

Abstract
A key stage in HIV-1 maturation towards an infectious virion requires sequential proteolytic cleavage of the Gag polyprotein leading to the formation of a conical capsid core that encloses the viral RNA genome and a small complement of proteins. The final step of this process involves severing the SP1 peptide from the CA-SP1 maturation intermediate, which triggers the condensation of the CA protein into the capsid shell. The details of the overall mechanism, including the conformation of the SP1 peptide in CA-SP1, are still under intense debate. In this report, we examine tubular assemblies of CA and the CA-SP1 maturation intermediates using Magic Angle Spinning NMR spectroscopy. At magnetic fields of 19.9 T and above, outstanding-quality 2D and 3D MAS NMR spectra were obtained for tubular CA and CA-SP1 assemblies yield, permitting resonance assignments for subsequent detailed structural characterization. Dipolar- and scalar-based correlation experiments unequivocally indicate that SP1 peptide is in a random coil conformation and mobile in the assembled CA-SP1. Analysis of two CA protein sequence variants reveals that, unexpectedly, the conformations of the SP1 tail, the functionally important CypA loop, and the loop preceding helix 8 are modulated by residue variations at distal sites. These findings provide support for the role of SP1 as a trigger of the disassembly of the immature CA capsid for its subsequent de novo reassembly into mature cores, and establish the importance of sequence-dependent conformational plasticity in CA assembly.
INTRODUCTION
Maturation of HIV-1 into an infectious virion involves sequential proteolytic cleavages of Gag polyprotein into its constituent domains (Figure 1A).1,2 The final step is the cleavage of the 14-residue SP1 peptide from the CA domain, followed by the condensation of the 231-residue CA protein into a conical capsid.3 In mature virions, the capsids are composed of ~1200 copies of the CA protein that encloses two copies of the viral RNA genome and a small complement of proteins.3,4 The capsid architecture was described using an idealized “fullerene cone” model, where 12 pentameric CA units are incorporated into the predominantly hexagonal surface lattice, to form a closed structure.5 The recent cryo-electron tomography (cryo-ET) studies of native HIV-1 cores revealed considerable variability in the number of hexameric CA units comprising the capsid, and all-atom molecular dynamics models were generated for capsids consisting of 166 or 216 CA hexamers that together with 12 pentamers assemble into a closed structure (Figure 1B).6
Figure 1.
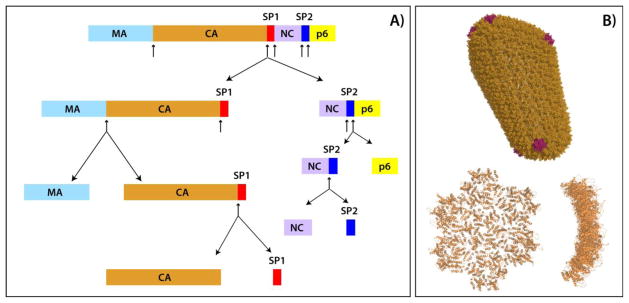
A) Domain organization of the Gag polyprotein and schematic representation of the sequential proteolytic cleavage sites in Gag during the HIV-1 virus maturation, based on in vitro cleavage rates.2 The proteolytic cleavage sites are indicated by arrows. Gag domains comprise: MA, matrix; CA, capsid, SP1, spacer peptide 1; NC, nucleocapsid; SP2, spacer peptide 2; p6, p6-gag. B) Top: All-atom molecular dynamics derived model of mature HIV-1 capsid constructed on the basis of cryo-ET studies. The capsid is comprised of 216 CA hexamers (depicted in orange) and 12 CA pentamers (depicted in magenta), PDB ID 3J3Y. Bottom: the CA hexamer of hexamers (HOH) building block, viewed from the top (left) and from the side (right), PDB ID 3J34. The structures were generated in MacPyMOL.43
Recent attention has focused on the maturation process as a potential target for a novel class of inhibitors termed maturation inhibitors.7,8 These include Bevirimat (BVM) and its derivatives, which inhibit the viral maturation by binding to the CA-SP1 cleavage site.9 Despite intense studies into the mechanisms of the maturation process and the action of maturation inhibitors, key aspects remain obscure at the molecular level. For example, the structure of the HIV-1 CA-SP1 maturation intermediate is unknown at atomic or subatomic resolution, neither in vitro nor in a cellular context. In addition, it still remains unknown how maturation inhibitors interact with SP1: neither are the interaction sites known, nor is there any information about possible conformational changes, induced in SP1 upon binding. An early solution NMR study of the SP1 peptide in the context of CACT D-SP1-NC suggested the presence of a minor α-helical conformation that is in equilibrium with a random coil conformation.10 Two subsequent studies conducted on an isolated SP1 peptide11 and on two SP1-containing peptides encompassing portions of CACT D and NC domains12 indicated that the helical structure in solution is promoted in 30% trifluoroethanol, and the coil- to-helix transition also occurs in aqueous media at high SP1 concentrations.11 A recent cryo-ET study of viral particles constructed from Gag and from various maturation intermediates revealed that, morphologically, the CA-SP1 maturation intermediate exhibits significant variability, and does not show the striated CA layer seen in the immature Gag and in the early-stage maturation intermediates.13 The inherent resolution of cryo-ET is such that the detailed structure of the SP1 peptide could not be delineated and it was not clear whether any of the electron density observed corresponded to the SP1 peptide in any of the constructs examined. A brand new cryo-ET study of CA-SP capsids from Rous sarcoma virus (RSV) assembled in vitro revealed that the RSV SP peptide in that virus core is unstructured.14
Another open question in the field is what serves as the trigger for the cleavage of SP1 and the subsequent HIV-1 capsid assembly. One hypothesis proposes that upon proteolytic cleavage of SP1 peptide, the CA lattice changes in shape from spherical to conical. The second hypothesis posits that during the retroviral maturation process, the immature lattice disassembles during the course of proteolytic cleavages of Gag and that the mature core reassembles de novo. The role of the SP1 cleavage during the final maturation step is thought to mediate disassembly of the immature CA.14 This most recent hypothesis is corroborated by several pieces of evidence, predominantly provided by cryo-ET results: (i) the removal of the RSV SP peptide is not required for the CA assembly into capsids in vitro, and (ii) the disassembly of the immature-like lattice appears to be required for correct conical core assembly. The above study also revealed that the SP peptide is unstructured in the assembled RSV CA-SP cores.
