

Abstract
Simvastatin is the active pharmaceutical ingredient of the blockbuster cholesterol lowering drug Zocor. We have previously developed an Escherichia coli based whole-cell biocatalytic platform towards the synthesis of simvastatin sodium salt (SS) starting from the precursor monacolin J sodium salt (MJSS). The centerpiece of the biocatalytic approach is the simvastatin synthase LovD, which is highly prone to misfolding and aggregation when overexpressed from E. coli. Increasing the solubility of LovD without decreasing its catalytic activity can therefore elevate the performance of the whole-cell biocatalyst. Using a combination of homology structural prediction and site-directed mutagenesis, we identified two cysteine residues in LovD that are responsible for nonspecific intermolecular crosslinking, which leads to oligomer formation and protein aggregation. Replacement of Cys40 and Cys60 with alanine residues resulted in marked gain in both protein solubility and whole-cell biocatalytic activities. Further mutagenesis experiments converting these two residues to small or polar natural amino acids showed that C40A and C60N are the most beneficial, affording 27% and 26% increase in whole cell activities, respectively. The double mutant C40A/C60N combines the individual improvements and displayed ~50% increase in protein solubility and whole-cell activity. Optimized fed-batch high-cell-density fermentation of the double mutant in an E. coli strain engineered for simvastatin production quantitatively (>99%) converted 45 mM MJSS to SS within 18 hours, which represents a significant improvement over the performance of wild type LovD under identical conditions. The high efficiency of the improved whole-cell platform renders the biocatalytic synthesis of SS an attractive substitute over the existing semisynthetic routes.
Keywords: Acyltransferase, simvastatin, site-directed mutagenesis, whole-cell biocatalyst, protein solubility
Introduction
Simvastatin is a highly effective cholesterol lowing drug (Freeman 2006; Tobert 2003) and is the active pharmaceutical ingredient (API) of the blockbuster drug Zocor (and its generic versions). Simvastatin is a semisynthetic derivative of the fungal natural product lovastatin (Askin et al. 1991; Hoffman et al. 1986), which is synthesized by Aspergillus terreus via the polyketide biosynthetic pathway (Hendrickson et al. 1999; Kennedy et al. 1999). We recently developed an Escherichia coli based whole-cell biocatalytic process towards the conversion of monacolin J sodium salt (MJSS), the C8-hydrolyzed intermediate of lovastatin, to simvastatin sodium salt (SS) using the simvastatin synthase LovD (Xie and Tang 2007) (Figure 1). LovD is a 46kDa acyltransferase found in the lovastatin biosynthetic pathway and catalyzes the final step of lovastatin biosynthesis (Xie et al. 2006). We showed the whole-cell platform can readily convert MJSS to SS using the cost-effective α-dimethylbutyryl-S-methyl-3-mercaptopropionate (DMB-S-MMP) as the thioester acyl donor (Figure 1) (Xie and Tang 2007). Further optimization of the E. coli expression host through deletion of a competing esterase bioH led to significant improvement in the kinetics of the biocatalyst (Xie et al. 2007), and rendered the process economically competitive with the optimized synthetic methods currently used to manufacture simvastatin (Schmid et al. 2001).
Figure 1.
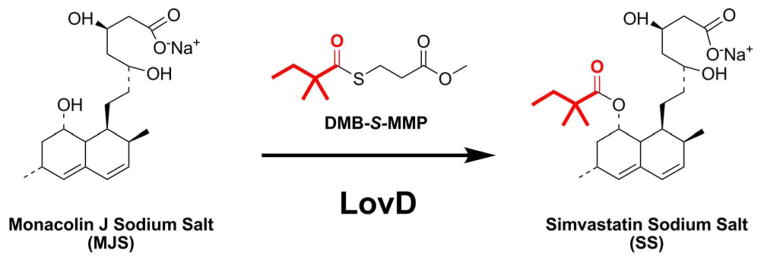
LovD catalyzes the acylation of monacolin J sodium salt (MJSS) to simvastatin sodium salt (SS) using the synthetic substrate DMB-S-MMP as acyl donor. An E. coli strain overexpressing LovD has been developed as whole-cell biocatalyst towards the synthesis of SS (Xie and Tang 2007).
The total activity of the whole-cell biocatalyst can be considered as the product of the specific activity of LovD and the expression level of soluble LovD. Enhancement of either property of LovD without negatively affecting the other will lead to further improvement in biocatalyst performance. We were especially interested in improving the solubility of LovD in E. coli. As shown in Figure 2, LovD is highly prone to misfolding and aggregation when expressed in the heterologous host E. coli. At higher temperatures (30°C and 37°C), nearly all of the overexpressed LovD was found in the insoluble fractions of the cell lysate. While a significant amount of soluble protein was obtained when the expression temperature was decreased to 25°C or below, more than 50% of total LovD remained in the insoluble pellets. Therefore, rationally improving the solubility of LovD is an attractive starting point in elevating the whole-cell biocatalyst activity.
Figure 2.
