

Abstract
Thiazole/oxazole-modified microcins (TOMMs) comprise a structurally diverse family of natural products with varied bioactivities linked by the presence of posttranslationally installed thiazol(in)e and oxazol(in)e heterocycles. The detailed investigation of the TOMM biosynthetic enzymes from Bacillus sp. Al Hakam (Balh) has provided significant insight into heterocycle biosynthesis. Thiazoles and oxazoles are installed by the successive action of an ATP-dependent cyclodehydratase (C- and D-protein) and a FMN-dependent dehydrogenase (B-protein), which are responsible for azoline formation and azoline oxidation, respectively. Although several studies have focused on the mechanism of azoline formation, many details regarding the role of the dehydrogenase (B-protein) in overall substrate processing remain unknown. In this work, we evaluated the involvement of the dehydrogenase in determining the order of ring formation, as well as the promiscuity of the Balh and microcin B17 cyclodehydratases to accept a panel of noncognate dehydrogenases. In support of the observed promiscuity, a fluorescence polarization assay was utilized to measure binding of the dehydrogenase to the cyclodehydratase using the intrinsic fluorescence of the FMN cofactor. Ultimately, the noncognate dehydrogenases were shown to possess cyclodehydratase-independent activity. A previous study identified a conserved Lys-Tyr motif to be important for dehydrogenase activity. Using the tools developed in this study, the Lys-Tyr motif was shown to not alter complex formation with the cyclodehydratase nor the reduction potential. Taken with the known crystal structure of a homolog, our data suggest that the Lys-Tyr motif is of catalytic importance. Overall, this study provides a greater level of insight into the complex orchestration of enzymatic activity during TOMM biosynthesis.
TOMMs represent a subset of the larger ribosomally synthesized and post-translationally modified peptide class of natural products.1 Characterized TOMMs feature a diverse range of biological activities including DNA gyrase inhibitors, translation inhibitors, and hemolytic toxins.1–5 The defining feature of TOMMs are the presence of thiazol(in)e and oxazol(in)e heterocycles derived from cysteine, serine, and threonine residues on a ribosomally produced precursor peptide.6 Thiazole and oxazole biosynthesis occurs over two steps, the first being an ATP-dependent cyclodehydration to afford azoline heterocycles, which is catalyzed by the collective efforts of the C- and D-proteins (cyclodehydratase) encoded in TOMM biosynthetic gene clusters (Figure 1).7–10 Two-electron oxidation of the azoline by a FMN-dependent dehydrogenase (B-protein) affords the azole heterocycle.11 In addition to these core modifications, many TOMMs contain a variety of other post-translational modifications catalyzed by ancillary enzymes found in the gene cluster.6, 12–14
Figure 1.
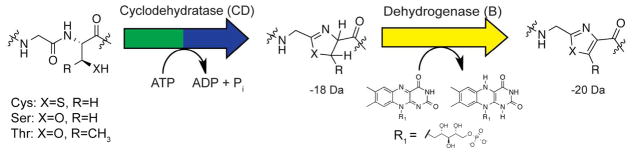
Synthesis of thiazole and oxazole heterocycles occurs over two distinct steps. First, the cyclodehydratase (C- and D- proteins) cyclizes cysteine, serine, or threonine residue into a thiazoline or oxazoline heterocycle through an ATP-dependent mechanism. Subsequently, a FMN-dependent dehydrogenase (B-protein) catalyzes the 2-electron oxidation to the azole heterocycle, resulting in a 20 Da mass loss from the unmodified amino acid.
Early studies on microcin B17 biosynthesis laid a foundation for understanding the mechanistic underpinnings of thiazole and oxazole installation.10, 11, 15 While recent work has delved into the mechanistic enzymology and substrate processing details of the Balh and cyanobactin cyclodehydratases, relatively little is known regarding any potential role played by the dehydrogenase in orchestrating substrate processing.7–9, 16–18 Upon its initial heterologous expression and co-purification with a visibly yellow FMN cofactor, the B-protein was postulated to oxidize azolines to the corresponding azole.11, 15 In microcin B17 biosynthesis, the dehydrogenase (McbC, whose letter designation derives from its position within the gene cluster, not its function) formed a complex with the cyclodehydratase (McbB and McbD) and proved to be an essential component for peptide processing.15 In contrast, the dehydrogenase has been shown to be dispensable for azoline formation with the Balh and cyanobactin cyclodehydratases.7, 17, 18 Using both native and artificial substrates, it was shown that the Balh cyclodehydratase (BalhC/D) cyclized particular cysteines, serines and threonines (BalhA1, 5 cysteines; BalhA2, 3 cysteines and 1 threonine).16, 17 However, the noncognate dehydrogenase from a highly similar biosynthetic gene cluster in Bacillus cereus 172560W, BcerB (78% identity/94% similarity to BalhB, which purifies with minimal FMN cofactor), was only capable of oxidizing thiazolines.17 The chemoselectivity for thiazolines was reminiscent of the dehydrogenase involved in patellamide biosynthesis, which oxidizes two thiazolines while leaving two oxazolines intact.19 Another unique feature of the Balh TOMM was revealed upon determining the order of thiazole biosynthesis; processing by the BcerB/BalhC/BalhD complex occurred in an overall C- to N-terminal direction for the two substrates, BalhA1 and BalhA2 (Figure S1). While this processing direction was unusual, it remained unclear if the cyclodehydratase or dehydrogenase governed the order of ring formation.17, 20
Bioinformatic analyses have now identified over 1000 TOMM gene clusters, which are found by conducting BLAST searches using the C- or D-proteins as the query sequence. Owing to the sequence similarity of the TOMM dehydrogenase to other non-TOMM dehydrogenases, BLAST searches using the B-protein as the query sequence to identify new TOMM clusters suffer from a high false-positive rate. Frequently, TOMM clusters lack a dehydrogenase altogether (e.g. trunkamide21) and such products contain exclusively azoline heterocycles, not azoles. A rare exception to this rule is illustrated by bottromycin, whose biosynthetic gene cluster lacks any recognizable dehydrogenase.22–25 Instead, it is postulated that the oxidation of the sole thiazoline to the thiazole occurs via an oxidative decarboxylation, which is plausible given that the thiazole derives from the most C-terminal cysteine of the core region of the peptide (Figure S2).22, 23, 25 TOMM dehydrogenases belong to the nitroreductase superfamily (PFAM PF00881), which also includes FMN-dependent dehydrogenases involved in polyketide synthesis among other processes.26, 27 TOMM dehydrogenases can be found as a single domain protein or as a fusion to another protein involved in peptide maturation (e.g. PatG, involved in patellamide biosynthesis).28, 29
The ability of the Balh TOMM synthetase to exhibit cyclodehydratase and dehydrogenase activity independent of each other afforded the unique opportunity to explore the finer details of dehydrogenase activity. By decoupling cyclodehydratase and dehydrogenase activity, we were positioned to address long-standing questions regarding the individual roles of the cyclodehydratase and dehydrogenase in the order of substrate processing. Furthermore, the tolerance of noncognate dehydrogenases was explored in detail, along with an investigation into the chemoselectivity of the dehydrogenase. Our study further implicates a previously identified Lys-Tyr motif, conserved among TOMM dehydrogenases, in catalysis.