Analysis of the maturation mechanism(s) has been challenging since assemblies of CA with various maturation inhibitors are not amenable for atomic-level structural studies by the well-established structural biology techniques. While solution NMR and X-ray diffraction provided a wealth of information on isolated protein molecules or domains of Gag, including CA, they cannot examine the proteins in the context of large-scale assemblies. Cryo-EM, having yielded critical knowledge about the structures of various HIV-1 proteins in the assembled states,5,15,16 in general does not provide atomic-resolution information, although recent advances are leading to breakthroughs toward this end.17 Furthermore, flash- freezing of specimens and cryogenic temperatures precludes the identification of dynamic regions in the assembled proteins.
Magic Angle-Spinning (MAS) NMR spectroscopy is an emerging and promising technique for studying HIV-1 protein assemblies, since it yields atomic-resolution information, does not require cryogenic temperatures and is operational at a wide range of sample conditions, including physiological or close to physiological temperatures and pH. Under such conditions, both structure and dynamics of assembled proteins can be readily probed. Indeed, our previous studies have already established that MAS NMR spectroscopy is a powerful tool for examining conical CA assemblies.18,19 Furthermore, through integration of MAS and solution NMR, comprehensive insights were gained into the structural plasticity of HIV-1 capsids. Specifically, we uncovered the key role of millisecond timescale motions in the hinge region of CA, coupled to a molecular switch mechanism that controls the number of accessible conformers and their populations in pleiotropic capsid assembly.18
In this report, we examine the structure of HIV-1 CA and CA-SP1 assemblies of tubular morphologies. Tubular assemblies resemble the authentic viral capsids and recapitulate their salient structural features: the curved hexagonal lattice formed by hexameric CA units, the intermolecular inter-hexamer CTD-CTD contacts across the local pseudo two- and three- fold axes, and the hexamer forming NTD-NTD and NTD-CTD interfaces.6,20 Despite critical structural information obtained from the cryo-EM studies of tubular assemblies and cryo- ET characterization of intact viral cores, the subnanometer resolution of these methods limits further structural analysis of HIV-1 maturation intermediates, of mature capsids, and of their interactions with maturation inhibitors and cellular host factors. We demonstrate here that tubular CA and CA-SP1 assemblies yield unprecedented high-resolution MAS NMR spectra at magnetic fields of 19.9 T and above, permitting resonance assignments that are a prerequisite for detailed structural characterization of the assembly. Our results for two amino acid sequence variants of CA, derived from strains HXB2 and NL4-3 (the primary amino acid sequences are shown in Figure 2D), reveal surprisingly large conformational differences in two functionally important loops, the CypA loop and the loop preceding helix 8. Remarkably, we find that SP1 peptide is dynamically disordered and exists in a random coil conformation in the assembled CA-SP1 from both constructs. Our results are in agreement with the recent findings by Keller et al.14 and provide further support for the hypothesis that capsid maturation proceeds through de novo reassembly and that the role of SP1 cleavage is to mediate the disassembly of the immature CA lattice for maturation. Taken together, our data highlight the importance of conformational plasticity in HIV-1 CA protein assembly and call for atomic- level scrutiny of the structures and dynamics of the various maturation intermediates.
Figure 2.

From left to right: TEM (A) and confocal (B) images of CA A92E, CA-SP1 A92E, CA WT and CA-SP1 WT tubular assemblies. (C) TEM image of the CA-SP1 WT tubular assemblies acquired at higher magnification and illustrating that highly regular hexameric lattice is clearly observed. (D) Primary sequences of the HXB2 and NL4-3 (A92E mutant) strains used in this work. The variations between the amino acid residues in HXB2 and NL4-3 strains are shown in red, and the 14-residue SP1 peptide is shown in blue.
RESULTS AND DISCUSSION
MAS NMR Spectroscopy of Tubular CA and CA-SP1 Assemblies
As shown in Figure 2, in the presence of 1 M NaCl, CA and CA-SP1 organize into homogeneous tubular assemblies, whose dimensions are in accord with previous observations.19–21 These conditions are fully compatible with sample requirements for MAS NMR spectroscopy, and Figure 2C illustrates the quality of these assemblies: a hexagonal lattice is readily discernible in the conventional negatively stained TEM images. As demonstrated by us previously, these assemblies are stable under MAS conditions.19
The tubular assemblies yield MAS NMR spectra of unprecedented resolution, as demonstrated in Figure 3. In contrast to previous studies of conical assemblies by us19 and of tubular assemblies by others,22 resonance assignments are relatively facile and can be performed for an extensive portion of all residues, even from the 2D spectra. In Figure 3, we provide a backbone walk for CA-SP1 (NL4-3/A92E), from 2D spectra. The corresponding backbone walk for CA (HXB2) tubular assemblies from 3D spectra is shown in Figure S1 of the Supporting Information. Similar-quality datasets have been acquired for the other two samples- CA-SP1 (HXB2) and CA (NL4-3/A92E). Analysis of the chemical shifts reveals that, with the exception of two flexible loops and several additional residues (vide infra), these shifts are in agreement with those previously reported by us as well as solution NMR chemical shifts of the CTD and NTD of the corresponding sequence variants (unpublished results and18–20,23). A detailed comparison and analysis of the resonance assignments will be presented in a forthcoming report; here we focus solely on three regions: the SP1 tail, the CypA loop, and the loop preceding helix 8.
Figure 3.

(A) Backbone walk for the stretch of residues R100-T110 of U-13C,15N tubular assemblies of HIV-1 CA-SP1 A92E (NL4-3 strain) acquired at 19.9 T and 4 C. The 1D trace at 112.4 ppm along the 15N dimension of the NCACX spectrum is shown on top. The following 2D data sets were used to establish this backbone walk: NCACX (top), NCOCX (middle right and bottom), and DARR (middle left and middle center). The DARR mixing time was 50 ms. (B) Expansion of the 2D DARR spectrum around the region containing the Cα-Cβ resonances of Ser and Thr residues. The acquisition parameters are given in the Supporting Information.