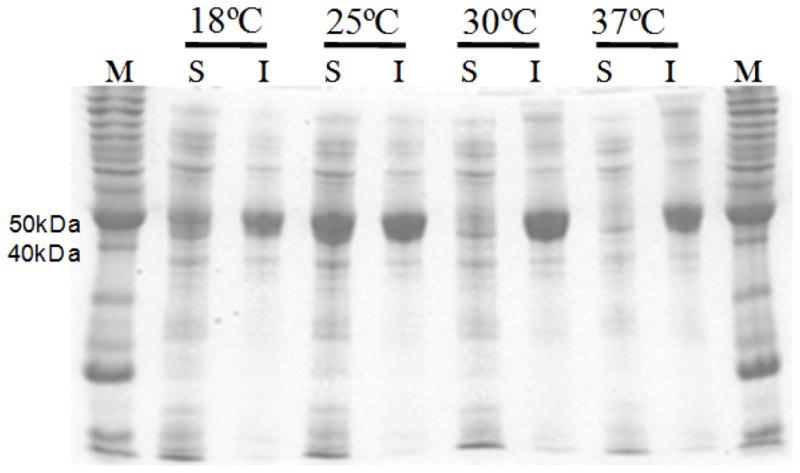
Expression of LovD from E. coli BL21(DE3)/pAW31. The soluble and insoluble portions of LovD expressed from E. coli at different temperatures were examined using SDS-PAGE. M: Invitrogen Benchmark Ladder; S: soluble fraction; I: insoluble fraction. The gel shows that LovD is prone to misfolding and aggregation, and even at lower expression temperatures, more than 50% of LovD remains insoluble.
A number of strategies have been developed to increase the solubility of a protein in order to improve protein production (Baneyx and Mujacic 2004; Fisher et al. 2006; Fowler et al. 2005) or to enable structural studies (Pedelacq et al. 2002). A commonly used approach is fusing the insoluble target to a highly soluble protein, such as the maltose-binding protein (MBP), thioredoxin, or glutathione-S-transferase (GST) (LaVallie et al. 2000; Sachdev and Chirgwin 1998; Smith 2000). Based on X-ray crystal or homology structural information, replacing surface-exposed hydrophobic residues with positively charged (Das and Georgiadis 2001; Jenkins et al. 1995; Mosavi and Peng 2003), negatively charged (Nieba et al. 1997; Trevino et al. 2007), or neutral polar amino acids (Trevino et al. 2007) can significantly improve the solubility of target proteins. Directed evolution approaches have been used to create highly soluble variants of target proteins. By using fused reporter proteins, more soluble variants can be obtained via fluorescent screening (Waldo et al. 1999) or antibiotic selection (Fisher et al. 2006) in a few rounds. A drawback of this approach is that protein activities may be deleteriously compromised as a result of the random mutations (Pedelacq et al. 2002).
A rational and effective strategy to improve protein solubility is the systematic scanning and mutagenesis of cysteine residues. Cysteine is the only natural amino acid that contains a reactive sulfhydryl group that can form intra- and inter-molecular disulfide bonds. Nonspecific disulfide bond formation between surface-exposed cysteines can lead to oligomerization and accumulation of insoluble proteins and aggregates. Global or site-specific replacement of cysteine residues with amino acids of comparable size or polarity has led to noteworthy improvement in recombinant protein solubility (Avramopoulou et al. 2004; Cozzolino et al. 2008; Ems-McClung et al. 2002), activity (Iwakura et al. 1995; Iwakura et al. 2006; Suemori and Iwakura 2007) and stability (Slusarczyk et al. 2000; Tishkov et al. 1993).
In this paper, we studied the effects of the nine wild-type cysteine residues on the solubility of LovD in E. coli. We found that replacing Cys40 and Cys60 can considerably increase LovD solubility without attenuating its catalytic activities. A double mutant C40A/C60N exhibited ~50% increase in soluble protein levels and a corresponding increase in whole-cell biocatalyst activity. Using this mutant in a high–cell density fermentation, we were able to synthesize >18 g/L of SS (99% MJSS conversion) in 18 hours.
Materials and Methods
General procedures
All reagents were purchased from standard sources. E. coli XL-1 Blue was used as the host for recombinant DNA cloning. The substrates MJSS and DMB-S-MMP were prepared as described previously (Xie and Tang 2007). E. coli BL21(DE3) was used as expression host for LovD and mutants. The engineered strain YT2 was used for evaluating whole-cell biocatalyst performance. Kinetic assays and reaction conversion were monitored with a Beckman Gold HPLC using a reverse phase C18 column (Alltech Apollo 5u, 150 mm × 4.6 mm) and a linear gradient: 60% CH3CN in water (0.1% trifluoroacetic acid [TFA]) to 95% CH3CN in water (0.1% TFA) for 10 min, 1 mL/min.
Site-directed mutagenesis
All lovD single site mutations were introduced using PCR-directed mutagenesis. The wild type expression plasmid pAW31 (based on pET28a) was used as the template (Xie et al. 2006). The double mutant C40A/C60N was created using LovD C40A expression plasmid as the template. Most of the PCR primers used for mutagenesis were designed by directly changing the cysteine codon to the desired amino acid codon. Several primers also introduce new restriction sites via silent mutation for restriction verification. All mutations were verified by DNA sequencing (McLab).
