biswas S.K., Ramji S., Prabhakaran D., Reddy K.S. Relation of serial changes in childhood body-mass index to impaired glucose tolerance in young adulthood. N. Engl. J. Med. 2004;350:865–875. doi: 10.1056/NEJMoa035698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lamon-Fava S., Wilson P.W., Schaefer E.J. Impact of body mass index on coronary heart disease risk factors in men and women. The Framingham Offspring Study. Arterioscler. Thromb. Vasc. Biol. 1996;16:1509–1515. doi: 10.1161/01.atv.16.12.1509. [DOI] [PubMed] [Google Scholar]
- 12.Jousilahti P., Tuomilehto J., Vartiainen E., Pekkanen J., Puska P. Body weight, cardiovascular risk factors, and coronary mortality. 15-year follow-up of middle-aged men and women in eastern Finland. Circulation. 1996;93:1372–1379. doi: 10.1161/01.cir.93.7.1372. [DOI] [PubMed] [Google Scholar]
- 13.Wing R.R., Bolin P., Brancati F.L., Bray G.A., Clark J.M., Coday M., Crow R.S., Curtis J.M., Egan C.M., Espeland M.A., Look AHEAD Research Group Cardiovascular effects of intensive lifestyle intervention in type 2 diabetes. N. Engl. J. Med. 2013;369:145–154. doi: 10.1056/NEJMoa1212914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lawlor D.A., Harbord R.M., Sterne J.A., Timpson N., Davey Smith G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat. Med. 2008;27:1133–1163. doi: 10.1002/sim.3034. [DOI] [PubMed] [Google Scholar]
- 15.Keating B.J., Tischfield S., Murray S.S., Bhangale T., Price T.S., Glessner J.T., Galver L., Barrett J.C., Grant S.F., Farlow D.N. Concept, design and implementation of a cardiovascular gene-centric 50 k SNP array for large-scale genomic association studies. PLoS ONE. 2008;3:e3583. doi: 10.1371/journal.pone.0003583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.IBC 50K CAD Consortium Large-scale gene-centric analysis identifies novel variants for coronary artery disease. PLoS Genet. 2011;7:e1002260. doi: 10.1371/journal.pgen.1002260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clarke R., Peden J.F., Hopewell J.C., Kyriakou T., Goel A., Heath S.C., Parish S., Barlera S., Franzosi M.G., Rust S., PROCARDros. Inf. Serv. Consortium Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N. Engl. J. Med. 2009;361:2518–2528. doi: 10.1056/NEJMoa0902604. [DOI] [PubMed] [Google Scholar]
- 18.Asselbergs F.W., Guo Y., van Iperen E.P., Sivapalaratnam S., Tragante V., Lanktree M.B., Lange L.A., Almoguera B., Appelman Y.E., Barnard J., LifeLines Cohort Study Large-scale gene-centric meta-analysis across 32 studies identifies multiple lipid loci. Am. J. Hum. Genet. 2012;91:823–838. doi: 10.1016/j.ajhg.2012.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ganesh S.K., Tragante V., Guo W., Guo Y., Lanktree M.B., Smith E.N., Johnson T., Castillo B.A., Barnard J., Baumert J., CARDIOGRAM, METASTROKE. LifeLines Cohort Study Loci influencing blood pressure identified using a cardiovascular gene-centric array. Hum. Mol. Genet. 2013;22:1663–1678. doi: 10.1093/hmg/dds555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson T., Gaunt T.R., Newhouse S.J., Padmanabhan S., Tomaszewski M., Kumari M., Morris R.W., Tzoulaki I., O’Brien E.T., Poulter N.R., Cardiogenics Consortium. Global BPgen Consortium Blood pressure loci identified with a gene-centric array. Am. J. Hum. Genet. 2011;89:688–700. doi: 10.1016/j.ajhg.2011.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cappola T.P., Li M., He J., Ky B., Gilmore J., Qu L., Keating B., Reilly M., Kim C.E., Glessner J. Common variants in HSPB7 and FRMD4B associated with advanced heart failure. Circ Cardiovasc Genet. 2010;3:147–154. doi: 10.1161/CIRCGENETICS.109.898395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saxena R., Elbers C.C., Guo Y., Peter I., Gaunt T.R., Mega J.L., Lanktree M.B., Tare A., Castillo B.A., Li Y.R., Look AHEAD Research Group. DIAGRAM consortium Large-scale gene-centric meta-analysis across 39 studies identifies type 2 diabetes loci. Am. J. Hum. Genet. 