Sequence Variability and Conformational Plasticity of CA
HIV-1 strains exhibit significant sequence variability24 and different Gag sequence variants are associated with differential viral infectivity.25–31 In laboratory settings, several naturally occurring sequence variants have been used for structural and functional studies, and mutants have been engineered to yield stable CA assemblies and/or force formation of particular type of oligomeric structures.26–31 This naturally begs two questions: i) to what extent do sequence variations impact the conformation and the dynamics of CA? and ii) which structural and/or dynamic features in CA are affected in the different sequence variants and how does this modulate the viral infectivity?
To address the above questions, we investigated two closely related strains of HIV-1, HXB2 and NL4-3. The latter also contains an A92E mutation in the functionally important CypA loop; mutations in this loop often result in defective virus particles.32–34 The A92E mutant is an escape mutant: its viral infectivity is ~10% of wild type infectivity, but is restored by the inhibition of Cyclophilin A (CypA).25,28 The amino acid sequence of CA proteins used in our study differs by five residues (Figure 2D). In Figure 4, a superposition of the NCA and NCACX spectra is shown for these two variants. From these spectra, the resonances corresponding to the residues differing between the two variants are easily assigned by inspection, except for the resonances of A208, which are missing. A large number of chemical shift changes are observed in residues that are conserved across the two sequence variants (Table 1). Remarkably, all of these changes are mapped to residues in the CypA loop (NTD) and in the loop preceding helix 8 (CTD), as illustrated in Figure 4C. Given that the amino acid sequence variations A92/E92 and V83/L83 both occur in the CypA loop, it may not be too surprising that the chemical and electronic environment is changed resulting in chemical shift differences for loop residues. On the other hand, the finding of changes for residues in the loop preceding helix 8 is puzzling, given that the closest sequence differences are N120/H120 and A208/G208, ca. 50 residues away in the primary sequence from this loop region. Inspection of the 3D structure of CA, however, reveals that the A208/G208 is spatially close to this loop, as illustrated in Figure 4C, and this may explain why the substitution of A208 by G208 influences the conformation and the chemical shifts of the loop preceding helix 8.
Figure 4.

The superposition of the NCA (A) and NCACX (B) spectra of CA NL4-3 A92E (orange) and CA HXB-2 (black). For the NCACX spectra, an expansion of the region for aliphatic sidechain resonances is shown. The red and magenta labels represent signals from the distinct residues in the NL4-3 A92E and HXB-2 variants, respectively. (C) A view of the 3D structure of CA (HXB2 variant, PDB file 3NTE44). Colored magenta are the residues that vary between the two constructs; in dark grey shown are the residues that are identical in the two variants but whose chemical shifts are perturbed.
Table 1.
Chemical shift differences in HXB2 and NL4-3 A92E variants
| Residue | N | Cα | Cβ | C′ |
|---|---|---|---|---|
| HXB2/NL4-3 | HXB2/NL4-3 | HXB2/NL4-3 | HXB2/NL4-3 | HXB2/NL4-3 |
| I6/L6 | 124.0/116.8 | 63.6/56.1 | 38.3/42.6 | 177.0/ND |
| V83/L83 | 112.5/116.7 | 62.4/55.5 | 32.3/42.7 | 175.4/ND |
| A92/E92 | 131.8/127.9 | 50.7/54.6 | 17.7/29.3 | ND/ND |
| N120/H120 | 122.7123.0 | 53.3/57.1 | 39.4/28.3 | 173.2/ND |
| A208/G208 | 125.3/110.0 | 51.5/45.3 | 18.6/ND | 175.0/ND |
| H84 | 116.7/118.7 | 52.9/53.2 | ND/27.0 | ND/ND |
| V86 | 121.3/121.5 | 62.4/62.5 | 33.1/33.0 | 176.5/ND |
| H87 | 124.1/125.3 | 54.6/55.7 | 29.8/29.4 | 175.3/ND |
| A88 | 128.1/127.8 | 52.2/52.1 | 19.4/19.0 | 178.6/ND |
| G89 | 109.3/109.6 | 44.5/44.5 | ND/ND | ND/ND |
| I91 | 121.8/121.5 | 60.2/60.1 | 38.6/38.5 | 176.0/ND |
| G94 | 112.5/112.4 | 45.1/45.3 | ND/ND | ND/ND |
| Q95 | 119.2/119.5 | 54.5/54.4 | 29.9/30.0 | 175.2/ND |
| M96 | 121.0/120.5 | 54.7/55.0 | 34.7/34.8 | 177.0/ND |
| R97 | 123.7/122.9 | 55.4/55.3 | 30.0/29.6 | 174.9/ND |
| E98 | 123.7/123.9 | 54.5/54.5 | 29.8/29.7 | 178.8/ND |
| R100 | 121.4/121.3 | 52.6/52.3 | 32.8/31.4 | 178.8/ND |
| N121 | 120.1/116.7 | 48.9/49.7 | 39.5/40.1 | ND/ND |
| R154 | ND/125.7 | ND/54.3 | ND/32.7 | 175.9/ND |
| Q155 | ND/128.2 | ND/55.9 | ND/27.2 | ND/ND |
| G156 | 117.3/117.5 | 45.0/44.9 | ND/ND | ND/ND |
| K158 | ND/115.3 | ND/54.3 | ND/32.5 | 176.1/ND |
| E159 | ND/126.1 | ND/53.7 | ND/32.2 | ND/ND |
| C198 | 116.5/ND | 63.5/ND | ND/ND | ND/ND |
| C218 | 110.0/ND | 60.2/ND | ND/ND | ND/ND |
SP1 is a Dynamic Random Coil in Assembled CA-SP1 (NL4-3/A92E)
The conformation of the SP1 peptide in CA-SP1 assemblies was investigated by a combination of dipolar and scalar correlation methods, as these provide complementary information on the rigid and mobile regions in a molecular structure, respectively.