Comparing Expression Levels of Soluble LovD
Each expression plasmid encoding lovD mutant was transformed into BL21(DE3). The transformant was cultured in 50 mL Luria-Bertani (LB) medium containing 35 mg/L kanamycin at 37°C to optical density (OD600) value of 0.4~0.6. Protein expression was induced with 0.1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) and the subsequent expression was performed at 25°C for 16 hours. Cells were collected by centrifugation (2000 × g, 4°C, 15 minutes), resuspended in 7 mL Buffer A (50 mM Tris-HCl, pH 8.0, 2 mM DTT, 2 mM EDTA), and lysed by sonication. Cell debris and insoluble proteins were removed by centrifugation (20,000 × g, 4°C, 1 hr). To the cleared cell lysate, 0.5 mL of Ni-NTA resin (Qiagen) was added to each sample. The mutants were then purified using a step gradient of Buffer A with increasing concentration of imidazole (10 mM, 20 mM and 250 mM). LovD variants were eluted with 5 mL Buffer A containing 250 mM imidazole. The protein concentrations were qualitatively assessed by SDS-PAGE and quantitatively determined by the Bradford protein assay using bovine serum albumin (BSA) as the standard.
Kinetic assay of wild type and mutant LovD
To obtain Km values for MJSS and kcat, the DMB-S-MMP concentration was fixed at 2 mM, while the concentration of MJSS was varied from 0.25 mM to 5 mM. To obtain Km values for DMB-S-MMP, the MJSS concentration was fixed at 2 mM, while the concentration of DMB-S-MMP was varied from 0.25 mM to 5 mM. Dimethyl sulfoxide (DMSO) was added to a final concentration of 10% to facilitate the solubilization of DMB-S-MMP. At different time points of the kinetic assay, an aliquot of the reaction mixture was removed, quenched with 1% TFA and extracted with ethyl acetate (EA) containing 1% acetic acid. The organic phase was separated, dried, resolubilized by ACN and analyzed by HPLC. Conversion of MJSS to SS was measured by integration of the peaks at 238 nm.
Measuring Whole-cell Biocatalysis Activity
Small scale shake flask whole-cell bioconversions of MJSS to SS were performed as described (Xie and Tang 2007). Wild type LovD and all mutants were cultured in parallel for comparison. A single colony of the freshly transformed YT2 strain was used to inoculate a 5 mL LB culture supplemented with 35 mg/L kanamycin. Following overnight growth at 37°C, 100 μl (0.2% vol/vol) of the overnight culture was inoculated into 50 mL LB medium supplemented with 35 mg/L kanamycin. When OD600 reached 0.4~0.6, 0.1 mM IPTG was added to the cultures and expression of all LovD variants was performed at 25°C for 16 hrs. To mimic the high density fermentation conditions, the cells were then concentrated 10-fold before addition of substrates. A 14 mL aliquot of each culture was collected by centrifugation (4°C, 2000 × g, 10 min). The cell pellet was gently resuspended in 1.33 mL of the medium supernatant, followed by addition of 70 μL of a MJSS (300 mM in water) to a final concentration 15 mM. The concentrated culture was then divided into seven 200 μL aliquots and 1 μL of pure DMB-S-MMP was added to each sample to a final concentration of ~20 mM. The small cultures were then shaken at 300 rpm at 25°C. At each time point, a complete extraction of one culture aliquot was performed by adding 10 μL of 20% SDS for cells lysis, followed by extraction with 500 μL EA containing 2% TFA. The organic phase was removed, evaporated, and redissolved in 500 μl ACN for HPLC analysis. The whole-cell activity was determined by fitting the linear regions of the conversion time course plot.
High-cell-density Fed-Batch Fermentation
High cell density fermentation and media recipes were adopted from previously published work by Pfeifer et al. with modifications (Pfeifer et al. 2002). The supplemental vitamin solution, with exception of biotin, was excluded from both inoculation and fermentation media. The strain YT2/pAW31 or YT2/C40A/C60N was first grown overnight in 5 mL LB medium supplemented with 35 mg/L kanamycin at 37°C and 250 rpm. One mL of the overnight culture was used to inoculate a 100 mL F1 medium supplemented with 35 mg/L kanamycin and this seed culture was grown for 12 hour at 37°C. A 25 mL aliquot of the seed culture was centrifuged (2000 × g, 10 min), and the cell pellet was resuspended in 10 mL F1 medium and transfered into a 2.5-Liter Applikon Biobundle vessel containing 1 L of F1 medium. The cells were grown at 37°C and the pH was maintained at 7.1 throughout the experiment with 2 M HCl and 50% NH4OH. The agitation rate and air flow rate were adjusted to maintain the dissolved oxygen (DO) level above 20%. Typically, the agitation was maintained at 1200 rpm and the aeration was controlled at 0.4~0.5 L/min. When glucose was depleted as indicated by a rapid rise in DO level, a peristaltic pump delivered 0.3 mL/min of a glucose feed solution (Pfeifer et al. 2002) to the fermentor. When the OD600 reading reached 20, the temperature of the fermentation broth was decreased to 25°C by cooling water and 0.5 mM IPTG was added to induce protein expression. The glucose feed and DO level control was maintained throughout the expression phase, which typically lasts 12 hours. At the end of the 12 hours, feeding and aeration were stopped, agitation was decreased to 250 rpm and pH was gradually adjusted to 7.8. Both substrates MJSS and DMB-S-MMP were added to the fermentor gradually over 10 hours to initiate the bioconversion. Stock MJSS was dissolved in water to a final concentration of 500 mM and DMB-S-MMP was added as a neat liquid. HPLC was used during the conversion process to quantify MJSS disappearance and SS formation.