2012;90:410–425. doi: 10.1016/j.ajhg.2011.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo Y., Lanktree M.B., Taylor K.C., Hakonarson H., Lange L.A., Keating B.J., IBC 50K SNP array BMI Consortium Gene-centric meta-analyses of 108 912 individuals confirm known body mass index loci and reveal three novel signals. Hum. Mol. Genet. 2013;22:184–201. doi: 10.1093/hmg/dds396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.The Atherosclerosis Risk in Communities (ARIC) Study: design and objectives. The ARIC investigators. Am. J. Epidemiol. 1989;129:687–702. [PubMed] [Google Scholar]
- 25.Fried L.P., Borhani N.O., Enright P., Furberg C.D., Gardin J.M., Kronmal R.A., Kuller L.H., Manolio T.A., Mittelmark M.B., Newman A. The Cardiovascular Health Study: design and rationale. Ann. Epidemiol. 1991;1:263–276. doi: 10.1016/1047-2797(91)90005-w. [DOI] [PubMed] [Google Scholar]
- 26.Friedman G.D., Cutter G.R., Donahue R.P., Hughes G.H., Hulley S.B., Jacobs D.R., Jr., Liu K., Savage P.J. CARDIA: study design, recruitment, and some characteristics of the examined subjects. J. Clin. Epidemiol. 1988;41:1105–1116. doi: 10.1016/0895-4356(88)90080-7. [DOI] [PubMed] [Google Scholar]
- 27.Beulens J.W., Monninkhof E.M., Verschuren W.M., van der Schouw Y.T., Smit J., Ocke M.C., Jansen E.H., van Dieren S., Grobbee D.E., Peeters P.H., Bueno-de-Mesquita H.B. Cohort profile: the EPIC-NL study. Int. J. Epidemiol. 2010;39:1170–1178. doi: 10.1093/ije/dyp217. [DOI] [PubMed] [Google Scholar]
- 28.Feinleib M., Kannel W.B., Garrison R.J., McNamara P.M., Castelli W.P. The Framingham Offspring Study. Design and preliminary data. Prev. Med. 1975;4:518–525. doi: 10.1016/0091-7435(75)90037-7. [DOI] [PubMed] [Google Scholar]
- 29.Cannon C.P., Curtis S.P., FitzGerald G.A., Krum H., Kaur A., Bolognese J.A., Reicin A.S., Bombardier C., Weinblatt M.E., van der Heijde D., MEDAL Steering Committee Cardiovascular outcomes with etoricoxib and diclofenac in patients with osteoarthritis and rheumatoid arthritis in the Multinational Etoricoxib and Diclofenac Arthritis Long-term (MEDAL) programme: a randomised comparison. Lancet. 2006;368:1771–1781. doi: 10.1016/S0140-6736(06)69666-9. [DOI] [PubMed] [Google Scholar]
- 30.Bild D.E., Bluemke D.A., Burke G.L., Detrano R., Diez Roux A.V., Folsom A.R., Greenland P., Jacob D.R., Jr., Kronmal R., Liu K. Multi-ethnic study of atherosclerosis: objectives and design. Am. J. Epidemiol. 2002;156:871–881. doi: 10.1093/aje/kwf113. [DOI] [PubMed] [Google Scholar]
- 31.Anderson G., Cummings S., Freedman L., Furberg C., Henderson M., Johnson S., Kuller L., Manson J., Oberman A., Prentice R., The Women’s Health Initiative Study Group Design of the Women’s Health Initiative clinical trial and observational study. Control. Clin. Trials. 1998;19:61–109. doi: 10.1016/s0197-2456(97)00078-0. [DOI] [PubMed] [Google Scholar]
- 32.Musunuru K., Lettre G., Young T., Farlow D.N., Pirruccello J.P., Ejebe K.G., Keating B.J., Yang Q., Chen M.H., Lapchyk N., NHLBI Candidate Gene Association Resource Candidate gene association resource (CARe): design, methods, and proof of concept. Circ Cardiovasc Genet. 2010;3:267–275. doi: 10.1161/CIRCGENETICS.109.882696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burgess S., Thompson S.G., CRP CHD Genetics Collaboration Avoiding bias from weak instruments in Mendelian randomization studies. Int. J. Epidemiol. 2011;40:755–764. doi: 10.1093/ije/dyr036. [DOI] [PubMed] [Google Scholar]
- 34.Harbord R.M., Higgins J.P.T. Meta-regression in Stata. Stata J. 2008;8:493–519. [Google Scholar]
- 35.Higgins, J.P., and Green, S. (2011). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0. The Cochrane Collaboration, http://www.cochrane-handbook.org.