To our surprise, the dipolar-based NCA and CP-DARR spectra of CA and CA-SP1 NL4-3/A92E assemblies are identical (Figure S3 of the Supporting Information). No new resonances were observed, indicating that the SP1 region is either unstructured or mobile, with the latter causing dynamic averaging of the NMR signals. Next, direct-DARR experiments were carried out in an attempt to detect the SP1 resonances. In these experiments, replacement of the 1H-13C cross-polarization transfer step by the direct 13C excitation using a π/2 pulse can restore signals in the cases where motions that occur on the time scale of the 1H-13C dipolar interactions may result in cross peak disappearance. As shown in Figure 5A–D, several new peaks emerged in the direct-DARR spectra of CA-SP1, all consistent with the residue types that are present in the SP1 tail. Site-specific resonance assignments were derived based on a J-INADEQUATE spectrum (Figure 5E–J) that revealed that these resonances are indeed associated with the SP1 tail (vide infra). No other differences in the spectra could be identified, indicating that the CA protein exhibits identical or very similar structures in the CA or CA-SP1 assemblies. The emergence of the new resonances from SP1 in CA-SP1 in the direct-DARR experiments is associated with the removal of the cross polarization step, in the pulse sequence, suggesting that the associated SP1 residues are intrinsically mobile. This notion was borne out by the J-based INADEQUATE experiments.
Figure 5.

The superposition of the direct-DARR (A–D), INADEQUATE (E–J), and CP-DARR (K) spectra of the tubular assemblies of CA (orange) and CA-SP1 (black) NL4-3 A92E acquired at 19.9 T and 4 °C. Expansions of different regions, illustrating the presence of new peaks in the spectra of the CA-SP1 assemblies that are not present in the corresponding spectra of the CA assemblies. In K), an expansion of the DARR spectrum is shown for the region (ω2: 50–57 ppm/ω1: 34–40 ppm) to illustrate the absence of the Cα -Cβ correlation of N240 in CA-SP1. In contrast, this cross peak is present in the direct-DARR and INADEQUATE spectra (expansions C and I, respectively). The DARR mixing time in the direct-DARR and CP-DARR experiments was 50 ms.
A superposition of the INADEQUATE spectra of CA and CA-SP1 is displayed in Figure 5E–J. Similar to the findings in the direct-DARR spectra (Figure 5A–D), resonances associated with SP1 emerge as new peaks in the CA-SP1 data set. All of these new peaks were assigned unambiguously to the SP1 residues, and the corresponding chemical shifts are provided in Table 2. Analysis of the Chemical Shift Index (CSI) again revealed that the SP1 residues are in a random coil conformation.
Table 2.
Chemical shifts of the SP1 tail and nearby residues of CA-SP1 assemblies in NL4-3/A92E and HXB2 variant). The CSI column is the secondary structure predicted by CSI method: H-α helix, B-β sheet, C-random coil.
| NL4-3/A92E | HXB2 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||
| Residue | Cα | Cβ | Cγ | Cδ | C′ | CSI | Residue | Cα | Cβ | Cγ | Cδ | C′ | CSI |
| G225 | G225 | ||||||||||||
| H226 | 55.6 | 29.8 | 131.6 | C | H226 | 55.3 | 29.4 | 131.5 | C | ||||
| K227 | 56.5 | 33.3 | 22.0 | C | K227 | 56.5 | 33.3 | C | |||||
| A228 | 52.9 | 17.3 | 173.4 | C | A228 | ||||||||
| R229 | 56.3 | 30.9 | 27.4 | C | R229 | 56.3 | 30.9 | C | |||||
| V230 | 63.6 | 33.4 | 20.0 | 181.2 | C | V230 | |||||||
| L231 | 56.7 | 43.4 | 182.4 | C | L231 | 56.7 | 43.4 | C | |||||
| 56.2 | 42.5 | C | |||||||||||
| A232 | 53.2 | 19.0 | C | A232 | 55.6 | 19.0 | H | ||||||
| E2331 | 56.8 | 27.9 | C | E2331 | 57.2 | 33.7 | 177.4 | C | |||||
| 56.1 | 29.7 | C | |||||||||||
| A234 | 53.2 | 19.0 | C | A234 | 48.4 | 16.8 | |||||||
| M 2352 | 56.9 | 32.4 | C | M 235 | |||||||||
| S236 | 58.8 | 63.7 | C | S236 | |||||||||
| Q2371 | 56.8 | 27.9 | C | Q2371 | 57.2 | 33.7 | 177.4 | C | |||||
| 56.1 | 29.7 | C | |||||||||||
| V238 | C | V238 | |||||||||||
| T239 | 61.8 | 70.3 | 174.3 | C | T239 | 61.8 | 70.3 | 174.3 | C | ||||
| 62.3 | 69.6 | 173.8 | C | 62.3 | 69.6 | 174.3 | C | ||||||
| N240 | 51.2 | 38.9 | 177.2 | 173.2 | C | N240 | |||||||
| 51.5 | 39.0 | ||||||||||||
| P241 | 27.6 | 51.9 | P241 | ||||||||||
| A242 | 53.2 | 19.0 | C | A242 | 55.6 | 19.0 | H | ||||||
| T243 | 70.5 | 69.4 | 19.3 | C | T243 | ||||||||
| I244 | 61.1 | 38.6 | 14.1 | C | I244 | ||||||||
| M 2452 | 57.2 | 33.5 | C | M 245 | |||||||||
The assignments for E233 and Q237 residues are ambiguous.
The assignments for M235 and M245 are based on solution chemical shifts.