Results and Discussion
Homology Model of LovD Shows Surface-Exposed Cysteines
To determine whether LovD can oligomerize through the nine cysteine residues via intermolecular disulfide bonds, we first analyzed a highly purified LovD sample using native gel electrophoresis. As shown in Figure 3A, LovD displayed single-band purity on SDS-PAGE after three successive chromatography steps (in the order of Ni-NTA affinity, anion-exchange and gel-filtration chromatography). However, when the same protein sample was analyzed by native gel electrophoresis in the absence of reducing agents, we observed the presence of significant amount of dimers and higher molecular weight aggregates. Analyzing the purified sample using gel filtration chromatography yielded a similar result, in which multiple peaks corresponding to an assortment of oligomers were observed. Following repurification and electrophoresis of LovD under highly reducing conditions (i.e. 10 mM DTT), a single monomeric species was observed; lower concentrations of DTT were not completely effective in preventing undesirable formation of oligomers (data not shown). We therefore attribute the formation of oligomers to spontaneous, nonspecific disulfide bond formations. The requirement of high DTT concentration to keep LovD in the monomeric form hints that even under the naturally reducing condition of the E. coli cytoplasm (Ritz and Beckwith 2001) where LovD is overexpressed, a fraction of LovD may still exist as aggregates as a result of cysteine crosslinking. Hence, rationally replacing the cysteine residues that are responsible for the observed oligomerization may lead to improvement in LovD solubility in E. coli.
Figure 3.
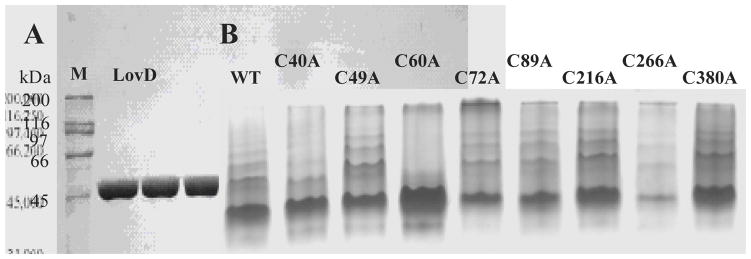
(A) SDS-PAGE of highly purified LovD (46kDa); (B) Analysis of oligomeric properties of LovD wild type and mutants using native gel electrophoresis under nonreducing conditions. The wild type LovD exists as a mixture of different oligomers. Both C40A and C60A variants of LovD have increased monomer content upon cysteine replacement, while no improvement was observed for other mutants compared to wild type LovD.
The cysteines in LovD are located at positions C40, C49, C60, C72, C89, C216, C266, C380, and C395. To map the potential positions of the nine residues, we constructed a homology structure of LovD using the program PHYRE (Protein Homology/analogY Recognition Engine) (Bennett-Lovsey et al. 2008) (Figure 4A). The predicted structure is based on the known crystal structure of Burkholderia gladioli esterase EstB (Wagner et al. 2002) (PDB ID 1CI8, 20% sequence identity), which is the most homologous enzyme to LovD of which the X-ray crystal structure has been solved. EtstB is an esterase with specific activity towards short chain fatty acid esters and triglycerides (Petersen et al. 2001) and displays a class C β-lactamase fold (Wagner et al. 2002). A sequence alignment between LovD and EstB is shown in Figure 4B. We note that there is a single cysteine residue (C217) in EstB and it is not aligned with any of the nine cysteines in LovD. As shown in Figure 4A, LovD is predicted to contain a central core of seven anti-parallel β-strands covered by two α-helixes on both sides of the sheets. The predicted locations of the cysteines are show in Figure 4A. While most of the cysteines are predicted to be buried in the protein interior of LovD (not shown), three residues (C40, C60 and C89) were surface-exposed in the model. In particular, C40 and C60 are located at the opposite ends of the β-sheet core of LovD, and both of the sulfhydryl moieties are predicted to be highly exposed. Hence, these two residues were tentatively implicated in the formation of the crosslinked oligomers observed in Figure 3. To validate the homology structure predictions and to reduce the undesirable spurious aggregation, we first performed alanine scanning mutagenesis of each of the cysteine residues. Nine single-site mutants (C40A, C49A, C60A, C72A, C89A, C216A, C266A, C380A, and C395A) were constructed.
Figure 4.

(A) Homology model structure of LovD generated by the program PHYRE based on the crystal structure of EstB from B. gladioli (1CI8)(Wagner et al. 2002). The backbone of the structure is represented in ribbon form, while the nine cysteine residues are shown in space-filled models. The homology model predicts that C40 and C60 are solvent exposed. (B) Pairwise sequence alignment between LovD and EstB. The alignment was performed using ClusterW and output was generated by ESPript.