Experimental Procedures
General Methods
Unless otherwise specified, all chemicals were purchased from Fisher Scientific or Sigma-Aldrich. Oligonucleotides were purchased from Integrated DNA Technologies. Restriction enzymes were purchased from New England BioLabs (NEB). dNTPs were purchased from Genscript. PfuTurbo DNA polymerase was purchased from Agilent. Sequencing grade trypsin was from Promega. DNA sequencing was performed by the Carver Biotechnology Center (University of Illinois at Urbana-Champaign) or ACGT, Inc.
Molecular Biology Techniques
The plasmids containing the Mcb and Balh/Bcer proteins have been described previously.15, 17 All other proteins were cloned into a modified pET28b plasmid containing MBP, followed by a thrombin and TEV protease cleavage sites, as described previously.3 Briefly, the dehydrogenases were amplified from genomic DNA using cloned Pfu DNA polymerase and the relevant primers (Table S1). The amplicons were then PCR purified prior to a double restriction enzyme digest using BamHI and NotI. After gel purification of the amplicons, along with a similarly digested pET28b-MBP plasmid, the DNAs were ligated with T4 DNA Ligase (NEB). Site-directed mutagenesis was conducted using the Quikchange method (Agilent).
Overexpression and Purification of Fusion Proteins
All proteins were expressed and purified as fusions to maltose binding protein (MBP) as described previously.9, 17 Proteolytical removal of MBP from was also performed as per previous reports.9, 17
General Synthetase Reactions
For Balh synthetase reactions, the precursor peptide (20–100 μM), B-protein (0 or 5–50 μM), C-protein (1–10 μM), and D-protein (1–10 μM) were combined in synthetase buffer (50 mM Tris, pH 7.4, 125 mM NaCl, 20 mM MgCl2, 2 mM ATP, and 10 mM DTT) with concurrent TEV protease treatment to remove the MBP fusion partner (1 μg protease: 50 μg protein). Reactions were allowed to proceed for 16 h at 25 °C. In reactions containing the B-protein, FMN (50 μM) was added to compensate for incomplete or inconsistent FMN loading during heterologous expression and purification. Precursor peptides containing only azoline heterocycles were generated from reactions that omitted the dehydrogenase. Synthetase reactions with the microcin B17 synthetase proteisn were conducted in a similar fashion as in the case of Balh, except those reactions had 20 μM McbA, 20 μM dehydrogenase, 5 μM McbB, and 5 μM McbD.
Azoline Localization
Synthetase reactions lacking the B-protein (100 μM BalhA1, 5 μM BalhC and 5 μM BalhD) were quenched after 30, 150, or 210 min by the addition of trypsin (1 μg trypsin to 25 μg total protein) for 1 h at 30 °C. Subsequently, iodoacetamide was added to a final concentration of 20 mM, and the labeling of uncyclized Cys residues was allowed to proceed for at least 30 min at 25 °C with minimal exposure to light. The samples were then acidified using formic acid (6% v/v) and the azolines localized via LC-MS/MS as described below.
HPLC MS/MS
All reverse-phase LC-FTMS was performed using an Agilent 1200 HPLC system with an autosampler connected directly to a ThermoFisher Scientific LTQ-FT hybrid linear ion trap, operating at 11 T. The mass spectrometer was calibrated weekly following the manufacturer’s protocol. Trypsin digested samples were separated using a 1 × 150 mm Jupiter C18 column (300 Å, 5 μM, Phenomenex). After a full scan detected in the FTMS, the MS/MS parent ions were manually selected and ion detection was carried out in the ion-trap mass spectrometer. The following parameters were used: isolation width, 3 m/z; normalized collision energy, 35; activation q value, 0.25; activation time, 30 ms. Data analysis was conducted using the Qualbrowser application of Xcalibur (Thermo-Fisher Scientific).
Determination of the Order of Azole Ring Formation
Generation of the penta-thiazoline BalhA1 (BalhA1-5Tzn) was accomplished using synthetase reactions lacking the B-proteins (50 μM BalhA1, 5 μM BalhC, 5 μM BalhD, and 1 μg TEV protease: 25 μg total protein). Upon complete formation of BalhA1-5Tzn (as judged by MALDI-TOF-MS), BcerB (5 μM) and FMN (50 μM) were then added to the reaction and allowed to proceed for either 4 or 45 minutes. Reactions were quenched by the addition of trypsin (1 μg trypsin: 25 μg total protein) for 1 h at 30 °C and subsequently acidified using formic acid (6% v/v) prior to HPLC MS/MS.
MALDI-TOF MS Analysis
All samples for MALDI-TOF-MS were desalted and concentrated by C18 ZipTip (Millipore) according to the manufacturer’s protocol. Samples were eluted in a saturated solution of CHCA matrix in 50:50 H2O: acetonitrile with 0.1% (v/v) trifluoroacetic acid. After the samples were allowed to dry under ambient conditions, they were analyzed using a Bruker Daltonics UltrafleXtreme MALDI-TOF/TOF using positive reflector mode. The instrument was calibrated using a peptide calibration kit (AB SCIEX). Data were analyzed using FlexAnalysis.
Fluorescence Polarization Assay for Dehydrogenase-Cyclodehydratase Complexation
Fluorescence polarization was conducted using a FilterMax F5 Multi-Mode Microplate Reader (Molecular Devices). The dehydrogenase (1 μM) and varying concentration of the C- and D-proteins were added to a 384-well black polystyrene microplate (Corning) and allowed to equilibrate for 30 min prior to measurement. For all samples, the autofluorescence of the C- and D-proteins were used for background correction. A sigmoidal dose-response curve fitting was conducted using OriginPro9.