Interestingly, comparison of CA-SP1 INADEQUATE and direct-DARR spectra revealed that for N240, the INADEQUATE spectrum contained two signals, separated by 0.3 ppm, while only single peaks were present in the direct-DARR spectrum at the same position. The resonances are strong and distinct and given that there is only one Asn residue in the sequence, those peaks are assigned to two N240 conformers. Additional information was inferred about the chemical exchange rates between the two conformers taking into account that the 13Cα-13Cβ J-coupling is ~30–40 Hz, and the cross polarization rf field in CP-DARR experiment is ~30 kHz. The N240 signal appears as a broadened pair of resonances in the INADEQUATE (Figure 5I), as a single peak in the direct-DARR (Figure 5C), and is completely absent in the CP-DARR spectrum (Figure 5K). The difference in the resonance frequencies for the two conformers is ~60 Hz. Therefore, the INADEQUATE experiment detects both conformers, whereas in the direct-DARR experiment only one of the two conformers, a more rigid one, is seen. Similarly, we find that T239 also exhibits two resonances in the INADEQUATE spectra, that are separated by 0.5 ppm (~100 Hz), with again these resonances completely missing in the CP-DARR spectra, just as noted for the N240 resonances. This finding suggests that motions on the tens of milliseconds time scale are averaging out the 1H-13C dipolar couplings for these residues.
Evidence for motions is also provided by the lack of signals in the CP-INADEQUATE spectra. In this experiment, a 1H-13C cross polarization period is incorporated in the pulse sequence prior to the INADEQUATE step (see Figure S2 for the pulse sequence diagrams). For rigid residues, the CP step will yield sensitivity enhancement while for residues that are mobile on the time scale of the 1H-13C dipolar interactions, ~23 kHz or tens of microseconds, the CP signal will be motionally averaged. Figure 6 displays a superposition of the CP-INADEQUATE and INADEQUATE spectra of CA assemblies, demonstrating that resonances associated with the stretch of residues H226–L231 are present in the INADEQUATE spectrum, but missing in the CP-INADEQUATE data set. These data, together with the lack of these signals in the CP-DARR spectra prove that these residues are mobile on the timescales of tens of microseconds. Furthermore, as seen in Figure 5, the signals from the H226–L231 residue stretch that are present in the INADEQUATE spectra of CA are not present in the spectra of CA-SP1. It therefore appears that presence of the SP1 tail introduced motions into the H226–L231 stretch of residues, similar to those observed for SP1 residues T239 and N240.
Figure 6.

The superposition of the CP-INADEQUATE (orange) and INADEQUATE (black) spectra of the tubular assemblies of CA NL4-3 A92E acquired at 19.9 T and 4 °C. A) and B) The expansion of the aliphatic regions containing extra INADEQUATE peaks. C) and D) The Cβ-Cγ region of His residues. E) and F) the Cα-C′ region.
In summary, resonances for twelve out of fourteen residues in the SP1 peptide are seen in the INADEQUATE spectra of the CA-SP1 NL4-3/A92E tubular assemblies, and CSI chemical shift analysis indicates a random coil conformation. The SP1 tail is mobile on the timescale of tens of microseconds, and some of the residues in SP1, such as T239 and N240, are also mobile on slower timescales, tens of milliseconds. Furthermore, the SP1 tail also modulates the motions of spatially close residues, in particular the H226–L231 stretch, which exhibits motions on both the microsecond and millisecond timescales in CA-SP1, while in CA the same residues are only mobile on microsecond timescale.
SP1 Conformation and Motions are Modulated by Distal Residues in HXB2
Comparison of the CP-DARR spectra of CA and CA-SP1 HXB2 reveals that two intense resonances are present in the CA-SP1 spectra, one in the Ala and another in the Thr region (Figure S4 of the Supporting Information). Using a combination of other types of spectra, these two peaks were assigned to the SP1 peptide residues, as discussed below.
The INADEQUATE spectra of CA and CA-SP1 HXB2 assemblies superimpose very well (Figure 7), indicating that the overall structure is very similar. Unlike the CA-SP1 NL4-3/A92E, only a few new signals from SP1 appear in the CA-SP1 HXB2, and the combined resonance assignments are summarized in Table 2. Two conformers for T239 were observed, similar to the findings for NL4-3/A92E, and the CSI analysis indicates that the corresponding residues in the SP1 tail are in a random coil conformation, similar to our findings for the CA-SP1 NL4-3/A92E assemblies. Resonances associated with other residues in SP1 are either averaged or shifted to the crowded areas of the spectrum, precluding their assignment.
Figure 7.

The superposition of the INADEQUATE spectra of the tubular assemblies of CA (orange) and CA-SP1 (black) HXB2 acquired at 21.1 T and 4 °C. A)–C) the aliphatic region; D) the Cβ-Cγ region of His residues; E) and F) the Cα-C′ region.
It is instructive to examine the resonances arising from the H226–L231 stretch of residues in HXB2. H226, K227, R229, and L231 resonances are seen in the CA (but not CA-SP1) spectra, and their chemical shifts are in excellent agreement with those from the NL4-3/A92E sample. Interestingly, the resonances for A228 and V230 are missing, possibly averaged out by motions.
Comparison of the INADEQUATE and CP-INADEQUATE spectra of CA and CA-SP1 HXB2 reveals that resonances for K227, R229, and L231 appear exclusively in the INADEQUATE data set of CA, similar to our findings for NL4-3/A92E. As discussed above, this observation indicates that these residues are mobile on the time scale of tens of microseconds. Interestingly, the peak corresponding to H226 appeared in both the INADEQUATE and CP-INADEQUATE spectra of CA HXB2, in contrast to the finding for CA NL4-3/A92E, and resonances for A228 and V230 in the CP-INADEQUATE spectrum of CA HXB2 sample are not at the same frequencies as in the CA NL4-3/A92E spectra.
Taken together, these findings indicate that the conformation of SP1 and of the spatially close residues is identical in HXB2 and NL4-3/A92E assemblies but the dynamics is different.
SP1 Modulates the Motions in the CypA Loop
To our surprise, we noted that in the NCA spectra of CA and CA-SP1 HXB2 a number of signals are different and that more resonances overlapped in the CA-SP1 data set; at the same time, no distinct new resonances appear (Figure S4 of the Supporting Information). Most of the different resonances are associated with residues in the functionally important CypA loop region, such as R97 and E98. Interestingly, substantial variations in the peak intensities between the CA and CA-SP1 spectra are seen: a number of intense resonances are present in the CA spectrum, while the corresponding peaks in the CA-SP1 data set are severely attenuated in intensity or completely missing. Notably, this behavior is observed for resonances associated with residues in the CypA loop, such as G89, A92, and G94. Interestingly, the A92 resonances are also missing in the CA-SP1 CP-DARR spectrum.