LovD C40A and LovD C60A Displayed Enhanced Solubility and Whole-cell Activity
Each of the nine mutants was expressed from the expression host BL21(DE3). To quantify the amounts of soluble proteins, we subjected the cleared lystate from these expression hosts to Ni-NTA purification. The purity of each LovD mutant was visually assessed by SDS-PAGE to be > 95%, and the amount of recovered protein was quantified using the Bradford assay (Bradford 1976). Figure 5 shows the relative amounts of soluble protein recovered for the alanine-scanning mutants compared to wild type LovD, which was determined to be 92 mg/L under identical expression conditions. As evident in the figure, the different Cys → Ala mutations have drastically different effects on the levels of soluble LovD. Interestingly, only the C40A and C60A mutants displayed higher levels of soluble protein. While the C60A mutant displayed a slight gain in solubility (2%), the C40A mutant was 23% more soluble than the wild type LovD. This result is remarkably in line with the prediction of the homology model in Figure 4A, and suggests that both Cys40 and Cys60 may indeed contribute to LovD being partially insoluble in E. coli. In contrast, the solubilities of the remaining point mutants were markedly lower than that of the wild type. In particular, soluble forms of C72A and C266A were present at levels < 20% of the wild type LovD, while nearly no soluble C395A was recovered. Examining the pellet portion of the lysate confirmed that most of these mutant proteins are located in the insoluble fraction, confirming the deleterious effects of these Cys → Ala mutations on the solubility of LovD. These cysteines residues may therefore play important roles in maintaining the three-dimensional structural integrity of LovD through optimized internal packing; there are no data to suggest the presence of native intramolecular disulfide bonds in LovD, and the homology model did not provide strong predictions for any such bonds.
Figure 5.

Comparison of soluble protein levels (grey) and whole cell biocatalytic activities (black) of cysteine point mutants of LovD. The soluble protein levels and whole cell activities of wild type LovD are 92 mg/L and 1.5 mM/hr, respectively, and are normalized to 100%.
We then studied the biochemical properties of the purified mutants. We incubated the mutants under nonreducing conditions and analyzed the oligomerization properties of the mutants using native gel electrophoresis (Figure 3B). While most of the mutants retained the same ability to form spurious oligomers as the wild type LovD, both C40A and C60A variants of LovD showed increased monomer content upon eliminating the sulfhydryl side chain. The contrast with wild type LovD is especially striking for the C60A mutant, in which nearly no crosslinked oligomers can be detected by electrophoresis. The results clearly indicated that Cys40 and Cys60 in LovD are involved in forming undesirable intermolecular disulfide bonds, and further validated the mapping predicted by the homology structural model. Combined with the enhancement in solubility of C40A and C60A, we conclude that attenuating oxidative oligomerization can indeed increase LovD solubility in vivo.
Given that C40 and C60 are predicted to be located away from the active site and the substrate binding pockets of LovD, we did not anticipate the mutations of C40A or C60A to significantly affect the kinetic properties of LovD. Indeed, when the mutants were assayed in vitro using MJSS and DMB-S-MMP as substrates, we observed nearly identical Michaelis-Menten kinetic parameters to those of the wild type LovD. The kcat and Km values of the two mutants are essentially the same as the wild type enzyme, confirming that these single point mutations do not negatively impact the catalytic efficiency of the acylation reaction.
We further tested the whole-cell activities of the nine mutants relative to that of wild type LovD. The ΔbioH version of BL21(DE3), YT2 (Xie et al. 2007), was used as the host strain. After overnight protein expression, we concentrated cells 10-fold to mimic the conditions in a high cell-density fermentation. Conversion of MJSS to SS was monitored by HPLC. Figure 5 shows the comparison of the linear portions of the conversion time course. The wild type conversion rate was 1.5 mM/hr and was normalized to 100%. As expected, YT2/C40A displayed the highest whole-cell activity and is 27% faster than that of the wild type. The increase in reaction rate is consistent with the 23% increase in soluble protein levels of the mutant. YT2/C60A displayed comparable whole-cell activity to that of wild type, reflecting its small increase in soluble protein levels. For most of the other mutants, decreased protein solubilities led to more dramatic decreases in whole-cell activities, possibly indicating these Cys → Ala mutations may lead to decrease in both protein solubility and specific enzyme activities.
Double mutation C40A/C60N showed highest whole-cell activity
Although mutating Cys60 was the most effective in attenuating oligomerization tendencies of LovD under reducing conditions (Figure 3B), the C60A mutation only afforded a minimal gain in protein solubility, and in turn whole-cell activity. We reasoned that the Cys → Ala mutation may not be the most optimal substitution, considering the solvent exposed thiol functional group is replaced with a nonpolar methyl group. To test whether inserting other amino acids at either residue 40 or residue 60 may lead to further improvement in protein solubility, we constructed additional point mutants at these two positions. We introduced other polar but uncharged natural amino acids side chains, including serine, threonine, asparagine and glutamine. Eight additional mutants (C40S, C60S, C40T, C60T, C40N, C60N, C40Q, and C60Q) were constructed by site-directed mutagenesis and were expressed and purified from BL21(DE3). Interestingly, while the C40A mutation remained the most soluble among the Cys40 variants, we found C60N to be the most soluble among the Cys60 variants. The expression level of soluble C60N was approximately 20% higher than that of the wild type. Whole-cell bioactivities studies further confirmed the robustness of the C60N mutant. As shown in Figure 6, the C60N mutant showed a 26% gain in whole-cell bioactivity compared to that of the wild type, comparable to that displayed by C40A.
Figure 6.

Comparison of whole-cell biocatalytic activities of LovD mutants at Cys40 and Cys60 positions. . The whole cell activities of wild type LovD is 1.5 mM/hr and is normalized to 100%. C40A and C60N are the most beneficial mutations at each position, affording 27% and 26% increase in activities, respectively. The double mutant C40A/C60N combines the individual improvements and displayed ~50% increase in the whole-cell activity.