Measurement of TOMM Dehydrogenase Reduction Potentials
The reduction potential for various TOMM dehydrogenases were determined as described by Massey.30 The dehydrogenase (300 μM BcerB and 100 μM other dehydrogenases), benzyl viologen (2 μM) and phenosafranine (10 μM) were added to a 10 mL round bottom flask and the final volume was brought to 2 mL using protein storage buffer [50 mM HEPES pH 7.5, 0.3 M NaCl, 2.5 % glycerol (v/v)]. The protein solution was degassed using a Schlenk line and subsequently introduced into an anaerobic chamber where the protein was allowed to equilibrate for 30 minutes while stirring. UV/Vis spectra were acquired using an Agilent 8453 spectrometer (Agilent Technologies). Sodium dithionite (1–5 μL of 10 mM) was employed to chemically reduce FMN and phenosafranine. After sodium dithionite addition, the solution was stirred for 5 minutes prior to recording another UV/Vis spectrum. This process was repeated until both FMN and phenosafranine were fully reduced. Using the absorbance measurements of FMN (440–450 nm) and phenosafranine (520 nm), a plot of log(ox/red) phenosafranin versus log(ox/red) dehydrogenase was used to calculate the reduction potential of the dehydrogenases.
Results
Order of Azoline and Azole Ring Formation
Azole formation by the Balh TOMM synthetase proceeds in an overall C- to N-terminal direction.17 An unresolved question was whether this processing direction was governed by the cyclodehydratase, the dehydrogenase, or by their collaboration. Due to chemical instability, routine LC-MS/MS cannot be employed to localize peptidic azolines, as such heterocycles rapidly hydrolyze and revert to the unmodified amino acid.31, 32 As an alternative approach, any unprocessed cysteines could be labeled using iodoacetamide, which would then be distinguishable from processed cysteines by LC-MS/MS.20, 33 To generate azoline intermediates, the BalhA1 precursor peptide was treated with the BalhC/D cyclodehydratase (dehydrogenase omitted). After a specified time, the reactions were quenched by the addition of trypsin and the unmodified cysteines were labeled with iodoacetamide. Subsequent acidification with formic acid hydrolyzed all thiazolines to cysteine. The modified and unmodified cysteines were then identified using LC-MS/MS (Figure 2). By this method, residues that were converted to thiazolines would be detected as an unmodified cysteine, while the residues labeled by iodoacetamide would indicate cysteines that did not undergo cyclodehydration. The order of azoline formation precisely followed the overall order of azole formation previously determined when the substrate was treated with the full consortium of TOMM synthetase proteins (BCD together).17 Irrespective of the presence of the dehydrogenase, Cys40 was cyclized first, followed by Cys31 and Cys34 in equal amounts, Cys45, and finally, Cys28 (Figure 3).
Figure 2.

Iodoacetamide labeling for azoline localization. In an ordered fashion, the cyclodehydratase installs azoline heterocycles onto the precursor peptide. Cysteines that have not undergone cyclodehydration were labeled with iodoacetamide. Acid-catalyzed hydrolysis reverts thiazolines back to cysteines. Localizing the unmodified and acetamide-labeled cysteines allowed the order of processing to be determined.
Figure 3.
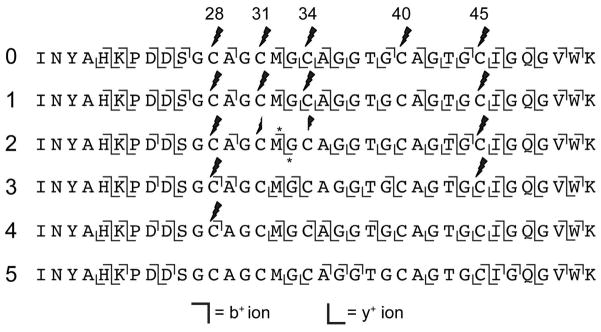
MS/MS fragmentation of BalhA1 azoline intermediates. BalhA1 reactions with BalhC/D to produce thiazoline-containing intermediates were treated with trypsin and iodoacetamide to identify unreacted cysteines. Subsequent addition of formic acid returned all thiazolines back to cysteine, at which point the samples were subjected to LC-FTMS/MS. The fragment ions observed for various ring intermediates are denoted by the b+ and y+ ion symbols. The leftmost numbers indicate ring state. Across the top, cysteines are numbered based on the sequence of full length BalhA1. Lightning bolts signify locations labeled with iodoacetamide. Asterisks denote ions containing a mixed population of two peptides.
The ability to decouple cyclodehydratase and dehydrogenase activity not only allowed the investigation of azoline order but also permitted the dissection of azole order of the dehydrogenase, BcerB. Upon adding BcerB to the penta-thiazoline of BalhA1 (BalhA1-5Tzn, pre-formed by BalhC/D), the order of oxidations en route to BalhA1-5Tzn could be monitored as in previous studies using LC-MS/MS17. Intriguingly, the processing order failed to follow azoline biogenesis. The first position to be oxidized was Tzn34, followed by an outward series of modifications simultaneously radiating towards the N- and C-termini (Figure 4).
Figure 4.
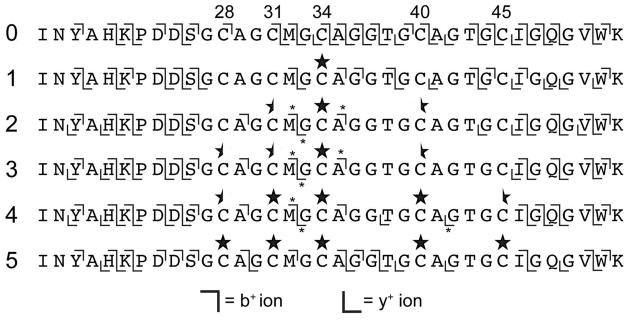
MS/MS fragmentation of BalhA1 azole intermediates. Synthetase reactions containing BalhA1-5Tzn treated with BcerB were quenched by the addition of trypsin and subjected to LC-FTMS/MS. The fragment ions observed for various ring intermediates are denoted by the b+ and y+ ion symbols. The leftmost numbers indicate ring state. Across the top, cysteines are numbered based on the sequence of full length BalhA1. Stars signify thiazole locations, while a half star signifies that there are ions indicative of a thiazole as well as a free cysteine at that position. Asterisks denote ions containing a mixed population of two peptides.