The observation of substantial resonance intensity variations for the CypA loop residues is intriguing. Our findings suggest that the SP1 tail induces motion in the CypA loop region rendering it more flexible in the CA-SP1 HXB2 tubular assemblies compared to the corresponding CA assemblies. The fact that SP1 has significant effects on the residues that are 150 amino acids away in the primary sequence and that belong structurally to the two separate domains of CA seemed puzzling. Even more surprising was the finding that for the CA NL4-3/A92E, no such effect on the CypA loop by the SP1 tail was observed. One possible explanation for this discrepancy may be that the A92E substitution in the CypA loop stabilizes the loop, such that SP1 no longer influences its dynamics. This notion is supported by our recent findings that the CypA loop dynamics, as measured via the H–N dipolar order parameters, is profoundly different in the HXB2 and NL4-3/A92E CA variants, with the A92E mutant exhibiting greatly attenuated motions on the nano to microsecond timescales (to be reported in a forthcoming manuscript).
CONCLUSIONS AND FUTURE OUTLOOK
Tubular assemblies of HIV-1 CA and CA-SP1 proteins yielded unprecedented-quality MAS NMR spectra. Results from the combined dipolar- and scalar-based experiments unequivocally show that the SP1 peptide tail is in a random coil conformation, dynamically disordered, and that conformational plasticity in functionally important loop regions of CA and CA-SP1 is induced by the primary sequence.
Our current results that the SP1 peptide is unstructured in the CA-SP1 assemblies, presented in this report, provide support for the hypothesis that capsid maturation proceeds through disassembly-reassembly and that the role of SP1 cleavage is to mediate the disassembly of immature CA lattice during final maturation.
The current work sets the stage for further investigations into the mechanism of HIV-1 maturation and into evaluation the interactions of HIV-1 protein assemblies with maturation inhibitors at the detailed structural level. MAS NMR methods are envisioned to become a central tool in these studies as they offer unprecedented atomic-resolution insight into the structure and dynamics of the various HIV-1 protein complexes.
METHODS
Sample preparation
Expression and purification of CA and CA-SP1 (NL4-3/A92E and HXB variants) were performed as reported previously with a slight modification.21 CA and CA-SP1, expressed in E. coli Ros2 (DE3) cells, were purified with cation exchange column chromatography with a 0–1 M NaCl gradient in a buffer containing 25 mM sodium phosphate pH 5.8, 1 mM DTT and 0.02% NaN3. Proteins were further purified using size-exclusion column chromatography equilibrated with a buffer containing 25 mM sodium phosphate, pH 7.5, 1 mM DTT and 0.02% NaN3. Tubular assemblies of CA and CA-SP1 were prepared from 32 mg/ml protein solutions in 25 mM phosphate buffer (pH 5.5) containing 1 M NaCl. The solutions were incubated at 37 ºC for 1 hour and stored at 4 ºC for subsequent experiments.
Transmission electron and laser confocal microscopy
The sample morphologies were characterized by TEM and laser confocal microscopy. TEM analysis was performed in a Zeiss CEM 902 transmission electron microscope operating at 80 kV. Samples were stained with ammonium molybdate (5 %w/v), deposited onto 400 mesh, formval/carbon-coated copper grids, and dried for 40 min. Some of the assemblies were analyzed using a Zeiss Libra 120 transmission electron microscope operating at 120 kV. Samples were stained with uranyl acetate (5% w/v), deposited onto 400 mesh, formval/carbon-coated copper grids, and dried for 40 min in the air. The copper grids were pre-treated with Pelco easiGlow Discharge Unit to deposit a charge, so that the tubular assemblies are uniformly spread on the grid surface and adhere to it.
Confocal images were acquired on a Zeiss LSM 510 NLO laser scanning microscope (Carl Zeiss, Germany) using a Zeiss 40× (NA 1.3) oil immersion objective lens. All data were collected using the 584 nm laser line of a 25 mW argon and the 633 nm laser line of a 35 mW HeNe lasers (LASOS, Germany) with a rhodamine reflector and the transmitted-light channel when appropriate. Protein solutions were stained with the Nile Blue A dye (10 ppm) solution being applied on the sample stage at 1:1 volume ratio.
Solid-state NMR spectroscopy
MAS NMR spectra of CA and CA-SP1 HXB2 were acquired at the National High Magnetic Field Laboratory (NHMFL) at 21.1 T on an ultrawide bore instrument35 outfitted with a 3.2 mm HXY sensitivity-enhanced Low-E probe designed and built at NHMFL.36,37 Sample was packed inside a 3.2 mm Pencil rotor (sample volume 36 uL). Larmor frequencies were 899.1 MHz (1H), 226.1 MHz (13C), and 91.1 MHz (15N). The temperature was 4±1 °C throughout the experiments. Spectra were collected at the MAS frequency of 14.000±0.001 kHz. 13C and 15N chemical shifts were referenced with respect to DSS and adamantane. The pulse sequence diagrams are shown in Figure S2 of the Supporting Information. The typical pulse lengths were 2.5 us (1H), 3 us (13C), and 5 us (15N). 1H-13C(15N) cross polarization employed a linear amplitude ramp; the 1H radio- frequency (rf) field was 62 kHz with the center of the ramp Hartmann-Hahn matched to the first spinning sideband. The 1H-13C and 1H-15N contact times were 1 ms. The 15N-13C DCP contact time was 5.0 ms. The 1H DARR field strength was 14 kHz; the DARR mixing time was 50 ms. Small phase incremental alternation (SPINAL-64)38 1H decoupling (100 kHz) was used during the direct and indirect acquisition periods. Recycle delay in all experiments was 2 s. Time-proportional phase incrementation (TPPI)39 was used for phase-sensitive detection in the indirect dimensions. We note that in the direct-DARR experiment, the phase cycle for the 13C pulses follows that of the NOESY experiment, and two magnetization transfer mechanisms may occur during the mixing: spin diffusion and cross-relaxation (NOESY). The cross-relaxation transfer is expected to be much less efficient and the observed cross-peaks are expected to be predominantly due to rotary-assisted spin diffusion.