We then tested whether the two beneficial cysteine mutations can be combined to give an additive gain in LovD solubility. A double mutant C40A/C60N was therefore constructed using site-directed mutagenesis. The expression level of soluble C40A/C60N was approximately ~140 mg/L, which represents a 50% increase in LovD solubility. When C40A/C60N mutant was expressed from YT2 and assayed as a whole-cell biocatalyst, we observed a corresponding 50% increase in the rate of SS synthesis (Figure 6). Taken together, these results suggest that the double mutant combines the solubility gains of the point mutants to afford the most soluble version of LovD to date, leading to significant increases in the total activities of the whole-cell biocatalyst. We tested the kinetic properties of the double mutant and showed that both the kcat and Km values remained nearly identical to that of the wild type, C40A and C60A single mutants (Table 1). The gain in activity is therefore completely attributed to the higher solubility of the mutant.
Table 1.
Michaelis-Menten kinetic parameters for LovD wild type, C40A, C60A, and C40A/C60N
| Wild Type | C40A | C60A | C40A/C60N | |
|---|---|---|---|---|
| Km of MJSS (mM) | 0.78 ± 0.12 | 0.80 ± 0.21 | 0.81 ± 0.19 | 0.82 ± 0.10 |
| Km of DMB-S-MMP (mM) | 0.67 ± 0.04 | 0.63 ± 0.06 | 0.58 ± 0.03 | 0.61 ± 0.03 |
|
| ||||
| kcat (min−1) | 0.62 ± 0.03 | 0.62 ± 0.05 | 0.60 ± 0.04 | 0.60 ± 0.02 |
High Cell-Density Fermentation Using the C40A/C60N Double Mutant
The ultimate goal of the protein engineering approach is to construct an E. coli based strain that can be used towards high throughput SS production. Towards this end, we developed and tested a fed-batch fermentation process using the more soluble C40A/C60N mutant. We used a glucose-limiting fermentation processes and maintained the dissolved oxygen (DO) level to >20%. The glucose-limiting process consistently gave significantly higher total LovD activity when compared to a DO-limiting fermentation process. The fed batch process was adopted from that described by Pfeifer et al (Pfeifer et al. 2002) with the following changes: 1) Protein induction was initiated when OD600 reached 20, at which time the temperature of the fermentation was reduced to 25°C to maximize LovD solubility; 2) Protein expression was allowed to proceed for 12 hours, at which time a typical OD600 reading of 70–80 can be attained. Subsequently, the glucose feeding was terminated and the pH of the fermentation broth was gradually increased from 7.1 to 7.8. The more basic condition was essential for optimal performance of LovD; and 3) Addition of MJSS and DMB-S-MMP was then initiated and both substrates were added over the period of 10 hours to minimize toxicity to the E. coli cells.
We tested the fermentative conversion of MJSS to SS using YT2 strain expressing either the wild type or the C40A/C60N LovD. We added MJSS to a final concentration of 45 mM (~14 g/L) and monitored the synthesis of SS using HPLC. The comparison between the two strains is shown in Figure 7. The more soluble variant of LovD clearly outperformed the wild type enzyme in this process and is remarkablely efficient in MJSS to SS conversion. Quantitative (99%) synthesis of SS (18 g/L) was achieved in ~ 18 hours after substrate addition using YT2/C40A/C60N. In contrast, YT2/pAW31 reached a maximum conversion of 91% in 26 hours and was not able to achieve higher conversion upon further fermentation, possibly due to loss of enzyme activity under prolonged reaction time. The average reaction rate was 2.5 mM/hr for YT2/C40A/C60N and 1.6 mM/hr for YT2/pAW31, indicating that the double mutant C40A/C60N was ~50% more efficient than the wild type during large scale high-cell-density fermentation (Figure 7).
Figure 7.

Conversion of MJSS to SS using high-cell-density fermentation. The strain YT2 overexpressing either wild type LovD or the C40A/C60N mutant was used. YT2/C40A/C60N was able to synthesize 18 g/L of SS in ~ 18 hours. The average reaction rate was 2.5 mM/hr for YT2/C40A/C60N and 1.6 mM/hr for YT2/pAW31, indicating the gain in protein solubility has significant effect on the performance of the whole cell biocatalyst.
In conclusion, we have successfully enhanced the solubility of the LovD by replacing two cysteine residues that are responsible for oxidative crosslinking. Our results further demonstrate the utilities of homology modeling and cysteine replacement in rational protein engineering. As a biocatalyst, the C40A/C60N double mutant is significantly more soluble when expressed in E. coli, and represents a significant upgrade over the wild type LovD as a simvastatin synthase. The improved mutant also serves as the starting point for further protein engineering efforts towards optimizing enzyme activity and thermostability.
Acknowledgments
This work was supported by the American Heart Association (YT: 0535069N) and National Institute of Health (YT/TOY: 1R21HL091197-01). JLG was supported by URC-CARE undergraduate research program.