Permissive Nature of TOMM Dehydrogenases
As we have noted in our earlier reports,9, 17 the BalhB dehydrogenase purifies with minimal bound FMN cofactor (predominately apo), thus the above studies were conducted using a noncognate dehydrogenase from Bacillus cereus 172560W. Dubbed BcerB, this protein co-purifies with near stoichiometric amounts of FMN and shares significant sequence similarity with BalhB (78% identity/94% similarity).9, 17 It has also been shown with the TOMM from Clostridium botulinum that a noncognate dehydrogenase, SagB from Streptococcus pyogenes (41% identity/80% similarity to ClosB), can successfully substitute for the cognate dehydrogenase, ClosB, during in vitro reconstitution of clostridiolysin S.34 In both of the cases mentioned above, the substituted dehydrogenase was closely related to the cognate dehydrogenase. To test the ability of more distant dehydrogenases to successfully complement thiazole/oxazole biosynthesis, a series of diverse dehydrogenases were cloned, expressed, purified, and tested for activity using the Balh and Mcb TOMM cyclodehydratases (Table S2, Figures 5, 6 and S1). All noncognate dehydrogenases formed thiazoles in conjunction with the Balh cyclodehydratase (Figure 5). The order of azole formation was also determined for reactions containing the TOMM dehydrogeneases from Bacillus amyloliquefaciens FZB42 and Clavibacter michiganensis subsp. sepedonicus (BamB and CmsB, respectively). Both dehydrogenases followed a highly similar pathway as BcerB when processing BalhA1-5Tzn (Figures 4 and S3). In stark contrast to the flexible nature of the Balh enzymes, only reactions with the cognate dehydrogenase (McbC) produced heterocycles when testing the Mcb cyclodehydratase (McbB/D). The observed dehydrogenase intolerance of the Mcb cyclodehydratase suggests that the noncognate dehydrogenases tested are not capable of forming a product trimeric complex with McbB/D, which has been shown to be essential for thiazole/oxazole biosynthesis.15
Figure 5.

The Balh cyclodehydratase tolerates noncognate dehydrogenases. The reactions containing the indicated reagents were allowed to react for 16 h at 25 °C and then analyzed using MALDI-TOF-MS after formic acid hydrolysis of thiazoline heterocycles. The numbers at the top indicate the number of azole heterocycles. The asterisk denotes a laser-induced artifact.
Figure 6.

Dehydrogenase intolerance of the Mcb cyclodehydratase. The reactions containing the indicated reagents were allowed to react for 16 h at 25 °C and then analyzed using MALDI-TOF-MS after formic acid hydrolysis of azoline heterocycles. The numbers at the top indicate the number of azole heterocycles. The asterisk denotes a laser-induced artifact.
Monitoring TOMM Synthetase Complex Formation by Fluorescence Polarization
Because noncognate dehydrogenases successfully oxidized BalhA1 in the presence of BalhC/D, while all the dehydrogenases (except McbC) tested failed to complement McbB/D, we hypothesized that the dehydrogenases might be able to form a complex with BalhC/D but not with McbB/D. To test this, we utilized a fluorescence polarization-based binding assay that capitalizes on the intrinsic fluorescence of the FMN cofactor. A similar assay has been employed to study the interaction of luciferase and lumazine.35, 36 An increase in the fluorescence polarization would occur upon binding of the dehydrogenase to the cyclodehydratase complex. Fluorescence polarization was measured using a fixed concentration of BcerB with increasing concentrations of BalhC/D (Figure 7). As the concentrations of BalhC/D were increased, significant fluorescence polarization was observed and the apparent Kd of 1.7 ± 0.2 μM aligned with the previously reported Kcomplex measured for McbC to McbB/D (1.8 ± 0.8 μM).15 All other non-cognate dehydrogenases were also subjected to this binding assay, yet no binding was observed (data not shown).
Figure 7.

Fluorescence polarization of BcerB and the Balh cyclodehydratase. The fluorescence polarization of the FMN cofactor of BcerB (1 μM) was measured with varying concentrations of the Balh cyclodehydratase. A dose-response fit of the data provided the apparent Kd. The error reported for each individual data point corresponds to the standard deviation of three technical replicates, while the error reported for the apparent Kd derives from the error associated with curve fitting.
Cyclodehydratase-Independent Dehydrogenation
To reconcile the dehydrogenase activity observed in the absence of detectable binding of noncognate dehydrogenases (with the exception of BcerB) to BalhC/D, the ability of such dehydrogenases to act upon BalhA1-5Tzn in a cyclodehydratase-independent fashion was measured. Initially, the requirements for efficient dehydrogenation were monitored using BcerB. After forming BalhA1-5Tzn with BalhC/D and concomitant TEV cleavage, acetonitrile was added to the reaction mixture to precipitate large proteins while leaving BalhA1-5Tzn in solution. After centrifugation, decantation, and evaporation of the acetonitrile supernatant by vacuum centrifugation, various combinations of BcerB, BalhC and BalhD were added (Figure S4). As a control, addition of BalhA1 to these samples did not result in any detectable enzymatic processing, indicating that the acetonitrile-based removal of BalhC/D was adequate and any residual enzymes were inactive (Figure S4). To deconvolute the mixture of heterocycles, formic acid was added to selectively hydrolyze the thiazolines, leaving thiazoles intact (Figure 8).16, 37 From these experiments, it became clear that dehydrogenation efficiently occurred only when the dehydrogenase was present with both cyclodehydratase proteins (i.e. BalhC/D), although thiazoline oxidation intermediates were observed when individual components of the cyclodehydratase were supplied separately (Figure 8). The cyclodehydratase-independent activity was then measured using the entire panel of noncognate dehydrogenases (Figure S5). Contrary to BcerB, all other noncognate dehydrogenases exhibited roughly the same processing efficiency, regardless of the presence of BalhC/D (Figure 5 and S5).
Figure 8.

Minimal requirement for dehydrogenase activity. BalhA1-5Tzn was treated with varying combinations of synthetase components. The reactions were allowed to react for 16 h at 25 °C and then analyzed using MALDI-TOF-MS after formic acid hydrolysis of thiazoline heterocycles. The numbers above the spectra indicate the number of azole heterocycles. Asterisks denote laser-induced artifacts.