MAS NMR spectra of CA and CA-SP1 NL4-3 A92E variant were acquired at the University of Delaware on a Bruker 19.9 T spectrometer outfitted with a 3.2 mm BioMAS HXY probe. Sample was packed into a 3.2 mm thin-wall Bruker rotor. Larmor frequencies were 850.400 MHz (1H), 213.855 MHz (13C), and 86.181 MHz (15N). Spectra were collected at the MAS frequency of 13.000±0.001 kHz regulated by a Bruker MAS controller, and the temperature was maintained at 4.0±0.5 °C throughout the experiments using a Bruker temperature controller. The typical pulse lengths were 3.3 μs (1H), 3.8 μs (13C), and 4.2 μs (15N), and 1H-13C cross polarization was performed with a linear amplitude ramp (80–100%), with the 1H rf field of 80 kHz matched to the second spinning sideband. The 1H-13C and 1H-15N contact times were 1 and 1.5 ms, respectively. SPINAL-64 decoupling (80 kHz) was applied during the evolution and acquisition periods. The 1H field strength during DARR was 13 kHz, and the DARR mixing time was 50 ms. Under these conditions predominantly one-bond correlations are observed in the spectra. States-TPPI40 was used for frequency discrimination in the indirect dimension.
Other relevant experimental conditions are indicated in the figure captions and in the Supporting Information.
Processing and analysis of solid-state NMR spectra
All spectra were processed with NMRpipe41 and analyzed using Sparky.42 For 2D and 3D data sets, 60° or 30° shifted sine bell apodization followed by a Lorentzian-to-Gaussian transformation was applied in all dimensions. Forward linear prediction to twice the number of the original data points was used in the indirect dimension in some data sets followed by zero filling to twice the total number of points.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (NIGMS Grant P50GM082251-03 and NCRR grant P20RR015588-10) and is a contribution from the Pittsburgh Center for HIV Protein Interactions. We thank Jennifer Mehrens and Maria DeLucia for their technical assistance with cloning and purification of the proteins used in this work. We are grateful to Kirk Czymmek and Jeffrey Caplan at the Delaware Biotechnology Institute Bio-Imaging Center at the University of Delaware for their kind assistance with acquiring TEM and confocal images. We acknowledge the support of the National Science Foundation (NSF Grant CHE0959496) and of the University of Delaware for the acquisition of the 850 MHz NMR spectrometer at the University of Delaware. The 21.1 T spectra were acquired at the following high- field NMR centers: i) National High Magnetic Field Laboratory supported through the National Science Foundation Cooperative Agreement (DMR-0084173) and by the State of Florida; ii) Environmental Molecular Sciences Laboratory, a national scientific user facility sponsored by the United States Department of Energy’s Office of Biological and Environmental Research and located at the Pacific Northwest National Laboratory in Richland, WA; and iii) the MIT-Harvard Center for Magnetic Resonance supported by NIH grant P41EB002026.
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
ASSOCIATED CONTENT
Supporting information. Backbone walk for residues R110-G116 in tubular assemblies of CA WT HXB2 strain extracted from 3D NCACX and NCOCX spectra (Figure S1); diagrams of pulse sequences (Figure S2); 2D NCA, CP-DARR, and direct-DARR spectra of CA and CA-SP1 NL4-3 A92E (Figure S3); comparison of CP-DARR spectra of CA and CA-SP1 HXB2 (Figure S4). Acquisition and processing parameters for the 19.9 and 21.1 T MAS NMR spectra of CA and CA-SP1 NL4-3 A92E and HXB2 (Table S1). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Gottlinger HG. Aids. 2001;15:S13–S20. doi: 10.1097/00002030-200100005-00003. [DOI] [PubMed] [Google Scholar]
- 2.Pettit SC, Moody MD, Wehbie RS, Kaplan AH, Nantermet PV, Klein CA, Swanstrom R. J Virol. 1994;68:8017–8027. doi: 10.1128/jvi.68.12.8017-8027.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sundquist WI, Krausslich HG. CSH Perspect Med. 2012;2:a006924. doi: 10.1101/cshperspect.a006924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Briggs JA, Wilk T, Welker R, Krausslich HG, Fuller SD. EMBO J. 2003;22:1707–15. doi: 10.1093/emboj/cdg143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ganser-Pornillos BK, Cheng A, Yeager M. Cell. 2007;131:70–9. doi: 10.1016/j.cell.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 6.Zhao GP, Perilla JR, Yufenyuy EL, Meng X, Chen B, Ning JY, Ahn J, Gronenborn AM, Schulten K, Aiken C, Zhang PJ. Nature. 2013;497:643–646. doi: 10.1038/nature12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salzwedel K, Martin DE, Sakalian M. AIDS Rev. 2007;9:162–172. [PubMed] [Google Scholar]