- 36.Nichols A. Causal inference with observational data. Stata J. 2007;7:507–541. [Google Scholar]
- 37.Palmer T.M., Sterne J.A., Harbord R.M., Lawlor D.A., Sheehan N.A., Meng S., Granell R., Smith G.D., Didelez V. Instrumental variable estimation of causal risk ratios and causal odds ratios in Mendelian randomization analyses. Am. J. Epidemiol. 2011;173:1392–1403. doi: 10.1093/aje/kwr026. [DOI] [PubMed] [Google Scholar]
- 38.Hindorff L.A., Sethupathy P., Junkins H.A., Ramos E.M., Mehta J.P., Collins F.S., Manolio T.A. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. USA. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Belalcazar L.M., Haffner S.M., Lang W., Hoogeveen R.C., Rushing J., Schwenke D.C., Tracy R.P., Pi-Sunyer F.X., Kriska A.M., Ballantyne C.M., Look AHEAD (Action for Health in Diabetes) Research Group Lifestyle intervention and/or statins for the reduction of C-reactive protein in type 2 diabetes: from the look AHEAD study. Obesity (Silver Spring) 2013;21:944–950. doi: 10.1002/oby.20431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fall T., Hägg S., Mägi R., Ploner A., Fischer K., Horikoshi M., Sarin A.P., Thorleifsson G., Ladenvall C., Kals M., European Network for Genetic and Genomic Epidemiology (ENGAGE) consortium The role of adiposity in cardiometabolic traits: a mendelian randomization analysis. PLoS Med. 2013;10:e1001474. doi: 10.1371/journal.pmed.1001474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harris R., Bradburn M., Deeks J., Harbord R., Altman D., Steichen T., Sterne J.A. Boston College Department of Economics; Chestnut Hill: 2006. METAN: Stata module for fixed and random effects meta-analysis. [Google Scholar]
- 42.Higgins J.P., Thompson S.G., Deeks J.J., Altman D.G. Measuring inconsistency in meta-analyses. BMJ. 2003;327:557–560. doi: 10.1136/bmj.327.7414.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Welsh P., Polisecki E., Robertson M., Jahn S., Buckley B.M., de Craen A.J., Ford I., Jukema J.W., Macfarlane P.W., Packard C.J. Unraveling the directional link between adiposity and inflammation: a bidirectional Mendelian randomization approach. J. Clin. Endocrinol. Metab. 2010;95:93–99. doi: 10.1210/jc.2009-1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Timpson N.J., Harbord R., Davey Smith G., Zacho J., Tybjaerg-Hansen A., Nordestgaard B.G. Does greater adiposity increase blood pressure and hypertension risk?: Mendelian randomization using the FTO/MC4R genotype. Hypertension. 2009;54:84–90. doi: 10.1161/HYPERTENSIONAHA.109.130005. [DOI] [PubMed] [Google Scholar]
- 45.Kivimäki M., Smith G.D., Timpson N.J., Lawlor D.A., Batty G.D., Kähönen M., Juonala M., Rönnemaa T., Viikari J.S., Lehtimäki T., Raitakari O.T. Lifetime body mass index and later atherosclerosis risk in young adults: examining causal links using Mendelian randomization in the Cardiovascular Risk in Young Finns study. Eur. Heart J. 2008;29:2552–2560. doi: 10.1093/eurheartj/ehn252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Timpson N.J., Nordestgaard B.G., Harbord R.M., Zacho J., Frayling T.M., Tybjærg-Hansen A., Smith G.D. C-reactive protein levels and body mass index: elucidating direction of causation through reciprocal Mendelian randomization. Int J Obes (Lond) 2011;35:300–308. doi: 10.1038/ijo.2010.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Freathy R.M., Timpson N.J., Lawlor D.A., Pouta A., Ben-Shlomo Y., Ruokonen A., Ebrahim S., Shields B., Zeggini E., Weedon M.N. Common variation in the FTO gene alters diabetes-related metabolic traits to the extent expected given its effect on BMI. Diabetes. 2008;57:1419–1426. doi: 10.2337/db07-1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nordestgaard B.G., Palmer T.M., Benn M., Zacho J., Tybjaerg-Hansen A., Davey Smith G., Timpson N.J. The effect of elevated body mass index on ischemic heart disease risk: causal estimates from a Mendelian randomisation approach. PLoS Med. 2012;9:e1001212. doi: 10.1371/journal.pmed.1001212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Palmer T.M., Lawlor D.A., Harbord R.M., Sheehan N.A., Tobias J.H., Timpson N.J., Davey Smith G., Sterne J.A. Using multiple genetic variants as instrumental variables for modifiable risk factors. Stat. Methods Med. Res. 2012;21:223–242. doi: 10.1177/0962280210394459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Knowler W.C., Barrett-Connor E., Fowler S.E., Hamman R.F., Lachin J.M., Walker E.A., Nathan D.M., Diabetes Prevention Program Research Group Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N. Engl. J. Med. 2002;346:393–403. doi: 10.1056/NEJMoa012512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Knowler W.C., Fowler S.E., Hamman R.F., Christophi C.A., Hoffman H.J., Brenneman A.T., Brown-Friday J.O., Goldberg R., Venditti E., Nathan D.M., Diabetes Prevention Program Research Group 10-year follow-up of diabetes incidence and weight loss in the Diabetes Prevention Program Outcomes Study. Lancet. 2009;374:1677–1686. doi: 10.1016/S0140-6736(09)61457-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Allison D.B., Faith M.S., Heo M., Kotler D.P. Hypothesis concerning the U-shaped relation between body mass index and mortality. Am. J. Epidemiol. 1997;146:339–349. doi: 10.1093/oxfordjournals.aje.a009275. [DOI] [PubMed] [Google Scholar]
- 53.Law M.R., Morris J.K., Wald N.J. Use of blood pressure lowering drugs in the prevention of cardiovascular disease: meta-analysis of 147 randomised trials in the context of expectations from prospective epidemiological studies. BMJ. 2009;338:b1665. doi: 10.1136/bmj.b1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pierce B.L., VanderWeele T.J. The effect of non-differential measurement error on bias, precision and power in Mendelian randomization studies. Int. J. Epidemiol. 2012;41:1383–1393. doi: 10.1093/ije/dys141. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.