Universal Importance of the Conserved Lys-Tyr Motif in TOMM Dehydrogenases
Sequence alignment of TOMM dehydrogenases reveals a highly conserved Lys-Tyr motif (Figure S6).16 Based on Protein DataBank entry 3EO7 (a putative nitroreductase from Anabaena variabilis ATCC 29413), which is the most similar protein with a published structure (12% identity/51% similarity to McbC), the Lys-Tyr motif is located near the FMN cofactor (closest distance is 5.6 Å, Figure S7). Mutagenesis of the Lys-Tyr motif failed to alter the FMN loading, further suggesting that the Lys-Tyr motif is not directly involved in FMN binding (Figure S8). A previous study, which replaced the Lys201 and Tyr202 of McbC to Ala resulted in inactive protein during microcin B17 in vitro reconstitution reactions, suggestive of a potential catalytic role.16 McbC-K201A and -Y202A were also inactive towards BalhA1-5Tzn (Figure S9). The equivalent Lys-Tyr motifs in the TOMM dehydrogenases from the Gram-positive Bacillus cereus 172560W (BcerB) and the thermoacidophilic archaeon Sulfolobus acidocaldarius DSM 639 (SaciB) were mutated to Ala and screened for activity. BcerB-K185A exhibited reduced activity, as indicated by the formation of up to four thiazoles in an end-point essay, while BcerB-Y186A and the double mutant (K185A/Y186A) were catalytically inactive (Figure 9). Similar to McbC, the single and double mutants to the Lys-Tyr motif in SaciB abolished activity (Figure 9). Increased protein concentrations (5–50 μM dehydrogenase) and reaction times (24 h) failed to restore detectable activity for the BcerB, SaciB, and McbC mutants. In further support for a catalytic role, BcerB-K185A and -Y186A were tested for their ability to form a complex with BalhC/D. Using the FMN-based fluorescence polarization assay, BcerB-K185A/Y186A was found to bind with similar affinity as wild type BcerB (Figure S10). Taken together, our data suggest that the Lys-Tyr motif plays a role in catalysis by the dehydrogenase.
Figure 9.

Dehydrogenase activity of Lys-Tyr mutants. A. Synthetase reactions containing BcerB and the indicated mutants were conducted and allowed to react for 16 h at 25 °C. Subsequently, all azoline heterocycles were hydrolyzed using formic acid prior to MALDI-TOF-MS analysis. B. Same as in A, except reactions contained SaciB and its Lys-Tyr mutants. The numbers above the spectra indicate the number of azole heterocycles. Asterisks denote laser-induced artifacts.
Comparison of TOMM Dehydrogenase Reduction Potentials
It was shown previously that while BcerB was an efficient thiazoline oxidase, this protein was unable to convert oxazolines to oxazoles, irrespective of the presence of BalhC/D.17 A possible explanation for the lack of activity towards oxazolines could arise from a difference in reduction potential compared to other B-proteins that produce oxazoles. To measure reduction potentials, UV/Vis spectra were collected while the FMN cofactor was chemically reduced using sodium dithionite (Figure S11). Addition and subsequent monitoring of the redox indicator dye, phenosafranine, allowed calculation of the dehydrogenase reduction potentials.30 Using this method, initial studies were focused on BcerB and McbC (oxidizes four oxazolines during microcin B17 maturation), which gave identical reduction potentials (Table 1).11 The reduction potential of BamB provided further evidence that BcerB does not harbor significantly different electrochemical properties compared to other TOMM dehydrogenases and as such, does not explain the observed selectivity towards thiazolines. Similarly, an altered reduction potential of the Lys-Tyr mutants could provide an explanation for the dearth of catalytic activity observed. However, the reduction potential of McbC-Y202A was not statistically different from BcerB (Table 1), further implicating the Lys-Tyr motif in enzymatic catalysis (Figure S2).
Table 1.
Reduction potentials of TOMM dehydrogenases. The redox potential of the listed TOMM dehydrogenases were measured by chemically reducing the FMN cofactor with sodium dithionite in the presence of phenosafranine, an indicator dye, and monitoring the reduction of both by UV/Vis spectroscopy.30 The error reported is the standard deviation of three independent measurements.
| Protein | Redox Potential (MV) |
|---|---|
| BcerB | −277 ± 4 |
| McbC | −277 ± 2 |
| BamB | −280 ± 3 |
| McbC Y202A | −272 ± 1 |
Discussion
During microcin B17 biosynthesis, the full trimeric complex of McbBCD is required for thiazole/oxazole ring formation.15 Fortunately, the cyclodehydratase from Balh does not require the dehydrogenase for activity, which allowed us to examine cyclodehydration separately from dehydrogenation. While the order of thiazoline formation by BalhC/D (Figure 3) followed the previously reported order for azole formation (generated from a full BCD reaction), the order of azole formation when the dehydrogenase was presented with a penta-azoline substrate (BalhA1-5Tzn) proved drastically different. During BalhA1-5Tzn processing, BcerB first oxidized the thiazoline at Cys34 (Tzn34) and subsequently proceeded outward toward the C- and N-termini simultaneously (Figure 4). Other dehydrogenases processed the BalhA1-5Tzn substrate in a similar fashion (Figure S3). A potential explanation could be that the dehydrogenases recognize a specific motif within the precursor peptide, which based on distance constraints, orients Tzn34 closest to the active site. However, both the microcin B17 and streptolysin S TOMM dehydrogenases (McbC and SagB, respectively) were shown to have little affinity for their cognate precursor peptides.15, 38 The aberrant order of cyclodehydratase-independent dehydrogenase activity also demonstrated that while BalhA1-5Tzn was a competent intermediate, it did not follow the native biosynthetic processing order. When the full biosynthetic complex was present, azole order proceeded in an overall C- to N-terminal direction and exhibited a distributive behavior, which was determined by the detection of intermediates with varying numbers of thiazole heterocycles.17 As such, we propose a biosynthetic pathway where dehydrogenation immediately follows cyclodehydration at every modified position.