- 8.Adamson CS, Salzwedel K, Freed EO. Expert Opin Ther Tar. 2009;13:895–908. doi: 10.1517/14728220903039714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Waheed AA, Freed EO. AIDS Res Hum Retrov. 2012;28:54–75. doi: 10.1089/aid.2011.0230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Newman JL, Butcher EW, Patel DT, Mikhaylenko Y, Summers MF. Protein Sci. 2004;13:2101–2107. doi: 10.1110/ps.04614804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Datta SAK, Temeselew LG, Crist RM, Soheilian F, Kamata A, Mirro J, Harvin D, Nagashima K, Cachau RE, Rein A. J Virol. 2011;85:4111–4121. doi: 10.1128/JVI.00006-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morellet N, Druillennec S, Lenoir C, Bouaziz S, Roques BP. Protein Sci. 2005;14:375–386. doi: 10.1110/ps.041087605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Marco A, Muller B, Glass B, Riches JD, Krausslich HG, Briggs JAG. PLoS Pathog. 2010;6:e1001215. doi: 10.1371/journal.ppat.1001215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keller PW, Huang R, England M, Waki K, Cheng N, Heymann JB, Craven RC, Freed EO, Steven AC. J Virol. 2013 doi: 10.1128/JVI.01408-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pornillos O, Ganser-Pornillos BK, Kelly BN, Hua Y, Whitby FG, Stout CD, Sundquist WI, Hill CP, Yeager M. Cell. 2009;137:1282–92. doi: 10.1016/j.cell.2009.04.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meng X, Zhao GP, Yufenyuy E, Ke DX, Ning JY, DeLucia M, Ahn J, Gronenborn AM, Aiken C, Zhang PJ. PLoS Pathog. 2012;8:e1002886. doi: 10.1371/journal.ppat.1002886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bammes BE, Rochat RH, Jakana J, Chen DH, Chiu W. J Struct Biol. 2012;177:589–601. doi: 10.1016/j.jsb.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Byeon IJL, Hou GJ, Han Y, Suiter CL, Ahn J, Jung J, Byeon CH, Gronenborn AM, Polenova T. J Am Chem Soc. 2012;134:6455–6466. doi: 10.1021/ja300937v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Han Y, Ahn J, Concel J, Byeon IJL, Gronenborn AM, Yang J, Polenova T. J Am Chem Soc. 2010;132:1976–1987. doi: 10.1021/ja908687k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Byeon IJL, Meng X, Jung JW, Zhao GP, Yang RF, Ahn JW, Shi J, Concel J, Aiken C, Zhang PJ, Gronenborn AM. Cell. 2009;139:780–790. doi: 10.1016/j.cell.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun S, Han Y, Paramasivam S, Yan S, Siglin AE, Williams JC, Byeon IJ, Ahn J, Gronenborn AM, Polenova T. Methods Mol Biol. 2012;831:303–31. doi: 10.1007/978-1-61779-480-3_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen B, Tycko R. Protein Sci. 2010;19:716–730. doi: 10.1002/pro.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jung J, Byeon IJL, Ahn J, Concel J, Gronenborn AM. Biomol NMR Assign. 2010;4:21–23. doi: 10.1007/s12104-009-9198-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Price AJ, Fletcher AJ, Schaller T, Elliott T, Lee K, KewalRamani VN, Chin JW, Towers GJ, James LC. PLoS Pathog. 2012;8:e1002896. doi: 10.1371/journal.ppat.1002896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aberham C, Weber S, Phares W. J Virol. 1996;70:3536–3544. doi: 10.1128/jvi.70.6.3536-3544.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qi ML, Yang RF, Aiken C. J Virol. 2008;82:12001–12008. doi: 10.1128/JVI.01518-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamashita M, Perez O, Hope TJ, Emerman M. PLoS Pathog. 2007;3:1502–1510. doi: 10.1371/journal.ppat.0030156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ylinen LMJ, Schaller T, Price A, Fletcher AJ, Noursadeghi M, James LC, Towers GJ. J Virol. 2009;83:2044–2047. doi: 10.1128/JVI.01876-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dismuke DJ, Aiken C. J Virol. 2006;80:3712–3720. doi: 10.1128/JVI.80.8.3712-3720.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.von Schwedler UK, Stray KM, Garrus JE, Sundquist WI. J Virol. 2003;77:5439–5450. doi: 10.1128/JVI.77.9.5439-5450.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pornillos O, Ganser-Pornillos BK, Kelly BN, Hua YZ, Whitby FG, Stout CD, Sundquist WI, Hill CP, Yeager M. Cell. 2009;137:1282–1292. doi: 10.1016/j.cell.2009.04.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saphire ACS, Bobardt MD, Gallay PA. J Virol. 2002;76:4671–4677. doi: 10.1128/JVI.76.9.4671-4677.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Braaten D, Franke EK, Luban J. J Virol. 1996;70:3551–3560. doi: 10.1128/jvi.70.6.3551-3560.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dietrich L, Ehrlich LS, LaGrassa TJ, Ebbets-Reed D, Carter C. J Virol. 2001;75:4721–4733. doi: 10.1128/JVI.75.10.4721-4733.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fu R, Brey WW, Shetty K, Gor’kov P, Saha S, Long JR, Grant SC, Chekmenev EY, Hu J, Gan Z, Sharma M, Zhang F, Logan TM, Bruschweller R, Edison A, Blue A, Dixon IR, Markiewicz WD, Cross TA. J Magn Reson. 2005;177:1–8. doi: 10.1016/j.jmr.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 36.Gor’kov PL, Chekmenev EY, Li CG, Cotten M, Buffy JJ, Traaseth NJ, Veglia G, Brey WW. J Magn Reson. 2007;185:77–93. doi: 10.1016/j.jmr.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 37.McNeill SA, Gor’kov PL, Shetty K, Brey WW, Long JR. J Magn Reson. 2009;197:135–144. doi: 10.1016/j.jmr.2008.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fung BM, Khitrin AK, Ermolaev K. J Magn Reson. 2000;142:97–101. doi: 10.1006/jmre.1999.1896. [DOI] [PubMed] [Google Scholar]
- 39.Marion D, Wuthrich K. Biochem Bioph Res Comm. 1983;113:967–974. doi: 10.1016/0006-291x(83)91093-8. [DOI] [PubMed] [Google Scholar]
- 40.Marion D, Ikura M, Tschudin R, Bax A. J Magn Reson. 1989;85:393–399. [Google Scholar]
- 41.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 42.Goddard TD, Kneller DG. SPARKY 3. University of California; 2004. [Google Scholar]
- 43.DeLano WL. MacPyMOL. DeLano Scientific LLC; 2006. [Google Scholar]
- 44.Du S, Betts L, Yang R, Shi H, Concel J, Ahn J, Aiken C, Zhang P, Yeh JI. J Mol Biol. 2011;406:371–86. doi: 10.1016/j.jmb.2010.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.