References
- Askin D, Verhoeven TR, Liu TM-H, Shinkai I. Synthesis of Synvinolin: Extremely High Conversion Alkylation of an Ester Enolate. J Org Chem. 1991;56 [Google Scholar]
- Avramopoulou V, Mamalaki A, Tzartos SJ. Soluble, oligomeric, and ligand-binding extracellular domain of the human alpha 7 acetylcholine receptor expressed in yeast - Replacement of the hydrophobic cysteine loop by the hydrophilic loop of the ACh-binding protein enhances protein solubility. J Biol Chem. 2004;279(37):38287–38293. doi: 10.1074/jbc.M402533200. [DOI] [PubMed] [Google Scholar]
- Baneyx F, Mujacic M. Recombinant protein folding and misfolding in Escherichia coli. Nat Biotechnol. 2004;22(11):1399–408. doi: 10.1038/nbt1029. [DOI] [PubMed] [Google Scholar]
- Bennett-Lovsey RM, Herbert AD, Sternberg MJ, Kelley LA. Exploring the extremes of sequence/structure space with ensemble fold recognition in the program Phyre. Proteins. 2008;70(3):611–25. doi: 10.1002/prot.21688. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Cozzolino M, Amori I, Pesaresi MG, Ferri A, Nencini M, Carri MT. Cysteine 111 affects aggregation and cytotoxicity of mutant Cu,Zn-superoxide dismutase associated with familial amyotrophic lateral sclerosis. J Biol Chem. 2008;283(2):866–874. doi: 10.1074/jbc.M705657200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das D, Georgiadis MM. A directed approach to improving the solubility of Moloney murine leukemia virus reverse transcriptase. Protein Sci. 2001;10(10):1936–1941. doi: 10.1110/ps.16301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ems-McClung SC, Benmoussa M, Hainline BE. Mutational analysis of the maize gamma zein C-terminal cysteine residues. Plant Sci. 2002;162(1):131–141. [Google Scholar]
- Fisher AC, Kim W, DeLisa MP. Genetic selection for protein solubility enabled by the folding quality control feature of the twin-arginine translocation pathway. Protein Sci. 2006;15(3):449–58. doi: 10.1110/ps.051902606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler SB, Poon S, Muff R, Chiti F, Dobson CM, Zurdo J. Rational design of aggregation-resistant bioactive peptides: Reengineering human calcitonin. Proc Natl Acad Sci U S A. 2005;102(29):10105–10110. doi: 10.1073/pnas.0501215102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman MW. Statins, cholesterol, and the prevention of coronary heart disease. Faseb J. 2006;20(2):200–1. doi: 10.1096/fj.06-0202ufm. [DOI] [PubMed] [Google Scholar]
- Hendrickson L, Davis CR, Roach C, Nguyen DK, Aldrich T, McAda PC, Reeves CD. Lovastatin biosynthesis in Aspergillus terreus: characterization of blocked mutants, enzyme activities and a multifunctional polyketide synthase gene. Chem Biol. 1999;6(7):429–39. doi: 10.1016/s1074-5521(99)80061-1. [DOI] [PubMed] [Google Scholar]
- Hoffman WF, Alberts AW, Anderson PS, Chen JS, Smith RL, Willard AK. 3-Hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors. 4 Side chain ester derivatives of mevinolin. J Med Chem. 1986;29(5):849–52. doi: 10.1021/jm00155a040. [DOI] [PubMed] [Google Scholar]
- Iwakura M, Jones BE, Luo JB, Matthews CR. A Strategy for Testing the Suitability of Cysteine Replacements in Dihydrofolate-Reductase from Escherichia-Coli. J Biochem. 1995;117(3):480–488. doi: 10.1093/oxfordjournals.jbchem.a124733. [DOI] [PubMed] [Google Scholar]
- Iwakura M, Maki K, Takahashi H, Takenawa T, Yokota A, Katayanagi K, Kamiyama T, Gekko K. Evolutional design of a hyperactive cysteine- and methionine-free mutant of Escherichia coli dihydrofolate reductase. J Biol Chem. 2006;281(19):13234–46. doi: 10.1074/jbc.M508823200. [DOI] [PubMed] [Google Scholar]
- Jenkins TM, Hickman AB, Dyda F, Ghirlando R, Davies DR, Craigie R. Catalytic Domain of Human-Immunodeficiency-Virus Type-1 Integrase - Identification of a Soluble Mutant by Systematic Replacement of Hydrophobic Residues. Proc Natl Acad Sci U S A. 1995;92(13):6057–6061. doi: 10.1073/pnas.92.13.6057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy J, Auclair K, Kendrew SG, Park C, Vederas JC, Hutchinson CR. Modulation of polyketide synthase activity by accessory proteins during lovastatin biosynthesis. Science. 1999;284(5418):1368–72. doi: 10.1126/science.284.5418.1368. [DOI] [PubMed] [Google Scholar]
- LaVallie ER, Lu ZJ, Diblasio-Smith EA, Collins-Racie LA, McCoy JM. Thioredoxin as a fusion partner for production of soluble recombinant proteins in Escherichia coli. Method Enzymol. 2000;326:322–340. doi: 10.1016/s0076-6879(00)26063-1. [DOI] [PubMed] [Google Scholar]
- Mosavi LK, Peng ZY. Structure-based substitutions for increased solubility of a designed protein. Protein Eng. 2003;16(10):739–745. doi: 10.1093/protein/gzg098. [DOI] [PubMed] [Google Scholar]