The ability of noncognate dehydrogenases to substitute for the native dehydrogenase does not appear to be universal in TOMM biosynthesis. With the Balh cyclodehydratase, every dehydrogenase tested could install thiazoles onto the BalhA1 substrate in the presence of BalhC/D (Figure 5), but only the native McbC dehydrogenase was tolerated during McbA processing (Figure 6). During in vitro reconstitution of microcin B17 biosynthesis, the three McbBCD proteins are each essential; omission of any protein abrogates all detectable enzymatic activity.15 Co-immunoprecipitation studies demonstrated that McbBCD form a heterotrimeric complex in which each component performs a unique function during azole formation.9, 11 Therefore, if a noncognate dehydrogenase fails to successfully form a productive heterotrimeric complex with McbB/D (cyclodehydratase), no heterocycle formation would be expected occur. The ability of the noncognate dehydrogenases to complement the Balh cyclodehydratase suggested a catalytically active trimeric complex was formed between the dehydrogenases and cyclodehydratase. To provide evidence for complex formation between noncognate dehydrogenases and the Balh cyclodehydratase, a fluorescence polarization assay utilizing the intrinsic fluorescence of the FMN cofactor was employed to monitor binding. Surprisingly, only BcerB, a dehydrogenase from a nearly identical biosynthetic gene cluster, in conjunction with BalhC/D produced a fluorescence polarization curve indicative of complex formation (Figure 7). The instability of McbBCD at high concentration without their MBP fusion partner precluded the monitoring of fluorescence polarization between McbC and McbB/D. The inability to detect an increase in fluorescence polarization using noncognate dehydrogenases suggested that there was cyclodehydratase-independent dehydrogenase activity.
To garner further support for the dehydrogenase activity in the absence of a cyclodehydratase, we monitored dehydrogenase activity towards BalhA1-5Tzn in the absence of BalhC/D. To prepare this substrate, synthetase reactions were initiated with BalhA1 and BalhC/D. After completion of the reaction, acetonitrile was added to precipitate large polypeptides (i.e. BalhC and BalhD) while leaving BalhA1 derivatives in solution. After removal of the acetonitrile, BalhA1-5Tzn was subjected to various enzymatic reactions. As expected, efficient dehydrogenation only occurred when all the biosynthetic enzymes were added (BCD), but a minimal amount of activity was detected when BcerB was present with either BalhC or BalhD (Figure 8). BcerB alone had no detectable activity when presented with BalhA1-5Tzn (Figures 8 and S4). However, the other TOMM dehydrogenases were able to catalyze azole formation in the absence of BalhC/D (Figure S5). This result provided an explanation for the inability to detect binding between the noncognate dehydrogenases (with the exception of BcerB) and BalhC/D while still being able to catalyze azole formation (Figure S5). It appears that all the dehydrogenases tested, excluding BcerB, exhibit nonspecific azoline oxidation.
A previous study identified a conserved Lys-Tyr motif (Figure S6) that abolished the dehydrogenase activity of McbC.16 In the process of evaluating the biochemical determinants of azoline oxidation, the importance of the Lys-Tyr motif was further established in two other distantly related TOMM dehydrogenases (BcerB and SaciB). Mutation of either the Lys or the Tyr within the SaciB motif eradicated enzymatic activity in our assays (Figure 9). In BcerB, the K185A mutation had reduced activity as indicated by the detection of predominantly a three thiazole heterocycle species of BalhA1. However, BcerB-Y186A and -K185A/Y186A were devoid of activity (Figure 9). In all cases, mutation of the Lys-Tyr motif did not alter the FMN content of the dehydrogenase (Figure S8). Using the fluorescence polarization assay, BcerB-K185A/Y186A was found to form a complex with BalhC/D (Figure S10), indicating that the Lys-Tyr motif did not play a critical role in the protein-protein interaction. Another possible explanation for the loss of activity was an altered reduction potential; however, the potentials of McbC-Y202A and BcerB were not significantly different (p > 0.1). Based on the crystal structure of the closest homolog (PDB: 3EO7), it appeared the Lys-Tyr motif was poised to participate in catalysis, given its proximity to the active site (Figure S2 and S7). Taken together with our fluorescence polarization assays, FMN loading, and electrochemical titrations, the loss of activity in three distinct TOMM dehydrogenases supports a catalytic role for these residues (Figure S2).16 A more detailed kinetic analysis of the Lys-Tyr mutants will be necessary to unequivocally establish a catalytic role for these residues.
Interestingly, the Balh cyclodehydratase can cyclize cysteine, serine, and threonine residues to their respective azoline heterocycles16, 17, yet BcerB lacks the capability to convert (methyl)oxazolines to (methyl)azoles. Owing to the greater aromatic character of a thiazole relative to an oxazole, the oxidation of a thiazoline is more thermodynamically favorable than oxazoline oxidation.39 Therefore, one way to chemoselectively oxidize thiazolines would be to finely tune the electrochemical potential of the dehydrogenase. However, our measurement of TOMM dehydrogenase reduction potentials fails to explain the observed chemoselectivity of BcerB, as the values measured were not significantly different than dehydrogenases known to form oxazoles (Table 1). The observed chemoselectivity must then derive from the ability of BcerB to discriminate between thiazolines and oxazoline by some other mechanism. A possible explanation is a difference in pKa between the α-hydrogens of a thiazoline versus an oxazoline. Base-catalyzed removal of the α-hydrogen may be rate determining in dehydrogenation reactions (Figure S2).11
In conclusion, the ability to discriminate between cyclodehydratase and dehydrogenase function afforded the opportunity to study the order of both azoline and azole ring formation, which, in the case of Balh, was dictated by the cyclodehydratase. Testing the limit of noncognate dehydrogenase swapping with the Balh cyclodehydratase demonstrated that most noncognate dehydrogenases do not bind the cyclodehydratase and nonspecifically oxidize the azoline-containing substrate. Furthermore, a conserved Lys-Tyr motif within the dehydrogenase was found to be critical for activity in distantly related TOMM dehydrogenases, which is postulated to play a role in acid/base catalysis. Through measurement of the reduction potential of a panel of dehydrogenases, the chemoselectivity of BcerB for thiazoline heterocycles does not result from a tuned redox potential and instead discriminates thiazoline from oxazoline at the substrate level.
Supplementary Material
Acknowledgments
We are grateful for the gift of Clavibacter michiganensis subsp. sepedonicus from Carol Ishimaru (University of Minnesota). We thank Kyle Dunbar, Courtney Cox, Robbie Dowen and Joyce Limm for technical assistance. Members of the Mitchell Lab carried out critical review of this manuscript. This work was supported by the US National Institutes of Health (NIH) (1R01 GM097142 to D.A.M.). X.L. was in part supported by a fellowship from the Department of Chemistry at the University of Illinois at Urbana-Champaign. The Bruker UltrafleXtreme MALDI TOF/TOF mass spectrometer was purchased in part with a grant from the National Center for Research Resources, National Institutes of Health (S10 RR027109 A).
Abbreviations
- ATP
adenosine-5′-triphosphate
- Balh
Bacillus sp. Al Hakam
- CHCA
α-cyano-4-hydroxycinammic acid
- CV
column volumes
- DTT
dithiothreitol
- FMN
flavin mononucleotide
- HEPES
2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid
- IPTG
isopropyl-β-D-thiogalactopyranoside
- LC-FTMS
liquid chromatography-Fourier transform mass spectrometry
- MALDI-TOF-MS
matrix-assisted laser desorption/ionization time-of-flight mass spectrometry
- MBP
maltose-binding protein
- TEV
tobacco etch virus
- TOMM
thiazole/oxazole-modified microcin
Footnotes
Supporting information contains additional figures described in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Arnison PG, Bibb MJ, Bierbaum G, Bowers AA, Bugni TS, Bulaj G, Camarero JA, Campopiano DJ, Challis GL, Clardy J, Cotter PD, Craik DJ, Dawson M, Dittmann E, Donadio S, Dorrestein PC, Entian KD, Fischbach MA, Garavelli JS, Goransson U, Gruber CW, Haft DH, Hemscheidt TK, Hertweck C, Hill C, Horswill AR, Jaspars M, Kelly WL, Klinman JP, Kuipers OP, Link AJ, Liu W, Marahiel MA, Mitchell DA, Moll GN, Moore BS, Muller R, Nair SK, Nes IF, Norris GE, Olivera BM, Onaka H, Patchett ML, Piel J, Reaney MJ, Rebuffat S, Ross RP, Sahl HG, Schmidt EW, Selsted ME, Severinov K, Shen B, Sivonen K, Smith L, Stein T, Sussmuth RD, Tagg JR, Tang GL, Truman AW, Vederas JC, Walsh CT, Walton JD, Wenzel SC, Willey JM, van der Donk WA. Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat Prod Rep. 2013;30:108–160. doi: 10.1039/c2np20085f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Molloy EM, Cotter PD, Hill C, Mitchell DA, Ross RP. Streptolysin S-like virulence factors: the continuing sagA. Nat Rev Microbiol. 2011;9:670–681. doi: 10.1038/nrmicro2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee SW, Mitchell DA, Markley AL, Hensler ME, Gonzalez D, Wohlrab A, Dorrestein PC, Nizet V, Dixon JE. Discovery of a widely distributed toxin biosynthetic gene cluster. Proc Natl Acad Sci USA. 2008;105:5879–5884. doi: 10.1073/pnas.0801338105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vizan JL, Hernandez-Chico C, del Castillo I, Moreno F. The peptide antibiotic microcin B17 induces double-strand cleavage of DNA mediated by E. coli DNA gyrase. EMBO J. 1991;10:467–476. doi: 10.1002/j.1460-2075.1991.tb07969.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bagley MC, Dale JW, Merritt EA, Xiong X. Thiopeptide antibiotics. Chem Rev. 2005;105:685–714. doi: 10.1021/cr0300441. [DOI] [PubMed] [Google Scholar]
- 6.Melby JO, Nard NJ, Mitchell DA. Thiazole/oxazole-modified microcins: complex natural products from ribosomal templates. Curr Opin Chem Biol. 2011;15:369–378. doi: 10.1016/j.cbpa.2011.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koehnke J, Bent AF, Zollman D, Smith K, Houssen WE, Zhu X, Mann G, Lebl T, Scharff R, Shirran S, Botting CH, Jaspars M, Schwarz-Linek U, Naismith JH. The Cyanobactin Heterocyclase Enzyme: A Processive Adenylase That Operates with a Defined Order of Reaction. Angew Chem. 2013;125:14241–14246. doi: 10.1002/anie.201306302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McIntosh JA, Schmidt EW. Marine molecular machines: heterocyclization in cyanobactin biosynthesis. Chembiochem. 2010;11:1413–1421. doi: 10.1002/cbic.201000196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dunbar KL, Melby JO, Mitchell DA. YcaO domains use ATP to activate amide backbones during peptide cyclodehydrations. Nat Chem Biol. 2012;8:569–575. doi: 10.1038/nchembio.944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Milne JC, Eliot AC, Kelleher NL, Walsh CT. ATP/GTP hydrolysis is required for oxazole and thiazole biosynthesis in the peptide antibiotic microcin B17. Biochemistry. 1998;37:13250–13261. doi: 10.1021/bi980996e. [DOI] [PubMed] [Google Scholar]
- 11.Li YM, Milne JC, Madison LL, Kolter R, Walsh CT. From peptide precursors to oxazole and thiazole-containing peptide antibiotics: microcin B17 synthase. Science. 1996;274:1188–1193. doi: 10.1126/science.274.5290.1188. [DOI] [PubMed] [Google Scholar]
- 12.McIntosh JA, Donia MS, Nair SK, Schmidt EW. Enzymatic basis of ribosomal peptide prenylation in cyanobacteria. J Am Chem Soc. 2011;133:13698–13705. doi: 10.1021/ja205458h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee J, Hao Y, Blair PM, Melby JO, Agarwal V, Burkhart BJ, Nair SK, Mitchell DA. Structural and functional insight into an unexpectedly selective N-methyltransferase involved in plantazolicin biosynthesis. Proc Natl Acad Sci USA. 2013;110:12954–12959. doi: 10.1073/pnas.1306101110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McIntosh JA, Donia MS, Schmidt EW. Ribosomal peptide natural products: bridging the ribosomal and nonribosomal worlds. Nat Prod Rep. 2009;26:537–559. doi: 10.1039/b714132g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Milne JC, Roy RS, Eliot AC, Kelleher NL, Wokhlu A, Nickels B, Walsh CT. Cofactor requirements and reconstitution of microcin B17 synthetase: a multienzyme complex that catalyzes the formation of oxazoles and thiazoles in the antibiotic microcin B17. Biochemistry. 1999;38:4768–4781. doi: 10.1021/bi982975q. [DOI] [PubMed] [Google Scholar]