- Nieba L, Honegger A, Krebber C, Pluckthun A. Disrupting the hydrophobic patches at the antibody variable/constant domain interface: Improved in vivo folding and physical characterization of an engineered scFv fragment. Protein Eng. 1997;10(4):435–444. doi: 10.1093/protein/10.4.435. [DOI] [PubMed] [Google Scholar]
- Pedelacq JD, Piltch E, Liong EC, Berendzen J, Kim CY, Rho BS, Park MS, Terwilliger TC, Waldo GS. Engineering soluble proteins for structural genomics. Nat Biotechnol. 2002;20(9):927–32. doi: 10.1038/nbt732. [DOI] [PubMed] [Google Scholar]
- Petersen EI, Valinger G, Solkner B, Stubenrauch G, Schwab H. A novel esterase from Burkholderia gladioli which shows high deacetylation activity on cephalosporins is related to beta-lactamases and DD-peptidases. Journal of Biotechnology. 2001;89(1):11–25. doi: 10.1016/s0168-1656(01)00284-x. [DOI] [PubMed] [Google Scholar]
- Pfeifer B, Hu Z, Licari P, Khosla C. Process and metabolic strategies for improved production of Escherichia coli-derived 6-deoxyerythronolide B. Appl Environ Microbiol. 2002;68(7):3287–92. doi: 10.1128/AEM.68.7.3287-3292.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz D, Beckwith J. Roles of thiol-redox pathways in bacteria. Annu Rev Microbiol. 2001;55:21–48. doi: 10.1146/annurev.micro.55.1.21. [DOI] [PubMed] [Google Scholar]
- Sachdev D, Chirgwin JM. Solubility of proteins isolated from inclusion bodies is enhanced by fusion to maltose-binding protein or thioredoxin. Protein Expr Purif. 1998;12(1):122–32. doi: 10.1006/prep.1997.0826. [DOI] [PubMed] [Google Scholar]
- Schmid A, Dordick JS, Hauer B, Kiener A, Wubbolts M, Witholt B. Industrial biocatalysis today and tomorrow. Nature. 2001;409(6817):258–68. doi: 10.1038/35051736. [DOI] [PubMed] [Google Scholar]
- Slusarczyk H, Felber S, Kula MR, Pohl M. Stabilization of NAD-dependent formate dehydrogenase from Candida boidinii by site-directed mutagenesis of cysteine residues. Eur J Biochem. 2000;267(5):1280–1289. doi: 10.1046/j.1432-1327.2000.01123.x. [DOI] [PubMed] [Google Scholar]
- Smith DB. Generating fusions to glutathione S-transferase for protein studies. Applications of Chimeric Genes and Hybrid Proteins, Pt A. 2000;326:254–270. doi: 10.1016/s0076-6879(00)26059-x. [DOI] [PubMed] [Google Scholar]
- Suemori A, Iwakura M. A systematic and comprehensive combinatorial approach to simultaneously improve the activity, reaction specificity, and thermal stability of p-hydroxybenzoate hydroxylase. J Biol Chem. 2007;282(27):19969–78. doi: 10.1074/jbc.M610320200. [DOI] [PubMed] [Google Scholar]
- Tishkov VI, Galkin AG, Marchenko GN, Egorova OA, Sheluho DV, Kulakova LB, Dementieva LA, Egorov AM. Catalytic Properties and Stability of a Pseudomonas Sp 101 Formate Dehydrogenase Mutants Containing Cys-255-Ser and Cys-255-Met Replacements. Biochem Bioph Res Co. 1993;192(2):976–981. doi: 10.1006/bbrc.1993.1511. [DOI] [PubMed] [Google Scholar]
- Tobert JA. Lovastatin and beyond: the history of the HMG-CoA reductase inhibitors. Nat Rev Drug Discov. 2003;2(7):517–26. doi: 10.1038/nrd1112. [DOI] [PubMed] [Google Scholar]
- Trevino SR, Scholtz JM, Pace CN. Amino acid contribution to protein solubility: Asp, Glu, and Ser contribute more favorably than the other hydrophilic amino acids in RNase Sa. J Mol Biol. 2007;366(2):449–460. doi: 10.1016/j.jmb.2006.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner UG, Petersen EI, Schwab H, Kratky C. EstB from Burkholderia gladioli: A novel esterase with a beta-lactamase fold reveals steric factors to discriminate between esterolytic and beta-lactam cleaving activity. Protein Sci. 2002;11(3):467–478. doi: 10.1110/ps.33002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldo GS, Standish BM, Berendzen J, Terwilliger TC. Rapid protein-folding assay using green fluorescent protein. Nature Biotechnol. 1999;17(7):691–695. doi: 10.1038/10904. [DOI] [PubMed] [Google Scholar]
- Xie X, Tang Y. Efficient Synthesis of Simvastatin Using Whole-Cell Biocatalysis. Appl Environ Microbiol. 2007;73:2054–2060. doi: 10.1128/AEM.02820-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Watanabe K, Wojcicki WA, Wang CC, Tang Y. Biosynthesis of lovastatin analogs with a broadly specific acyltransferase. Chem Biol. 2006;13(11):1161–9. doi: 10.1016/j.chembiol.2006.09.008. [DOI] [PubMed] [Google Scholar]
- Xie X, Wong W, Tang Y. Improving simvastatin bioconversion in Escherichia coli by deletion of bioH. Metab Eng. 2007;9(4):379–386. doi: 10.1016/j.ymben.2007.05.006. [DOI] [PubMed] [Google Scholar]