- 16.Dunbar KL, Mitchell DA. Insights into the mechanism of peptide cyclodehydrations achieved through the chemoenzymatic generation of amide derivatives. J Am Chem Soc. 2013;135:8692–8701. doi: 10.1021/ja4029507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Melby JO, Dunbar KL, Trinh NQ, Mitchell DA. Selectivity, directionality, and promiscuity in peptide processing from a Bacillus sp. Al Hakam cyclodehydratase. J Am Chem Soc. 2012;134:5309–5316. doi: 10.1021/ja211675n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McIntosh JA, Donia MS, Schmidt EW. Insights into heterocyclization from two highly similar enzymes. J Am Chem Soc. 2010;132:4089–4091. doi: 10.1021/ja9107116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmidt EW, Nelson JT, Rasko DA, Sudek S, Eisen JA, Haygood MG, Ravel J. Patellamide A and C biosynthesis by a microcin-like pathway in Prochloron didemni, the cyanobacterial symbiont of Lissoclinum patella. Proc Natl Acad Sci USA. 2005;102:7315–7320. doi: 10.1073/pnas.0501424102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kelleher NL, Hendrickson CL, Walsh CT. Posttranslational heterocyclization of cysteine and serine residues in the antibiotic microcin B17: distributivity and directionality. Biochemistry. 1999;38:15623–15630. doi: 10.1021/bi9913698. [DOI] [PubMed] [Google Scholar]
- 21.Donia MS, Ravel J, Schmidt EW. A global assembly line for cyanobactins. Nat Chem Biol. 2008;4:341–343. doi: 10.1038/nchembio.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crone WJK, Leeper FJ, Truman AW. Identification and characterisation of the gene cluster for the anti-MRSA antibiotic bottromycin: expanding the biosynthetic diversity of ribosomal peptides. Chem Sci. 2012;3:3516–3521. [Google Scholar]
- 23.Gomez-Escribano JP, Song LJ, Bibb MJ, Challis GL. Posttranslational beta-methylation and macrolactamidination in the biosynthesis of the bottromycin complex of ribosomal peptide antibiotics. Chem Sci. 2012;3:3522–3525. [Google Scholar]
- 24.Hou Y, Tianero MD, Kwan JC, Wyche TP, Michel CR, Ellis GA, Vazquez-Rivera E, Braun DR, Rose WE, Schmidt EW, Bugni TS. Structure and biosynthesis of the antibiotic bottromycin D. Org Lett. 2012;14:5050–5053. doi: 10.1021/ol3022758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huo L, Rachid S, Stadler M, Wenzel SC, Muller R. Synthetic biotechnology to study and engineer ribosomal bottromycin biosynthesis. Chem Biol. 2012;19:1278–1287. doi: 10.1016/j.chembiol.2012.08.013. [DOI] [PubMed] [Google Scholar]
- 26.Sonnhammer EL, Eddy SR, Durbin R. Pfam: a comprehensive database of protein domain families based on seed alignments. Proteins: Struct Funct Bioinf. 1997;28:405–420. doi: 10.1002/(sici)1097-0134(199707)28:3<405::aid-prot10>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 27.Finking R, Marahiel MA. Biosynthesis of nonribosomal peptides. Annu Rev Microbiol. 2004;58:453–488. doi: 10.1146/annurev.micro.58.030603.123615. [DOI] [PubMed] [Google Scholar]
- 28.Agarwal V, Pierce E, McIntosh J, Schmidt EW, Nair SK. Structures of cyanobactin maturation enzymes define a family of transamidating proteases. Chem Biol. 2012;19:1411–1422. doi: 10.1016/j.chembiol.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koehnke J, Bent A, Houssen WE, Zollman D, Morawitz F, Shirran S, Vendome J, Nneoyiegbe AF, Trembleau L, Botting CH, Smith MC, Jaspars M, Naismith JH. The mechanism of patellamide macrocyclization revealed by the characterization of the PatG macrocyclase domain. Nat Struct Mol Biol. 2012;19:767–772. doi: 10.1038/nsmb.2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Massey V. A simple method for the determination of redox potentials. In: Curti B, Ronchi S, Zanetti G, editors. Flavins and Flavoproteins. Walter de Gruyter; Como, Italy: 1991. pp. 59–66. [Google Scholar]
- 31.Martin RB, Lowey S, Elson EL, Edsall JT. Hydrolysis of 2-Methyl-Delta-2-Thiazoline and Its Formation from N-Acetyl-Beta-Mercaptoethylamine - Observations on an N-S Acyl Shift. J Am Chem Soc. 1959;81:5089–5095. [Google Scholar]
- 32.Martin RB, Parcell A. Hydrolysis of 2-Methyl-Delta-Oxazoline - an Intramolecular O-N-Acetyl Transfer Reaction. J Am Chem Soc. 1961;83:4835–4838. [Google Scholar]
- 33.Lee MV, Ihnken LA, You YO, McClerren AL, van der Donk WA, Kelleher NL. Distributive and directional behavior of lantibiotic synthetases revealed by high-resolution tandem mass spectrometry. J Am Chem Soc. 2009;131:12258–12264. doi: 10.1021/ja9033507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gonzalez DJ, Lee SW, Hensler ME, Markley AL, Dahesh S, Mitchell DA, Bandeira N, Nizet V, Dixon JE, Dorrestein PC. Clostridiolysin S, a post-translationally modified biotoxin from Clostridium botulinum. J Biol Chem. 2010;285:28220–28228. doi: 10.1074/jbc.M110.118554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Petushkov VN, Gibson BG, Lee J. Properties of recombinant fluorescent proteins from Photobacterium leiognathi and their interaction with luciferase intermediates. Biochemistry. 1995;34:3300–3309. doi: 10.1021/bi00010a020. [DOI] [PubMed] [Google Scholar]
- 36.Visser AJ, Lee J. Association between lumazine protein and bacterial luciferase: direct demonstration from the decay of the lumazine emission anisotropy. Biochemistry. 1982;21:2218–2226. doi: 10.1021/bi00538a034. [DOI] [PubMed] [Google Scholar]
- 37.Deane CD, Melby JO, Molohon KJ, Susarrey AR, Mitchell DA. Engineering Unnatural Variants of Plantazolicin through Codon Reprogramming. ACS Chem Biol. 2013;8:1998–2008. doi: 10.1021/cb4003392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mitchell DA, Lee SW, Pence MA, Markley AL, Limm JD, Nizet V, Dixon JE. Structural and functional dissection of the heterocyclic peptide cytotoxin streptolysin S. J Biol Chem. 2009;284:13004–13012. doi: 10.1074/jbc.M900802200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eicher T, Hauptmann S, Speicher A. The chemistry of heterocycles: structure, reactions, syntheses, and applications. 2. Wiley-VCH; Weinheim: 2003. p. 149. completely rev., and enl. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.